

Summary
Adult neurogenesis in the olfactory epithelium is often depicted as a unidirectional pathway during homeostasis and repair. We challenge the unidirectionality of this model by showing that epithelial injury unlocks the potential for Ascl1+ progenitors and Neurog1+ specified neuronal precursors to dedifferentiate into multipotent stem/progenitor cells that contribute significantly to tissue regeneration in the murine olfactory epithelium (OE). We characterize these dedifferentiating cells using several lineage tracing strains and single-cell mRNA-seq, and we show that Sox2 is required for initiating dedifferentiation and that inhibition of Ezh2 promotes multipotent progenitor expansion. These results suggest that the apparent hierarchy of neuronal differentiation is not irreversible, and that lineage commitment can be overridden following severe tissue injury. We elucidate a previously unappreciated pathway for endogenous tissue repair by a highly regenerative neuroepithelium and introduce a system to study the mechanisms underlying plasticity in the OE that can be adapted for other tissues.
Keywords: Dedifferentiation, reprogramming, olfactory epithelium, adult neurogenesis
Graphical abstract
Lin et al. demonstrate that Ascl1+ and Neurog1+ neuronal progenitors can acquire cell fate plasticity after injury using genetic lineage trace and transplantation assays. Injury-induced multipotency occurs through a developmentally reminiscent endogenous up-regulation of Sox2, KLF4, and Pax6, and dedifferentiation efficiency can be enhanced by Ezh2 inhibition.
Introduction
The adult stem cell field has been strongly influenced by groundbreaking experiments in the hematopoietic system beginning in the mid-20th century. From these experiments emerged a model in which a multipotent, self-renewing hematopoietic stem cell differentiates along a stereotypical, unidirectional, progressively branching set of pathways to generate all of the terminally differentiated cell types in blood (reviewed in Ramalho-Santos and Willenbring 2007). This sequential, hierarchical lineage model has long been the archetype applied to all self-renewing adult tissues. However, based on an increased understanding of development and regeneration, this traditional model of a single multipotent stem cell generating all downstream progeny is proving to be a flawed metaphor, particularly in the case of solid tissues. For example, in the lung, gut, pancreas, mammary gland, skin, and liver, more than one cell type, defined by location, mitotic activity and marker profile, is capable of multipotency; in many cases, a population of reserve, or relatively dormant, stem cells that activate only in response to severe tissue damage exists alongside a pool of active stem cells that normally support tissue homeostasis. Furthermore, recent studies have identified dedifferentiation in mammalian cells previously thought to be terminally differentiated. These cells are also able to assume multipotency and other “stemness” properties (Kopp et al., 2016; Kumar et al., 2011; Rios et al., 2014; Rompolas et al., 2013; Stange et al., 2013; Tata et al., 2013; Tetteh et al., 2016).
The mammalian olfactory epithelium, a neuroepithelium that renews itself normally and reconstitutes itself after injury, contains olfactory sensory neurons and multiple, easily characterized non-neuronal cell types, as well as clear readouts for testing progenitor cell capacity (Fig. 1A). Previous observations by our lab and many others demonstrate an apparently hierarchical and unidirectional progression of progenitor types culminating in the birth and differentiation of neurons and also non-neuronal cells when epithelial repair requires them as well. At the apex of that hierarchy are two multipotent stem cell populations: reserve horizontal basal cells (HBCs) and active globose basal cells (GBCs) (Chen et al., 2004; Guo et al., 2010; Huard et al., 1998; Leung et al., 2007; Schnittke et al., 2015; Reviewed in Schwob et al. 2017) (Fig. 1A-B). HBCs are mitotically quiescent, molecularly and morphologically homogeneous, and express the transcription factor Trp63, which serves as the master regulator of HBC identity and reserve status; elimination of p63 in response to epithelial injury is necessary and sufficient to turn HBCs into active progenitors (Fletcher et al., 2011; Leung et al., 2007; Schnittke et al., 2015). The GBCs are heterogeneous with respect to molecular profile and progenitor capacity; they effectuate both the life-long neurogenesis that replenishes the population of olfactory sensory neurons (OSNs) (Jang et al., 2003) in the absence of epithelial injury, as well as wholesale tissue regeneration, in which context they generate not only neurons, but also non-neuronal cells, such as sustentacular (Sus) and duct/gland cells (Huard et al., 1998; Chen et al., 2004) (Fig. 1B).
Figure 1. Ascl1+ and Neurog1+ Progenitors are Neuronally Specified until Injury Unlocks a Multipotent State.
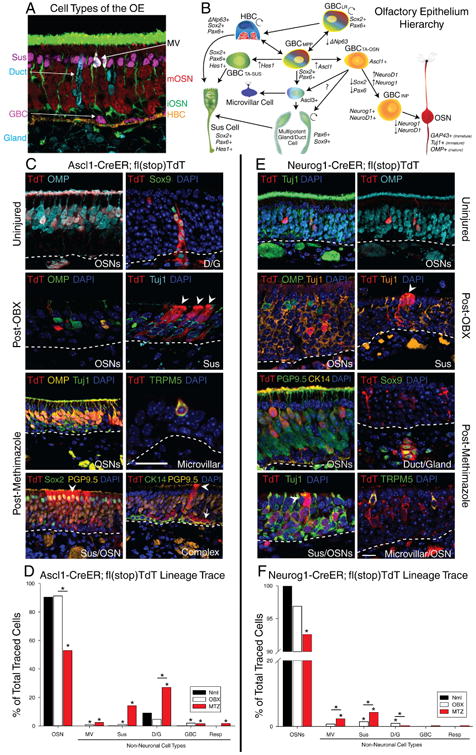
(A) IHC of major cell types of the OE. OMP (Red) – Mature OSN; Tuj1 (Green) – Immature OSN; TRPM5 (White) – Microvillar cell; apical Sox2 (Purple) – Sus cell; Sox9/Aqp5 (Cyan) – D/G cells; basal Sox2+ (Cyan)/CK14- (Gold) – GBCs; CK14 (Gold) – HBCs. (B) Stem cell hierarchical model of olfactory epitheliopoiesis. (C) IHC of clones generated by Ascl1-CreER GBCs in uninjured, 14 days post-OBX, and 14 days post-Methimazole conditions. The arrowheads indicate non-neuronal Sus cells, while the arrow points to a GBC. The scale bar in the microvillar panel equals 10 μm and applies only to the panel it is in. (D) Counts of total cells of each type identified under each type of Ascl1 lineage trace, depicted as a percentage of total traced cells across replicates (n = 6). (E) IHC of clones generated by each type of Neurog1-CreER GBCs in all three conditions. The scale bar equals 10 μm and applies to all other panels. (F) Counts of total cells of each type identified under each type of Neurog1 lineage trace, depicted as before (n = 6). * p < 0.05, Kruskal-Wallis ANOVA on Ranks. Unless otherwise noted, comparisons are to uninjured controls (Nml). See also Figure S1.
During the course of neurogenesis, multipotent GBCs, which express Sox2/Pax6 and closely resemble the cells of the olfactory placode/pit, progress through multiple stages marked by the successive expression of several neurogenic basic helix-loop-helix transcription factors. Importantly, these factors, starting with Ascl1, followed by Neurog1 and NeuroD1, have been shown to indicate neuronal specification in the epithelium, as in the central nervous system (CNS) (Chen et al., 2014; Goldstein et al., 2015; Huard et al., 1998; Jang et al., 2003; Joiner et al., 2015; Krolewski et al., 2012; Leung et al., 2007; Manglapus et al., 2004; Packard et al., 2011a, 2016; Shaker et al., 2012; Xie et al., 2016). These neurogenic factors act in opposition to drivers of non-neuronal cell differentiation such as Hes1, which directs the formation of Sus cells(Cau et al., 1997; Guillemot and Joyner, 1993; Krolewski et al., 2012; Packard et al., 2011a). These observations and others provide a clear delineation of all the major cell types in the OE, including progenitor cells, directly facilitating study of cell fate decisions (Fig. 1A,B)
Furthermore, injury models of differing specificity and severity allow a detailed examination of OE regeneration and probe progenitor cell capacity in settings where epithelial reconstitution imposes different demands. Surgical ablation of the olfactory bulb (OBX) transects the axons of and kills mature OSNs and, going forward, abbreviates neuronal lifespan, such that newly born OSNs die around their transition to full maturity (Schwob et al., 1992). Direct injury to the OE in response to the administration of selective olfactotoxins (e.g., inhalation of methyl bromide [MeBr] or injection of methimazole [MTZ]) kills neurons, Sus cells, and duct/gland cells while sparing some of the GBCs and HBCs in the basal compartment (Bergström et al., 2003; Schwob et al., 1995). Following direct epithelial lesion, both stem cell pools contribute to regeneration. Dormant HBCs lose p63 expression, activate into GBCs, and contribute to epithelial repair (Fletcher et al., 2011; Leung et al., 2007; Packard et al., 2011b; Schnittke et al., 2015).
The regenerative capacity of the OE is perhaps not surprising considering that it is a critical and ancient sensory system that has undergone only modest changes during evolution (Eisthen, 1997) and given its vulnerability to damage as a consequence of its exposure to the external environment (Mackay-Sim and Kittel, 1991). In light of the complex, contextual contributions of stem and progenitor cells to tissue repair and regeneration in multiple organs, elucidating the mechanisms underlying plasticity in the OE will provide insight into the regenerative processes operating elsewhere, including the nervous system. To that end, we used in situ genetic lineage tracing, transplantation assays, and single-cell RNAseq transcriptomics to demonstrate that the current hierarchy of fate specification in the OE is merely a preference that can be overridden following severe injury to the tissue. Reprogramming via the expression of multiple Yamanaka factors looks to be the responsible mechanism.
Results
Ascl1+ and Neurog1+ Progenitors are Neuronally Specified until Injury Unlocks a Multipotent State
To study the differentiative capacity of progenitors in the OE that are ostensibly neuronally specified, we first used Ascl1-CreER; Ai9 flox(Stop)TdTomato mice (hereafter referred to as Ascl1-CreER) for Tamoxifen-induced conditional lineage tracing. By three days after Tam induction in the uninjured epithelium, labeled cells had begun to express immature neuronal markers and had extended processes (Fig. S1A). By the end of a 2-week chase period following Tam injections on two consecutive days, Ascl1 progeny had promoted out of the GBC population. As predicted, Ascl1+ GBCs gave rise to OSNs that matured and expressed olfactory marker protein (OMP), which is associated with fully mature OSNs. Unexpectedly, the progeny included a small, but not insignificant number of Bowman's duct/gland cells, positively identified by their expression of Sox9 (Fig. 1C, Uninjured). Ascl1 has also been associated with both neuroendocrine and secretory cell development (Kokubu et al., 2008; Roach and Wallace, 2013), which is concordant with the generation of the non-neuronal progeny here. More surprising, however, were the cell types generated by Ascl1+ GBCs after injury. Two weeks after OBX we detected Ascl1-derived Sus cells, marked by their distinctive morphology and by the absence of Tuj1/OMP expression, suggesting that injury is capable of unlocking a broadened repertoire for the Ascl1+ GBCs (Fig. 1C, Post-OBX).
Furthermore, after MTZ injection, a harsher injury model that ablates non-neuronal cells as well as OSNs, the emergent multipotency of Ascl1+ GBCs was much more pronounced. Neurons, duct/gland units, microvillar cells, and Sus cells were generated, marked by OMP, Sox9, TRPM5, and intense apical Sox2 labeling, respectively (Fig. 1C). Furthermore, a small but significant number of respiratory cells were generated after the harsher MTZ injury (Fig. 1D). Of note, clones were usually accompanied by OSNs. In addition, Ascl1-derived GBCs persisted through the chase period, in contrast to uninjured tissue; GBC self-renewal is exemplified by their presence within large complex clones – where they are identified as GBCs by the exclusionary criteria of remaining unlabeled by either the robust neuronal marker PGP9.5 or the equally robust HBC marker KRT14 (Fig. 1C). Quantification of the Ascl1-generated cell types revealed statistically significant increases in the generation of non-neuronal cell types as the injury becomes more severe (Fig. 1D).
Given the extent of multipotency after injury, we sought to rule out the possibility that a population of non-Ascl1 multipotent cells underwent Tam-induced recombination when transitioning to Ascl1 expression and became the source of the non-neuronal progeny. To delimit the potential timing of recombination after injection, we first demonstrated that the nuclear localization of CreER and labeling with TdT are both lost by 3 days post-Tam (Fig. S1C). Thus, Tam is capable of activating recombination in this setting for only 2 days after injection. Second, we varied the time of MTZ lesioning relative to Tam labeling (Fig. S1D). We saw a tight, less than 48-hour window during which the recombined cells are capable of acting as multipotent progenitors with this paradigm (Fig. S1E). Thus, any upstream, previously multipotent cells would need to activate Ascl1 expression within the time limit of 2 days while still retaining multipotency.
The HBC reserve stem cells are apparently incapable of reaching the Ascl1 stage within that time limit, based on the following. First, we used a trigenic Ascl1-CreER;fl(stop)TdT;Trp63-eGFP animal and the same Tam protocol to perform simultaneous double lineage trace of the two populations, relying on eGFP perdurance to trace HBC progeny up through 5 days after Mtz injury (Romano et al., 2012). HBC-derived eGFP and Ascl1-driven TdT did not overlap during that period (Fig. S1F). Second, we used Keratin5-CreER to trace the HBC lineage at doses of Tam that result in near 100% recombination and TdT expression by HBCs; the progeny of labeled HBCs did not express Ascl1 until at least 5 days post-MTZ injury (Figs. S1G).
To investigate whether GBCs downstream of the Ascl1 population were also capable of multipotency after injury, we generated Neurog1-CreER;Ai9 flox(Stop)TdTomato mice (hereafter referred to as Neurog1-CreER) and performed similar experiments as above. First, the kinetics of the Neurog1+ GBCs were determined in the context of the uninjured epithelium: at one day following the second Tam injection the overwhelming majority of TdT+ cells were immature OSNs that were strongly labeled by Tuj1 and had begun to elaborate an axon and dendrite. Over the course of 3 days, most of the Neurog1 progeny transitioned into OMP+ mature neurons (Fig. S1B).
By comparison with uninjured tissue, where Neurog1+ GBCs generated only neurons, bulbectomy unlocked the potential of Neurog1+ GBCs to generate non-neuronal cell types such as Sus cells, while MTZ injury that was timed to coincide with the second Tam injection induced a significantly higher percentage of non-neuronal cells (Fig. 1E, Uninjured vs. Post-OBX vs. Methimazole). Quantification showed that the extent of multipotency exhibited by Neurog1+ GBCs post-injury, defined as the percentage of non-neuronal cells, was less than that seen with Ascl1+ GBCs. Nevertheless, the statistically significant increase in lineage plasticity is remarkable, given that the Neurog1+ GBCs act as immediate neuronal precursors and promote out of the GBC population within a few days absent injury (Fig. 1F). Like the Ascl1 GBCs, plasticity was sharply limited to a two-day window of labeling centered on the day of injury, suggesting that Tam perdurance is an unlikely explanation for the multipotency (Fig. S1E). We repeated our analysis using KRT5-CreER mediated lineage trace using immunolabeling for NeuroD1, a surrogate for Neurog1 that is also expressed by the immediate precursor GBCs, and again saw that HBC-derived cells did not express NeuroD1 until at least 5 days post-MTZ (Fig. S1H). Thus, in aggregate, these cells, which were previously specified to make neurons, were capable of generating all of the cell types of the OE after injury, except for the dormant HBC stem cells.
Induced Multipotent Progenitors Transplant and Maintain Their Multipotent State
Transplantation of olfactory epithelial stem and progenitor cells has proven to be a sensitive and effective method for assaying and understanding the regulation of differentiative potency (Reviewed in Schwob et al. 2017). For example, HBC stem cells must experience the effects of epithelial damage in situ prior to isolation and transplantation in order to evince active multipotency even when engrafted into the MeBr-lesioned OE (Schnittke et al., 2015). Accordingly, we tested whether or not the Neurog1+ GBCs require preconditioning by the timing and the in situ milieu of injury to exhibit multipotency following transplantation into the lesioned epithelium as do HBCs, using a Neurog1-eGFP;CAG-TdTomato line for post-transplantation tracing.
Neurog1-eGFP+ GBCs were FACS-isolated 5 and 14 days after OBX and transplanted into host animals 1 day post-MeBr. Isolation at 5 days post-OBX corresponds to maximal GBC proliferation, while at 14 days post-OBX the epithelium is beginning to stabilize (Schwartz Levey et al., 1991). The tissue was harvested two weeks after transplantation (Fig. 2A). Transplantation of multipotent Sox2+ GBCs FACS-isolated from uninjured Sox2-eGFP;CAG-TdTomato animals served as a positive control for multipotency. FACS strategies were designed to remove contaminating HBCs, and, in the case of Sox2, to eliminate the Sus cells on the basis of their many-fold higher level of GFP expression, as compared to the Sox2+ GBCs (Krolewski et al., 2013) (Figs. S2A, S2B). Enrichment of each target population was tested using qRT-PCR for Neurog1 and Sox2, respectively. Unexpectedly, Sox2 mRNA was significantly increased in the Neurog1+ population 5 days post-OBX by comparison with uninjured or 14 days post-OBX (Fig. S2C).
Figure 2. Induced Multipotent Progenitors Transplant and Maintain Their Multipotent State.
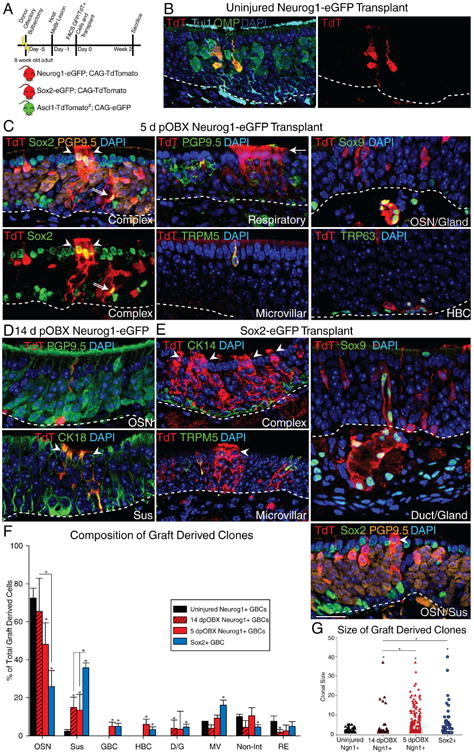
(A) Experimental paradigm for challenging progenitor cell capacity by transplantation into lesioned hosts and schematic of donor reporter mice used. (B) IHC of clones generated 14 days post-engraftment of uninjured Neurog1+ GBCs. (C) IHC of clones generated 14 days post-engraftment of 5 days post-OBX Neurog1+ GBCs. The two left-most panels are different images of the same field to highlight the complexity of the clone. In all panels, the arrowheads mark non-neuronal Sus cells, the double arrow marks a Sox2+ GBC, and the arrow marks the ciliary mat characteristic of columnar respiratory cells. The asterisks mark TRP63+ HBCs. (D) IHC of clones generated 14 days post-engraftment of 14 days post-OBX Neurog1+ GBCs. (E) IHC of clones generated 14 days post-engraftment from positive control Sox2+ GBCs. The scale bar equals 10 μm and applies to all panels. (F) Counts of cells of each type of graft-derived cells, as a percentage of total (n=3). * p < 0.05, Kruskal-Wallis ANOVA on Ranks. (G) Clonal size analysis of engrafted cells (n = 3). *p < 0.05, Mann-Whitney Rank Sum Test. See also Figure S2
Transplanted Neurog1-eGFP+ cells from uninjured donors generated small, marker-positive neuron-only clones (Fig. 2B). Transplanted Neurog1-eGFP+ cells harvested 5 days post-OBX recapitulated the induction of multipotency. In addition to the cell types seen after in situ lineage trace, engrafted Neurog1+ progenitors also gave rise to TRP63+ HBCs, albeit at low numbers. Finally, they also generated columnar respiratory epithelial cells, which were recognized by their distinctive ciliary brush border (Fig. 2C). Transplanted Neurog1+ cells isolated 14 days post-OBX injury were limited to generating Sus cells and OSNs, apparently having lost the capacity to make several other cell types, including HBCs and GBCs (Fig. 2D). The transplantation of bona fide Sox2+ GBCs from uninjured OE yielded the expected full breadth of lineages (Fig. 2E). Quantification of transplant-derived cells confirmed the correlation between extent of multipotency and immediacy of injury. Of note, the levels of plasticity in the Neurog1 population were greater after transplantation as compared to in situ lineage trace (Figs. 1F, 2F).
We also compared the sizes of engrafted clones, and found that Neurog1+ GBCs from uninjured OE were very limited in their proliferative capacity, while those harvested 5 days post-OBX cells proliferated to a degree that was indistinguishable from the Sox2+ controls, based on clone size. Interestingly, despite the persistent though reduced multipotency of Neurog1+ GBCs isolated 14 days post-OBX injury cells, clone size was significantly diminished, regressing toward that of Neurog1+ GBCs from the uninjured epithelium, suggesting that multipotency and proliferative capacity may not be tightly linked (Fig. 2G); as noted above, expression of Sox2 mRNA by Neurog1+ GBCs had also declined at 14 days by comparison with its high expression at 5 days post-bulbectomy (Fig. S2C).
The transplantation of Ascl1+ GBCs required the development of a new strain of reporter mice, as the existing ΔAscl1-eGFP mice (Kim et al., 2011; Wildner et al., 2006) could not be used for isolating cells from the uninjured adult epithelium (negative data not shown). In order to generate a FACSable reporter, we used CRISPR/Cas9 to knock-in two copies of TdT separated from the intact native Ascl1 locus and the other by 2A sequences to ensure high level expression from this normally low copy-number transcript (see Supplemental Materials and Methods) (Fig. S2D-E). The preservation of the native locus achieves detectable and faithful expression under both resting and post-injury conditions by IHC, and is useful for FACS isolation (Fig. S2F).
This Ascl1-TdT2 line was then bred to a pan-eGFP line for transplantation, which showed that even Ascl1-TdT+ cells isolated from the uninjured epithelium were capable of generating non-neuronal cell types, in contrast to the progeny observed with in situ lineage tracing in that setting (Fig. S2G). We were unable to detect a broadening of differentiative potency in Ascl1+ GBCs that were isolated following bulbectomy. However, clonal size was significantly increased as a consequence of injury prior to isolation (Fig. S2H).
Expression Profiling of OSKM Reprogramming Factors
The induction of Sox2 mRNA in cells acquiring multipotency, prompted us to investigate directly whether other members of the OSKM reprogramming set, Oct4, KLF4, or c-Myc were induced in these cells (Takahashi et al., 2007). Neurog1-eGFP+ cells, which do not normally express OSKM factors, express Sox2 and KLF4 mRNA after injury (Fig. 3A). They also begin to express Pax6, a transcription factor that is also expressed in the multipotent stem cells intrinsic to the OE (Fig. 3A). Finally, we profiled major known factors involved in OE neurogenesis and observed a sharp down-regulation of the late neurogenic factors NeuroD1 and then Neurog1, and an acute up-regulation of Ascl1 (Fig. 3B), reversing the order in which they are expressed during normal development (Cau et al., 1997).
Figure 3. Expression Profiling of OSKM Reprogramming Factors and Other Likely Mediators.
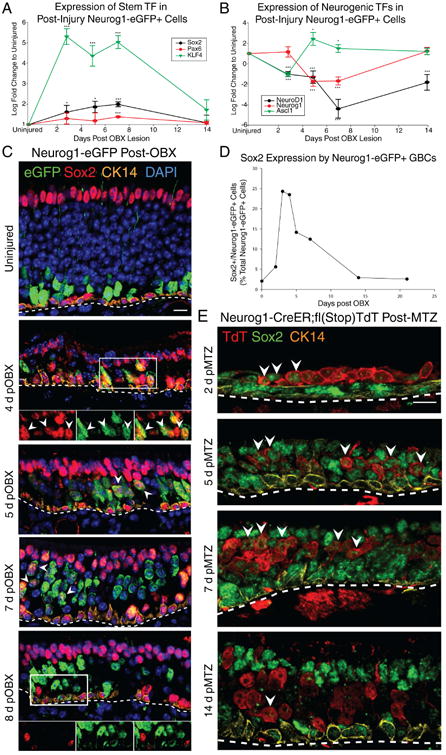
(A) qRT-PCR of Sox2, KLF4 and Pax6 in FACS-isolated Neurog1+ GBCs as a function of survival time after OBX. (B) qRT-PCR of neurogenic transcription factors in the same samples. * p<0.05, *** p < 0.001, ANOVA. (C) IHC for Sox2, Neurog1-eGFP and CK14 as a function of time after bulbectomy. Arrowheads mark examples of eGFP+/Sox2+ cells. The scale bar equals 10 μm. (D) Counts of double positive Sox2+/Neurog1-eGFP+ cells by IHC, depicted as a percentage of all Neurog1-eGFP+ cells as a function of time after bulbectomy. (E) IHC for Sox2 following Neurog1-CreER-mediated lineage tracing as a function of time post-methimazole (MTZ) injury. The scale bar equals 10 μm. See also Figure S3
With respect to the expression of KLF4 protein, the small subset of Sox2+ GBCs that expressed KLF4 in the uninjured epithelium expanded greatly following bulbectomy, such that it became expressed in not only basal stem cells, but also in a subset of apical cells (Fig. S3A).
We chose to focus on Sox2 and Pax6 instead of KLF4, as their expression and roles are far better characterized in the OE (Guo et al. 2010; Packard et al. 2016). IHC analysis compared the induction of Sox2 mRNA with Sox2 protein expression at various time points post-OBX by monitoring GFP+ cells in the Neurog1-eGFP reporter mouse (Fig. 3C). Quantification of Neurog1-eGFP+/Sox2+ cells revealed a rapid induction of Sox2-positivity that peaked 3-4 days post-OBX, after which it dropped slowly to baseline levels by 14 days (Fig. 3D); the increase was confirmed with a second, independent Sox2 antibody (Fig. S3C). Pax6 expression also increased post-injury although it lagged somewhat (Fig. 3A, S3B).
To determine whether the induction of Sox2 persisted, we traced the progeny of Neurog1-CreER-expressing cells labeled prior to MTZ lesion for the first two weeks after injury, and saw that Sox2 up-regulation was nearly universal for as long as 7 days post-MTZ, before dropping back to baseline by 14 days post-MTZ (Fig. 3E). This level of robust Sox2 induction during the post-lesion period correlated with the generation of non-neuronal cells by Neurog1+ GBC-derived progenitors (Fig. 1F). Given this Sox2 induction, we assessed whether a very small Neurog1+/Sox2+ population that previously existed during normal homeostasis might undergo massive expansion to account for the increase in their prevalence during the first few days after lesion. We used the presence of CreER in both the Ascl1 and Neurog1-CreER driver lines to assay the size of this putative population and to demonstrate that this is very unlikely to be the case – Ascl1-CreER+ GBCs account for the vast majority of the Sox2+ GBCs in normal tissue, but do not predominate to the same extent after injury. In contrast, the Neurog1-CreER+/Sox2+ GBCs were very few in normal tissue and increased by greater than an order of magnitude post-injury, which cannot be due to geometric amplification of a pre-existing population given the cell cycle length of GBCs (Huard and Schwob, 1995). The overall decrease in the prevalence of Ascl1-CreER+ GBCs is a result of multiple factors: the increase in the Neurog1-derived Sox2+ GBCs, the activation of HBCs to become multipotent GBCs (Figs. S1F and S1G), the proliferation of spared Ascl1-CreER-/Sox2+ upstream GBCs, and the loss of Ascl1 and retention of Sox2 by a proportion of the previously Ascl1-CreER+ GBCs (Fig. S3F-H). The latter fits with the reversal of the normal neurogenic expression pattern suggested by qRT-PCR, establishing them as multipotent GBCs (Fig. S3H).
We also tested whether the induction of Sox2 in Neurog1-eGFP+ cells recapitulated the early development of the OE, when GBCs are highly plastic (Gokoffski et al., 2011; Krolewski et al., 2011). At postnatal day 3, there was a significant number of Sox2 and Neurog1-eGFP double positive cells, but by PND5, double-labeled cells had dropped to levels comparable to adult (Fig. S3D-E).
Sox2 is Required for Induced Multipotency
Given the role of Sox2 during the induction of somatic cell pluripotency, we tested whether the initiation of high level Sox2 expression in GBCs that are ostensibly limited to generating neurons is responsible for their assumption of multipotency after injury. Thus, we drove conditional knockout of Sox2 by Neurog1-CreER or Ascl1-CreER, using the same labeling and injury protocols as before.
In the unlesioned OE, Neurog1-CreER driven genetic excision of Sox2 had no detectable effect, in keeping with the absence of measurable Sox2 mRNA or protein expression in the vast majority of Neurog1+/NeuroD1+ GBCs absent injury (Figs. 3C, S4A, S4C) (Guo et al., 2010). In contrast, Ascl1-CreER-driven excision of Sox2, which is normally expressed in Ascl1+ GBCs (Fig. S4A) (Guo et al., 2010), reduced proliferation in the recombined GBCs and resulted in a lower absolute number of progeny per clone (Fig. S4D), although both neurons and D/G cells were still generated (Fig. S4C); the reduction in clone size following recombination in Ascl1+ GBCs fits well with the effects of Sox2 knockout in HBCs that subsequently undergo lesion-induced activation (Packard et al., 2016).
Following MTZ injury, Sox2 knockout had a marked effect on progenitor potency with either driver. With the Ascl1-CreER driver, conditional knockout was efficacious since the vast majority of the TdT+ cells in mice homozygous for the floxed allele lacked detectable Sox2 shortly after MTZ-lesion (Fig. 4A) in contrast to the normal universality of Sox2 expression by spared GBCs and HBCs after injury (Guo et al., 2010; Packard et al., 2016). Knockout of Sox2 with either the Ascl1 or Neurog1 driver prevented the generation of non-neuronal cells when assayed two weeks after recombination and injury (Fig. 4B, 4D), thus preventing Neurog1+ or Ascl1+ GBCs from becoming multipotent. The small number of TdT+ Sus cells in these cases retained Sox2 immunostaining, indicating that they originated from a progenitor in which recombination at the Sox2 locus was incomplete (Fig. 4C). Furthermore, the loss of Sox2 from Ascl1 GBCs decreased the ability of the tissue to regenerate after injury (Fig. 4E).
Figure 4. Sox2 is Required for Induced Multipotency.
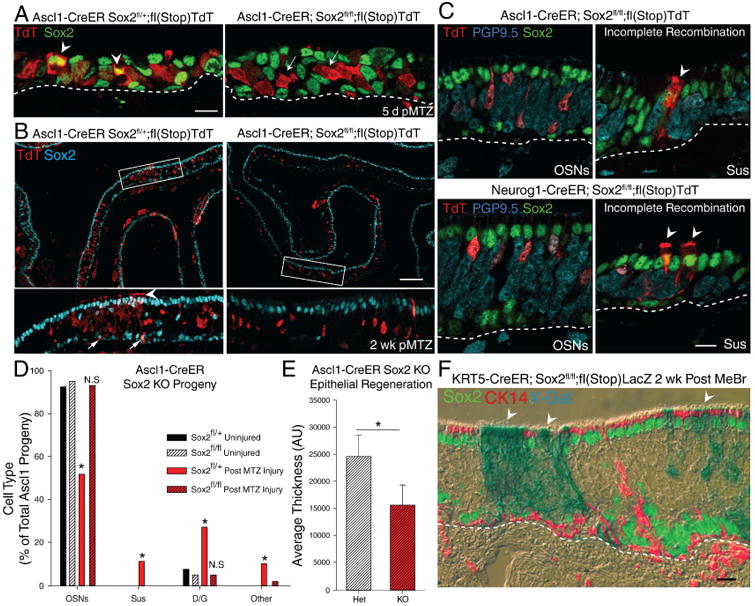
IHC of Sox2 protein expression following Ascl1-CreER-driven Sox2 excision in mice that are either heterozygous or homozygous for the floxed Sox2 allele 5 days post-methimazole (MTZ). The arrowheads indicate TdT+/Sox2+ cells, while the arrows indicate TdT+/Sox2- cells. The scale bar represents 10 μm. Low-magnification image of Ascl1-CreER-driven excision in floxed Sox2 heterozygotes vs. homozygotes with lineage trace 2 weeks post-MTZ. The arrowhead marks Sus cells, the arrows mark self-renewed Sox2+ GBCs. The scale bar represents 100 μm. (C) High magnification of IHC of clones generated in the floxed Sox2 homozygous animals co-stained with Sox2. Arrowheads indicate Sus cells that failed to completely recombine and still express Sox2 demonstrating that recombination is occasionally incomplete. The scale bar represents 10 μm. (D) Counts of cell types generated in heterozygous vs homozygous floxed Sox2 mice (n = 3) after verifying Sox2 IHC negativity. * p < 0.05, Kruskal-Wallis ANOVA on Ranks. (E) Average tissue thickness 2 weeks after injury (n=3); * p < 0.001 two-tailed T-test. (F) IHC of K5-CreER-mediated Sox2 knockout and lineage trace post-injury, X-gal (blue in phase contrast) overlaid with fluorescent IHC for Sox2 (green) and CK14 (red). The scale bar equals 10 μm. See also Figure S4
Conditional knockout of Sox2 correlated with high levels of immunodetectable Ezh2, which is known to play a role in Sox2 regulation as well as restricting cell lineage potential (Snitow et al., 2015). The reciprocity between Sox2 and Exh2 was evident by examining neighboring cells that still express Sox2 in the floxed homozygote or in mice that are heterozygous for the floxed allele (Fig. S4E).
Finally, we verified that elimination of Sox2 does not block Sus cell differentiation per se when they arise from an alternative progenitor population; in this case, a Krt5-CreER driver was used to excise Sox2 in HBCs, which were then activated by epithelial injury. Under these circumstances, Sox2-deficient Sus cells were evident and otherwise indistinguishable from wild-type HBC-derived ones (Fig. 4F).
Single Cell Transcriptional Profiling and Validation of Functional Pathways
Given the lesion-induced induction of multipotency, we performed single-cell mRNAseq (scRNAseq) analysis of Neurog1-eGFP cells that were FACS-isolated from uninjured epithelium and from tissue harvested 5 days and 14 days post-OBX. We captured and sequenced 140 Neurog1-eGFP+ cells that were pruned to 120 that passed QC filtering (Fig. S5A). t-distributed stochastic neighbor embedding (t-SNE) dimension-reduction plotting of the cells converged to three distinct populations (Fig. S5B). To place the Neurog1+ cells into cellular context, we captured and analyzed 210 additional single cells harvested from uninjured olfactory epithelium as well as FACS-isolated OMP-eGFP+ mature olfactory neurons. Batch-to-batch variation normalization was assessed by overlaying run identities on top of the t-SNE plot (Fig. S5D). Each cluster in the data set was identified as a known cell type based on published and validated cell markers, including the identification of Sus cells by their expression of KRT18 and Cyp2g1 and the identification of mature and immature olfactory neurons by their expression of OMP and GAP43, respectively (data not shown). We overlaid the Neurog1-eGFP+ sorted cells on the whole epithelial profile. Some of the Neurog1+ cells were on a normal neurogenic trajectory, but injury shifted some of the Neurog1+ cells to overlay a cluster containing heterogeneous, stem-like cells (Fig. 5A).
Figure 5. Single Cell Transcriptional Profiling and Validation of Functional Pathways.
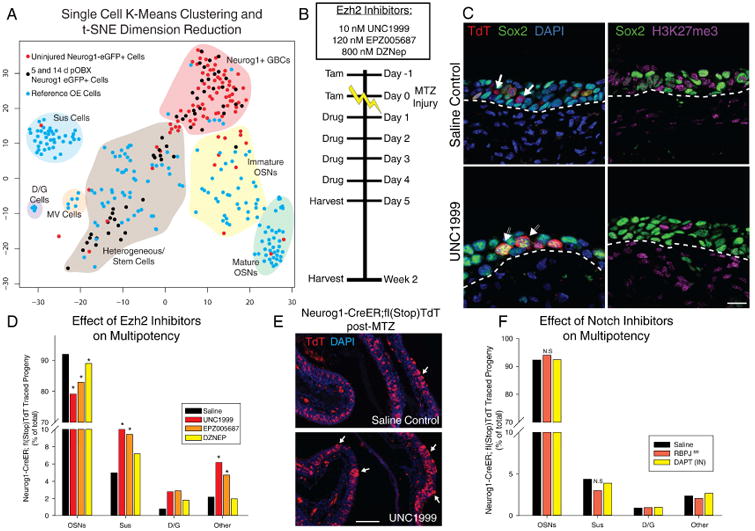
(A) t-distributed stochastic neighbor embedding (t-SNE) dimension reduction of single cell transcriptomes. Blue cells were cells sampled from uninjured OE; red cells are Neurog1-eGFP+ FACS isolated cells from uninjured tissue; black cells are isolated from 5 and 14 dpOBX tissue. Shaded areas and labels represent generalized, manually-identified cell groups. (B) Experimental paradigm of dosing with interventional small molecules. (C) IHC of H3K27me3 and Sox2 with vehicle or Ezh2 inhibitor UNC1999, 5 days after methimazole (MTZ) injury. The scale bar equals 10 μm. (D) Quantification of the effect of Ezh2 inhibition on progeny after injury using three inhibitors with different potencies plotted as a total percentage of cell types across replicates (n = 3). (E) Low mag image of saline treated vs. UNC1999 treated Neurog1 lineage trace. Arrows mark locations of assumed multipotency yielding Sus cells. The scale bar equals 100 μm (F) Quantification of Notch inhibition using DAPT or RBPJ genetic knockout depicted as in (D) (n = 3). * p < 0.05, Kruskal-Wallis ANOVA on Ranks. See also Figure S5
We also combined our single cell data with a recently published dataset describing the trajectory of the reserve HBC population following artificial TRP63 knockout, which forces them to activate and differentiate (Fletcher et al., 2017). This dataset contains a high number of what we identified as multipotent GBCs, and the Neurog1-eGFP+ cells merge with this cluster, suggesting that these cells are reverting to an endogenous natural multipotent state after injury at a transcriptional level (Fig. S5E).
FASTProject and WebGestalt analysis, using Broad Institute Molecular Signatures, identified pathways that were significantly enriched over baseline expectations at both times after bulbectomy (Fig. S5F). Transcription factor hubs that were highly enriched in each condition, such as MYC and Sox family transcription factors at 5 days post-OBX, validated previous qRT-PCR and immunohistochemical results (Figs. S2C, 3, S5G).
Analysis at the single cell level identified Ezh2 as a potential epigenetic regulator of induced multipotency, in keeping with the reciprocity in its expression by comparison with Sox2 (Fig. S4E). In contrast, canonical Notch signaling was not differentially regulated, despite its critical role in transdifferentiation elsewhere (Lafkas et al., 2015; Yimlamai et al., 2014). Accordingly, we set out to investigate these two pathways experimentally, and also SHH, IGF, and TGFβ signaling cascades using available mouse models and small molecule interventions.
Several well characterized Ezh2 small molecule inhibitors of varying IC50s were tested for their effect on dedifferentiation and potency (Campbell et al., 2015; Xu et al., 2015; Zhang et al., 2015); in decreasing order of potency, they are UNC1999, EPZ005687, and DZNEP. Four sequential days of intranasal delivery of either saline vehicle or inhibitor, starting the day after MTZ injection, was appended to the same labeling and injury paradigm used for Neurog1-CreER in-situ lineage tracing (Figs. 1, 5B). The efficacy of intranasal inhibitor was confirmed by the reduction in staining for H3K27me3 immediately after the last day of drug dosing; there was also an increase in general Sox2 levels compared to saline (Fig. 5C). After recoveries of two weeks, IHC-classified non-neuronal cell types were significantly increased as a consequence of Ezh2 inhibition with each of the three inhibitors (Figs. 5D-E).
The effect of intranasal Notch inhibitor DAPT was also assessed as well as the consequences of knocking out RBPJ, the main effector for canonical Notch signaling. In keeping with the scRNAseq observations, both approaches failed to demonstrate any apparent role for Notch signaling in modulating induced multipotency (Fig. 5F).
Finally, we tested the effects of SHH, IGF1R, and TGFβ inhibition using a similar intranasal delivery described above and saw small, non-significant variations in clonal composition and efficiency of induced multipotency (Figs. S5H and S5I).
Discussion
Previous models of olfactory neurogenesis depict a unidirectional, consecutive loss of cell potency finally yielding mature neurons. With the data presented here, we believe that such a model is too simplistic. Instead, we propose the following: adult olfactory epithelial progenitors that are normally fated and specified to make only neurons, in the case of the Neurog1+ GBCs, or only neurons and duct/gland cells, in the case of Ascl1+ GBCs, have the potential to acquire stem-like multipotency, expanded proliferative capacity, and a greater degree of self-renewal in response to epithelial damage. Moreover, progenitor plasticity becomes more extensive when the damage to the epithelium is more severe.
The mechanism driving this increase in progenitor potency is at least partially cell-intrinsic, since it depends on the environment from which the progenitors are isolated. For example, Neurog1-eGFP+ GBCs from uninjured mice cannot gain multipotency when engrafted into the directly lesioned epithelium, where the surrounding endogenous progenitors are actively multipotent (Chen et al., 2004; Huard et al., 1998; Leung et al., 2007; Schnittke et al., 2015), in contrast to the multipotency of Neurog1+ GBCs harvested following epithelial damage.
scRNAseq characterization demonstrated that many signaling pathways and signatures were differentially regulated during the GBC response to injury. In particular, up-regulation of Sox2 mRNA and protein expression correlated with enhanced progenitor cell capacity, and conditional excision of Sox2 prevented that plasticity. Sox2-dependence corresponds well with reports that it plays roles in dedifferentiation of cancer and in EMT (Herreros-Villanueva et al., 2013). On the other hand, expanded progenitor cell capacity did not require the ongoing elevation of Sox2 expression, since multipotency persists, for example, at 14 days post-OBX by which time Sox2 protein and transcript levels had returned to baseline. The return to baseline 2 weeks after bulbectomy anticipates the very near normal mRNA profile of Neurog1+ GBCs harvested 3 weeks post-OBX, when the epithelium has stabilized albeit at a higher rate of GBC proliferation (Krolewski et al., 2013; Schwartz Levey et al., 1991; Schwob et al., 1992). Sox2 is also expressed in Neurog1-eGFP+ cells during development, which may explain the report that the embryonic OE demonstrates a high degree of lineage plasticity such that downstream GBCs are also able to generate non-neuronal Sus cells (Gokoffski et al., 2011). Thus, dedifferentiation seen after injury in the adult OE may also be a return to a normal developmental state.
The reciprocity between Sox2 vs. Ezh2 expression also linked the down-regulation of epigenetic repression, mediated by Ezh2 and the Polycomb repressor complex, to the initiation and, possibly, maintenance of expanded progenitor capacity. The participation of Ezh2 in restraining multipotency was substantiated by the expansion of progenitor capacity in situ following infusion of small molecule inhibitors of Ezh2. In addition, we observed the up-regulation of several non-coding RNAs known to interact with Ezh2, such as MALAT1, suggesting a role for non-coding RNAs in specifying epigenetic targets during this acquired multipotency (Holoch and Moazed, 2015). In aggregate, our data show that injury induces signaling differences in a number of pathways, which may inhibit Ezh2 and thereby modulate Sox2 epigenetically in the spared Neurog1+ and Ascl1+ GBCs. The combined signals appear to push these neuronally specified cells back up the hierarchy to a multipotent GBC state. This state, however, only lasts briefly, allowing these cells to participate in tissue regeneration without altering stem cell balance over the long term (Fig. 6). A similar role has been ascribed to Ezh2 in lung and muscle development, where Ezh2 normally restricts basal cell lineage potential while its depletion results in increased potency (Snitow et al., 2016, 2015). Given the usual opposing roles of Polycomb-group vs. Trithorax-group proteins, our data suggest that it may be possible to enhance dedifferentiation via the Trithorax axis (Smith et al., 2016).
Figure 6. Working Model.
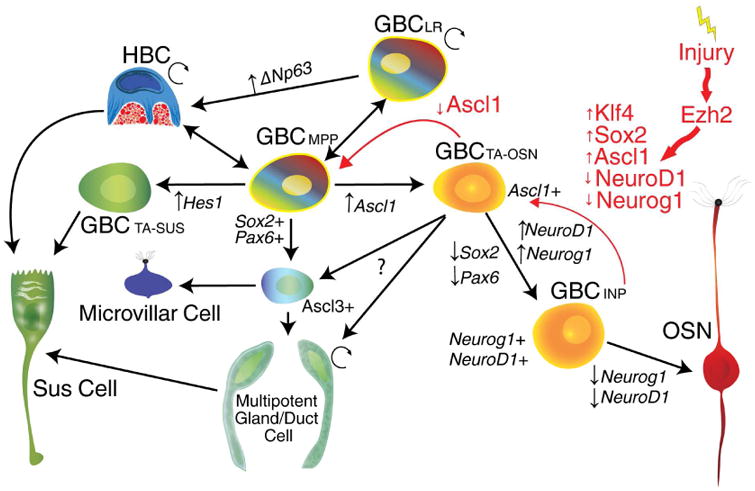
(A) Compiled working model with black arrows representing the bulk flow of cell fate during regeneration with red arrows representing the proposed path taken by dedifferentiating cells during the initial response to injury.
In addition, there were significant changes in pathways modulating mRNA capping, stability and degradation. We also identified significant differences in metabolic processes, which comport with studies showing that altered metabolism is required and plays an active role in cell fate conversion (Folmes et al., 2012; Gascón et al., 2015; Ryall et al., 2015). However, testing other upstream signaling pathways via small molecule inhibition yielded mixed results. Most chemically mediated somatic cell reprogramming paradigms use inhibition of TGF-β or its downstream mediator SMAD, which in effect can replace c-MYC or Sox2 in the reprogramming process (Masserdotti et al., 2016; Smith et al., 2016). However, TGF-β inhibition in the setting of the lesioned OE had an attenuated effect. We were also surprised to find that Notch signaling did not play a role in regulating dedifferentiation in OE, as it participates in progenitor cell plasticity in the pancreas, liver, and airway.
The existence of tissue and context-specific mechanisms for dedifferentiation raises several questions regarding the process itself. For example, while it appears that initiation signals may be different, the underlying fundamental mechanisms may be conserved. We draw parallels between the events in the OE and the need for Pax6 induction in fish and chick retina Müller glia during dedifferentiation after injury (Karl et al., 2008; Marquardt et al., 2001). Even more a propos, lens removal in the newt eye results in dedifferentiation of pigmented epithelial cells, marked by expression of Sox2 and KLF4 (Maki et al., 2009). Additionally, it is known that hair cells of the auditory system undergo robust regeneration, specifically in non-mammalian vertebrates, such as chick, using dedifferentiation/trans-differentiation mechanism that is remarkably similar to the OE. There, the supporting cells can either dedifferentiate or directly trans-differentiate into hair cells after injury by inducing Atoh1 expression. Strikingly, these dedifferentiative processes in non-mammalian vertebrates can, in fact occur in mice—where hair cell regeneration is negligible—but only during the first 2 weeks of postnatal development, which mirrors the Sox2 expression we see in both postnatal and injured adult OE (reviewed in Bermingham-McDonogh and Reh, 2011). Here, we show not only the up-regulation of Pax6, KLF4, and Sox2, but also that Sox2 is required for this dedifferentiation. Taken together, these commonalities suggest that this form of tissue specific, endogenous dedifferentiation may be conserved across tissue types.
Our findings demonstrate a previously unappreciated pathway for tissue regeneration naturally utilized by a highly regenerative neuroepithelium. While “dedifferentiation” has been widely used to describe mature, quiescent cells re-entering mitosis to generate more cells of the same type, we believe it also describes the phenomenon shown here, with lineage-specified cells adopting a more primitive, multipotent stem cell state that then actively participates in tissue regeneration, recapitulating a process observed during normal development. The term “trans-differentiation” also describes this phenomenon, as a neuronally fated cell is converted to other cell lineages; however, the phrase lacks emphasis on the existence of developmentally equivalent states. Finally, “reprogramming”, which has been almost exclusively used to describe the assumption of pluripotency by mature somatic cells in response to manipulation, is highly relevant in this case, as olfactory progenitors are utilizing a subset of Yamanaka reprogramming factors (Jopling et al., 2011). Indeed, given reports that POU transcription factor family members can adequately substitute for canonical OKSM factors, and our finding that c-MYC downstream targets were differentially regulated in the scRNAseq, it is not unreasonable to hypothesize that a natural, controlled form of Yamanaka reprogramming occurs in olfactory progenitors (Sarkar and Hochedlinger, 2013; Soufi, 2014). Further investigation into in vivo reprogramming via transient combinations of OSKM factors may yield important insight into natural modulatory mechanisms at play during tissue regeneration and tumorigenesis, allowing a more nuanced use of reprogramming.
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact James Schwob (jim.schwob@tufts.edu)
Experimental Model and Subject Details
Animals and breeding
Healthy, non-immune compromised mice that had not undergone any other procedure except those explicitly stated in experimental designs were used for all experiments. All mice were nulliparous and drug naïve until dosed according to experimental designs. Eight-10 week-old wild-type mice used as hosts for the transplantation studies were F1-generation males from C57/B6J and 129S1/Sv1MJ cross that was bred in house or ordered from Jackson Labs (Bar Harbor, ME, Stock #101043). Neurog1-eGFP BAC transgenic mice generated through the GENSAT Project were obtained from Jackson Labs (Bar Harbor, ME, Stock #017306). ΔSox2-eGFP mice were generously provided by Dr Mahendra Rao (Ellis et al., 2004). ΔOMP-eGFP mice were generously provided by Dr. Peter Mombaerts (Potter et al., 2001). Neurog1-CreER driver mice were generously provided by Dr. Lisa Goodrich (Kim et al., 2011). Ascl1-CreER driver mice; stock #012882, Cre reporter strain R26R(TdTomato)(B6.Cg-Gt(ROSA)26Sortm9(CAG-TdTomato)Hez/J; stock #007909, and fl(Sox2); stock #013093 were obtained from Jackson Labs. The constitutive CAG-TdTomato animal was generated in-house by breeding the R26R(TdTomato) strain with the germ-line constitutive Sox2-Cre recombinase. For transplant studies, the Sox2 and Neurog1 eGFP reporter mice were bred to this constitutive CAG-TdTomato strain for in-vivo tracing after engraftment. Mixed sex adult mice (8-10 weeks) were used for harvesting cells. The Ascl1-TdTomato2 mouse was generated in-house, described below. For genetic lineage tracing, Neurog1 and Ascl1-CreER driver mice were bred to the R26R(TdTomato) strain. Finally, for all genetic excision studies, these bi-genic animals were bred to fl(Sox2) animals to yield tri-genic lineage traced knockouts. For all genetic lineage tracing experiments, mixed sex adult mice (8-10 weeks) were used, and littermates were randomly assigned to experimental groups.
All mice were maintained on ad libitum rodent chow and water. All animals were housed in a heat and humidity controlled, AALAC-accredited vivarium operating under a 12:12-hour light-dark cycle. All protocols of the use of vertebrate animals were approved by the Committee for the Humane Use of Animals at Tufts University School of Medicine, where the animals were housed and the experimented conducted.
Method Details
Generation of the Ascl1-TdTomato2 mouse line
To generate the Ascl1-TdTomato2 line, we created a knock-in vector using 2 kb homologous arms flanking the cassette. The 5′ arm contains a portion of the Ascl1 promoter sequence, as well as the protein coding sequence, while the 3′ end begins immediately following the stop codon of the native Ascl1 coding sequence. To generate a high signal reporter, we intended to express two copies of TdTomato at 1:1 of near 1:1 stoichiometry with the native Ascl1 protein. TdTomato was chosen as the fluorescent protein as the Ascl1-eGFP reporter mouse is insufficient for signal detection in the adult OE tissue, and TdTomato is approximately six times brighter. Significant effort was invested to reduce the chance of self-mediated recombination, as TdTomato is itself a tandem repeat. Thus, we fused the Ascl1 protein coding exon to a 60 bp 2A skip peptide identified from the porcine teschovirus-1 (P2A), preceeded by a 9 bp “GSG” motif to increase skip efficiency. The P2A sequence is followed by the coding sequence of a codon-optimized TdTomato, a different, T2A skip peptide, isolated from Thoseaasigna, and a second, original, non-optimized TdTomato sequence, flanked by the 3′ homology arm. In summary, alterations to the coding sequence were able to reduce sequence similarity by 22%. To construct the targeting vector, the codon optimized TdTomato was synthesized de novo from IDT as gBlocks with 35 bp mini-homology overhangs to its neighbors. The remaining homology arms, and unmodified TdTomato sequences were generated using PCR from template or genomic DNA with similar mini-homology overhangs, except these also incorporated the 2A peptides. These 100 bp megaprimers were synthesized from IDT with HPLC purification. These primers also included silent point-mutations to remove gRNA binding sites for later downstream CRISPR application. PCRs was done using NEB Q5 hot-start polymerase, and significantly optimized using Taguchi array design, modulating dNTP, primer, template, MgCl2, Q5 polymerase, 1,2 propanol, and betaine additives. Amplicons were then gel extracted on a blue/green LED light to prevent UV damage, and then extracted using silica-matrix bead technology for stringent removal of contaminants (Macherey-Nagel NucleoSpin Gel and PCR Clean-up Kit, 740609.50). Finally, individual pieces along with a minimal, linearized puc19 backbone were ligated together using Gibson assembly (NEB Gibson Assembly Master Mix, E2611S) following kit instructions with the following modifications: reaction size was 5 ml, 0.15 pmol per fragment was added, and a 1 hour Dpn1 digest was included at the end of the incubation to remove contaminating plasmid. For CRISPR/Cas9 editing, we used the p×330 plasmid (Addgene, 42230) to in-vitro transcribe gRNA targeted for the Ascl1 locus, designed through the CRISPR Design tool, as described in Supplemental Table 3 (crispr.mit.edu). The top six gRNA designs were generated, and used in a mix. Microinjections into FVB background embryos were performed in the Maine Medical Center Research Institute, Mouse Transgenic & In Vivo Imaging Core Facility under the directorship of Dr. Lucy Liaw. This was supported by grant number P30GM103392, Phase III COBRE in Vascular Biology (Robert E. Friesel PhD, P.I.), a grant supported by the National Institute of General Medical Sciences. 60 founders were generated, 23% were homozygotes and 25% were heterozygotes for positive insertion using long-range PCR and Southern blot. This mouse has been deposited to MMRRC, Stock number: 043552.
Transplantation procedure
Transplantations were performed as follows. Donor cells for transplantation and infection were dissociated from donor animals anaesthetized with IP injection of a triple cocktail of ketamine (37.5 mg/kg), xylazine (7.5 mg/kg), and acepromazine (1.25 mg/kg) and perfused with cold Low Ca2+ Ringers solution (140mM NaCl, 5mM KCl, 10mM HEPES, 1mM EDTA, 10mM glucose, 1mM sodium pyruvate, pH7.2). The olfactory epithelium was separated from respiratory tissue, finely minced, and incubated in 0.05% Trypsin-EDTA for 10 minutes on an orbital shaker at 37°C where it forms a sticky ball. The supernatant was replaced with an enzyme cocktail containing (100 U/ml collagenase, 250 U/ml hyaluronidase, 75 U/ml DNase I, 0.1 mg/ml trypsin inhibitor, 2.5 U/ml Dispase II, 350 U/ml Collagenase Type I, 5U/ml papain; from Worthington Biochemical, Roche, and Sigma) in Regular Ringers solution (140mM NaCl, 5mM KCl, 10mM HEPES, 1mM EDTA, 10mM glucose, 1mM sodium pyruvate, 1mM CaCl2, 1mM MgCl2, pH7.2). The tissue was incubated in the enzyme cocktail on an orbital shaker at 37°C for 15 minutes, after which the supernatant was replaced with fresh enzyme cocktail and incubated once again for 15 minutes. The collected supernatant was stored on ice in the meantime. After the second incubation, the pooled supernatant is filtered through 30 μm filter and centrifuged at < 500g on a swinging bucket rotor for 5 minutes. The pellet was resuspended in 1× HBSS containing 25 mM HEPES, 10 mM EDTA for FACS purification based on eGFP positivity. The resulting cells are pelleted at < 500g on a swinging bucket rotor for 5 minutes and resuspended in 50 ml of DMEM for transplantation.
The host mice were prepared to accept cell engraftment by lesioning the epithelium through passive exposure to methyl bromide gas (MeBr) at 180 ppm for 8 hours. 24 hours later, the mice were anesthetized with an induction cocktail containing 37.5 mg/kg ketamine, 7.5 mg/kg xylazine, and 1.25 mg/kg acepromazine and maintained at a surgical anesthetic plane with a cocktail consisting of 47.5 mg/kg ketamine and 0.9 mg/kg acepromazine as needed. Their neck was shaved and disinfected with iodine and ethanol. A tracheotomy was then performed to maintain a patent airway while the palate was raised with a 3 cm piece of PE-100 tubing to close to nasopharyngeal passage. Approximately 50 ml of cells prepared earlier were infused into one naris through a cannula made from PE-10 tubing and filled the nasal cavities until fluid exited the contralateral naris. The mice were then positioned at a 45° angle and rotated to the alternate side every 45 minutes to allow filling of the lateral regions of the nasal cavity and the cul-de-sacs. After 3 hours, the solution was aspirated from the nasal cavity, the trachea was sutured with 10-0 Ethilon, and the skin was closed with 5-0 suture. Mice were given 1 ml of sterile saline subcutaneously and placed on a warm pad overnight, at a maximum of two mice per cage.
Olfactory bulbectomy and methimazole lesion
Olfactory bulbectomy was performed as described previously (Schwob et al., 1992). Mice were anesthetized and maintained as described above while immobilized in a stereotactic mount above a 37°C heating pad for thermoregulation. A single incision was made on shaved, sterilized skin to expose the overlying frontal bone. A bone drill exposes the bulb on each hemisphere which is removed by gentle aspiration. Sterile Oxycel was placed within the cavity to achieve hemostasis and the overlying skin was sutured. Mice are allowed to recover as described above.
Methimazole lesion was performed by IP injection of a single dose at 75 mg/kg using a solution of 10 mg/ml methimazole. Mice were placed on a heat pad overnight to accelerate recovery.
Tamoxifen preparation and administration
Tamoxifen was purchased from Sigma (#T5648), dissolved in sterile USP grade corn oil at 30 mg/ml by gentle inversion at 60°C for 10 minutes. For all lineage tracing experiments (Neurog1-CreER, Ascl1-CreER, and KRT5-CreER), this solution was injected I.P. at 150 mg/kg once a day for two days.
Tissue processing
At indicated time points, mice were anaesthetized with a lethal dose of triple cocktail as described earlier. Animals were transcardially flushed with 10 ml of PBS, then perfused at a constant rate slowly with 40 ml of ice cold freshly prepared 1% PLP solution (1% PFA, 0.1M monobasic and dibasic phosphates, 90 mM lysine, 0.1M meta-sodium periodate). The tissue was dissected and then post-fixed in 10 ml of 1% PLP under vacuum for 1 hour and decalcified in saturated EDTA overnight at 4°C. They were then cryoprotected in 30% sucrose in PBS overnight at 4°C, embedded in OCT compound (Miles Inc., Elkhard, IN), and snap frozen in liquid nitrogen. Depending on the application, either 9 mm or 20 mm coronal sections (imaging or transplant screening, respectively) were cut on a Leica cryostat, mounted on Ultraclear Plus charged slides (Denville Scientific) and stored at -20°C until needed.
Immunohistochemistry
The primary antibody dilutions and details of their working conditions and detection methodologies are listed in Supplemental Table 1. Before immunostaining, tissue sections were first rinsed in PBS to remove OCT and then subjected to antibody-specific pretreatments which consist of all combinations of no pretreatment, 5-minute incubation in 3% H2O2 in MeOH, or heated for 10 minutes when overlaid with 0.01M pH 6.0 citrate buffer in a commercial food steamer. Sections were incubated with primary antibody diluted in 10% donkey serum, 5% nonfat dry milk, 4% BSA, 0.1% TritonX-100 in PBS for 1 hour at room temperature. Detection method of each antibody is specified in Supplemental Table 1 and described below. All washes are done 3×5 min at RT in PBS.
Secondary detection
A variety of fluorophores conjugated to secondary antibodies were obtained from Jackson Immunological and resuspended according to manufacturer's specifications. After primary antibodies were washed off, these secondary antibodies were then incubated for one hour at room temperature at 1:150.
Tertiary amplification
Biotinylated secondary antibodies were obtained from Jackson Immunological and resuspended according to manufacturer's specifications. After primary antibodies were washed off, biotinylated secondaries were then incubated for one hour at room temperature at 1:150, followed by washing. Finally, fluorescently- conjugated streptavidins were then used at 1:400 for one hour at room temperature.
Tyramide signal amplification
Biotinylated secondaries were used as described above, however after washing, horseradish peroxidase conjugated streptavidin (HRP-SA) diluted in 1% casein in Tris buffer was used at 1:400 for one hour at room temperature. After washing, FITC, Cy3, Cy5-tyramide diluted in 0.1M Borate, 0.003% H2O2, was applied for 15-30 minutes at room temperature.
For visualizing β-galactosidase expression, tissue sections were incubated overnight in 5-bromo-4-chloro-3-indolyl-d-D-galactopyranoside (X-gal, Sigma) as described previously (Huard et al., 1998) followed by pretreatments and fluorescent staining as described above.
After staining and nuclear counterstain, slides were coverslipped in 0.5% N-propyl gallate suspended in 90% glycerol, sealed with nail polish and stored at -20°C prior to imaging or counting.
Imaging
Stained sections were imaged on either a Zeiss LSM510 or LSM800 confocal microscope in multi-track mode, with the exception of the 6 channel image in Figure 1, which was imaged in spectral scanning Lambda Mode followed by linear unmixing. Image preparation was first processed using Fiji software only to adjust color palette, balance and contrast, which were applied to the entire image prior to figure assembly in Adobe Illustrator CS5.1 where images were cropped and set.
qRT-PCR
FACS purified cells are lysed and RNA isolated using a commercial DNA-free RNA purification kit (ZymoResearch). cDNA was generated using PrimeScript RT Master Mix (Takara, #RR036A) qRT-PCR was performed using a Biorad CFX96 using RT2 SYBR Green qPCR Mastermix (Qiagen #330523) and primers described in Supplemental Table 2, designed from PrimerBank.
Single Cell mRNAseq
Two fluorescent reporter mouse lines were used to isolate single cells of interest: Neurog1-eGFP, and Neurog1-eGFP;pan-TdTomato. The presence of TdTomato as a genetic marker allowed for the mixture of two conditions into a single Fluidigm C1 Autoprep IFC chip, and effective elimination of batch effects through two runs. Uninjured, 5 days post-OBX, and 14 days post-OBX animals were dissociated according to protocols described above, and FACS-isolated for either double or single positive cells. Isolated cells were then captured on a 5-10 um mRNA Seq IFC (Fluidigm, 100-5759) and visually inspected to positively identify single cell capture prior to lysis and cDNA synthesis according to manufacturer's instructions. Three RNA spike-ins were added to the lysis buffer for further batch effect correction. cDNA was quantified using Invitrogen's Quant-iT high sensitivity DNA Assay kit (Q33120) prior to library construction using Illumina Nextera XT Library Prep Kit (FC-131-1024), following manufacturer's instructions and Fluidigm modifications. Single cell libraries were tagged with unique barcodes using Nextera XT Index Kit V2 B,C,D (FC-131-2002, FC-131-2003, FC-131-2004), and pooled prior to Agencourt AMPure XP Bead cleanup. The library was quantified using Quant-iT and size distribution determined using a Fragment Analyzer (Advanced Analytical Technologies, Inc) prior to sequencing at ∼3M 100bp paired end reads per cell on an Illumina HiSeq2500 running in High Output v4 mode. Demultiplexed reads were FASTQC trimmed prior to mapping using STAR on the Tufts High Performance Computing Cluster. Single cell samples that expressed an outlying level of either beta-actin or GAPDH were excluded from further analysis. We processed the filtered single cell data using standard hierarchical clustering and visualization of clusters using t-SNE dimension reducing algorithms. FASTProject was loaded with MSigDB Hallmark gene sets generated enriched molecular signatures. WebGestalt was used using default settings to generate “Transcription Factor Target Analysis” and “Wikipathways Analysis.” To analyze additional data in Fletcher et al., 2017, we accessed data using SRA toolkit and esearch for PRJNA377622. This data was processed identically as data generated within this study, as described above. All visualizations were generated in R.
Quantification and Statistical Analysis
Cell counts were done on a Nikon 800E epifluorescent microscope with a dual red/green filter set to allow direct observation and simultaneous counting of Tdtomato (red) lineage-traced cells with markers of choice stained to green. For transplantation experiments, consecutive 20 μm sections of the olfactory epithelium through middle levels along the anteroposterior axis of the epithelium, were stained and completely counted systematically to identify all clones that engrafted. A clone was defined as being a tight group of cells that do not have other labeled cells within ten cell bodies in any of the x, y, or z (consecutive slides) directions. For lineage trace experiments and all other quantifications, all graft-derived cells were counted in a minimum of 9 evenly spaced 9 μm sections through middle levels along the anteroposterior axis of the epithelium. For all experiments at least three animals were counted per transplant or condition. Counts were performed blinded across conditions. Statistical analysis and graphs were done using either SigmaPlot or R software with a minimum cutoff of p<0.05 for statistical significance. All data first was tested for normality using the Shapiro-Wilk Normality test. Datasets passing normality were then tested using ANOVA and Holm-Sidak methods for testing significance, while datasets that failed were subjected to non-parametric Kruskal-Wallis ANOVA, using Dunn's method. Clonal size distributions were tested using the Mann-Whitney Rank Sum test. Tissue regeneration after Sox2 knockout passed normality and was tested using a two-tailed T-test. In all graphs, mean values and standard deviation are reported.
Data and Software Availability
Data Resources
Raw data files for single cell RNAseq analysis have been deposited in the NCBI Gene Expression Omnibus under accession numbers GEO: GSE92842
Supplementary Material
Highlights.
Injury unlocks multipotency of Ascl1+ and Neurog1+ neuronally specified progenitors
Induced multipotent progenitors transplant and maintain their multipotency
Single-cell RNA-seq reveals Ezh2 as an epigenetic regulator of multipotency
Sox2 is required for initiation of dedifferentiation but not maintenance
Acknowledgments
The authors thank Po Kwok-Tse for her excellent technical contributions. This work was supported by funding from the NIH NIDCD, R01 DC002167 (JES), F31 DC014637 (BL), F31 DC014398 (JHC), F30 DC013962 (DBH)
Footnotes
Author Contributions: Conceptualization, BL, JHC, JP, MZ, DBH, WJ, JES; Methodology, BL, JHC, JP, WJ; Software, BL, MZ; Formal Analysis BL, MZ; Investigation, BL, JHC, JP, MZ, DBH, WJ; Resources, BL, JHC, DBH, JES; Writing – Original Draft, BL; Writing – Review and Editing, BL, JES; Visualization, BL, MZ, JES; Supervision, JES; Funding Acquisition, BL, JHC, DBH, JES.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bergström U, Giovanetti A, Piras E, Brittebo EB. Methimazole-Induced Damage in the Olfactory Mucosa: Effects on Ultrastructure and Glutathione Levels. Toxicol Pathol. 2003;31:379–387. doi: 10.1080/01926230390201101. [DOI] [PubMed] [Google Scholar]
- Bermingham-McDonogh O, Reh TA. Regulated reprogramming in the regeneration of sensory receptor cells. Neuron. 2011;71:389–405. doi: 10.1016/j.neuron.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, et al. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust in Vivo Activity. ACS Med Chem Lett. 2015;6:491–495. doi: 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cau E, Gradwohl G, Fode C, Guillemot F. Mash1 activates a cascade of bHLH regulators in olfactory neuron progenitors. Development. 1997;124:1611–1621. doi: 10.1242/dev.124.8.1611. [DOI] [PubMed] [Google Scholar]
- Chen M, Tian S, Yang X, Lane AP, Reed RR, Liu H. Wnt-Responsive Lgr5+ Globose Basal Cells Function as Multipotent Olfactory Epithelium Progenitor Cells. J Neurosci. 2014;34:8268–8276. doi: 10.1523/JNEUROSCI.0240-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Fang H, Schwob JE. Multipotency of purified, transplanted globose basal cells in olfactory epithelium. J Comp Neurol. 2004;469:457–474. doi: 10.1002/cne.11031. [DOI] [PubMed] [Google Scholar]
- Eisthen HL. Evolution of vertebrate olfactory systems. Brain Behav Evol. 1997;50:222–233. doi: 10.1159/000113336. [DOI] [PubMed] [Google Scholar]
- Fletcher RB, Prasol MS, Estrada J, Baudhuin A, Vranizan K, Choi YG, Ngai J. P63 Regulates Olfactory Stem Cell Self-Renewal and Differentiation. Neuron. 2011;72:748–759. doi: 10.1016/j.neuron.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher RB, Das D, Gadye L, Street KN, Baudhuin A, Wagner A, Cole MB, Flores Q, Choi YG, Yosef N, et al. Deconstructing Olfactory Stem Cell Trajectories at Single-Cell Resolution. Cell Stem Cell. 2017;20:817–830.e8. doi: 10.1016/j.stem.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CDL, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascón S, Murenu E, Masserdotti G, Ortega F, Russo GL, Petrik D, Deshpande A, Heinrich C, Karow M, Robertson SP, et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell. 2015:396–409. doi: 10.1016/j.stem.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Gokoffski KK, Wu HH, Beites CL, Kim J, Kim EJ, Matzuk MM, Johnson JE, Lander AD, Calof AL. Activin and GDF11 collaborate in feedback control of neuroepithelial stem cell proliferation and fate. Development. 2011;138:4131–4142. doi: 10.1242/dev.065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BJ, Goss GM, Hatzistergos KE, Rangel EB, Seidler B, Saur D, Hare JM. Adult c-Kit(+) progenitor cells are necessary for maintenance and regeneration of olfactory neurons. J Comp Neurol. 2015;523:15–31. doi: 10.1002/cne.23653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F, Joyner aL. Dynamic expression of the murine Achaete-Scute homologue Mash-1 in the developing nervous system. Mech Dev. 1993;42:171–185. doi: 10.1016/0925-4773(93)90006-j. [DOI] [PubMed] [Google Scholar]
- Guo Z, Packard A, Krolewski RC, Harris MT, Manglapus GL, Schwob JE. Expression of pax6 and sox2 in adult olfactory epithelium. J Comp Neurol. 2010;518:4395–4418. doi: 10.1002/cne.22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreros-Villanueva M, Zhang JS, Koenig a, Abel EV, Smyrk TC, Bamlet WR, de Narvajas aaM, Gomez TS, Simeone DM, Bujanda L, et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis. 2013;2:e61. doi: 10.1038/oncsis.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71–84. doi: 10.1038/nrg3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard JMT, Schwob JE. Cell cycle of globose basal cells in rat olfactory epithelium. Dev Dyn. 1995;203:17–26. doi: 10.1002/aja.1002030103. [DOI] [PubMed] [Google Scholar]
- Huard JM, Youngentob SL, Goldstein BJ, Luskin MB, Schwob JE. Adult olfactory epithelium contains multipotent progenitors that give rise to neurons and non-neural cells. J Comp Neurol. 1998;400:469–486. [PubMed] [Google Scholar]
- Jang W, Youngentob SL, Schwob JE. Globose basal cells are required for reconstitution of olfactory epithelium after methyl bromide lesion. J Comp Neurol. 2003;460:123–140. doi: 10.1002/cne.10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner aM, Green WW, McIntyre JC, Allen BL, Schwob JE, Martens JR. Primary Cilia on Horizontal Basal Cells Regulate Regeneration of the Olfactory Epithelium. J Neurosci. 2015;35:13761–13772. doi: 10.1523/JNEUROSCI.1708-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling C, Boue S, Izpisua Belmonte JC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nat Rev Mol Cell Biol. 2011;12:79–89. doi: 10.1038/nrm3043. [DOI] [PubMed] [Google Scholar]
- Karl MO, Hayes S, Nelson BR, Tan K, Buckingham B, Reh Ta. Stimulation of neural regeneration in the mouse retina. Proc Natl Acad Sci. 2008;105:19508–19513. doi: 10.1073/pnas.0807453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Ables JL, Dickel LK, Eisch AJ, Johnson JE. Ascl1 (Mash1) defines cells with long-term neurogenic potential in subgranular and subventricular zones in adult mouse brain. PLoS One. 2011;6:e18472. doi: 10.1371/journal.pone.0018472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubu H, Ohtsuka T, Kageyama R. Mash1 is required for neuroendocrine cell development in the glandular stomach. Genes to Cells. 2008;13:41–51. doi: 10.1111/j.1365-2443.2007.01146.x. [DOI] [PubMed] [Google Scholar]
- Kopp JL, Grompe M, Sander M. Stem cells versus plasticity in liver and pancreas regeneration. Nat Cell Biol. 2016;18:238–245. doi: 10.1038/ncb3309. [DOI] [PubMed] [Google Scholar]
- Krolewski RC, Jang W, Schwob JE. The generation of olfactory epithelial neurospheres in vitro predicts engraftment capacity following transplantation in vivo. Exp Neurol. 2011;229:308–323. doi: 10.1016/j.expneurol.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krolewski RC, Packard A, Jang W, Wildner H, Schwob JE. Ascl1 (mash1) knockout perturbs differentiation of nonneuronal cells in olfactory epithelium. PLoS One. 2012;7:e51737. doi: 10.1371/journal.pone.0051737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krolewski RC, Packard A, Schwob JE. Global expression profiling of globose basal cells and neurogenic progression within the olfactory epithelium. J Comp Neurol. 2013;521:833–859. doi: 10.1002/cne.23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Pa, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011;147:525–538. doi: 10.1016/j.cell.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafkas D, Shelton A, Chiu C, de Leon Boenig G, Chen Y, Stawicki SS, Siltanen C, Reichelt M, Zhou M, Wu X, et al. Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature. 2015;528:127–131. doi: 10.1038/nature15715. [DOI] [PubMed] [Google Scholar]
- Leung CT, Coulombe Pa, Reed RR. Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat Neurosci. 2007;10:720–726. doi: 10.1038/nn1882. [DOI] [PubMed] [Google Scholar]
- Mackay-Sim A, Kittel P. Cell dynamics in the adult mouse olfactory epithelium: a quantitative autoradiographic study. J Neurosci. 1991;11:979–984. doi: 10.1523/JNEUROSCI.11-04-00979.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki N, Suetsugu-Maki R, Tarui H, Agata K, Del Rio-Tsonis K, Tsonis PA. Expression of stem cell pluripotency factors during regeneration in newts. Dev Dyn. 2009;238:1613–1616. doi: 10.1002/dvdy.21959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglapus GL, Youngentob SL, Schwob JE. Expression patterns of basic helix-loop-helix transcription factors define subsets of olfactory progenitor cells. J Comp Neurol. 2004;479:216–233. doi: 10.1002/cne.20316. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Masserdotti G, Gascón S, Götz M. Direct neuronal reprogramming: learning from and for development. Development. 2016;143:2494–2510. doi: 10.1242/dev.092163. [DOI] [PubMed] [Google Scholar]
- Packard A, Giel-Moloney M, Leiter A, Schwob JE. Progenitor cell capacity of NeuroD1- expressing globose basal cells in the mouse olfactory epithelium. J Comp Neurol. 2011a;519:3580–3596. doi: 10.1002/cne.22726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard A, Schnittke N, Romano RA, Sinha S, Schwob JE. DeltaNp63 regulates stem cell dynamics in the mammalian olfactory epithelium. J Neurosci. 2011b;31:8748–8759. doi: 10.1523/JNEUROSCI.0681-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard AI, Lin B, Schwob JE. Sox2 and Pax6 Play Counteracting Roles in Regulating Neurogenesis within the Murine Olfactory Epithelium. PLoS One. 2016;11:e0155167. doi: 10.1371/journal.pone.0155167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho-Santos M, Willenbring H. On the Origin of the Term “Stem Cell. Cell Stem Cell. 2007;1:35–38. doi: 10.1016/j.stem.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Rios AC, Fu NY, Lindeman GJ, Visvader JE. In situ identification of bipotent stem cells in the mammary gland. Nature. 2014;506:322–327. doi: 10.1038/nature12948. [DOI] [PubMed] [Google Scholar]
- Roach G, Wallace RH. Loss of ascl1a prevents secretory cell differentiation within the zebrafish intestinal epithelium resulting in a loss of distal intestinal motility. Dev Biol. 2013:1–16. doi: 10.1016/j.ydbio.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano Ra, Smalley K, Magraw C, Serna Va, Kurita T, Raghavan S, Sinha S. Np63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–782. doi: 10.1242/dev.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Greco V. Spatial organization within a niche as a determinant of stem-cell fate. Nature. 2013;502:513–518. doi: 10.1038/nature12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall JG, Cliff T, Dalton S, Sartorelli V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell. 2015;17:651–662. doi: 10.1016/j.stem.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, Hochedlinger K. The Sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15–30. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittke N, Herrick DB, Lin B, Peterson J, Coleman JH, Packard AI, Jang W, Schwob JE. Transcription factor p63 controls the reserve status but not the stemness of horizontal basal cells in the olfactory epithelium. Proc Natl Acad Sci. 2015;201512272 doi: 10.1073/pnas.1512272112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz Levey M, Chikaraishi DM, Kauer JS. Characterization of potential precursor populations in the mouse olfactory epithelium using immunocytochemistry and autoradiography. J Neurosci. 1991;11:3556–3564. doi: 10.1523/JNEUROSCI.11-11-03556.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob JE, Szumowski KE, Stasky Aa, Mieleszko Szumowski KE, Stasky Aa. Olfactory sensory neurons are trophically dependent on the olfactory bulb for their prolonged survival. J Neurosci. 1992;12:3896–3919. doi: 10.1523/JNEUROSCI.12-10-03896.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob JE, Youngentob SL, Mezza RC. Reconstitution of the rat olfactory epithelium after methyl bromide-induced lesion. J Comp Neurol. 1995;359:15–37. doi: 10.1002/cne.903590103. [DOI] [PubMed] [Google Scholar]
- Schwob JE, Jang W, Holbrook EH, Lin B, Herrick DB, Peterson JN, Hewitt Coleman J. Stem and progenitor cells of the mammalian olfactory epithelium: Taking poietic license. J Comp Neurol. 2017;525:1034–1054. doi: 10.1002/cne.24105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaker T, Dennis D, Kurrasch DM, Schuurmans C. Neurog1 and Neurog2 coordinately regulate development of the olfactory system. Neural Dev. 2012;7:28. doi: 10.1186/1749-8104-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Sindhu C, Meissner A. Molecular features of cellular reprogramming and development. Nat Rev Mol Cell Biol. 2016;17:139–154. doi: 10.1038/nrm.2016.6. [DOI] [PubMed] [Google Scholar]
- Snitow M, Lu M, Cheng L, Zhou S, Morrisey EE. Ezh2 restricts the smooth muscle lineage during mouse lung mesothelial development. Development dev. 2016 doi: 10.1242/dev.134932. 134932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitow ME, Li S, Morley MP, Rathi K, Lu MM, Kadzik RS, Stewart KM, Morrisey EE. Ezh2 represses the basal cell lineage during lung endoderm development. Development. 2015;142:108–117. doi: 10.1242/dev.116947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A. Mechanisms for enhancing cellular reprogramming. Curr Opin Genet Dev. 2014;25:101–109. doi: 10.1016/j.gde.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stange DE, Koo BK, Huch M, Sibbel G, Basak O, Lyubimova A, Kujala P, Bartfeld S, Koster J, Geahlen JH, et al. Differentiated troy(+) chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell. 2013;155:357–368. doi: 10.1016/j.cell.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, Law BM, Vinarsky V, Cho JL, Breton S, Sahay A, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–223. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetteh PW, Basak O, Farin HF, Wiebrands K, Kretzschmar K, Begthel H, van den Born M, Korving J, de Sauvage F, van Es JH, et al. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell. 2016;18:203–213. doi: 10.1016/j.stem.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Wildner H, Müller T, Cho SH, Bröhl D, Cepko CL, Guillemot F, Birchmeier C. dILA neurons in the dorsal spinal cord are the product of terminal and non-terminal asymmetric progenitor cell divisions, and require Mash1 for their development. Development. 2006;133:2105–2113. doi: 10.1242/dev.02345. [DOI] [PubMed] [Google Scholar]
- Xie B, Zhang H, Wei R, Li Q, Weng X, Kong Q, Liu Z. Histone H3 lysine 27 trimethylation acts as an epigenetic barrier in porcine nuclear reprogramming. Reproduction. 2016;151:9–16. doi: 10.1530/REP-15-0338. [DOI] [PubMed] [Google Scholar]
- Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, Allison DF, Cai L, Rockowitz S, Liu S, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125:346–357. doi: 10.1182/blood-2014-06-581082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–1338. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Yang X, Ma X, Ingram DR, Lazar AJ, Torres KE, Pollock RE. Antitumor effects of pharmacological EZH2 inhibition on malignant peripheral nerve sheath tumor through the miR-30a and KPNB1 pathway. Mol Cancer. 2015;14:1–12. doi: 10.1186/s12943-015-0325-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.