

Abstract
The completion of the human genome project at the beginning of the 21st century, along with the rapid advancement of sequencing technologies thereafter, has resulted in exponential growth of biological data. In genetics, this has given rise to numerous variation databases, created to store and annotate the ever-expanding dataset of known mutations. Usually, these databases focus on variation at the sequence level. Few databases focus on the analysis of variation at the 3D level i.e. mapping, visualizing, and determining the effects of variation in protein structures. Additionally, these web servers seldom incorporate tools to help analyze this data. Here, we present the Human Mutation Analysis (HUMA) web server and database. HUMA integrates sequence, structure, variation and disease data into a single, connected database. A user-friendly interface provides click-based data access and visualization, while a RESTful web API provides programmatic access to the data. Tools have been integrated into HUMA to allow initial analyses to be carried out on the server. Furthermore, users can upload their private variation datasets, which are automatically mapped to public data and can be analyzed using the integrated tools. HUMA is freely accessible at https://huma.rubi.ru.ac.za.
Keywords: HUMA, SNP, variation database, downstream variant analysis, protein variants, structural bioinformatics
Introduction
The advent of Next Generation Sequencing (NGS) technologies has resulted in the exponential growth of biological sequence data. This is partially thanks to initiatives, such as the International HapMap Project (The International Hapmap Consortium 2003) and the 1000 Genomes Project (The 1000 Genomes Project Consortium 2015), that have taken advantage of NGS technologies to generate comprehensive datasets of human genetic variation.
The overarching aim of these sequencing projects is to investigate the link between heredity and disease/phenotypes. This is done by comparing the genomes of individuals who suffer from a disease with those of healthy individuals. When successful, these projects can associate mutations in a population with disease susceptibility or resistance. This has given rise to the possibility of personalized medicine, where treatments are tailored to individuals based on the existence (or lack) of certain mutations.
Unfortunately, African populations are underrepresented in most genetic datasets. As such, Africa is in danger of falling further behind in the battle against disease. To address this imbalance, the Human Heredity and Health in Africa (H3Africa) Initiative was founded (The H3Africa Consortium 2014). The H3Africa initiative aims to advance genomic studies on the continent by sequencing thousands of individuals across numerous African populations. Additionally, H3Africa hopes to build research capacity on the continent by training scientists and building infrastructure.
Projects such as H3Africa, HapMap, and 1000 Genomes, result in enormous genetic variation datasets. Finding ways to store and analyze these datasets remains one of the great challenges of the 21st century. To this end, several variation databases have been developed. The most well-known of these databases is arguably dbSNP (Sherry et al. 2001), which was created and managed by the National Center for Biotechnology Information (NCBI). The dbSNP database acts as a central repository for all known short variation and incorporates data from HapMap, 1000 Genomes, and user submissions.
The NCBI has also developed various other variation databases including dbVAR (Lappalainen et al. 2013), which stores structural variation, dbGaP (Mailman et al. 2007), which is focused on the relationship between genotype and phenotype, and ClinVar (Landrum et al. 2014), which stores details on the clinical significance of variation. Similarly, the European Bioinformatics Institute (EBI) has developed several variation databases including the European Variation Archive (EVA) , the Database of Genomic Variants archive (DGVa) (Lappalainen et al. 2013), and the European Genome-phenome Archive (EGA) (Lappalainen et al. 2015). Data is regularly shared between EBI and NCBI databases.
A joint-venture between the EBI and National Human Genome Research Institute (NHGRI) resulted in the NHGRI-EBI GWAS Catalog (Welter et al. 2014), a manually-curated collection of published genome-wide association studies (GWAS) containing over 36 000 Single Nucleotide Polymorphisms (SNPs).
Where the previously mentioned databases are solely focused on variation, the Ensembl database (Hubbard et al. 2002) provides comprehensive coverage of biological data, including genes, transcripts, proteins, exons, coding sequences (CDS), and phenotypes. Ensembl maps variation, aggregated from several sources, to this data.
Additionally, a number of locus specific variation databases have been developed over the years (Kuntzer et al. 2010). These databases focus on variations located in a specific gene and, as they are usually curated by experts on the particular gene, often offer a higher degree of quality than general databases (although this varies greatly from database to database) (Johnston and Biesecker 2013). Examples of such databases include the IARC TP53 database (Bouaoun et al. 2016) and BRCA-Share (Béroud et al. 2016), which cater to TP53 (MIM# 191170) and BRCA (MIM# 113705 and MIM# 600185) variants, respectively.
Although the above databases (many more exist) have proven to be useful repositories for genetic variation, they all focus on the analysis of variation at the sequence level. Most datasets containing disease-associated SNPs have been obtained from GWAS or candidate gene association studies (CGAS). Although powerful, these statistical experiments do not provide information on the functional effects of variation and, as such, do not provide insight into why variants might be damaging. To gain an understanding of the functional effects of coding variation (i.e. variation that occurs in protein CDS), studies must be conducted that analyze the effects of these variants on protein structure. This is important for drug design and discovery.
Unfortunately, databases and web servers that focus on variant analysis at the protein structure level are few and far between (Brown and Tastan Bishop 2017). One such database, PinSnps (Lu et al. 2016), allows users to visualize the locations of variations in a protein structure. Similarly, LS-SNP/PDB (Ryan et al. 2009), is a database of annotated variations, pre-mapped to protein structures. These structure-based databases are useful, but tend to neglect sequence level data. On the other hand, sequence level databases, other than the Ensembl database, do not link particularly well to other types of data i.e. they focus solely on variation data. In addition, the web interfaces provided for these databases often leave a lot to be desired, offering no meaningful way of visualizing and interacting with the underlying data.
It is in this context that we have developed the Human Mutation Analysis (HUMA) web server. HUMA was developed as part of the H3ABioNet project (Mulder et al. 2016), a bioinformatics network that forms part of the H3Africa Consortium. One of the goals of H3ABioNet is to build bioinformatics research capacity on the African content, with the end goal being the ability to store, manage, and analyze datasets from H3Africa projects within Africa, rather than sending them abroad.
HUMA aggregates data, including gene and protein sequences, protein structures, variation, diseases, and literature, from several existing public databases into a single, connected database. Access to the data is provided via a user-friendly, web interface and RESTful web API. Although focused around analyzing variation at the protein structure level, HUMA does not neglect sequence level details, mapping variation to gene and protein sequences as well as protein structures.
Rather than being a simple database, HUMA aims to provide a platform for analyzing variation. To this end, modern web technologies have been used to provide a slick interface and smooth data visualization. Additionally, HUMA provides several tools to analyze variation in protein sequence and structures.
To analyze new data being produced by H3Africa projects (and others), HUMA also gives users the ability to upload their own private datasets. This data is stored separately from public data, and can be shared between users and groups, thereby facilitating collaboration.
Like Ensembl, HUMA incorporates and links multiple different types of data. However, HUMA differentiates itself with its focus on variation at the protein level, its incorporation of protein-level analysis tools, and the ability to upload private datasets.
Material and Methods
Implementation
HUMA makes use of a MySQL relational database. Elasticsearch (Elastic 2017) was used to provide intelligent and fast searching of the database. Parser scripts to populate the database were written in C++ and Python. The HUMA web server was developed using the Django web framework, and the Bootstrap and Knockout.js JavaScript frameworks were used to develop the web interface. Additionally, the PV and MSA JavaScript plugins were used to further develop our own PV-MSA plugin, previously used in the PRIMO homology modeling web server (Hatherley et al. 2016).
Data sources
HUMA aggregates data from various sources into a single, connected database. Protein data was obtained from UniProt (Apweiler 2004) and Ensembl. This data includes protein identifiers, names and descriptions, and related literature, as well as features such as binding sites, secondary structure, and modified residues. Supporting sequences such as the transcripts, exons, and CDS for proteins were also obtained from Ensembl. These supporting sequences provided chromosomal co-ordinates for proteins.
Protein family and domain data was obtained from Pfam (Finn et al. 2014). This data was linked to proteins and includes the Pfam accession, the family or domain name, and the co-ordinates of the domain in the protein.
All protein structures were obtained from the Protein Data Bank (PDB) (Berman et al. 2000). Human PDB files were parsed and the relevant details, such as identifiers, the names and UniProt accession numbers of the chains within the PDB files, and the sequences of the solved structures, were extracted.
Gene sequences and meta-data were downloaded from Ensembl and the HUGO Gene Nomenclature Committee (HGNC) (Gray et al. 2015). These details include gene names, identifiers and symbols, as well as the chromosomal co-ordinates for the gene sequence. Ensembl also provided phenotypes linked to genes. To improve the “searchability” of the database, gene name and identifier synonyms and alternatives were obtained from HGNC.
All human variation in dbSNP was downloaded in Variant Calling Format (VCF) from the dbSNP FTP site. In addition, variants from the UniProt humsavar.txt file were also incorporated into the database. Although variant data from UniProt did not include chromosomal co-ordinates and most of the variants overlapped with those from dbSNP, the humsavar.txt file included data linking variants to diseases.
Disease data was obtained from UniProt, ClinVar (Landrum et al. 2014), and the Online Mendelian Inheritance in Man (OMIM) database (Hamosh et al. 2000). UniProt and ClinVar disease data included links to variation. OMIM data was used to provide the names of the diseases when ClinVar only provided the identifiers.
Database design
HUMA data is split between two separate databases. The public database stores data that has been aggregated from the various public data sources described above. The private database stores data that should not automatically be shared between users. This data includes user account details, user groups, private datasets, and job results.
Public Database
A simplified design of the public section of the HUMA database is depicted in Figure 1. A. The database is designed around four types of data: proteins (blue); genes (purple); diseases (red); and variants (orange). These data types are tightly coupled, allowing for quick access to related data types when the database is searched.
Figure 1.
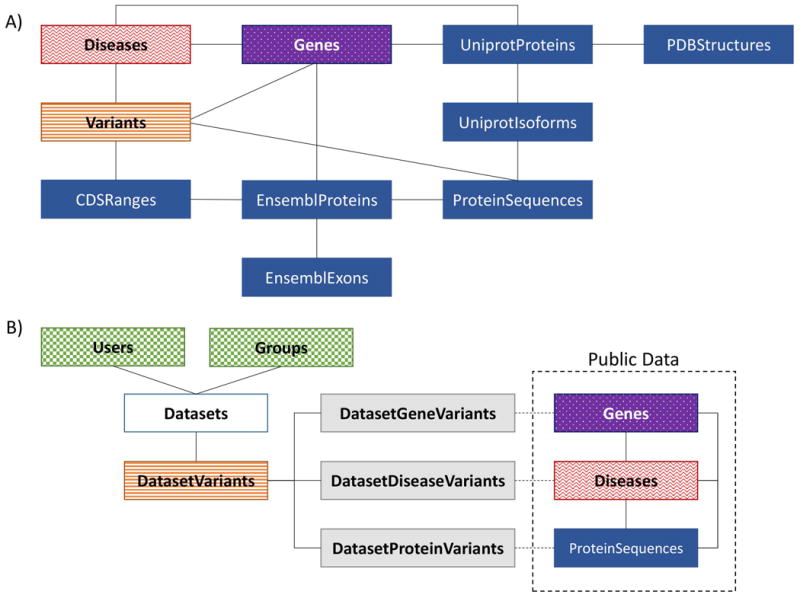
A) Public database design – a simplified design of the public HUMA database. The database is divided into four sections: proteins (blue/dark solid), variants (orange/horizontal stripes), diseases (red/waves), and genes (purple/dotted). B) Private database design – private data is stored in a separate database. Public proteins, genes, and disease are linked to private variants (orange/horizontal stripes) during the mapping process when the variants are first uploaded. These links are stored in the “connector” tables (grey/light solid). Dataset (white) ownership and sharing is managed via the account tables (green/checkered).
The protein section of the database is focused around the UniprotProteins table. This table links to additional tables, not depicted in Figure 1, that store alternate names, synonyms, identifiers, features, families, domains, and literature related to a given protein. For a given UniProt protein, there may be several additional sequences stored, known as isoforms. As such, HUMA allows more than one sequence for any one protein to be stored – hence the need for the UniprotIsoforms table. Because protein sequences are also obtained from Ensembl, a separate ProteinSequences table was created to reduce redundancy. Sequences from both Uniprot and Ensembl are stored in this table. A hash of the sequence is used as the primary key to ensure that duplicate sequences from UniProt and Ensembl are not stored twice. In addition, the CDS for each protein are stored in the EnsemblProteins table, and are used together with the protein exons to calculate CDS ranges on the chromosome (this is discussed further in the ‘mapping variants to protein sequences’ section).
The Genes table also links to additional tables not depicted in Figure 1. These tables store alternative gene identifiers, symbols and names. Genes link to proteins in a “many-to-many” relationship, where one gene may code for more than one protein and one protein may be produced by more than one gene. This is compounded by the fact that there are duplicate proteins stored in the UniProt database (and thus in HUMA). Similarly, the Genes table links to the Variants and Diseases tables via many-to-many relationships.
As with the Genes table, the Diseases table links directly to the Variants and UniprotProteins tables via “many-to-many” relationships. The relationship between variants and proteins differs, however, as the Variant table links to the ProteinSequences table, rather than directly to the UniprotProteins table. This is because variants are mapped to a specific position in a protein sequence.
By closely linking the four data types, this design ensures that there is quick access from any one of the data types to any other data type. This proves useful when it comes to loading large pages containing various different data types, as it allows for efficient database queries.
Private database
To allow users to upload their own variant data, and to ensure that that data is kept secure, private datasets are stored in a separate MySQL database (Figure 1. B), where they are linked to the Users and Groups tables (green), which provide the means for owning and sharing datasets. Nevertheless, private data is linked to public data such as genes, diseases, and proteins by storing the identifiers for those data types in “connector” tables (grey). The identifiers in these tables can be used to look up the related data in the public database.
Private datasets are also linked to user accounts to ensure that no unauthorized users can access them. Additional users can be given access to datasets by sharing the datasets with user-defined groups.
Similarly, HUMA stores the details and results from any tools that are run via the HUMA web server. As with datasets, these jobs are linked to user accounts and can also be shared with user groups.
Populating the database
The HUMA database has been populated using a semi-automated pipeline containing a mixture of C++ and Python scripts (Figure 2). The C++ scripts were written to parse large files that took too long to do with Python. The compute intensive mapping of variants to proteins and genes, described in the next section, was also performed using C++ scripts.
Figure 2. Database population workflow.
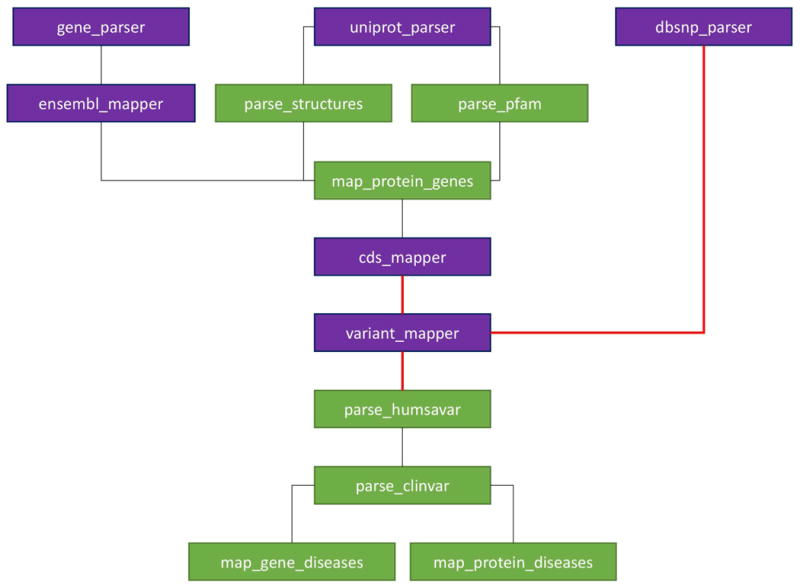
The workflow for populating the database. Blocks represent scripts that parse data files and populate the database with that data. Dark (purple) blocks represent parsers written in C++ while light (green) blocks represent Python scripts. Red/bold lines represent a part of the workflow that cannot be automated i.e. manual intervention is required.
Each block in Figure 2 depicts a script used to populate the database. Purple blocks depict C++ scripts, while green blocks depict Python scripts. The lines between the scripts depict dependencies i.e. the order in which the scripts must run. All of a script's dependencies must be satisfied before it can be executed. Red/bold lines between blocks depict non-automatic dependencies i.e. the pipeline cannot automatically move to the next stage as some sort of manual intervention is required.
The gene_parser script parses gene data obtained from Ensembl and HGNC. This data includes gene identifiers, symbols, names, and chromosomal co-ordinates, as well as the gene sequences themselves. As gene sequences can be relatively long, each sequence is stored as a file and the path to the file is stored in the database. Data linking genes to diseases (as well as the diseases themselves) are also inserted by this script.
The uniprot_parser script extracts relevant data from the SwissProt and Trembl “.dat” files, as well as a Fasta file containing all human Uniprot protein sequences, obtained from the Uniprot FTP site. Data from these files includes identifiers, names, sequences, features, and literature related to the protein.
The dbsnp_parser script parses and inserts all variation from a dbSNP VCF file into a temporary VariantStore table, where it waits to be mapped to proteins and genes.
The ensembl_mapper script inserts proteins and exons from Ensembl into the database. The exon data includes the chromosomal co-ordinates and the protein data includes the CDS. This data is required when mapping variants to protein sequences. In addition, the script maps exons to the relevant protein, and proteins to the relevant genes (hence gene_parser must first have completed). The Ensembl proteins are also mapped to Uniprot proteins during this step by adding the Ensembl protein sequences to the ProteinSequences table, where they overlap with the Uniprot sequences.
Once the uniprot_parser script has finished executing, the parse_structures and parse_pfam Python scripts are free to run. The parse_structures script inserts all human PDB structures into the database. Each protein chain in a PDB structure has an associated UniProt accession number, which is used to link protein structures to their respective UniProt entries.
The parse_pfam script parses data downloaded from the Pfam FTP site and inserts it into the HUMA database. This data includes protein families and domains.
The map_protein_genes script links UniProt proteins to genes from Ensembl by parsing the ID mapping file that can be obtained from the Uniprot FTP site. This file contains mapping between accession numbers and various identifiers from other public databases. For proteins that cannot be mapped via this method, the script attempts to link the UniProt proteins to Ensembl genes via the EnsemblProteins table.
HUMA calculates CDS ranges on the chromosome by concatenating exons to generate the coding DNA (cDNA) and finding the position at which the CDS starts within the cDNA. Combined with the chromosomal co-ordinates of the exons, this allows for the chromosomal co-ordinates of the CDS ranges to be calculated. Variants can then be mapped to the CDS based on chromosomal co-ordinates, and, from there, to the protein sequence. This process is carried out by the cds_mapper and variant_mapper scripts and is discussed further in the next section. Before the variant_mapper script is executed, all indices, primary keys, and foreign keys are manually removed from the table. This improves performance when inserting data into the table. Once the data has been inserted, the keys and indices are reintroduced to improve lookup performance.
The parse_humsavar and parse_clinvar scripts extract data from the UniProt humsavar.txt file and ClinVar variant_summary.txt file, respectively, to map variants to diseases. These files also add the diseases to the database as they go (if the disease does not already exist in the database).
Finally, the map_gene_diseases and map_protein_diseases scripts link genes and proteins, respectively, directly to diseases based on whether any variants in those genes or proteins are linked to the disease (keeping in mind that certain genes are already linked to diseases by the gene_parser).
Mapping variants to protein sequences
In the HUMA database, variants are mapped to gene and protein sequences based on chromosomal co-ordinates. This is a straight forward process for genes, but when mapping variants to proteins, the co-ordinate ranges of the protein's CDS must first be calculated. UniProt data does not incorporate chromosomal co-ordinates. On the other hand, co-ordinates of protein exons can be obtained from Ensembl, but Ensembl proteins do not map directly to PDB structures. As such, both UniProt and Ensembl protein data was required to allow variants to be mapped from the chromosome to protein structures.
The process by which variation is mapped to protein sequences is depicted in Supp. Figure S1. As mentioned earlier, protein sequences are obtained from UniProt and Ensembl, and mapped to one another based on 100% sequence identity. This results in a non-redundant protein dataset. Exons and CDSs are also obtained from Ensembl. Exons are concatenated to form the cDNA, and the coding start point in the cDNA is determined by mapping/aligning the CDS to the cDNA. The co-ordinate ranges on the chromosome that make up the CDS can then be calculated based on the chromosome co-ordinates of the exons that make up the cDNA, and the start point of the CDS in the cDNA. Variation in VCF format (in our case, from dbSNP 147), can then be substituted into CDS ranges based on chromosome co-ordinates and translated to determine the position and amino acid change in the protein sequence (if any).
Search
The HUMA web server provides intelligent and fast search by using an open-source technology called Elasticsearch. Elasticsearch is a document store, which runs as a separate database alongside the HUMA database. Data from the HUMA database is pre-indexed in the document store. As such, when a user performs a search via the Proteins, Genes, or Disease pages, the query never hits the MySQL database, but rather the Elasticsearch document store with the indexed data.
Elasticsearch is extremely fast and scalable. This allows HUMA to search across significantly more data than if it were to query a relational database, such as MySQL, directly. Elasticsearch is also able to perform fast, full-text searches that consider spelling mistakes and typos made by the user. This is also useful when there are different ways to spell words e.g. hemoglobin vs haemoglobin.
Elasticsearch produces ranked search results, where a hit is ranked higher depending on how closely it matches the search term. When calculating a score for a hit, certain data can be weighted more heavily. For example, when searching for a protein, if the search term used matches to a protein name, it will produce a higher score than if it matches text within the protein function description, which can be a paragraph long and contain the names of various other interacting proteins. Weighting such as this have been organized across the various data fields in the HUMA database in order to produce the best and most relevant results when a user performs a search. These weightings were chosen by trail-and-error and will evolve with time as we gather more data.
While we have found that our search works well for genes and proteins, our implementation is limited when it comes to disease searches. The reason for this is that the only disease data stored in the HUMA database is the disease ID and name i.e. no description or meta-data around diseases are stored. Ideally, searching for a term such as “cardiovascular disease” would return diseases related to the heart, but this would require that meta-data or tags were available for each disease, which could then be used to categorize them. That being said, diseases can be searched based on related data (e.g. searching based on a gene symbol (e.g. HBB) or protein accession (e.g. P68871) will find diseases related to that gene or protein, respectively). Future work that introduces more disease data will improve the disease search further.
Visualization
HUMA is largely focused on the analysis of variation at the protein level. As such, being able to visualize the location of variants in the protein structure was deemed important. To do this, two components were required. Firstly, a molecular visualizer was required to render the protein structure in 3D. Secondly, and a little less obviously, an alignment viewer was required. This was due to the fact that the sequence obtained from UniProt/Ensembl and the sequence extracted from the PDB file does not necessarily match. For example, the PDB file may be missing residues where the structure couldn't be solved may contain mutations that the Uniprot sequence does not.
We have previously developed the PV-MSA plugin for the PRIMO web server. The plugin wraps two existing plugins (PV and MSA) into a single component, and allows structures to be visualized along with an alignment. Selecting residues in the structure highlights the residue in the alignment and vice versa. For HUMA, the plugin was extended to allow the alignment and molecular visualizer to be decoupled i.e. they can now be placed at separate locations on a webpage, rather than being tied together.
VAPOR
The Variant Analysis Portal (VAPOR) is a computational workflow, consisting of eight distinct tools, used to predict the effects of variants on protein function and stability (Figure 3). Of the eight tools, the first and last tools are simply used to format the inputs and outputs, respectively, of the six variant analysis tools.
Figure 3. VAPOR workflow.
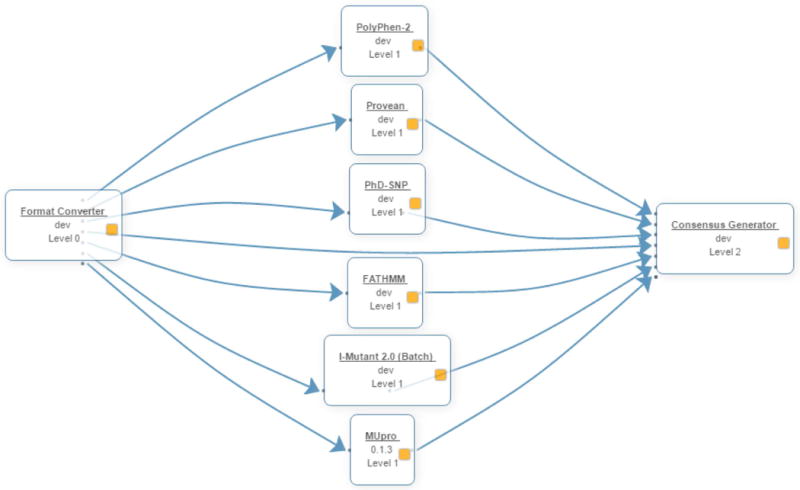
The VAPOR workflow consists of three stages. Firstly, user input is converted into the various formats required by the prediction tools. The predictions are then executed and the results are merged at the last stage.
The tools selected for VAPOR had to meet two criteria. Firstly, they had to be free to use and available for download so that they could be installed locally on our cluster.
Secondly, they had to accept a protein sequence and amino acid change (or list of amino acid changes) as input. Tools that only accepted variations in the form of nucleotide changes and chromosome co-ordinates were ruled out as they could not be accurately mapped to the protein sequence supplied to the other tools.
Due to the latter requirement, tools such as FoldX (Guerois et al. 2002) and Rosetta, which are commonly used for predicting the effect of mutations on protein stability, but accept a protein structure as input, were ruled out. In future, Rosetta predictions may be included in a structural version of VAPOR.
Given the above-mentioned criteria, six tools were selected to make up the initial VAPOR workflow. The tools are split into two categories. The first category consists of tools that predict the functional effect of variants on proteins i.e. whether the variant is damaging/deleterious or tolerated/benign. This category has PolyPhen-2 (Adzhubei et al. 2010), PROVEAN (Choi et al. 2012), PhD-SNP (Capriotti et al. 2006), and FATHMM (Shihab et al. 2013).
The second category predicts the effect of mutations on protein stability. This category is made up of the last two tools, I-Mutant 2.0 (Capriotti et al. 2005) and MuPRO (Cheng et al. 2006).
Input supplied to VAPOR is used by the six chosen analysis tools, each of which accepts this input in a slightly different format. Instead of requiring the user to enter the data in six different formats, the Format Converter tool automatically converts the input into the correct format for each of the tools and passes the converted inputs to the respective tools. Once the tools have finished running, their outputs are passed to the final tool, the Consensus Generator, which merges the results into a single text file.
It is important to note that VAPOR is not a meta-predictor. It does not use machine learning or any other method to generate a consensus. It simply runs the tools independently and merges the results into a convenient table, which can be downloaded by users. This is important to keep in mind, as simply combining the results of these tools could be misleading (Vihinen 2014).
On the other hand, VAPOR still plays an integral role as part of the HUMA web server. It provides a single input interface to execute variation effect prediction tools against variants in the HUMA database. Without VAPOR, users would have to download their variant datasets of interest, browse to the web servers for each of the tools, submit the datasets individually to each of the servers, and then download and collate the results from each of those servers. This requires significant time and effort, especially considering certain servers only allow you to submit a single variation at a time.
Integrating tools
Tools are integrated into the HUMA via Job Management System (JMS) (Brown et al. 2015) (Figure 4). JMS is a workflow management system and web-based cluster front-end that makes its functionality available via a RESTful web API. VAPOR is housed as a workflow within our JMS instance. HUMA provides an interface to this workflow, leveraging the RESTful web API of JMS to forward user input to the workflow management system. JMS manages the execution of the VAPOR workflow on the cluster and, on completion, returns the results to HUMA.
Figure 4. Tool integration via JMS.
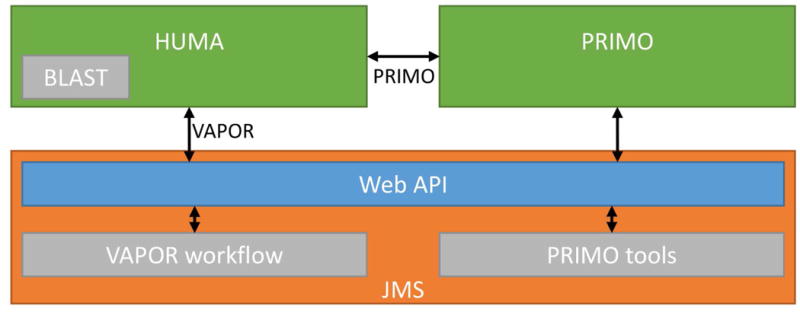
Tools are integrated via the JMS workflow management system, which provides a RESTful web API that allows external servers, such as HUMA, to access it. In the case of PRIMO, HUMA accesses the PRIMO API, which accesses the JMS API.
Similarly, the PRIMO homology modeling pipeline is housed in JMS. However, PRIMO has been integrated into HUMA by allowing users to link their HUMA and PRIMO user accounts. User input is forwarded from HUMA to PRIMO, which then runs the homology modeling job via JMS. As such, updates to PRIMO will automatically result in updates to HUMA. To use PRIMO via the HUMA interface, users must first link their PRIMO account to their HUMA account.
Protein BLAST has been integrated into HUMA to allow the database to be searched based on sequence similarity. Unlike VAPOR and PRIMO, BLAST can return results in real time. As such, there is no need to execute the job on the cluster via JMS. Instead, the tool is run directly by the HUMA web server and the results are returned immediately.
Web interface
The HUMA web interface has been built as a Single Page Application (SPA). This means that the entire website is made up of only one page. The illusion of multiple pages is created by showing and hiding different sections of the page when links are clicked, searches are conducted, or jobs submitted. SPAs provide improved performance by decreasing the bandwidth usage between the client browser and the server. Normally, when a request is sent to the server, it requires that the entire page be reloaded. With SPAs, requests are sent using Asynchronous JavaScript And XML (AJAX), negating the need to reload the page. The server then responds with only the data that should be displayed on the page, rather than an entirely new webpage. JavaScript is used to arrange the returned data on the page. In our case, a JavaScript framework called Knockout.js is used to bind data to elements on the web page, automatically updating the page when new data is received. This process also allows for a more fluid and pleasing user experience as loading screens can be displayed while waiting for data from the server and smooth transitions can be applied when moving between “virtual pages”.
Results and Discussion
Accessibility
The HUMA web server is accessible at https://huma.rubi.ru.ac.za. Both data and tools can be accessed via a user-friendly web interface and RESTful web API. To run tools, upload private datasets, and join groups, users must first register for a free account. To run PRIMO jobs, these accounts must be linked to an account on the PRIMO web server.
Public data
A large amount of data from various sources has been aggregated in the HUMA database (Table 1). This data can be divided into the four types or categories discussed previously. Using various methods, each of these categories has been linked together, so that users can search based on a selected category and easily find all the related data as well. For example, searching for a variant will also display and link to the genes and/or proteins that the variant is found in, as well as any diseases that might be associated with it.
Table 1. HUMA by the numbers – a depiction of the amount of data retrieved from each source and stored in the HUMA database.
| Category: Proteins | ||
| Uniprot | Proteins | 154 527 |
| Isoforms | 176 494 | |
| Unique protein sequences | 156 406 | |
| Families and domains | 6 256 | |
| Ensembl | Unique protein sequences | 83 414 |
| Unique CDS | 101 619 | |
| Exons | 735 779 | |
| Uniprot + Ensembl | Unique protein sequences | 157 392 |
| PDB | Structures | 32 113 |
| Chains | 66 383 | |
| Unique chains (protein sequences) | 5 683 | |
| Section: Genes | ||
| Ensembl | Genes | 22 097 |
| HGNC | Genes | 19 187 |
| Category: Diseases | ||
| All sources | Diseases | 14 224 |
| Category: Variation | ||
| dbSNP | Variants | 152 345 291 |
| Protein variants | 14 452 754 | |
| Gene variants | 74 835 201 | |
| Disease variants | 71 259 | |
Although variants are also mapped to gene sequences, HUMA mostly focuses on variation at the protein level. Nevertheless, HUMA can be searched from the angle of any of the four data types described previously. Previously, HUMA was used to study the effects of variation on the Renin-Angiotensin System (RAS) (Brown et al. 2017). To illustrate this functionality, we will now present a case study of how a user might go about analyzing the disease, “beta-thalassemia” (MIM# 613985), via the HUMA web interface.
Given that this case study is focused on a disease, we will start our analysis on the disease search page on the HUMA website. Searching for the term, “beta-thalassemia”, yields a table with 100 results. The most relevant result is the “BETA-THALASSEMIA” entry from OMIM. A unique feature of HUMA search results is that, along with the diseases returned by the search term, HUMA also returns figures depicting how many much data in other categories is associated with the disease. For example, in the results table, we can see that the OMIM entry for BETA-THALASSEMIA has 1 gene, 65 variants and 4 proteins associated with it. Selecting this entry takes the user to the disease detail page . This page shows further details about the disease entry, as well as specific details about which genes, proteins, and variants have been associated with the disease.
From the disease detail page for BETA-THALASSEMIA, we can see that beta-thalassemia has been linked to the hemoglobin subunit beta (HBB) (MIM# 141900) gene. Clicking on the gene symbol in the “Genes” block on this page takes the user to the gene detail page for HBB. Like the disease details page, this page has more detailed information about the gene, including related proteins, diseases, and variants, as well as the gene sequence and the positions of variants in the sequence.
On the gene detail page, the “Proteins” block displays all the protein sequences that that are coded by HBB. From the dropdown box, users can select the different proteins, which, in this case, are all hemoglobin beta protein sequences. Reviewed sequences are sorted to the top of the dropdown box. The first sequence, P68871, has the most information associated with it and, as such, is a good option to select to continue with the analysis.
With P68871 selected in the dropdown, clicking on the accession number in the “Proteins” block will direct the user to the protein details page for P68871 (Figure 5). As is the theme in HUMA, the protein details page also contains details about related genes, diseases, and variants. However, as HUMA has a focus on analysis at the protein level, this page incorporates a large amount of additional data, including protein structures, supporting sequences, such as the cDNA, CDS, and exons, as well as literature linked to the protein. Users can visualize the protein structures, select variants and see them mapped to the proteins sequence and structure, and highlight the locations of features such as binding sites, secondary structure, and modified residues in the structure.
Figure 5. Protein detail page.
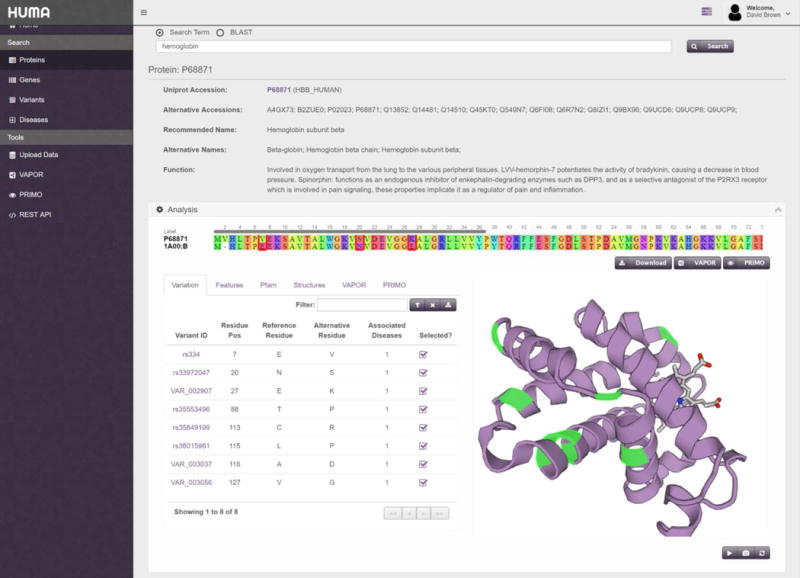
Detailed result page for P68871. Selected variants are highlighted in the protein structure and outlined in the alignment. Only the “Analysis” block is visible in this screenshot.
The “Analysis” block on the protein detail page displays the protein sequence aligned to the sequence of the structure that is being displayed in the box. Alongside the structure, in the “Variants” tab, is a table containing all variations that have been mapped to the protein. In Figure 5, the filter button above the table has been used to filter to show only non-synonymous variations that are linked to disease. From there, selecting the number in the “Associated Diseases” column (or hovering over it), displays which disease has been linked to that variation. All variations linked to beta-thalassemia have been selected. Selecting these variations highlights them in green in the protein structure. In the alignment at the top of the “Analysis” block, the variation is substituted into the sequence and the position in the alignment is outlined in red.
The “Analysis” block also contains several more tabs in addition to the “Variants” tab. The Features tab lists the features obtained from UniProt such as binding sites, modified residues, secondary structure, and chains. Similarly to selecting a variant, selecting a feature will highlight the location of the feature in the alignment and protein structure.
The Pfam tab lists the protein domains and families obtained from the Pfam database. As with features and variations, selecting an entry here will highlight its position in the structure and sequence.
The Structures tab allows the user to select different PDB structures that may have mapped to the Uniprot sequence. Selecting a different structure will replace the structure on the right with the newly selected structure and update the alignment at the top of the page.
Lastly, the VAPOR and PRIMO tabs are only visible when a user is logged in. Here, users can find the results of any VAPOR or PRIMO jobs that they have previously run and that are linked to the protein in question. The VAPOR and PRIMO buttons located underneath the sequence alignment let users submit a sequence with selected variations, if applicable, to the VAPOR and PRIMO tools, respectively. Jobs submitted in this fashion will be linked to the protein and will be accessible from these tabs.
Other than the “Analysis” block, the protein result page includes a “Genes” block, “Diseases” block, “Sequences” block, and “References” block, which show genes that code the protein, diseases associated with the protein, supporting sequences, and related literature, respectively.
Tools
HUMA has two main tools built into it. The first of these is the VAPOR workflow. Continuing with our beta-thalassemia example, the next step would be to analyze the P68871 variants that were previously selected. As mentioned previously, the VAPOR button, located below and to the right of the alignment, lets the user run a VAPOR job using the modified sequence as input. Clicking the button redirects to and automatically populates the VAPOR submission page with the P68871 protein sequence and the selected variants. Clicking the “Submit” button will then run the VAPOR job on the server before taking the user to the results page where they can monitor the progress of the job. Once complete, the VAPOR output will be displayed in the form of a table on the results page (Figure 6).
Figure 6. VAPOR results page.
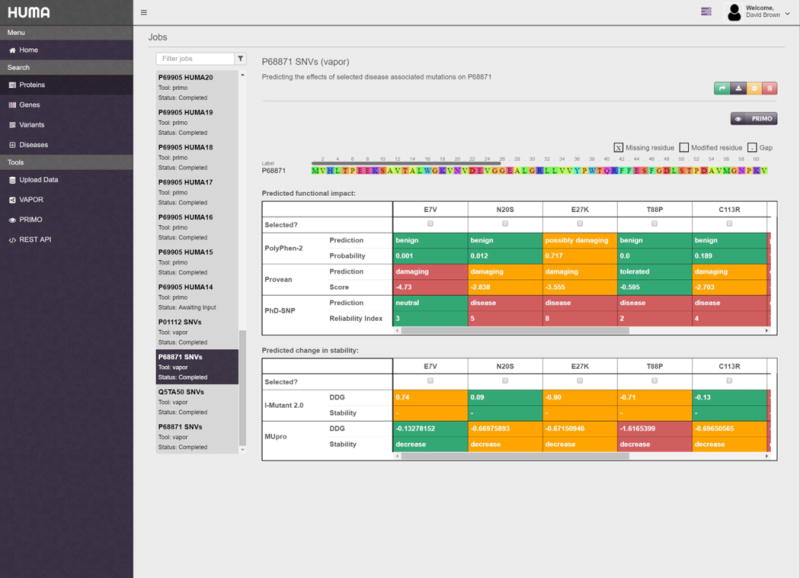
Results are split into two tables, the first of which contains functional predictions and the second of which contains stability predictions. From here, specific mutations can be selected and modeled into the protein structure using PRIMO.
To further analyze mutations that have been predicted to be harmful by VAPOR, these mutations can be selected in the results table and modeled into the structure of the protein sequence by clicking on the PRIMO button (Figure 6). Like before, this automatically populates the PRIMO submission page with the relevant data – in this case, the mutated protein sequence as well as a useful job name and description.
Both PRIMO and VAPOR have been integrated into the HUMA interface in two separate places (in addition to the actual submission pages). As shown in the case study, PRIMO and VAPOR have been integrated into the “Analysis” block on the protein detail page. Similarly, these tools have been integrated into the “Protein View” section of the dataset detail page, which will be discussed in the next section.
Private datasets
Registered users can upload their own datasets to HUMA via the datasets page. Uploads are accepted in VCF format and automatically mapped to gene and protein sequences using the process described in the Materials and Methods section above. In addition, dataset variants are compared to variants in the public database. If a dataset variant matches a public variant that has been associated with the disease, the dataset variant is also associated with that disease.
Once mapping is completed, users can view the results by selecting the relevant dataset. This will direct the user to the dataset detail page. This page contains three different views. The “Variants View” simply displays all variants in the dataset.
The “Genes View” displays the genes that variants in the dataset were mapped to. Selecting a gene will display all the variants from the dataset that mapped to that gene, along with the position in the gene sequence that the variant occurs at.
Similarly, the “Proteins View” displays the proteins that variants in the dataset were mapped to. Selecting a protein displays an analysis block where users can visualize variants in the protein structure and substitute variants into the protein sequence. PRIMO and VAPOR have been integrated here to allow users to analyze unique variants.
As with the previous views, the “Diseases View” displays the diseases associated with variants in the dataset. Selecting a disease will display all variants that were mapped to that disease.
Additional variants can be uploaded to the dataset via the “Uploads” tab. Similarly, variants can be removed from datasets by deleting individual variants via the “Variants View” or deleting genes, proteins, or diseases via their respective views.
Filtering and downloading data
On all pages containing lists of variants, the lists can be filtered and the resulting variant datasets can be downloaded. Filters depend on the page data type. For example, on the protein result page, variants can be filtered based on their residue position, amino acid change, or whether they are associated with any diseases. On the genes result page, variants can be filtered based on the nucleotide position in the gene or the allele change.
Variants can also be downloaded in several different formats. These formats include identifiers, VCF, and one- and three-letter codes (e.g. A7V or Ala7Val).
Collaboration
To facilitate collaboration and the sharing of data, HUMA allows users to create groups. Once a user has created a group, they may invite other users to join their groups. Users will only be added to the group when they accept the invitation.
Groups provide several collaborative features. The most important of these features is the ability to share datasets. Additionally, the results from VAPOR and PRIMO jobs can also be shared with groups. Any users in the group will then have permission to view, analyze, and download this shared data.
Groups also provide a shared forum, where datasets and jobs can be discussed. Providing a means to discuss group-related topics on HUMA, rather than via e-mail, for example, means that these discussions remain easily accessible and do not get lost in the long term.
Conclusions
The HUMA web server has been developed as part of H3ABioNet to provide a repository for variation data generated by H3Africa sequencing projects. HUMA aggregates data form various public sources into a single, connected database, and uses this data to enrich user-uploaded, private datasets. In addition, HUMA provides tools to visualize variation in protein structures, predict the effect of variation on protein function and stability, and model variation into protein structures. As such, HUMA is more than a simple database, but rather a platform for the analysis of genetic variation in humans.
In this paper, we have discussed the utility of HUMA. Users can either use the platform to analyze existing public data, or upload their own, private datasets. The VAPOR workflow incorporates 6 distinct prediction tools to determine the effects of variants on protein function and stability. From there, PRIMO can be used to model interesting variants into the protein structure.
HUMA's focus on variant analysis at the protein structure level, as well as the ability to allow users to upload their own datasets, distinguishes it from existing databases. Additionally, HUMA provides a modern, fast, and user-friendly web interface, as well as a powerful and comprehensive RESTful web API. This combination of data, tools, and modern web technologies makes HUMA a unique platform for structural bioinformaticians and biologists, as well as geneticists.
Future work on HUMA will focus on incorporating additional data into the public database, including protein interaction networks, pathways, and existing drugs that target proteins. In addition, new ways of analyzing this data will be built into the web server, such as a structural version of VAPOR, network analysis, principle component analysis, and molecular docking.
Supplementary Material
Acknowledgments
Thanks to Dinesh Trivedi for providing excellent technical advice and for his help optimizing the website performance.
This research is supported by the National Institutes of Health Common Fund [grant number U41HG006941] to H3ABioNet. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:115D–119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béroud C, Letovsky SI, Braastad CD, Caputo SM, Beaudoux O, Bignon YJ, Bressac-De Paillerets B, Bronner M, Buell CM, Collod-Béroud G, Coulet F, Derive N, et al. BRCA Share: A Collection of Clinical BRCA Gene Variants. Hum Mutat. 2016;37:1318–1328. doi: 10.1002/humu.23113. [DOI] [PubMed] [Google Scholar]
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat. 2016;37:865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- Brown DK, Penkler DL, Musyoka TM, Bishop ÖT. JMS: An Open Source Workflow Management System and Web-Based Cluster Front-End for High Performance Computing. PLoS One. 2015;10:e0134273. doi: 10.1371/journal.pone.0134273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DK, Sheik Amamuddy O, Tastan Bishop Ö. Structure-Based Analysis of Single Nucleotide Variants in the Renin-Angiotensinogen Complex. Glob Heart. 2017 doi: 10.1016/j.gheart.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DK, Tastan Bishop Ö. Role of Structural Bioinformatics in Drug Discovery by Computational SNP Analysis. Glob Heart. 2017 doi: 10.1016/j.gheart.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22:2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62:1125–32. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elastic. 2017.
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: The HGNC resources in 2015. Nucleic Acids Res. 2015;43:D1079–D1085. doi: 10.1093/nar/gku1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerois R, Nielsen JE, Serrano L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. Online Mendelian Inheritance in Man (OMIM) Hum Mutat. 2000;15:57–61. doi: 10.1002/(SICI)1098-1004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Hatherley R, Brown DK, Glenister M, Tastan Bishop Ö. PRIMO: An Interactive Homology Modeling Pipeline. PLoS One. 2016;11:e0166698. doi: 10.1371/journal.pone.0166698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Biesecker LG. Databases of genomic variation and phenotypes: existing resources and future needs. Hum Mol Genet. 2013;22:R27–R31. doi: 10.1093/hmg/ddt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntzer J, Eggle D, Klostermann S, Burtscher H. Human variation databases. Database. 2010;2010:baq015–baq015. doi: 10.1093/database/baq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen I, Almeida-King J, Kumanduri V, Senf A, Spalding JD, Ur-Rehman S, Saunders G, Kandasamy J, Caccamo M, Leinonen R, Vaughan B, Laurent T, et al. The European Genome-phenome Archive of human data consented for biomedical research. Nat Genet. 2015;47:692–695. doi: 10.1038/ng.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen I, Lopez J, Skipper L, Hefferon T, Spalding JD, Garner J, Chen C, Maguire M, Corbett M, Zhou G, Paschall J, Ananiev V, et al. dbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013;41:D936–D941. doi: 10.1093/nar/gks1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Herrera Braga J, Fraternali F. PinSnps: structural and functional analysis of SNPs in the context of protein interaction networks. Bioinformatics. 2016;32:2534–2536. doi: 10.1093/bioinformatics/btw153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–6. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder NJ, Adebiyi E, Alami R, Benkahla A, Brandful J, Doumbia S, Everett D, Fadlelmola FM, Gaboun F, Gaseitsiwe S, Ghazal H, Hazelhurst S, et al. H3ABioNet, a sustainable pan-African bioinformatics network for human heredity and health in Africa. Genome Res. 2016;26:271–277. doi: 10.1101/gr.196295.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M, Diekhans M, Lien S, Liu Y, Karchin R. LS-SNP/PDB: Annotated non-synonymous SNPs mapped to Protein Data Bank structures. Bioinformatics. 2009;25:1431–1432. doi: 10.1093/bioinformatics/btp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, Day INM, Gaunt TR. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The H3Africa Consortium. Research capacity. Enabling the genomic revolution in Africa. Science. 2014;344:1346–8. doi: 10.1126/science.1251546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Hapmap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Vihinen M. Majority Vote and Other Problems when using Computational Tools. Hum Mutat. 2014;35:912–914. doi: 10.1002/humu.22600. [DOI] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.