

Abstract
For polymeric nanoparticles (NPs) to deliver more drugs to tumors than free drug solution, it is critical that the NPs establish interactions with tumor cells and avoid removal from the tumors. Since traditional polyethylene glycol (PEG) surface layer interferes with the cell-NP interaction in tumors, we used a water-soluble and blood-compatible chitosan derivative called zwitterionic chitosan (ZWC) as an alternative surface coating for poly(lactic-co-glycolic acid) (PLGA) NPs. The ZWC-coated PLGA NPs showed pH-dependent surface charge profiles and differential cellular interactions according to the pH of the medium. The in vivo delivery of ZWC-coated NPs were evaluated in mice bearing LS174T-xenografts using magnetic resonance (MR) imaging and fluorescence whole body imaging, which respectively tracked iron oxide particles and indocyanine green (ICG) encapsulated in the NPs as tracers. MR imaging showed that ZWC-coated NPs were more persistent in tumors than PEG-coated NPs, in agreement with the in vitro results. However, the fluorescence imaging indicated that the increased NP retention in tumors by the ZWC coating did not significantly affect the ICG distribution in tumors due to the rapid release of the dye. This study shows that stable drug retention in NPs during circulation is a critical prerequisite to successful translation of the potential benefits of surface-engineered NPs.
Keywords: pH-responsive, drug delivery, PLGA nanoparticles, small molecules, in vivo imaging, encapsulation stability
Graphical abstract
1. Introduction
Polymeric nanoparticles (NPs) have widely been explored as a carrier of anti-cancer drugs in systemic chemotherapy. The premise of the NP-based chemotherapy is that NPs remain in the blood vessels of normal tissues but extravasate near the tumors due to the defects in peritumoral vasculature [1]. This allows NPs to gain preferential access to tumors compared to free drugs, potentially reducing side effects and increasing therapeutic efficacy. To realize this potential, it is necessary that NPs avoid non-specific interactions with the monocyte phagocyte system (MPS) and survive in circulation until they reach tumors. Moreover, NPs need to establish interactions with tumor cells once they reach the tumors so that the NPs avoid washout from the tissues. The polymeric layer traditionally used for protecting NPs in circulation, such as polyethylene glycol (PEG), does not meet these requirements because it generally interferes with the NP-cell interactions, irrespective of the circumstances, creating a ‘PEG dilemma’ [2]. These challenges may have collectively contributed to dismal efficiency of NP delivery to tumors, which is measured to be no higher than 1% of the total injected dose [3].
In an effort to improve NP retention in tumors, early studies have proposed the use of pH-sensitive surface modification [4–6], which enhances cell-NP interactions in the acidity of tumor microenvironment [7, 8]. For example, a recent article describes mesoporous silica NPs modified with a pH-sensitive peptide called pH (low) insertion peptide (pHLIP), which forms a hydrophobic transmembrane helix at acidic pH to efficiently enter cells [6]. The pHLIP-modified NPs showed greater anticancer effect than PEGylated counterpart with doxorubicin as the payload [6]. In the same vein, we previously proposed that a low molecular weight chitosan (LMWC) can serve as an alternative polymer for the protection of NP surface [9, 10]. Chitosan is a linear polyaminosaccharide with a pKa value close to 6.5, assumes positive charges in the acidic tumor microenvironment, and helps establish electrostatic interactions with the negatively charged cell membrane. Due to the low molecular weight (<6.5 kDa), LMWC remains hydrophilic in neutral pH and fulfills its role as a protective surface layer. LMWC-coated NPs show good cell interactions selectively at acidic pH sparing the cells in neutral pH. However, LMWC is not completely water-soluble in neutral pH and shows limited compatibility with blood components [11].
To address these limitations, we synthesized water-soluble derivatives of LMWC, called zwitterionic chitosans (ZWCs), by partial succinylation of the amine groups [11]. ZWC show unique pH-dependent charge profiles, where they are positively charged at acidic pH due to the inherent amine groups and negatively charged in relatively basic pH due to the newly introduced carboxyl groups. The pH at which the charge changes depends on the degree of succinylation, which can be easily controlled by the feed ratio of succinyl anhydride to chitosan (An/Am ratio). We reported that ZWC had excellent blood compatibility and anti-inflammatory activities [12] and formed an electrostatic complex with cationic polyamidoamine (PAMAM) dendrimers to reduce the cytotoxicity [13]. Owing to the ability to change charges according to the pH, ZWC prevented non-specific cellular interaction of PAMAM dendrimers at neutral pH but allowed them to interact with cells in acidic milieu [13].
On the basis of water solubility, cytocompatiblity, and pH-sensitive charge profile, we choose to use ZWC instead of LMWC for the modification of polymeric NP surface in this study. Here, the surface of poly(lactic-co-glycolic acid) (PLGA) NPs is coated with ZWC using dopamine as a mediator, which polymerizes in a weak alkaline solution to form a thin layer that accommodates ligands with nucleophilic groups [14]. With this method, various functional ligands can be conjugated onto NPs irrespective of the surface reactivity [14–16]. We test the pH-responsive cellular interaction of ZWC-coated NPs and compare with pH-independent PEG-coated NPs using different cell models. To evaluate the in vivo performance of the NPs, we encapsulate two distinct tracers – hydrophobic iron oxide (IO) particles and indocyanine green (ICG) - in the core NPs, where IO represents NPs and ICG does a small molecule drug (<1000 Da [17]) delivered by the NPs. We compare the distribution profiles of the two tracers using magnetic resonance (MR) imaging and near-infrared (NIR) fluorescence imaging, respectively, and critically analyze their difference. The results indicate that ZWC-coated NPs show the pH-dependent cell interactions and greater retention in tumors; however, the benefit may not translate to in vivo efficacy without stable encapsulation of the payload.
2. Materials and methods
2.1. Materials
All biochemical reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. Poly(lactic-co-glycolic acid (PLGA. LA:GA = 85:15, 150 kDa) and rhodamine-conjugated PLGA (Rho-PLGA, LA:GA = 50:50, 30 kDa) were purchased from Akina, Inc. (West Lafayette, IN). Methoxyl polyethylene glycol-NH2 (PEG-NH2, 2000 Da) was purchased from Nanocs (New York, NY). Iron oxide (IO) particles (5 nm) and gold (Au) particles (5 nm) were purchased from Ocean NanoTech (San Diego, CA). Indocyanine green (ICG) was purchased from MP biomedicals (Santa Ana, CA), dopamine HCl from Alfa Aesar (Ward Hill, MA), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) from Invitrogen (Eugene, OR). Deionized (DI) water was obtained from a Milli-Q ultrafiltration system (Millipore, MA). ZWC was synthesized by partial succinylation of chitosan with an anhydride to amine (An/Am) feed ratio of 0.1 and 0.3 and characterized as described previously [11].
2.2. Preparation of core PLGA NPs
PLGA NPs were prepared by the single emulsion method. Typically, 50 mg of PLGA was dissolved in 4 mL of dichloromethane (DCM). The polymer solution was added to 10 mL of 4% polyvinyl alcohol (PVA) solution and emulsified with a probe sonicator (Sonics Vibracell, Newtown, CT) for 2 min in ice, pulsing at a power level of 7 W and a 2:1 duty cycle every 6 s. The emulsion was transferred to 20 mL of DI water, stirred for 1 h, and evaporated with a rotary evaporator for another 1 h to remove DCM. A modeling study predicts that 99.9% DCM is extracted from the particle core in 0.01 s and >98% of the extracted DCM is evaporated in air in <40 min [18]. Therefore, the majority of DCM is likely to have been removed by the above procedure. The formed NPs were collected by centrifugation at 16,000 rcf for 20 min at 4 °C and washed twice with DI water.
PLGA NPs were labeled in four different ways according to the purpose of the experiment. For flow cytometry analysis of cell-NP interactions, NPs were fluorescently labeled by replacing10 mg of PLGA with Rho-PLGA. For magnetic resonance imaging of NP distribution in live animals, IO particles were added to the PLGA solution at the IO/PLGA weight ratio of 5% to produce IO-embedded NPs. For imaging the biodistribution of a small molecule payload, ICG was encapsulated as a model drug in PLGA NPs by the double emulsion method. First, 625 μL of an aqueous phase containing 19 mg of human serum albumin (HSA) and 5 mg of ICG was emulsified in 6.3 mL of PLGA solution (20 mg/mL) via probe sonication at a power level of 7 W and a 1:1 duty cycle every 2 s for 1 min, creating a primary (w1/o) emulsion. The emulsion was further emulsified in 24 mL of 2.5% PVA solution via probe sonication (2 min, 4s on/2s off) to form a secondary emulsion (w1/o/w2), which was dispersed in 42 mL of DI water and stirred for 1 h, followed by rotary evaporation for 2 h. For pharmacokinetics study, Au particles were encapsulated in the NPs by the double emulsion method. One mL of an aqueous phase containing 1 mg of Au particles was emulsified in 5 mL of PLGA solution (20 mg/mL) via probe sonication and further emulsified in PVA solution in the same way as ICG-containing NPs. The NPs were collected and washed in the same way as above. These NPs were called Rho-NP (PLGA NPs labeled with covalently conjugated rhodamine), IO@NP or Au@NP (PLGA NPs labeled with IO or Au particles physically embedded in the NP matrix), and ICG@NP (PLGA NPs labeled with ICG dye physically encapsulated in the NP matrix), according to the labeling method.
2.3. Surface modification of PLGA NPs with ZWC and PEG (Fig. 1)
Fig. 1.

(a) Schematic diagram of surface-modified NPs. (b) TEM images of surface-modified IO@NPs and ICG@NPs. Scale bars: 50 nm (top), 100 nm (bottom).
The core NPs were prime-coated with polydopamine (pD) as previously reported [14]. Briefly, the core NPs were incubated with dopamine in Tris buffer (10 mM, pH 8.5) for 3 h. The pD-coated NPs (NP-pD) were collected by centrifugation at 16,000 rcf for 20 min at 4 °C. Separately, ZWC0.1 (ZWC produced with an An/Am ratio of 0.1) was prepared as 0.5 mg/mL solution in DI water, which was acidified to pH 6.3. ZWC0.3 (ZWC produced with an An/Am ratio of 0.3) and PEG-NH2 were prepared as 2 mg/mL solution in Tris buffer (10 mM, pH 8.5). Twenty milligrams of NP-pD was dispersed in 20 mL of ZWC0.1 solution or 5 mL of ZWC0.3 or PEG-NH2 solution and vortex-mixed for 9 h. The NPs were then collected by centrifugation and freeze-dried with trehalose for storage. The surface-modified NPs were named NP-pD-ZWC0.1, NP-pD-ZWC0.3, and NP-pD-PEG, according to the surface material.
2.4. Characterization of surface-modified NPs
The hydrodynamic diameter and zeta potential of surface-modified NPs were measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS90 (Malvern, Worcestershire, UK). The particle size was measured with NPs dispersed in distilled water. The zeta potential was measured using NPs dispersed in buffers (10 mM) with different pHs. The NPs were imaged with a Tecnai F20 transmission electron microscope (FEI, Hillsboro, OR) after negative staining with 2% phosphotungstic acid (PTA). Fe and Au contents in NPs were determined by atomic absorption spectroscopy (AAS) using a Perkin-Elmer 3110 Spectrometer (Waltham, MA).
2.5. Cell culture
4T1 mouse breast cancer cells (ATCC, Manassas, VA) were grown in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), and LS174T human colon cancer cells (ATCC, Manassas, VA) in Eagle’s Minimal Essential Medium (EMEM) containing 10% FBS. NIH/3T3 mouse fibroblasts (ATCC, Manassas, VA) and J774A.1 macrophages (ATCC, Manassas, VA) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% calf bovine serum. All the media contained 100 units/mL of penicillin and 100 μg/mL of streptomycin. Cells were incubated at 37 °C in a humidified 5% CO2 atmosphere.
2.6. Cytocompatibilty of surface-modified NPs
Cytocompatibility of IO@NPs and ICG@NPs was tested in NIH/3T3 fibroblasts and J774A.1 macrophage using the MTT assay. Cells were plated in a 96-well plate at a density of 104 cells per well in 200 μL of complete medium, grown overnight, and treated with the NPs in a final concentration ranging from 0.05 to 0.5 mg/mL. After 24 h incubation, the medium was replaced with 100 μL of fresh medium containing 37.5 μg of MTT reagent and incubated for 3.5 h, followed by the addition of stop/solubilization solution. The absorbance of solubilized formazan was read with a SpectraMax M3 microplate reader (Molecular Devices, Sunnyvale, CA) at a wavelength of 562 nm. The measured sample absorbance was normalized to the absorbance of control cells without NP treatment.
2.7. NP-cell interactions
2.7.1. Confocal microscopy of LS174T cells incubated with surface-modified NPs
LS174T cells were seeded in a 35 mm glass bottomed Petri dish (MatTek). After 3 days, the cells were incubated with Rho-NPs (Rho-NP-pD, Rho-NP-pD-ZWC0.1, Rho-NP-pD-ZWC0.3, and Rho-NP-PEG) at pH 6.7 or 7.6 for 4 h and washed twice with fresh complete medium to remove free or loosely bound NPs. The cells were incubated in 2 μg/mL Hoechst for 5 min for nuclei staining and washed twice with medium. Live cells were imaged with a Nikon A1R confocal microscope (Nikon America Inc., Melville, NY).
2.7.2. Flow cytometry of 4T1 cells and J774A.1 macrophages incubated with surface-modified NPs
4T1 cells were seeded in a 12-well plate at a density of 104 cells per well in 1 mL of complete medium and grown to 90% confluence. The cells were incubated with Rho-NPs at pH 6.7, 7.0 or 7.6 for 4 h and washed twice with fresh medium to remove free or loosely bound NPs. J774A.1 macrophages were seeded in a 24-well plate at a density of 104 cells per well in 1 mL of complete medium and grown to 90% confluence. The cells were incubated with Rho-NPs at pH 7.4 for 4 h and washed twice with fresh medium. The cells were trypsinized and collected by 5 min centrifugation at 930 rcf, redispersed in 0.3 mL of medium, and analyzed with an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) equipped with an FL-1 detector (λex/λem = 488/525 nm).
2.8. Stability of NPs in 50% serum
To estimate the stability of ICG@NPs in blood, ICG@NPs were incubated in 50% FBS diluted with PBS at a concentration of 0.2 mg/mL. At predetermined time points, NPs were centrifuged at 9200 rcf for 15 min to separate a supernatant and a pellet. The supernatant and the pellet were resuspended in 50% FBS, placed in a black 96-well plate and imaged with an IVIS Lumina II system (PerkinElmer, MA). To evaluate the stability of Au or IO encapsulation, Au@NPs and IO@NPs were suspended in 50% FBS at a concentration of 1 mg/mL and incubated for 2–4 days at 37 °C. The NPs were imaged with TEM after 2% PTA negative staining.
2.9. Pharmacokinetics
All animal procedures were approved by Purdue Animal Care and Use Committee, in conformity with the NIH guidelines for the care and use of laboratory animals. Female Balb/c mice (6–7 weeks) were purchased from Envigo (Indianapolis, IN) and acclimatized for 1 week prior to the procedure. The mice were administered with 1 mg of Au@NPs dispersed in 5% dextrose injection solution via tail vein. At 1, 3, 6, 12, and 24 h post-injection, 3 mice were randomly selected from each group and sacrificed for blood sampling. Blood was drawn from the orbital sinus and collected in BD Vacutainer with EDTA as an anticoagulant. NP concentration in whole blood was determined by detecting Au by AAS.
2.10. NP administration in tumor-bearing animals
6–7 week old female athymic nude mice (Foxn1nu) were purchased from Envigo (Indianapolis, IN) and acclimatized for 1 week prior to the procedure. Each mouse received a subcutaneous injection of 3 × 106 LS174T cells in the upper flank of the right hind leg. When the average tumor volume reached 100–200 mm3, animals received a tail-vein injection of IO@NPs (n=5 per group) or ICG@NPs (n=3 per group, repeated twice) dispersed in phosphate buffer (10 mM, pH 7.4). Each animal received 2.5 mg of IO@NPs (equivalent to 5 mg Fe/kg) or 2 mg of ICG@NPs that showed comparable fluorescence intensity when suspended in 50% FBS. With another cohort of animals, tumors were grown to 150–1000 mm3 to measure the internal pH of tumor mass using an InLab solids electrode (Mettler Toledo, Columbus, OH).
2.11. Magnetic resonance imaging of NP-treated animals
MR images of IO@NP-treated animals were acquired under 3% isoflurane anesthesia, before and 2, 13, 36 and 60 h after the NP administration. Axial views of whole-body MR images were obtained using a BioSpec 7T small-animal MRI scanner (Bruker, Billerica, MA). T2*-weighted MR images were obtained using a gradient echo sequence (TR/TE = 1781/15 msec, FOV = 32 mm2, flip angle = 90°/180°, matrix size = 256 × 256, NEX = 4, number of slices = 20, and section thickness = 1 mm). T2*-weighted datasets were processed with ImageJ 1.47v (Bethesda, MD). The signal intensity (SI) of tumor was normalized to SI of muscle at each time point. Signal enhancement was defined as [Normalized SI of tumor (pre-injection) - Normalized SI of tumor (post-injection)]/Normalized SI of tumor (pre-injection) × 100%.
2.12. Near infrared fluorescence imaging of NP-treated animals
Animals receiving ICG@NPs were imaged under isoflurane anesthesia. The whole-body fluorescence was measured before and 2, 6, 10, 22, 32, 56 and 80 h after injection using the IVIS Lumina II system with 745/ICG filters, 3s exposure time and medium binning. The average radiant efficiency of tumor and shoulder skin was measured at each time point. The fluorescence intensity of tumor region of interest was expressed as radiant efficiency of tumor relative to radiant efficiency of skin.
2.13. Statistical Analysis
All data were analyzed using GraphPad Prism 7 (La Jolla, CA) with ANOVA to determine difference among the groups followed by the recommended multiple comparisons test. A value of p < 0.05 was considered statistically significant.
3. Result and Discussion
3.1. Synthesis and characterization of surface-modified NPs
Surface-modified NPs were created by coating the surface of PLGA NPs with ZWCs or PEG-NH2. Two types of ZWCs (ZWC0.1 and ZWC0.3) were produced with the An/Am ratio of 0.1 and 0.3 (Supporting Fig. 1). ZWC shows negative charges at relatively basic pH and positive charges at acidic pH, with the transition pH inversely proportional to the An/Am ratio [11]. Typically, ZWC0.3 shows a transition pH at 6.6–7.0 [11, 13], and ZWC0.1 at 6.9–7.2 (Supporting Fig. 1). The NPs coated with ZWC0.1 and ZWC0.3 (NP-pD-ZWC0.1 and NP-pD-ZWC0.3) showed pH-dependent surface charges similar to those of respective ZWC (Fig. 2d), which confirms successful modification of the NP surface. Unlike NP-pD-ZWCs, NPs modified with PEG-NH2 (NP-pD-PEG) were slightly negatively charged irrespective of the pH.
Fig. 2.

(a) Confocal microscopic images of LS174T cells incubated with surface-modified Rho-NPs at different pHs for 4 h. (b) Representative flow cytometry histograms of 4T1 cells incubated with surface-modified Rho-NPs at different pHs for 4 h. (c) Fluorescence intensity of Rho-NP-treated cells relative to untreated control cells (n=2–3 independently prepared samples, mean ± standard deviation). (d) Zeta potential of surface-modified NPs at different pH (n=3 measurements of a representative set of samples, average ± standard deviation). (e) Relationship between the fluorescence intensity of NP-incubated cells (indicating the extent of NP-cell interactions) and the zeta potential of NPs measured at different pHs (Pearson correlation coefficient: 0.9507).
The hydrodynamic diameter of core NPs measured by DLS was 220 nm on average with relatively monodisperse distribution (polydispersity index ~0.1). NP-pD-PEG maintained a similar size distribution as core NPs. NP-pD-ZWC0.1 and NP-pD-ZWC0.3 were slightly larger than the core NPs, although both remained below 300 nm (Table 1). On the other hand, TEM images show that all NPs were ~100 nm or smaller in diameter, indicating that the size measured by DLS reflect varying degrees of aggregation following the purification steps. It is uncertain whether the NPs will be resolved to the original size during circulation. Although the unresolved NPs are larger than typical NPs used in systemic applications (<100 nm) [19], their size is still smaller than the cutoff size of hyperpermeable tumor microvessels (1.2 μm) [20]; thus, we expect that NPs will be able to reach tumors via the leaky vasculature despite the increased chance of RES accumulation. All the surface-modified NPs showed wrinkled layers indicating pD coating [10], which however did not amount to a measurable thickness (Fig. 1b). Therefore, the relatively large size of NP-pD-ZWC is interpreted as slightly increased NP aggregation rather than the thickness of the surface layer.
Table 1.
Particle size of the surface-modified NPs
| Sample Name | Z-Average (d, nm) | Polydispersity index (PI)a | |
|---|---|---|---|
| Au@NPs | Au@NP-pD-ZWC0.3 | 260.1 ± 5.0b | 0.067 ± 0.027 |
| Au@NP-pD-PEG | 228.4 ± 3.5b | 0.079 ± 0.045 | |
| IO@NPs | IO@NP | 223.2 | 0.081 |
| IO@NP-pD-ZWC0.1 | 286.4 ± 16.7 (n=3)c | 0.127 ± 0.035 | |
| IO@NP-pD-ZWC0.3 | 244.6 ± 8.0 (n=3)c | 0.086 ± 0.006 | |
| IO@NP-pD-PEG | 218.7 ± 4.1 (n=3)c | 0.127 ± 0.067 | |
| ICG@NPs | ICG@NP | 225.0 ± 6.3 (n=2)c | 0.082 ± 0.033 |
| ICG@NP-pD-ZWC0.1 | 287.9 ± 14.4 (n=4)c | 0.145 ± 0.033 | |
| ICG@NP-pD-ZWC0.3 | 271.4 ± 12.7 (n=4)c | 0.103 ± 0.025 | |
| ICG@NP-pD-PEG | 242.7 ± 7.3 (n=4)c | 0.077 ± 0.039 |
PI, an estimate of the width of the particle size distribution, obtained from the cumulant analysis as described in the International Standard on DLS ISO 13321:1996 and ISO 22412:2008 (Malvern DLS technical note MRK1764-01). PI < 0.1 is considered monodisperse, and >0.7 very broad [22, 23].
Mean ± standard deviation of 3 measurements of a representative batch.
Mean ± standard deviation of identically and independently prepared batches.
Cytocompatibility of surface-modified NPs was tested with NIH3T3 and J774A.1 cell lines (Supporting Fig. 2). All three NPs showed comparable effects on these cells, causing no more than 20%–40% decrease in mitochondrial metabolic activity at concentrations as high as 0.5 mg/mL, which is comparable to core PLGA NPs [21]. This indicates that neither pD nor surface modifiers (ZWC0.1, ZWC0.3 and PEG-NH2) had additional toxicity to the cells. Based on this result, the following cell experiments were performed at 0.2 mg/mL.
3.2. pH-dependence of NP-cell interactions
To evaluate whether the pH-dependent charge variability translates to pH-dependent cell-NP interactions, the surface-modified Rho-NPs were first incubated with LS174T human colon cancer cells at pH 6.7 and 7.6, representing the pHs of hypoxic tumors and normal tissues, respectively. Cells treated with NP-pD-ZWC0.1 and NP-pD-ZWC0.3 at pH 6.7 showed higher NP signals than those treated at pH 7.6 (Fig. 2a). The NP signals were observed from the interior of the cell clusters. Cells treated with NP-pD-PEG showed weak NP signals at both pHs, comparable to those treated with NP-pD-ZWC at pH 7.6. This result demonstrates that NP-pD-ZWC interact with LS174T cells in a pH-dependent manner due to the pH-sensitive surface layer.
The extent of pH-sensitive NP-cell interactions was also evaluated by flow cytometry. Since LS174T cells grew in clusters, which were not readily dispersed into single cell suspension with mild treatment, 4T1 mouse breast cancer cell line was used for flow cytometry. 4T1 cells were treated with the Rho-NPs for 4 h, washed with fresh medium, and trypsinized for flow cytometry analysis. We noticed that NP-pD-ZWC0.1 resisted washing and remained with cells at pH 6.7 and pH 7.0 and NP-pD-ZWC0.3 persisted likewise at pH 6.7 (Supporting Fig. 3). In both cases, some of the cell-surface bound NPs appeared to be aggregated. In contrast, the cells treated with NP-pD-ZWC at pH 7.6 and those with NP-pD-PEG at all pHs had no apparent aggregation of NPs adherent to the cell surface. Consistent with the visual observation, 4T1 cells treated with NP-pD-ZWC0.1 showed a significant difference in fluorescence intensity according to the pH of the medium: the fluorescence intensity was highest for cells treated at pH 6.7, followed by those at pH 7 and 7.6 (Fig. 2b, c). The cells treated with NP-pD-ZWC0.3 showed little difference between pH 7.0 and 7.6 but showed significantly higher fluorescence intensity at pH 6.7. On the other hand, the cells treated with NP-pD-PEG did not show any difference in fluorescence intensity at all pHs. This result confirms that NP-pD-ZWCs interact with cells in a pH-dependent manner. Moreover, the difference between NP-pD-ZWC0.1 and NP-pD-ZWC0.3 demonstrates that it is possible to fine-tune the pH at which the NPs establish the interactions with cells. There was a positive correlation between the NP surface charge and fluorescence intensity of 4T1 cells (representing NPs associated with the cells) with a Pearson’s correlation coefficient of 0.9507, indicating that the cell-NP interaction is mediated by an electrostatic interaction (Fig. 2e).
To test if ZWC protects NPs from phagocytic uptake in neutral pH, Rho-NPs were incubated with J774A.1 macrophages, and their interactions were analyzed with flow cytometry. The fluorescence intensity of cells slightly increased upon the incubation with NPs, due to the basal level of non-specific interaction with NPs, common to all cell-NP combinations. Nevertheless, there was no significant difference between NP-pD-PEG and NP-pD-ZWCs (Supporting Fig. 4), suggesting that ZWC would provide similar protection as PEG [10, 24].
3.3. Pharmacokinetics of NP-pD-ZWC and NP-pD-PEG
Prior to imaging the distribution of NPs and the payload, we evaluated pharmacokinetics of NP-pD-ZWC and NP-pD-PEG in healthy female Balb/c mice. Au particles (5 nm) were used to label NPs due to the lack of interference in detection and stable encapsulation in NPs. Au@NPs were comparable to unlabeled NPs in the size (Table 1) and surface charge (Supporting Fig. 5a). As shown in TEM images, Au particles remained in the NPs after 48 h incubation in FBS (Supporting Fig. 5b). NP-pD-ZWC and NP-pD-PEG showed similar blood concentrations at all time points except for the first point (0.5 h), indicating that their pharmacokinetic behaviors were comparable (Fig. 3), consistent with the similar stealth effect observed in vitro (Supporting Fig. 4).
Fig. 3.
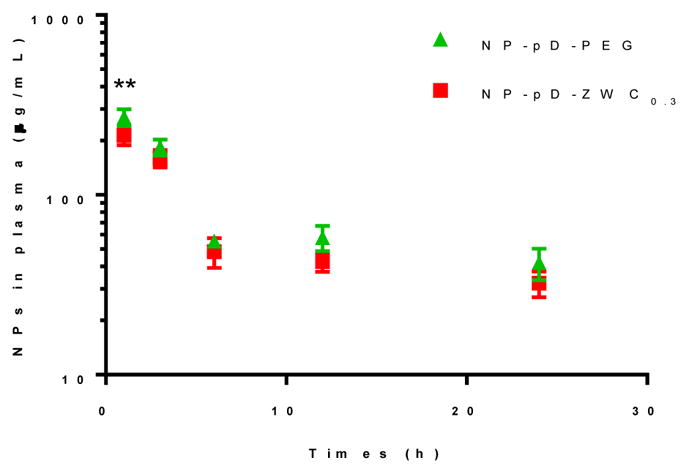
Pharmacokinetics of NP-pD-PEG and NP-pD-ZWC0.3 in health female Balb/c mice.
3.4. NP and dye distribution in tumor-bearing mice
We expect that NP-pD-ZWCs will show greater tumor distribution than NP-pD-PEG. Both NPs will depend on passive extravasation for initial accumulation. Given that both NP-pD-PEG and NP-pD-ZWCs show similar pharmacokinetic profiles (Fig. 3), the extent of initial accumulation may not differ much from each other. However, due to the ability to establish interactions with cells in acidic microenvironment, NP-pD-ZWCs will be better retained in tumors than NP-pD-PEG. Therefore, NP-pD-ZWCs are expected to show greater net tumor accumulation than NP-pD-PEG. To test this hypothesis, we compared the distribution of different NPs in tumor-bearing mice via non-invasive imaging techniques.
We chose the LS174T model based on the literature indicating acidic tumor microenvironment [25] and confirmed that LS174T xenografts in nude mice developed weakly acidic pH ranging from 6.7 to 7.2 (Supporting Table). NP distribution was observed using two imaging modalities: MR and NIR fluorescence imaging. For the MR imaging, NPs were loaded with 4 wt% IO particles (IO@NPs), which enables T2 weighted MR imaging. Due to the large size (5 nm), IO particles were expected to stay in NPs until significant degradation of NP matrix. For the NIR fluorescence imaging, NPs were loaded with 0.1 wt% ICG (ICG@NPs), which has fluorescence spectrum in the NIR range (Ex/Em =745/790 nm). ICG itself is an FDA-approved imaging agent [26, 27] and explored as a photothermal agent [28, 29], but as a compound with a molecular weight of 775 Da and high affinity for serum proteins [30] it also represents small molecule drugs (as opposed to biologics) with high protein binding. To predict the labeling stability of NPs in blood circulation, we incubated IO@NPs and ICG@NPs in 50% FBS, which simulates the protein content in blood [31], and examined IO retention in NPs and dye release kinetics at different time points. Similar to Au, most IO remained in the NPs with no stray IO after 4 day incubation (Fig. 4a). This indicates that MRI signals mainly come from IO@NPs and hence represent the NP distribution. On the other hand, the majority of ICG was detected in the supernatant of incubation medium after 2 h incubation, irrespective of the NP type, indicating that ICG was released from ICG@NPs in 50% FBS and the surface coating did not affect the dye release profile (Fig. 4b). This is a typical release behavior of small molecule drugs loaded in polymeric NPs in blood-like media [32]. Therefore, the fluorescence intensity of ICG in whole body imaging may well represent the distribution of payloads physically encapsulated in NPs.
Fig. 4.

Stability of IO@NPs and ICG@NPs in 50% FBS: (a) Representative TEM images of IO@NPs after 4 day-incubation in 50% FBS. (b) ICG release from ICG@NPs in 50% FBS over 24 h. n=3 identically prepared batches; average ± standard deviation.
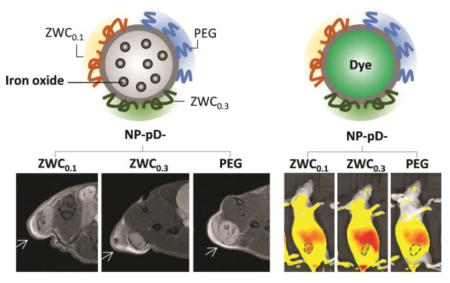
First, we observed animals injected with IO@NPs coated with ZWCs or PEG using MR imaging. All animals showed darkening of areas within the tumor relative to the pre-injection images from the first time point (2 h post-injection) and maintained the contrast over 60 h, indicating the NP accumulation in tumors (Fig. 5, Supporting Fig. 6). Animals treated with IO@NP-pD-ZWCs showed greater signals than IO@NP-pD-PEG at 2 h and 13 h post-injection, and the trend persisted over 60 h although statistical significance was not detected beyond 13 h. There was no significant difference between IO@NP-pD-ZWC0.1 and IO@NP-pD-ZWC0.3. This result indicates that NP-pD-ZWCs persist better than NP-pD-PEG due to the ability to interact with cells in an acidic tumor microenvironment, consistent with our hypothesis, although the fine difference between ZWC0.1 and ZWC0.3 was not reflected in vivo.
Fig. 5.

(a) Representative T2-weighted MR images of LS174T tumors before and after the treatment with surface-modified IO@NPs. Arrows indicate LS174T tumors. See Supporting Fig. 6 for all five animals per treatment. (b) Signal enhancement at tumor site after injection of IO@NPs in two different representations (n=5 mice per treatment; average ± standard deviation). Two graphs are different representations of the same data set: (left) time profile of signal change; (right) comparison among NP types at each time point. *: p<0.05; **: p<0.01 by Tukey’s multiple comparisons test.
Next, animals receiving ICG@NPs were observed with NIR fluorescence imaging. Prior to the injection, we confirmed that all ICG@NPs showed comparable fluorescence intensity in 50% FBS (Supporting Fig. 7). At 2 h post-injection, the ICG fluorescence signal was distributed throughout the body, mostly concentrated in the abdomen area corresponding to the small intestine. As the signal in the abdomen faded away over the next 4 to 8 h (6 and 10 h post-injection), tumors were identified with fluorescence signals. The tumor signals were detectable until 22 h post-injection (NP-pD-ZWC0.1) and 10 h post-injection (NP-pD-ZWC0.3 and NP-pD-PEG) (Fig. 6, Supporting Fig. 8). Most signals were cleared except for the intestine by 56 h post-injection. A repeated experiment showed a similar trend except that the difference of NP-pD-ZWC0.1 from the other two was not significant (Supporting Fig. 9).
Fig. 6.
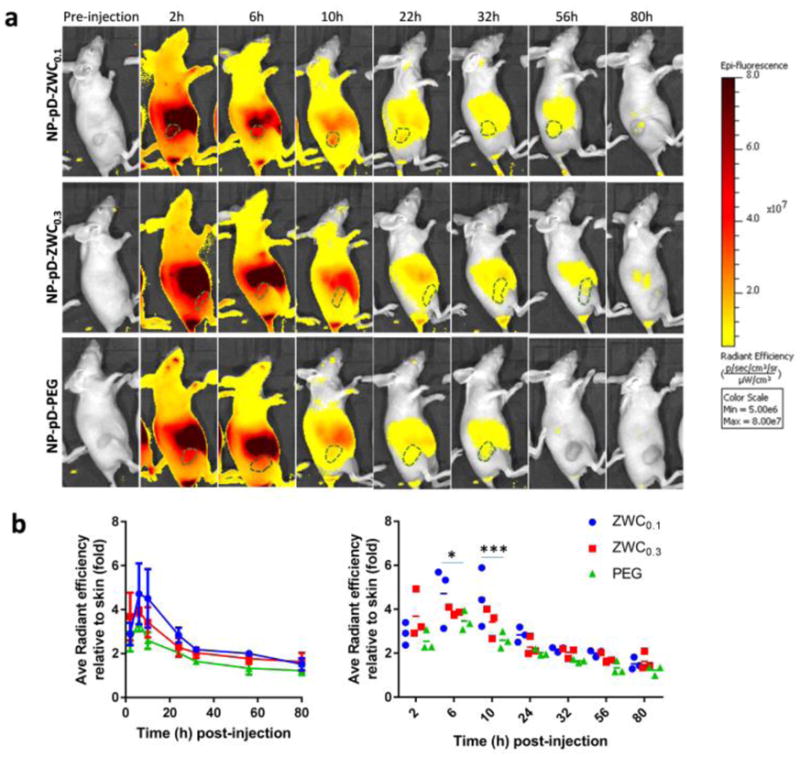
(a) Representative IVIS images of LS174T tumor-bearing animals treated with surface-modified ICG@NPs. See Supporting Fig. 8 for all three animals per treatment. (b) Radiant efficiency of tumor area (marked as dotted line in a) relative to the skin of the shoulder front at each time point in two different representations (n=3 per treatment; average ± standard deviation). Two graphs are different representations of the same data set: (left) time profile of signal change; (right) comparison among NP types at each time point. *: p<0.05; ***: p<0.001 by Tukey’s multiple comparisons test.
The NIR imaging results show two notable differences from those of MR imaging. First, the fluorescence signals of tumors declined more rapidly than the MR signals. Second, the difference between NP-pD-ZWCs and NP-pD-PEG in fluorescence imaging is smaller than that of MR imaging. These differences are attributable to differential stability of signal tracers (IO vs. ICG) in circulating NPs. Given the encapsulation stability in 50% FBS (Fig. 4), the hydrophobic 10 nm IO particles are likely to remain stable in NPs throughout the observation period, whereas ICG molecules would release quickly in blood due to the small size and hydrophilicity. Indeed, the fluorescence distribution followed the typical pattern of unencapsulated ICG distribution [33], where circulating ICG is taken up by the hepatocytes and transported to the small intestine by biliary excretion [34, 35].
These results have an implication relevant to clinical translation of NP drug carriers. The MR imaging shows that the NP accumulation can be improved by exploiting the acidity of tumor microenvironment and the pH-sensitive reversal of NP surface charges as expected from the cell studies. Despite the difference in NP distribution, all three NPs showed similar dye distribution patterns reminiscent of free dye in NIR imaging. This observation indicates that when the drug is not stably retained in NPs during circulation, only a minor fraction of the total payload enjoys the benefit of the NPs and the majority faces the same fate as free drugs. This may seem obvious given the results; however, the current practice of NP evaluation does not necessarily help predict such outcomes. In general, drug (or dye) release kinetics is tested in buffers with little relevance to the protein-rich nature of blood [32]. Drug or dye release in buffers is often much slower than in protein-rich medium and interpreted as stable drug encapsulation [32]. In fact, if the ICG release kinetics had been examined in a buffer with no added proteins, the results would have indicated stable retention of the dye over 48 h (Supporting Fig. 10) and led us to expect differential distribution of ICG reflecting the NP distribution patterns. Nevertheless, we find that drug and dye releases are significantly affected by the protein content in the medium. Several other in vivo events (e.g., protein corona formation [36]) can also lead to suboptimal clinical outcomes. Our observation and other recent studies [37, 38] suggest that stable drug retention in NPs during circulation is no less critical than any other issues. Given that the stability is the very first challenge that NPs face in vivo, it is worthwhile to make dedicated efforts to improve drug encapsulation stability in circulating NPs and validate it through rigorous testing in future NP development. Currently, various physical and chemical strategies are employed to increase the stability and drug retention of NP carriers [39].
4. Conclusion
To overcome the dilemma of PEG-coated NPs, we produced pH-sensitive NPs by modifying the surface of PLGA NPs with a chitosan derivative called ZWC. The ZWC-coated NPs showed different surface charge profile according to the pH, which allowed them to interact with cells in acidic pH and avoid the interaction at neutral pH. Similar to the in vitro cell interactions, the ZWC-coated NPs showed more persistent retention in tumors than PEG-coated NPs, as demonstrated by MR imaging of animals treated with IO-embedded NPs. However, fluorescence signals in tumors treated with ICG-loaded NPs declined more quickly than those of MR imaging with less difference between ZWC- and PEG-coated NPs, suggesting rapid ICG release in blood. For effective translation of the potential benefit of ZWC-coated NPs, future efforts need to be made to improve the stability of drug encapsulation in circulation and ensure the stability through rigorous testing.
Supplementary Material
Acknowledgments
This work was supported by a Lilly Innovation Fellowship Award, NIH R01 EB017791, NSF DMR-1410987, and the Purdue University Center for Cancer Research (NIH P30 CA023168). We thank Gregory Tamer for technical support with MR imaging and data analysis, Paul Bower for assistance with atomic absorption spectroscopy, and Dr. Wooin Lee (Seoul National University) for discussion of pharmacokinetics data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matsumura Y, Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 2.Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: A strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev. 2011;63:152–160. doi: 10.1016/j.addr.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014. [Google Scholar]
- 4.Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. Super pH-sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J Control Release. 2008;129:228–236. doi: 10.1016/j.jconrel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264–273. doi: 10.1016/j.jconrel.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Dang M, Tian Y, Zhu Y, Liu W, Tian W, Su Y, Ni Q, Xu C, Lu N, Tao J, Li Y, Zhao S, Zhao Y, Yang Z, Sun L, Teng Z, Lu G. Tumor Acidic Microenvironment Targeted Drug Delivery Based on pHLIP-Modified Mesoporous Organosilica Nanoparticles. ACS Appl Mater Interfaces. 2017;9:30543–30552. doi: 10.1021/acsami.7b10840. [DOI] [PubMed] [Google Scholar]
- 7.Volk T, Jaihde E, Fortmeyer HP, Glisenkamp KH, Rajewsky MF. pH in human tumour xenografts: effect of intravenous administration of glucose. Br J Cancer. 1993;68:492–500. doi: 10.1038/bjc.1993.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engin K, Leeper DB, Cater JR, Thistlethwaite AJ, Tupchong L, McFarlane JD. Extracellular pH distribution in human tumors. Int J Hyperthermia. 1995;11:211–216. doi: 10.3109/02656739509022457. [DOI] [PubMed] [Google Scholar]
- 9.Amoozgar Z, Park J, Lin Q, Yeo Y. Low Molecular-Weight Chitosan as a pH-Sensitive Stealth Coating for Tumor-Specific Drug Delivery. Mol Pharm. 2012;9:1262–1270. doi: 10.1021/mp2005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abouelmagd S, Ku YJ, Yeo Y. Low molecular weight chitosan-coated polymeric nanoparticles for sustained and pH-sensitive delivery of paclitaxel. J Drug Target. 2015;23:725–735. doi: 10.3109/1061186X.2015.1054829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu P, Bajaj G, Shugg T, Van Alstine WG, Yeo Y. Zwitterio nic Chitosan Derivatives for pH-Sensitive Stealth Coating. Biomacromolecules. 2010;11:2352–2358. doi: 10.1021/bm100481r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bajaj G, Van Alstine WG, Yeo Y. Zwitterionic Chitosan Derivative, a New Biocompatible Pharmaceutical Excipient, Prevents Endotoxin-Mediated Cytokine Release. PLoS ONE. 2012;7:e30899. doi: 10.1371/journal.pone.0030899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu KC, Yeo Y. Zwitterionic Chitosan-Polyamidoamine Dendrimer Complex Nanoparticles as a pH-Sensitive Drug Carrier. Mol Pharm. 2013;10:1695–1704. doi: 10.1021/mp300522p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park J, Brust TF, Lee HJ, Lee SC, Watts VJ, Yeo Y. Polydopamine-Based Simple and Versatile Surface Modification of Polymeric Nano Drug Carriers. ACS Nano. 2014;8:3347–3356. doi: 10.1021/nn405809c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gullotti E, Park J, Yeo Y. Polydopamine-Based Surface Modification for the Development of Peritumorally Activatable Nanoparticles. Pharm Res. 2013;30:1956–1967. doi: 10.1007/s11095-013-1039-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han N, Pang L, Xu J, Hyun H, Park J, Yeo Y. Development of Surface-Variable Polymeric Nanoparticles for Drug Delivery to Tumors. Mol Pharm. 2017;14:1538–1547. doi: 10.1021/acs.molpharmaceut.7b00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsson P, Kihlberg J. How Big Is Too Big for Cell Permeability? J Med Chem. 2017;60:1662–1664. doi: 10.1021/acs.jmedchem.7b00237. [DOI] [PubMed] [Google Scholar]
- 18.Katou H, Wandrey AJ, Gander B. Kinetics of solvent extraction/evaporation process for PLGA microparticle fabrication. Int J Pharm. 2008;364:45–53. doi: 10.1016/j.ijpharm.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 20.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc Nat Acad Sci USA. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J, Kadasala NR, Abouelmagd SA, Castanares MA, Collins DS, Wei A, Yeo Y. Polymer–iron oxide composite nanoparticles for EPR-independent drug delivery. Biomaterials. 2016;101:285–295. doi: 10.1016/j.biomaterials.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.M.I. Limited, editor. Dynamic light scattering - common terms defined. 2014. [Google Scholar]
- 23.I.O.f. Standardization, ISO 22412:2008(en) Particle size analysis - Dynamic light scattering (DLS), in.
- 24.Lee H, Lee E, Kim DK, Jang NK, Jeong YY, Jon S. Antibiofouling polymer-coated superparamagnetic iron oxide nanoparticles as potential magnetic resonance contrast agents for in vivo cancer imaging. J Am Chem Soc. 2006;128:7383–7389. doi: 10.1021/ja061529k. [DOI] [PubMed] [Google Scholar]
- 25.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 26.Schaafsma BE, Mieog JS, Hutteman M, van der Vorst JR, Kuppen PJ, Lowik CW, Frangioni JV, van de Velde CJ, Vahrmeijer AL. The clinical use of indocyanine green as a near-infrared fluorescent contrast agent for image-guided oncologic surgery. J Surg Oncol. 2011;104:323–332. doi: 10.1002/jso.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Vorst JR, Schaafsma BE, Verbeek FP, Keereweer S, Jansen JC, van der Velden LA, Langeveld AP, Hutteman M, Lowik CW, van de Velde CJ, Frangioni JV, Vahrmeijer AL. Near-infrared fluorescence sentinel lymph node mapping of the oral cavity in head and neck cancer patients. Oral Oncol. 2013;49:15–19. doi: 10.1016/j.oraloncology.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niu C, Xu Y, An S, Zhang M, Hu Y, Wang L, Peng Q. Near-infrared induced phase-shifted ICG/Fe3O4 loaded PLGA nanoparticles for photothermal tumor ablation. Sci Rep. 2017;7:5490. doi: 10.1038/s41598-017-06122-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel RH, Wadajkar AS, Patel NL, Kavuri VC, Nguyen KT, Liu H. Multifunctionality of indocyanine green-loaded biodegradable nanoparticles for enhanced optical imaging and hyperthermia intervention of cancer. J Biomed Opt. 2012;17:046003. doi: 10.1117/1.JBO.17.4.046003. [DOI] [PubMed] [Google Scholar]
- 30.Yoneya S, Saito T, Komatsu Y, Koyama I, Takahashi K, Duvoll-Young J. Binding properties of indocyanine green in human blood. Invest Ophthalmol Vis Sci. 1998;39:1286–1290. [PubMed] [Google Scholar]
- 31.Sherwood L. Human Physiology: From Cells to Systems. 3. Wadsworth Pub. Co; Belmont, California: 1997. [Google Scholar]
- 32.Abouelmagd SA, Sun B, Chang AC, Ku YJ, Yeo Y. Release kinetics study of poorly water-soluble drugs from nanoparticles: Are we doing it right? Mol Pharm. 2015;12:997–1003. doi: 10.1021/mp500817h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bahmani B, Lytle CY, Walker AM, Gupta S, Vullev VI, Anvari B. Effects of nanoencapsulation and PEGylation on biodistribution of indocyanine green in healthy mice: quantitative fluorescence imaging and analysis of organs. Int J Nanomedicine. 2013;8:1609–1620. doi: 10.2147/IJN.S42511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shinohara H, Tanaka A, Kitai T, Yanabu N, Inomoto T, Satoh S, Hatano E, Yamaoka Y, Hirao K. Direct measurement of hepatic indocyanine green clearance with near-infrared spectroscopy: separate evaluation of uptake and removal. Hepatology. 1996;23:137–144. doi: 10.1053/jhep.1996.v23.pm0008550033. [DOI] [PubMed] [Google Scholar]
- 35.Chijiiwa K, Watanabe M, Nakano K, Noshiro H, Tanaka M. Biliary indocyanine green excretion as a predictor of hepatic adenosine triphosphate levels in patients with obstructive jaundice. Am J Surg. 2000;179:161–166. doi: 10.1016/s0002-9610(00)00274-9. [DOI] [PubMed] [Google Scholar]
- 36.Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, Kelly PM, Aberg C, Mahon E, Dawson KA. Transferrin- functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8:137–143. doi: 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- 37.Xu P, Gullotti E, Tong L, Highley CB, Errabelli DR, Hasan T, Cheng JX, Kohane DS, Yeo Y. Intracellular Drug Delivery by Poly(lactic-co-glycolic acid) Nanoparticles, Revisited. Mol Pharm. 2009;6:190–201. doi: 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cook RL, Householder KT, Chung EP, Prakapenka AV, DiPerna DM, Sirianni RW. A critical evaluation of drug delivery from ligand modified nanoparticles: Confounding small molecule distribution and efficacy in the central nervous system. J Control Release. 2015;220:89–97. doi: 10.1016/j.jconrel.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y, Lammers T, Storm G, Hennink WE. Physico-Chemical Strategies to Enhance Stability and Drug Retention of Polymeric Micelles for Tumor-Targeted Drug Delivery. Macromol Biosci. 2017;17:1600160. doi: 10.1002/mabi.201600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.