

Abstract
GSD Ia (von Gierke Disease, Glycogen Storage Disease Type Ia) is a devastating genetic disorder with long-term sequelae, such as non-alcoholic fatty liver disease and renal failure. Down-regulated autophagy is involved in the development of hepatic metabolic dysfunction in GSD Ia; however, the role of autophagy in the renal pathology is unknown. Here we show that autophagy is impaired and endoplasmic reticulum (ER) stress is increased in the kidneys of a mouse model of GSD Ia. Induction of autophagy by rapamycin also reduces this ER stress. Taken together, these results show an additional role for autophagy down-regulation in the pathogenesis of GSD Ia, and provide further justification for the use of autophagy modulators in GSD Ia.
Keywords: autophagy, von Gierke’s disease, GSD Ia, rapamycin, kidney, ER stress, glucose-6-phosphatase, glucose-6-phosphate, glycogen storage disease type I
Introduction
GSD Ia (Glycogen Storage Disease Type Ia, also known as von Gierke disease) is a devastating disorder of glycogen metabolism caused by a loss-of-function of glucose-6-phospatase α (G6PC), an enzyme that catalyzes the terminal step of both gluconeogenesis and glycogenolysis, the conversion of glucose-6-phosphate (G6P) to glucose [1]. The failure to generate glucose from the liver leads to hypoglycaemia in infancy that can by prevented by appropriate dietary therapy [1]. However, even when normoglycaemia is maintained, patients develop secondary metabolic sequelae such as NAFLD and renal failure [1]. Since G6PC is primarily expressed in the liver and kidneys, these two tissues are affected most as the disease progresses [1]. In the case of the kidneys, patients with GSD Ia suffer a disease resembling diabetic nephropathy that can progress to renal failure [2].
(Macro)autophagy is a cellular process whereby which damaged or unneeded cellular components are sequestered in organelles called autophagosomes, which are then trafficked to lysosomes for destruction by digestive enzymes [3]. Recently, our group showed that an impairment in autophagy is key to the development of hepatic metabolic derangements in this disease [4]. Increased ER stress in neutrophils also has been shown to be pathogenic in the closely-related disease, GSD Ib [5]. In the kidney, autophagy occurred after exposure to insults that caused endoplasmic reticulum (ER) stress [6]. Moreover, treatment with a pharmacological of autophagy, rapamycin, alleviated the effects of ER stressors [7].
Since increased ER stress is a known cause of kidney damage [6], plays a role in the development of podocyte injury [8], as well as mediates the progression of chronic kidney disease in proteinuric mice [9], we hypothesized that increased ER stress may occur in the kidneys of patients suffering from GSD Ia and that treatment with autophagy-inducing agents such as rapamycin might relieve this ER stress. Accordingly, we examined ER stress markers and autophagy in the kidneys of young G6PC−/− mice before or after treatment with rapamycin.
Materials and Methods
Study Approval
All animal experiments were approved by the Institutional Animal Care and Use Committee of Duke University (protocol number: A070-14-03). All animals received care as per NIH publication 86-23, and all reasonable steps to prevent suffering were undertaken.
Antibodies
Primary antibodies recognizing phosphorylated-PERK (3179), PERK (3192), CHOP (2895), LC3 (2775), and α -tubulin were purchased from cell signaling technologies. Primary antibody recognizing ATF6 (09-069) was purchased from Merck Millipore. Primary antibody recognizing β-actin (sc-8432) and HRP-conjugated secondary antibodies raised against mouse (sc-2954) and rabbit (sc-2955) IgG were purchased from SantaCruz Biotechnologies.
Animal Models
G6PC−/− mice were identified as described previously [4]. Rapamycin treatment was performed as previously described [4].
Western Blotting
Western blotting was performed identically as per previously described methods [4].
Electron Microscopy
Samples were harvested, prepared and imaged following methods previously published [4].
Statistics
Animal experiments were performed with 3–5 mice per group. Results are expressed as mean ± SEM. In all parts, * represents p<0.05 as per Student’s T-test.
Histology
At sacrifice, kidney tissue was fixed in formalin, then sectioned and stained with haematoxylin and eosin. Images were acquired with a 20× objective lens (total magnification 200×).
Results
We first showed that similar to our findings in the liver [4], autophagy was inhibited in the kidneys of G6PC−/− mice by Western blotting for the LC3-II levels (Fig. 1 A), a commonly used marker for autophagosome number [10]. This decrease in LC3-II was associated with increases in the protein levels of phosphorylated Extracellular Signal-regulated Kinase (pERK), cleaved Activating Transcription Factor 6 (ATF6), and CCAAT-enhancer-binding Protein Homologous Protein (CHOP) (Fig. 1 B, C, D), that are known indicators of ER stress [11]. Ultrastructural analysis also displayed features of ER stress (Fig. 1 E), notably dilation of the endoplasmic reticulum [12]. Treatment of these mice with the autophagy-inducing drug rapamycin restored LC3-II levels (Fig. 1 F) and reduced the levels of the aforementioned ER stress markers (Fig. 1 G, H, I). Histological analysis revealed cytoplasmic vacuolation of kidney tubular cells in the G6PC−/− mice, implying storage of glycogen and lipids, which resolved when treated with rapamycin (Fig. 1 J). Rapamycin also reversed the ultrastructural changes observed in the KO mice (Fig. 1 K).
Figure 1.
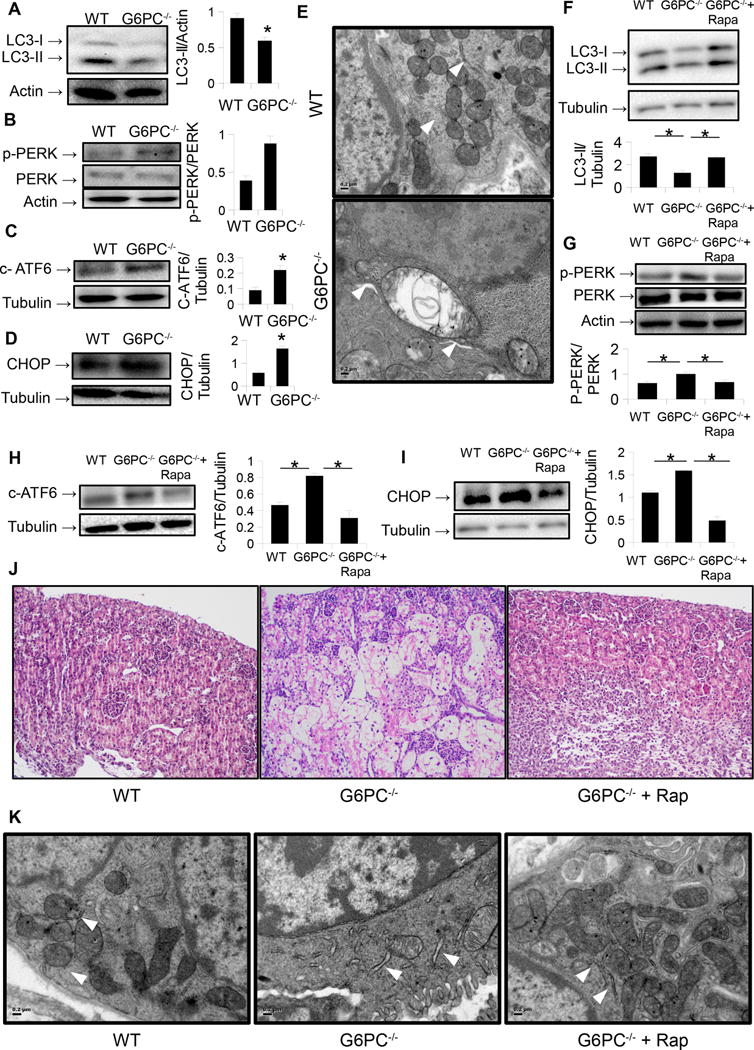
A.) Levels of autophagosome marker LC3-II are decreased in the kidneys of G6PC−/− mice. B-D.) Protein levels of phosphorylated PERK (B), cleaved ATF6 (c-ATF6) (C), and CHOP are increased in the kidneys of the same mice. E.) Ultrastructural analysis (20,000 ×) of G6PC−/− mouse kidneys reveals dilation of the endoplasmic reticulum (ER). ER noted as white arrowheads. Scale bar = 0.2 μm. F.) Treatment of G6PC−/− mice with rapamycin increases kidney levels of LC3-II. G-I.) Levels of ER stress markers p-PERK (G), cleaved ATF6 (H), and CHOP (I) in these same mice. J.) Histological analysis (20× objective) with H&E stain in the same mice reveals vacuolation and storage of clear material (lipid/glycogen) in the kidney tubular cells of G6PC−/− which resolves with rapamycin treatement. K.) Ultrastructural analysis (20,000 ×) of G6PC−/− mouse kidneys following rapamycin treatment reveals and improvement in ER morphology (white arrowheads).
Discussion
These data provide further new evidence for the importance of autophagy in the pathogenesis of GSD Ia, and extend our previous findings in the liver [4] to the kidneys. By two weeks after birth, levels of renal ER stress makers were elevated in the G6PC−/− mice, implying that renal damage already was taking place. Thus, this increase in ER stress may not only serve as an early marker for kidney damage in GSD Ia, but also could lead to the renal failure that eventually occurs. In this connection, a link between ER stress, autophagy, and the development of renal cysts had previously been postulated by Gjorgjieva and colleagues [13]. Our findings here, thus provide evidence to add GSD Ia to the list of many kidney diseases, including diabetic nephropathy [11], in which increased ER stress has been implicated to have a pathogenic role.
Interestingly, treatment with rapamycin for one week reverses many of the changes seen in the ER stress pathway in the kidneys of the G6PC KO mice. We previously showed that induction of autophagy by rapamycin leads to improvement in hepatic metabolism in this disorder [4]. Our current findings show that improvement in autophagy reduces renal ER stress in a mouse model of GSD Ia. Although we did not assess the benefits of long-term treatment with rapamycin on renal function in GSD Ia, our findings nonetheless provide further evidence that autophagy induction may be a viable therapeutic option in GSD Ia.
Acknowledgments
Financial Support: This work was supported by Singapore NMRC grant NMRC/CIRG/1340/2012 and NMRC/CSA/0054/2013 to PMY, as well as by support from the Alice and Y. T. Chen Center for Genetics and Genomics to DDK. This study was also supported by grant R01DK105434 from the National Institute of Diabetes and Digestive and Kidney Diseases. We would also like to acknowledge inspiration and support from Dr. Emory and Mrs. Mary Chapman and their son Christopher, and from Dr. John and Mrs. Michelle Kelly. We deeply appreciate the dedication shown by the staff of the Duke Department of Laboratory Animal Resources.
List of Abbreviations
- G6Pase α
Glucose-6-phosphatase α
- G6PC
Glucose-6-phosphatase α catalytic subunit
- GSD Ia
Glycogen Storage Disease Type Ia
- G6P
Glucose-6-Phosphate
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- Rap
Rapamycin
- KD
Knockdown
- KO
Knockout
Footnotes
Conflict of Interest: A patent application has been filed by Duke University and Duke-NUS Graduate Medical School related to the treatment of glycogen storage disease with rapamycin, and BLH, PMY, and DDK are inventors.
References
- 1.Chou JY, Jun HS, Mansfield BC. Glycogen Storage Disease Type I and G6pase-Beta Deficiency: Etiology and Therapy. Nat ReviEndocrinol. 2010;6:676–688. doi: 10.1038/nrendo.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yiu WH, Pan CJ, Ruef RA, Peng WT, Starost MF, Mansfield BC, et al. Angiotensin Mediates Renal Fibrosis in the Nephropathy of Glycogen Storage Disease Type Ia. Kid Intl. 2008;73:716–723. doi: 10.1038/sj.ki.5002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Komatsu M. Autophagy: Renovation of Cells and Tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Farah BL, Landau DJ, Sinha RA, Brooks ED, Wu Y, Fung SY, et al. Induction of Autophagy Improves Hepatic Lipid Metabolism in Glucose-6-Phosphatase Deficiency. J Hepatol. 2016;64:370–379. doi: 10.1016/j.jhep.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Jun HS, Lee YM, Cheung YY, McDermott DH, Murphy PM, De Ravin SS, et al. Lack of Glucose Recycling between Endoplasmic Reticulum and Cytoplasm Underlies Cellular Dysfunction in Glucose-6-Phosphatase-Beta-Deficient Neutrophils in a Congenital Neutropenia Syndrome. Blood. 2010;116:2783–2792. doi: 10.1182/blood-2009-12-258491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cybulsky AV. The Intersecting Roles of Endoplasmic Reticulum Stress, Ubiquitin- Proteasome System, and Autophagy in the Pathogenesis of Proteinuric Kidney Disease. Kid Intl. 2013;84:25–33. doi: 10.1038/ki.2012.390. [DOI] [PubMed] [Google Scholar]
- 7.Dong G, Liu Y, Zhang L, Huang S, Ding HF, Dong Z. Mtor Contributes to ER Stress and Associated Apoptosis in Renal Tubular Cells. Am J Physiol Ren Physiol. 2015;308:F267–274. doi: 10.1152/ajprenal.00629.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan Y, Xu X, Zhao C, Zhao M, Wang H, Zhang B, et al. The roles of oxidative stress, endoplasmic reticulum stress, and autophagy in aldosterone/mineralocorticoid receptor-induced podocyte injury. Lab Invest. 2015;95:1374–86. doi: 10.1038/labinvest.2015.118. [DOI] [PubMed] [Google Scholar]
- 9.El Karoui K, Viau A, Dellis O, Bagattin A, Nguyen C, Baron W, et al. Endoplasmic reticulum stress drives proteinuria-induced kidney lesions via Lipocalin 2. Nat Com. 2016;7:10330. doi: 10.1038/ncomms10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunard R, Sharma K. The Endoplasmic Reticulum Stress Response and Diabetic Kidney Disease. American journal of physiology Renal physiology. 2011;300:F1054–1061. doi: 10.1152/ajprenal.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asselah T, Bieche I, Mansouri A, Laurendeau I, Cazals-Hatem D, Feldmann G, et al. In Vivo Hepatic Endoplasmic Reticulum Stress in Patients with Chronic Hepatitis C. The Journal of pathology. 2010;221:264–274. doi: 10.1002/path.2703. [DOI] [PubMed] [Google Scholar]
- 13.Gjorgjieva M, Raffin M, Duchampt A, Perry A, Stefanutti A, Brevet M, et al. Progressive development of renal cysts in glycogen storage disease type I. Human molecular genetics. 2016;25:3784–3797. doi: 10.1093/hmg/ddw224. [DOI] [PubMed] [Google Scholar]