

Abstract
Introduction
The multifunctional serine protease thrombin exerts proinflammatory and profibrotic cellular effects that may contribute to cardiac remodeling. This study was designed to investigate whether direct thrombin inhibition with dabigatran attenuates myocardial injury in the setting of pressure overload-induced heart failure.
Material and Methods
Transverse aortic constriction (TAC) surgery was performed on C57Bl/6J male mice to elicit cardiac hypertrophy. TAC, or sham, mice were randomly assigned to receive chow supplemented with the oral anticoagulant, dabigatran etexilate, or placebo.
Results
Dabigatran did not affect cardiac hypertrophy, as measured by heart weight-to-body weight or the heart weight-to-tibia length, although a non-significant reduction in myocardial hypertrophic markers (ANP, BNP and MHC) occurred. Dabigatran reduced perivascular fibrosis by 25%, interstitial fibrosis by 54%, and the expression of myocardial fibrosis markers collagen I & III, MMP9, SMA, and PAR-1. These changes were associated with significant improvement in both coronary flow reserve and global left ventricular function. In cultured cardiac fibroblasts, dabigatran decreased thrombin and PAR-1-mediated collagen deposition by 30% and 37%, respectively.
Conclusions
Dabigatran attenuates cardiac fibrosis in the setting of pressure overload and improves coronary flow reserve and global cardiac function possibly by inhibiting thrombin activity and down-regulating PAR-1 expression in the absence of an effect on cardiomyocyte hypertrophy.
Keywords: Cardiac fibrosis, direct thrombin inhibitor, protease-activated receptor, thrombin
INTRODUCTION
Myocardial fibrosis, a key pathological process of extracellular matrix (ECM) remodeling in the heart leading to increased myocardial stiffness, (1) occurs with, left ventricular hypertrophy (LVH), myocardial infarction (MI), diabetes mellitus (DM), and advanced age (2). A dysregulation of the ECM synthesis along with a decreased degradation results in an heightened mechanical stiffness leading to diastolic dysfunction ultimately leading to heart failure (3). This fibrotic process is characterized by activation of cardiac fibroblasts (myofibroblasts), that synthesize and excrete excessive collagen-rich extracellular matrix (collagen I and III) into the cardiac interstitial and perivascular regions in response to a variety of stimuli such as hypertension, inflammation, hormones, cytokines, and growth factors (4) Progressive fibrotic remodeling may ultimately lead to cardiac dysfunction, abnormalities of coronary reserve, and fatal arrhythmias (5, 6).
Thrombin, a multifunctional serine protease, exerts pro-inflammatory and pro-fibrotic cellular effects in normal tissues (7) and in settings of pathological conditions such as liver fibrosis (8), pulmonary fibrosis (9), renal fibrosis (10), atherosclerotic plaque formation (11) and tumor angiogenesis (12–16). Accumulating evidence suggests a role for thrombin in cardiac hypertrophy/fibrosis, either through its proinflammatory and profibrotic functions, or by signaling via the cell-surface protease-activated receptors (PAR) (17, 18). Enhanced thrombin generation, inflammatory markers, and fibrinolytic activity are associated with hypertrophic cardiomyopathy and heart failure (19, 20)
The G protein-coupled receptor protease-activated receptor-1 (PAR-1) is fundamentally responsible for the cellular response to thrombin (13). It has been shown to be highly expressed in cardiac fibroblasts in adult rats and involved in regulating myofibroblast transformation activation and the increase in collagen (I and III) synthesis (18). Additionally increased intercellular calcium, extracellular signal-regulated kinase (ERK) 1/2, DNA synthesis and cellular proliferation have all been reported (21). In the profibrotic lung, ERK1/2 signaling, induced by PAR-1 expression, has been shown to be critical in the formation of pulmonary fibrosis, where PAR-1 is significantly increased in patients with severe lung disease (12, 14, 15). Additionally, increased PAR-1 expression has been observed in the hearts of patients with ischemic and idiopathic-dilated cardiomyopathy, and in animals with chronic heart failure (22).
In-vitro, thrombin treatment brings about phenotype modulation of vascular smooth muscle cells, myofibroblast transformation and proliferation, as well as matrix remodeling (23). TGF-b, responsible for cellular proliferation and differentiation, and procollagen secretion resulting from thrombin induced PAR-1 and ERK1/2 signaling and down regulation of matrix metalloproteinase (MMP, involved in the breakdown of extracellular matrix) demonstrates the importance of thrombin in this disease process (24). Prevention of thrombin associated stiffening and contraction of cardiac fibroblast tissue through the Inhibition of PAR-1 was shown to be effective. Therefore modulating thrombin’s action and its interaction with PAR-1 may represent a novel approach in targeting organ pathological fibrosis associated with a number of chronic diseases.
The pro-drug dabigatran etexilate undergoes esterase-mediated hydrolysis to dabigatran a selective direct thrombin inhibitor and thereby functions as an anticoagulant. Dabigatran’s mechanism of action functions by reversibly binding to the active site of thrombin, whereby it inhibits the Arg-Gly bond cleavage of fibrinogen, a process that is normally required for fibrin formation (11). In addition, when bound to thrombin, dabigatran appears to prevent cleavage at the extracellular N-terminal of PAR-1 resulting in an inhibition of the majority of profibrotic events induced by thrombin (11). Additionally, dabigatran may prevent thrombin’s inflammatory and fibrotic effects as recent studies demonstrated a beneficial effect of dabigatran in fibrotic lung disease, and a reduction in collagen production by lung fibroblasts (13, 25). In the current study, the authors investigated the role of thrombin in mediating cardiac fibrosis in the setting of pressure overload and in activation of cardiac fibroblasts in vitro. We report that dabigatran attenuates cardiac fibrosis and improves coronary flow reserve and global cardiac function in association with down-regulation of PAR-1 expression, without effects on cardiomyocyte hypertrophy.
MATERIALS AND METHODS
Animals
Male mice (C57BL/6J) aged 10- to 12-weeks, were purchased from the Jackson Laboratories (Bar Harbor, ME). Animals were housed in sterile micro-isolator cages (Lab Products, Maywood, NJ), fed drug supplemented or control food and acidified water ad libitum and were maintained on 14-hour day/10-hour night-light cycle. This study was conducted according to the National Institute of Health Guidelines for the Care and Use of Laboratory Animals, and was approved by the Institutional Animal Care and Use Committee of the University of Kentucky.
Transverse Aortic Constriction
Pressure overload of the Left Ventricle (LV) was induced via transverse aortic constriction (TAC) as described previously (22, 23). Sham control mice underwent a similar procedure in which a suture was passed around the aorta but removed without tying (24). Blood flow velocity between the innominate and left common carotid artery was measured by Doppler (Indus Instruments, Houston, TX) before and after surgery. The pressure gradients across the banded site of aorta arch were evaluated by the ratio of flow velocity in the right-to-left carotid artery; the increased ratio indirectly represents pressure overload to the left ventricle.
Drug treatment
Mice were randomly assigned into one of four groups (TAC + dabigatran, TAC + placebo, sham + dabigatran, sham + placebo), and fed chow supplemented with 10- mg/gm dabigatran etexilate or placebo (Boehringer Ingelheim, Biberach Germany) for up to five weeks. Dabigatran etexilate or placebo chow was provided to the animals one week prior to surgery (pre-treatment) or immediately after surgery (immediate treatment) and continued for the duration of the study.
Coagulation assays
At time points one and five weeks post-surgery, blood was collected into EDTA-coated tubes for complete blood count analysis (CBC) performed using a Hemavet 950FS (Drew Scientific Inc., Waterbury, CT). Plasma was prepared for thrombin clotting time (TT), thrombin activity assay, and plasma inflammatory biomarkers. The TT was determined by Hemoclot® thrombin inhibitor assay, according to the manufacturer’s protocol (ANIARA, Mason, OH). The diluted TT is particularly sensitive to the effects of dabigatran, and displays a linear dose-response over therapeutic concentrations; therefore, dabigatran plasma concentrationwas calculated from diluted TT (26). Plasma thrombin activity was detected using SensoLyte® 520 thrombin activity assay kit, according to the manufacturer’s protocol (AnaSpec, Fremont, CA). Plasma cytokines were quantified using a mouse 96-well multiplex assay kit (MPXMCYTO-70K; Millipore, Billerica, MA), according to the manufacturer’s protocol. Bead fluorescence was detected using a BioRad Bioplex 200 suspension array reader and analyzed using Bio-Plex Manager 4.0 software (BioRad Laboratories, Hercules, CA).
Echocardiography
Animals were anaesthetized with 1–1.5% isoflurane inhalation to maintain heart rate between 450 to 550 bpm, and imaged by transthoracic echocardiography (Vevo 770 Imaging System with a 15-MHz probe, VisualSonics, Toronto, Ontario, Canada) before surgery and at three, and five weeks post-surgery, as previously described (27). Cardiac ventricular dimensions were measured on M-mode images. LV function was evaluated by Tissue Doppler Imaging (TDI) with the sampling region within the LV posterior wall in short-axis view at the level of the papillary muscles, as previously described (28).
Doppler Studies
Velocity of blood flow in the carotid arteries and left coronary artery was measured using a 20 MHz Doppler probe (Indus Instruments, Houston, TX) as described previously (29) at five weeks post-surgery. A computer-based Doppler signal processor (Model DSPW, Indus Instruments) was used to store the Doppler signals for later analysis. The coronary flow velocities under basal (1% isoflurane) and hyperemic (2.5% isoflurane) conditions were recorded at five weeks after surgery. The coronary flow reserve (CFR) was calculated from the ratio of hyperemic-to-basal coronary flow velocity during the systolic and diastolic period.
Histological analysis
Hearts were excised and fixed in 4% paraformaldehyde, and then embedded in paraffin. Transverse sections of heart were cut in 4μm sections at the papillary muscle level and subsequently stained with hematoxylin and eosin, Masson-trichrome, and Picrosirius red. Approximately 10 visual fields on two randomly selected sections from each animal were visualized by light microscopy using an objective with a calibrated magnification (×400). Perivascular fibrosis (area of Picrosirius red-positive staining surrounding the vessel/total lumen area) in LV wall and interstitial fibrosis (area of interstitial Picrosirius red-positive staining/total myocardial area of the field) in papillary muscle, were quantified by MetaMorph imaging software (Molecular Devices, Sunnyvale, CA).
Cardiac fibroblast culture and collagen deposition
Cardiac fibroblasts were harvested from TAC or sham mice that had consumed dabigatran etexilate or placebo chow for five weeks after surgery, or from hearts of normal mice on regular chow. Hearts were removed under aseptic conditions and immediately retrograde perfused with calcium-free PBS and dispase solution. LV was isolated, minced, and digested at 37° C. Cells were collected by centrifugation and cultured in DMEM medium containing 10% FBS and 1% antibiotics (penicillin/streptomycin) at 37°C in 5%CO2. Cells were used at passages two through four.
Cells (5 × 104/well) were cultured in 96-well plates to confluence and incubated in serum-free DMEM overnight. The cells were then pre-incubated in a final volume of 100 μl/well DMEM with either the active drug dabigatran (400 ng/ml), or vehicle for one hour, followed by a 96-hour incubation with thrombin (1 unit/ml) or the thrombin receptor-activating peptide SFLLRN (10 μM). Insoluble collagen binding to Sirius red dye was measured in cells washed and fixed with Boiun’s solution (Sigma Aldrich, St. Louis MO) for one hour at room temperature. Unbound Sirius red was removed by washing four times with 0.1 N HCl. Bound dye was eluted with 0.5 N NaOH under mild shaking for 30 minutes, and optical density at 550 nm was recorded.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Comparison of multiple groups was performed by two-way ANOVA using SigmaPlot (version 10.0, Systat Software, Inc., Chicago, IL). All other statistical analyses were performed by t-test or one-way ANOVA; P < 0.05, was accepted as statistically significant.
RESULTS AND DISCUSSION
Dabigatran does not alter cardiac hypertrophy
Approximately 5% of the mice died immediately following TAC, and the remaining mice survived to five weeks. No overt bleeding complications or statistically significant body weight differences were observed between surgical and drug treatment groups. Mice were fed chow with placebo or dabigatran etexilate, which undergoes rapid conversion to the active drug dabigatran. Dabigatran dosing was monitored by diluted thrombin clotting time (dTT). When fed to animals for one week before (pre-treatment) or immediately after surgery (immediate treatment), dabigatran etexilate significantly extended dTT by 3.2-fold and 3.7-fold, respectively. The effect of dabigatran on dTT was independent of procedure (Table 1). The dTT is particularly sensitive to the effects of dabigatran and displays a linear dose-response over therapeutic concentrations. Therefore, plasma concentration of dabigatran can be calculated from dTT (26). Using this, plasma levels where found to reach approximately 0.4 μg/ml in animals fed chow with dabigatran etexilate, and was undetectable in animals fed placebo chow (Table 1). Plasma thrombin activity was 38.9% lower in plasma from dabigatran etexilate-fed mice than in placebo (p<0.001) (Table 1).
Table 1.
Diluted thrombin time and thrombin activity in mice fed placebo or dabigatran chow.
| Thrombin time (sec) | Dabigatran (μg/ml) | Thrombin activity (RFU) | ||||
|---|---|---|---|---|---|---|
| Sham | TAC | Sham | TAC | Sham | TAC | |
| Placebo | 32.3 ± 1.4 | 30 ± 0 | ND | ND | 571 ± 21 | 423 ± 53 |
| Dabigatran | 102 ± 18* | 98 ± 1.9* | 0.41 ± 0.06* | 0.39 ± 0.01* | 301 ± 12* | 285 ± 18* |
Diluted thrombin time and activity in mice fed placebo or dabigatran chow. Placebo: sham n=6, TAC=10; Dabigatran: sham n=7, TAC=8. Data are expressed as mean ± SEM, statistical analysis by two-way ANOVA with Holm-Sidak method;
P<0.05 versus corresponding placebo. ND = Not detectable.
Following TAC, the ratio of flow velocity in the right-to-left carotid artery increased significantly (3.9 ± 0.36 and 1.2 ± 0.12 in TAC and sham groups, respectively, p<0.001), and was similar in placebo and dabigatran TAC groups at five weeks (3.87± 0.38 and 4.03 ± 0.35; p>0.05), indicating a similar load on the LV. As expected, TAC resulted in cardiac hypertrophy as assessed by both an increase in LV mass and LVPWD at five weeks (Table 2). Dabigatran etexilate consumption either immediately (Supplemental Fig. 1) following surgery or as pretreatment did not affect cardiac hypertrophy as measured by heart weight-to-body weight ratio (Fig. 1A), heart weight-to-tibia length ratio (Fig. 1B), and LV mass or LV posterior wall thickness dimension, determined by echocardiography (LVPWD; Table 2). No statistically significant difference in the expression of myocardial hypertrophic markers BNP, ANP, or cardiac MHCβ was observed in mice in the pre-treatment groups (Figs. 1C – E). A non-significant reduction occurred in the expression of myocardial hypertrophic markers in the dabigatran immediate treatment group (Supplemental Fig. 1).
Table 2.
TAC-induced center ventricular hypertrophy in mice fed placebo or dabigatran etexilate chow.
| Baseline | Week 3 | Week 6 | |||||
|---|---|---|---|---|---|---|---|
| n | LV mass | LVPWD | LV mass | LVPWD | LV mass | LVPWD | |
| Placebo + sham | 6 | 60 ± 0.8 | 0.60 ± 0.02 | 67 ± 3.5 | 0.69 ± 0.05 | 62 ± 3.8 | 0.65 ± 0.04 |
| Placebo + TAC | 10 | 54 ± 5.6 | 0.53 ± 0.02 | 79 ± 2.9 | 0.81 ± 0.05 | 93 ± 5.8* | 0.88 ± 0.03* |
| Dabigatran + sham | 7 | 54 ± 2.8 | 0.49 ± 0.04 | 62 ± 1.8 | 0.62 ± 0.01 | 56 ± 3.2 | 0.62 ± 0.04 |
| Dabigatran + TAC | 8 | 62 ± 4.3 | 0.63 ± 0.01 | 73 ± 9.5 | 0.81 ± 0.09 | 78 ± 5.8* | 0.81 ± 0.05* |
TAC-induced center ventricular hypertrophy in mice fed placebo or dabigatran etexilate chow. LV mass presented in mg; LVPWd in mm. Data are expressed as mean ± SEM. Statistical analysis was performed by two-way ANOVA with Holm-Sidak method;
P<0.05 vs corresponding sham. No difference between placebo + TAC and dabigatran + TAC was observed.
Figure 1. Dabigatran lacks effect on pressure overload-induced cardiac hypertrophy.
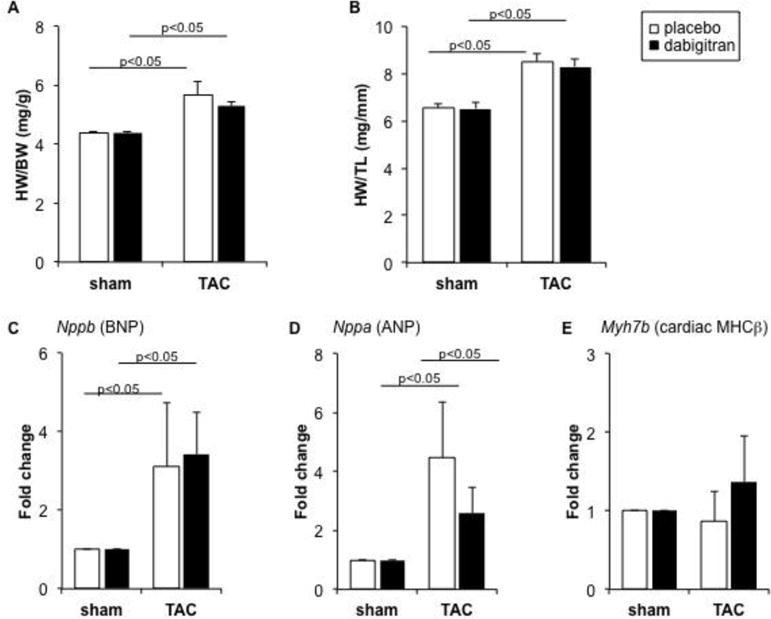
Mice were randomized to receive dabigatran or placebo chow prior to sham or TAC surgery. Five weeks after surgery organs were collected for analysis. (A) Heart weight/body weight (HW/BW) ratio; (B) HW/tibia-length ratio; (C-E) Relative gene expression levels (normalized to 18s RNA) of BNP, ANP, and ᵝ-MHC mRNA in tissue from LV apex. The fold change is in comparison to values obtained in placebo + sham group, which was set as 1. Data are expressed as mean ± SEM; placebo + sham n= 4; placebo + TAC, n= 7; dabigatran + sham, n=5; dabigatran + TAC, n=7.
Dabigatran attenuates myocardial fibrosis in the setting of LVH
TAC elicits remodeling of the coronary arteries, characterized by outward expansion of the vessel wall. Remodeling, as measured by artery thickness, was lower in animals pretreated with dabigatran (Fig 2A) with a trend towards more vimentin expression, although statistical significance was not achieved(Fig 2B). TAC increased collagen deposition in perivascular (Figs. 2A, C) and interstitial areas (Figs. 2A, B), and this was accompanied by increases in in expression of mRNA for collagen I, collagen III, MMP-9, and smooth muscle actin (SMA) (Figs 3B–3E). Dabigatran reduced TAC-induced interstitial fibrosis by 54% (Fig. 2D) and perivascular fibrosis by 25% (Fig. 2E). Interstitial fibrosis displayed a linear relationship with cardiac hypertrophy (heart: body-weight ratio). In mice treated with dabigatran, less fibrosis was observed at any given heart: body-weight ratio (Fig 2F). TAC-induced fibrosis is associated with connective tissue growth factor expression and SM α-actin expression by immunohistochemistry particularly around the coronary arteries (Fig 3A and B). Consistent with the histologic findings, fibrosis-related gene expression was lower in mice pretreated with dabigatran etexilate diet (Figs. 3C–F) and in mice fed dabigatran etexilate immediately after TAC (Supplemental Fig. 2). PAR-1 is highly expressed in the TAC heart and was reduced 42% by dabigatran pre-treatment (Fig. 3G), and 64% by dabigatran initiated after TAC (Supplemental Fig. 2D).
Figure 2. Dabigatran reduces cardiac fibrosis.
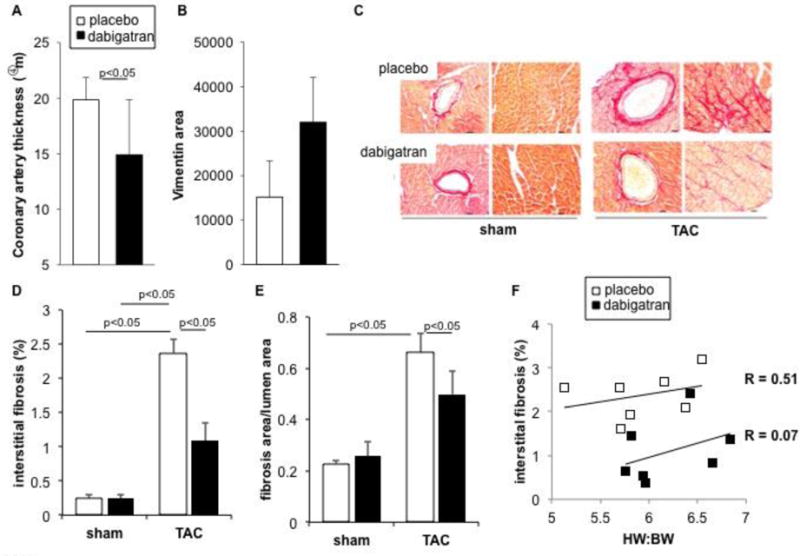
(A) Thickness of left coronary arteries and (B) average area of vimentin staining after TAC. (C) Picrosirius red-staining of collagen in transverse sections of LV five weeks after surgery in mice pretreated with dabigatran etexilate or placebo chow. Images of perivascular fibrosis were taken in LV free wall and interstitial fibrosis in papillary muscle. Scale bar: 20 μm. (D) Quantitative analysis of interstitial fibrosis (% of total myocardial area of the field), and (E) perivascular fibrosis (area of Picrosirius red-positive staining surrounding the vessel/total lumen area). Data are expressed as mean ± SEM; placebo + sham n= 4; placebo + TAC, n= 8; dabigatran + sham, n=5; dabigatran + TAC, n=7. (F) Linear regression anlysis of interstitial fibrosis as a function of HW:BW after TAC. Analysis of parallel lines indicates that while the slopes are not significantly different (P = 0. 0.488), the line y intercepts are significantly different (P = 0.008). Each point represents data from an individual animal.
Figure 3. Dabigatran reduces expression of markers of fibrosis.
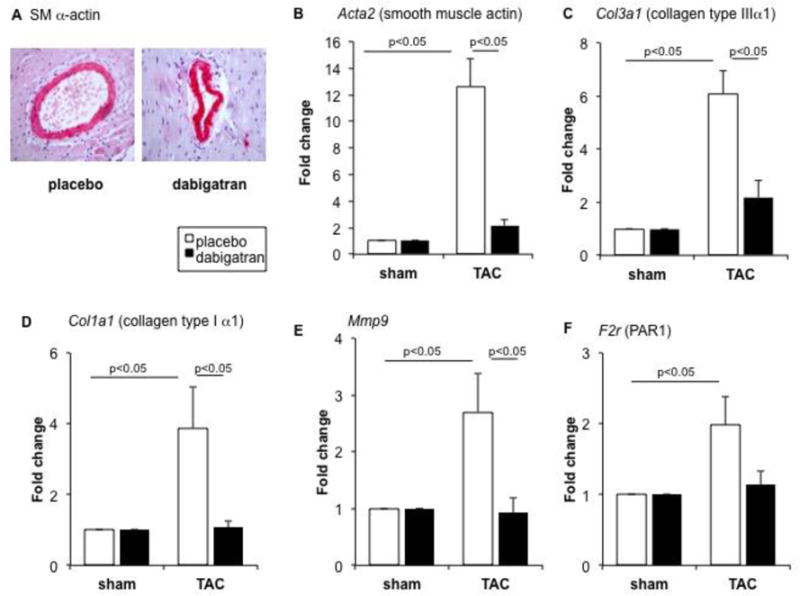
Mice were randomized to receive dabigatran etexilate or placebo chow prior to sham or TAC surgery. Sections after TAC surgery were stained for smooth muscle (SM) α-actin (A; red staining). Relative expression of smooth muscle actin (B), collagen III (C), collagen I (D), MMP-9 (E), X, PAR-1 (F) mRNA were measured in LV apex at five weeks after surgery. Expression level of each gene was normalized to 18s ribosomal RNA levels. The fold change is in comparison to values obtained in placebo + sham group, which was set as 1. Data are expressed as mean ± SEM; placebo + sham n= 3; placebo + TAC, n= 7; dabigatran + sham, n=6; dabigatran + TAC, n= 7.
Dabigatran improves coronary flow reserve and cardiac function
LV hypertrophic remodeling is associated with impairments in myocardial microcirculation and CFR. Therefore, we analyzed the effect of dabigatran on coronary artery flow. TAC decreased the coronary artery diastolic peak flow reserve in both placebo- and dabigatran etexilate-fed animals by 36.1% and 26.8 %, with respect to sham (Fig. 4A). There was no difference between placebo and dabigatran-treated groups (Fig. 4A), indicating that diastolic peak flow in the left coronary artery was unaffected by dabigatran, which was in keeping with similar lumen areas of the coronary arteries histologically (Supplemental Fig. 3). Next, we measured coronary flow velocity to estimate CFR during systolic and diastolic period and observed a 22% improvement in dabigatran etexilate-fed animals compared to placebo after TAC (Fig. 4B). Coronary perfusion reserve inversely correlated with fibrosis after TAC (Fig. 4C). Furthermore, dabigatran also improved global left ventricular function as evaluated by tissue Doppler Tei-index by 60%, in comparison to placebo (Fig. 4D).
Figure 4. Dabigatran improves coronary flow reserve and function and reduces thrombin-stimulated collagen production.
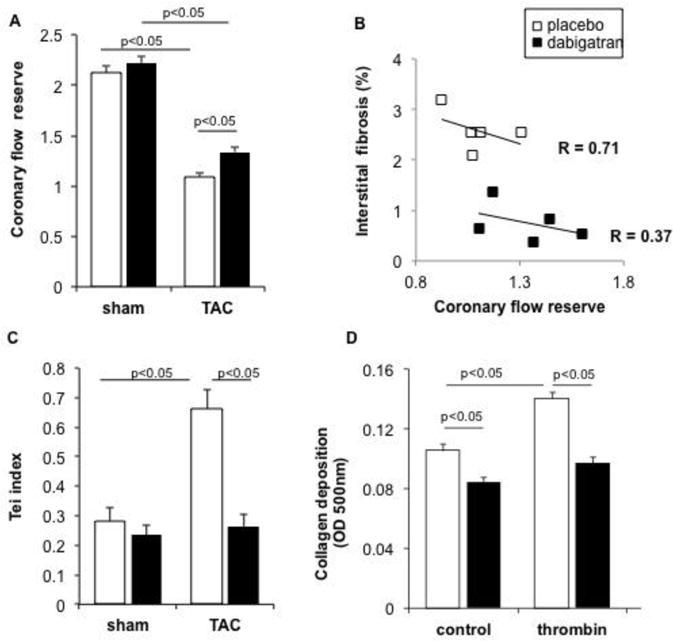
Mice were randomized to receive dabigatran etexilate or placebo chow prior to sham or TAC surgery. Five weeks after surgery, quantitative analysis was performed of coronary flow reserve (ratio of hyperemic to basal velocity) (A).). The flow velocities are representative of four-to–five animals per group. Coronary flow reserve inversely correlated with myocardium fibrosis after TAC by linear regression (BAnalysis of parallel lines indicates that while the slopes of the lines are not significantly different (P = 0.502), the line y intercepts are significantly different (P = 0.0016). Dabigatran also improved global left ventricular function as evaluated by tissue Doppler Tei index (C). Data are expressed as mean ± SEM; placebo + sham n= 6; placebo + TAC, n= 10; dabigatran + sham, n= 7; dabigatran + TAC, n= 8. Fibroblasts were isolated from normal adult mice with regular chow and collagen production measured as described in Methods (D). Data are mean ± SEM from four experiments performed in duplicate.
Dabigatran inhibits collagen production in cardiac fibroblasts
To confirm the anti-fibrotic effect of dabigatran in cardiac remodeling was via thrombin activity, we examined its effect on thrombin-induced cardiac fibroblast collagen deposition in vitro. In fibroblasts isolated from normal adult mice, dabigatran active drug used at concentrations achieved in plasma in vivo decreased by 30% collagen production stimulated by thrombin (Fig. 4D). Fibroblasts isolated from dabigatran etexilate-fed mice at five weeks after TAC, also displayed 15% lower collagen production than that observed in cells from placebo-treated mice, although the difference did not reach statistical significance.
Our results indicate that the oral administration of the direct thrombin-inhibitor dabigatran attenuates pressure overload-induced cardiac fibrosis and improves myocardial perfusion and function expression in the absence of effects on cardiomyocyte hypertrophy. The inhibitory effect on collagen production was recapitulated using cardiac myofibroblasts isolated from mice fed dabigatran etexilate, and by treating cells ex vivo with active dabigatran. Our findings are in keeping with other recent observations that thrombin inhibition blunts fibrosis in liver and lung. Interestingly, dabigatran reduces the development of atherosclerosis in mice, and in this context, appears to stabilize plaque by increasing SMC and collagen content (30), indicating a differential role for thrombin based on the setting. Whether this represents a direct effect of thrombin signaling or an indirect effect, for example due to less fibrin accumulation, remains unknown at this time.
Several of our observations merit additional discussion. We observed a direct correlation between fibrosis and myocardial hypertrophy and coronary flow reserve, suggesting the possibility of a causal relationship. Dabigatran reduced fibrosis and improved coronary flow reserve. decline after TAC, as has been previously reported (31). The decline was less with dabigatran treatment. Our findings suggest a correlation between coronary flow reserve, vascular remodeling, and fibrosis. Furthermore, the improvement in perfusion reserve with dabigatran may maintain global left ventricular function after TAC. This was supported by the favorable effect of dabigatran on Doppler Tei index, a useful index for assessing cardiac systolic and diastolic function in mice (28).
The plasma concentrations of the active metabolite of dabigatran etexilate are associated with dabigatran safety and efficacy. Plasma levels reached approximately 0.4 μg/ml in animals fed chow with dabigatran, and efficiently extended thrombin clotting time to threefold. The mice had no wounds or other overt bleeding. Of note, the plasma concentration achieved in mice is approximately two-fold higher than that observed in humans (32). Dabigatran is less potent in inhibiting murine versus human thrombin, and two-fold higher concentrations are required in mice to achieve similar thrombin inhibitory effects as those in humans (33).
CONCLUSIONS
Dabigatran attenuates pressure overload-induced cardiac fibrosis and improves myocardial perfusion and function by inhibiting thrombin activity, and possibly through down-regulation of PAR-1 expression, with little effect on cardiac hypertrophy. Our findings suggest that dabigatran could have clinically important benefits in preventing pathological myocardial fibrosis.
Supplementary Material
Highlights.
Direct thrombin inhibition had no effect on pressure-induced cardiac hypertrophy.
Direct thrombin inhibition reduces the development of cardiac fibrosis in the setting of pressure overload.
Reduction in cardiac fibrosis was associated with improvements in coronary flow reserve.
Acknowledgments
Anping Dong designed the research, performed the animal surgery, carried out plasma assay, ultrasound/Doppler, cardiac fibroblast culture, analyzed the data, and wrote the first draft of the manuscript. Paul Mueller made corrections of the first draft and contributed to the discussion of the paper. Liping Yang carried out cardiac fibroblast isolation and cell culture. Fanmuyi Yang performed histopathology of the heart tissue. Andrew Morris made important intellectual contributions to the study and manuscript. Susan Smyth designed the research, wrote, and made corrections to the manuscript, and contributed intellectually to the overall completion of all study.
Funding: This project was supported by an investigator-initiated grant from Boehringer Ingelheim and the Heart Lung and Blood Institute (R01HL078663 and T32 T32HL072743). This material is also based on work supported in part by resources at the Lexington VA Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
The authors have no financial disclosures to report.
References
- 1.Sugihara N, Genda A, Shimizu M, Suematsu T, Kita Y, Minamoto M, Kawagoshi H, Umeda K, Chin S, Takeda R. Diastolic dysfunction and its relation to myocardial fibrosis in essential hypertension. J cardiol. 1988;18:353–361. [PubMed] [Google Scholar]
- 2.Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. 2017;113:389–398. doi: 10.1093/cvr/cvx012. [DOI] [PubMed] [Google Scholar]
- 3.Chaturvedi RR, Herron T, Simmons R, Shore D, Kumar P, Sethia B, Chua F, Vassiliadis E, Kentish JC. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circ. 2010;121:979–988. doi: 10.1161/CIRCULATIONAHA.109.850677. [DOI] [PubMed] [Google Scholar]
- 4.Gyöngyösi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, González A, Thum T, Díez J, Jaisser F, Pizard A, Zannad F. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. 2017;19:177–191. doi: 10.1002/ejhf.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. 2002;27:92–98. doi: 10.1007/s00059-002-2354-y. [DOI] [PubMed] [Google Scholar]
- 7.Carney DH, Mann R, Redin WR, Pernia SD, Berry D, Heggers JP, Hayward PG, Robson MC, Christie Enhancement of incisional wound healing and neovascularization in normal rats by thrombin and synthetic thrombin receptor-activating peptides. J Clin Invest. 1992;89:1469–1477. doi: 10.1172/JCI115737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duplantier JG, Dubuisson L, Senant N, Freyburger G, Laurendeau I, Herbert JM, Desmoulière A, Rosenbaum J. A role for thrombin in liver fibrosis. Gut. 2004;53:1682–1687. doi: 10.1136/gut.2003.032136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernández-Rodríguez NA, Cambrey AD, Harrison NK, Chambers RC, Gray AJ, Southcott AM, duBois RM, Black CM, Scully MF, McAnulty RJ, et al. Role of thrombin in pulmonary fibrosis. Lancet. 1995;346:1071–1073. doi: 10.1016/s0140-6736(95)91744-6. [DOI] [PubMed] [Google Scholar]
- 10.Cunningham MA, Rondeau E, Chen X, Coughlin SR, Holdsworth SR, Tipping PG. Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exper Med. 2000;191:455–462. doi: 10.1084/jem.191.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelken NA, Coughlin SR, Gordon D, Wilcox JN. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. 1991;88:1121–1127. doi: 10.1172/JCI115411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogatkevich GS, Gustilo E, Oates JC, Feghali-Bostwick C, Harley RA, Silver RM, Ludwicka-Bradley A. Distinct PKC isoforms mediate cell survival and DNA synthesis in thrombin-induced myofibroblasts. American journal of physiology. Lung Cell Mol Physiol. 2005;288:L190–201. doi: 10.1152/ajplung.00448.2003. [DOI] [PubMed] [Google Scholar]
- 13.Bogatkevich GS, Ludwicka-Bradley A, Silver RM. Dabigatran, a direct thrombin inhibitor, demonstrates antifibrotic effects on lung fibroblasts. Arthritis and Rheum. 2009;60:3455–3464. doi: 10.1002/art.24935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howell DC, Laurent GJ, Chambers RC. Role of thrombin and its major cellular receptor, protease-activated receptor-1, in pulmonary fibrosis. Biochemical Soc Trans. 2002;30:211–216. doi: 10.1042/. doi: 10.1042/ [DOI] [PubMed] [Google Scholar]
- 15.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- 16.Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer cell. 2006;10:355–362. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Sabri A, Muske G, Zhang H, Pak E, Darrow A, Andrade-Gordon P, Steinberg SF. Signaling properties and functions of two distinct cardiomyocyte protease-activated receptors. Circ Res. 2000;86:1054–1061. doi: 10.1161/01.res.86.10.1054. [DOI] [PubMed] [Google Scholar]
- 18.Snead AN, Insel PA. Defining the cellular repertoire of GPCRs identifies a profibrotic role for the most highly expressed receptor, protease-activated receptor 1, in cardiac fibroblasts. FASEB J. 2012;26:4540–4547. doi: 10.1096/fj.12-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moshal KS, Tyagi N, Moss V, Henderson B, Steed M, Ovechkin A, Aru GM, Tyagi SC. Early induction of matrix metalloproteinase-9 transduces signaling in human heart end stage failure. J Cell Mol Med. 2005;9:704–713. doi: 10.1111/j.1582-4934.2005.tb00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moshal KS, Tyagi N, Henderson B, Ovechkin AV, Tyagi SC. Protease-activated receptor and endothelial-myocyte uncoupling in chronic heart failure. American journal of physiology. Heart Circ Physiol. 2005;288:H2770–2777. doi: 10.1152/ajpheart.01146.2004. [DOI] [PubMed] [Google Scholar]
- 21.Sabri A, Short J, Guo J, Steinberg SF. Steinberg, Protease-activated receptor-1-mediated DNA synthesis in cardiac fibroblast is via epidermal growth factor receptor transactivation: distinct PAR-1 signaling pathways in cardiac fibroblasts and cardiomyocytes. Circ Res. 2002;91:532–539. doi: 10.1161/01.res.0000035242.96310.45. [DOI] [PubMed] [Google Scholar]
- 22.Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, Andrade-Gordon P, Kotzsch M, Spring D, Luther T, Abe J, Pohlman TH, Verrier ED, Blaxall BC, Mackman N. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circ. 2007;116:2298–2306. doi: 10.1161/CIRCULATIONAHA.107.692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldsack NR, Chambers RC, Dabbagh K, Laurent GJ. Thrombin. Int J Biochem Cell Biol. 1998;30:641–646. doi: 10.1016/s1357-2725(98)00011-9. [DOI] [PubMed] [Google Scholar]
- 24.Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J, Strande JL. Protease-activated receptor 1 inhibition by SCH79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J Cardiovasc Pharmacol Ther. 2013;18:460–475. doi: 10.1177/1074248413485434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bogatkevich GS, Ludwicka-Bradley A, Nietert PJ, Akter T, van Ryn J, Silver RM. Antiinflammatory and antifibrotic effects of the oral direct thrombin inhibitor dabigatran etexilate in a murine model of interstitial lung disease. Arthritis Rheum. 2011;63:1416–1425. doi: 10.1002/art.30255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stangier J, Feuring M. Using the HEMOCLOT direct thrombin inhibitor assay to determine plasma concentrations of dabigatran. Blood Coagul Fibrinolysis. 2012;23:138–143. doi: 10.1097/MBC.0b013e32834f1b0c. [DOI] [PubMed] [Google Scholar]
- 27.Yang F, Dong A, Mueller P, Caicedo J, Sutton AM, Odetunde J, Barrick CJ, Klyachkin YM, Abdel-Latif A, Smyth SS. Coronary artery remodeling in a model of left ventricular pressure overload is influenced by platelets and inflammatory cells. PloS one. 2012;7:e40196. doi: 10.1371/journal.pone.0040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaefer A, Meyer GP, Hilfiker-Kleiner D, Brand B, Drexler H, Klein G. Evaluation of Tissue Doppler Tei index for global left ventricular function in mice after myocardial infarction: comparison with Pulsed Doppler Tei index. Eur J Echocardiogr. 2005;6:367–375. doi: 10.1016/j.euje.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Hartley CJ, Reddy AK, Madala S, Michael LH, Entman ML, Taffet GE. Doppler estimation of reduced coronary flow reserve in mice with pressure overload cardiac hypertrophy. Ultrasound Med Biol. 2008;34:892–901. doi: 10.1016/j.ultrasmedbio.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borissoff JI, Otten JJ, Heeneman S, Leenders P, van Oerle R, Soehnlein O, Loubele ST, Hamulyák K, Hackeng TM, Daemen MJ, Degen JL, Weiler H, Esmon CT, van Ryn J, Biessen EA, Spronk HM, ten Cate H. Genetic and pharmacological modifications of thrombin formation in apolipoprotein e-deficient mice determine atherosclerosis severity and atherothrombosis onset in a neutrophil-dependent manner. PloS one. 2013;8:e55784. doi: 10.1371/journal.pone.0055784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartley CJ, Reddy AK, Michael LH, Entman ML, Chintalagattu V, Khakoo AY, Taffet GE. Coronary flow reserve in mice: effects of age, coronary disease, and vascular loading. Conference proceedings : … Annual International Conference of the IEEE Engineering in Medicine and Biology Society. Conf Proc IEEE Eng Med Biol Soc. 2010:3780–3783. doi: 10.1109/IEMBS.2010.5627571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Ryn J, Stangier J, Haertter S, Liesenfeld KH, Wienen W, Feuring M, Clemens A. Dabigatran etexilate–a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost. 2010;103:1116–1127. doi: 10.1160/TH09-11-0758. [DOI] [PubMed] [Google Scholar]
- 33.van Ryn J, Goss A, Hauel N, Wienen W, Priepke H, Nar H, Clemens A. The discovery of dabigatran etexilate. Front Pharmacol. 2013;4:12. doi: 10.3389/fphar.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.