

Abstract
Purpose
Exosomes are small membrane vesicles (30–100 nm in diameter) secreted by cells into extracellular space. The present study evaluated the effect of chemotherapeutic agents on exosome production and/or release, and quantified the contribution of exosomes to intercellular drug transfer and pharmacodynamics.
Methods
Human cancer cells (breast MCF7, breast-to-lung metastatic LM2, ovarian A2780 and OVCAR4) were treated with paclitaxel (PTX, 2–1000 nM) or doxorubicin (DOX, 20–1000 nM) for 24–48 h. Exosomes were isolated from the culture medium of drug-treated donor cells (Donor cells) using ultra-centrifugation, and analyzed for acetylcholinesterase activity, total proteins, drug concentrations, and biological effects (cytotoxicity and anti-migration) on drug-naïve recipient cells (Recipient cells). These results were used to develop computational predictive quantitative pharmacology models.
Results
Cells in exponential growth phase released ~220 exosomes/cell in culture medium. PTX and DOX significantly promoted exosome production and/or release in a dose- and time-dependent manner, with greater effects in ovarian cancer cells than in breast cancer cells. Exosomes isolated from Donor cells contained appreciable drug levels (2–7 pmole/106 cells after 24 h treatment with 100–1000 nM PTX), and caused cytotoxicity and inhibited migration of Recipient cells. Quantitative pharmacology models that integrated cellular PTX pharmacokinetics with PTX pharmacodynamics successfully predicted effects of exosomes on intercellular drug transfer, cytotoxicity of PTX on Donor cells and cytotoxicity of PTX-containing exosomes on Recipient cells. Additional model simulations indicate that within clinically achievable PTX concentrations, the contribution of exosomes to active drug efflux increased with drug concentration and exceeded the p-glycoprotein efflux when the latter was saturated.
Conclusions
Our results indicate (a) chemotherapeutic agents stimulate exosome production or release, and (b) exosome is a mechanism of intercellular drug transfer that contributes to pharmacodynamics of neighboring cells.
Keywords: exosome, exocytosis, intercellular drug transfer, quantitative pharmacology, chemoresistance
Graphical abstract
Introduction
Inadequate drug delivery is a major cause of treatment failures in solid tumors [1]. After entering the systemic circulation, e.g., via an intravenous injection, the drug encounters multiple transport barriers before reaching and exerting its action on the intended targets. Recent intraoperative intravital microscopy findings in patients further show that about one-half of vessels in human tumors are not patent or functional [2, 3]. These issues highlight the need to better understand the mechanisms of interstitial drug transfer. The present study examined the potential role of exosomes.
Cells utilize exocytosis to sort intracellular substances into exosomes that are subsequently released to the extracellular space [4]. Exosomes are small membrane vesicles with an average diameter of between 30 and 100 nm. They originate from the inward budding of endosomal lumen layer and carry cellular components including lipids, proteins (e.g., heat shock proteins, transcription factors, enzymes, major histocompatibility receptors and tetraspanins), and nuclei acids (e.g. DNA, mRNA, microRNA and long non-coding RNA) [4–7].
The life-cycle of exosomes comprises endosome biogenesis, trafficking, release, and re-uptake via endocytosis [4, 7, 8]. Biogenesis begins with internalization of plasma membrane as early endosomes, which later become multivesicular bodies and form intraluminal vesicles (pre-exosomes) that mature into exosomes. Contents of exosomes are sorted and loaded through ESCRT-dependent and -independent mechanisms. In the latter, a sphingolipid ceramide is involved in the loading of microRNA and lipid rafts into endosomes, and the initiation of exosome biogenesis [9, 10]. Several Rab proteins, including Rab-27a/b, Rab-11 and Rab-35, are known molecular motors that drive multivesicular bodies towards plasma membrane [11–13]. Release of exosomes into extracellular space is mediated by exocytosis, which involves fusion of exosome membrane with plasma membrane using SNARE (soluble N-ethylmaleimide sensitive fusion protein attachment receptors)-dependent and -independent mechanisms. Re-uptake of exosomes into cells primarily uses receptor-mediated endocytosis, with plasma membrane fusion and phagocytosis as minor pathways [6, 14].
Cancer cells generally produce higher levels of exosomes compared to normal cells [15]. Exosomes derived from cancer cells are involved in distal metastatic niche initiation [16, 17], intercellular communications (e.g., during drug resistance development [18, 19]), and immune system modulation [20, 21]. Cancer cells enhance their exosome secretion in response to environmental changes including pH [22], ion [23], temperature [24], and treatment by cytotoxic agents [25]. For example, liver HepG2 cells, when treated with cytotoxics (PTX, etoposide, irinotecan, carboplatin), release exosomes containing elevated level of heat shock proteins [25] triggered as a response to stress and as a survival mechanism [26].
Most exosome studies have focused on characterizing their contents and biological functions [13, 15–18, 27, 28]. The current study used in vitro experiments and in silico studies to investigate the intercellular drug transfer via exosomes and the quantitative relationship between this process and pharmacodynamics (PD) in solid tumors. Paclitaxel (PTX) and doxorubicin (DOX) were the test drugs as they are commonly used in first-line therapy of multiple types of major solid tumors including, e.g., ovarian, breast, lung, and prostate cancers [29]. Our results indicate exosomes is a mechanism of intercellular drug transfer with significant pharmacological consequences.
Materials and methods
Reagents
PTX and DOX (purity >99.5%), and cell culture grade dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). Drug stock solutions were prepared in DMSO and diluted to desired concentrations such that the DMSO concentration was below 0.5%.
Cell culture
Human breast adenocarcinoma MCF7 cells were purchased from ATCC (Manassas, VA). LM2 cells, a highly breast-to-lung metastatic subline of human breast cancer MDA-MB-231 cells [30], were a gift from Dr. Y. Kang (Princeton University, NJ). Human ovarian cancer A2780 and OVCAR4 cells were provided by Dr. D. Dhanasekaran (University of Oklahoma Health Sciences Center, OK). MCF7 and LM2 cells were maintained in DMEM (Mediatech, VA), and A2780 and OVCAR4 cells in RPMI-1640 (ATCC). The medium for cell growth (Growth Medium) was supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, GA), whereas the medium for exosome isolation (Conditioned Medium) was supplemented with 10% exosome-depleted-FBS (System Biosciences, CA). All medium contained 100 IU/mL penicillin and 100 μg/mL streptomycin (Mediatech, VA). Cells were cultured in a humidified incubator at 37°C with 5% CO2.
Effects of PTX and DOX on exosomes
Effects of PTX and DOX on exosome production and release were studied using cytotoxic drug concentrations that produced 20, 50 and 80% of their maximal cytotoxicity (EC20, EC50 and EC80; see below for their determination). Exosomes collected from cells treated with drugs (Donor cells) are denoted as Drug-Exoconc,time, e.g., PTX-Exo500nM,24h denotes exosomes from Donor cells treated with 500 nM PTX for 24 h. PTX-Exo was used to study its pharmacological effects (Cytotoxicityrecipient and Anti-migrationrecipient) on drug-naïve recipient cells (Recipient cells).
Exosome isolation, characterization, and quantification
Exosomes were isolated using a previously reported serial centrifugation protocol [31], with the following modifications. In brief, cells were cultured in T175 flasks, without or with drug. The post-incubation exosome-containing medium (Conditioned Medium) was collected at preselected time points, centrifuged twice, first at 2,000 × g for 10 min and then 10,000 × g for 30 min, to remove dead cells and debris. The supernatant was transferred and ultracentrifuged at 100,000 × g for 18 h at 4°C using a type 50.2 Ti rotor (Optima™ L-100 XP, Beckman Coulter Instruments). The pellet was washed once with 4 mL phosphate-buffered saline (PBS, pH 7.4), then ultra-centrifuged at 100,000 × g for 70 min at 4°C. For studying biological activity of exosomes, the resulting pellet was re-suspended in Growth Medium supplemented with 10% exosome-depleted FBS after PBS wash. For the remaining studies, the pellet was re-suspended in 150 μL PBS.
Exosome morphology was characterized using transmission electron microscopy. In brief, an exosome sample was diluted 1:100 in PBS, loaded on a 400 mesh, Formvar-coated, glow discharged copper grid using the single drop method. The liquid was removed 60–90 s later by wicking with filter paper and the exosome-loaded grid washed with deionized water for 10 s. After removing the water again with filter paper, the sample was stained with 4% uranyl acetate (pH 4.4, negative staining solution) for 60 s, washed in deionized water and air dried at room temperature. The grid was then viewed on a transmission electron microscope (Hitachi H7600, equipped with a 2k × 2k AMT digital camera) at 80 kV. A second aliquot of exosome suspension was used to determine exosome size using nanoparticle tracking analysis (NanoSight NS300; Malvern Instruments, Inc., UK). A third aliquot of exosome suspension was used to determine the total protein amount by BCA assay (Pierce kit, ThermoFisher Scientific Inc., CA). Another aliquot was used to determine the activity of acetylcholinesterase (AChE), an enzyme found in exosomes [12, 23], by the Ellman colorimetric assay (Sigma-Aldrich) [32]. Briefly, exosome suspension (20 μL) was added to an aqueous reaction system (80 μL) containing 1 mM 5,5′-dithio-bis(2-nitrobenzoic acid) and 3 mM acetylthiocholine chloride (substrate for AChE), per well in a 96-well plate. After 40 min incubation at room temperature, the absorbance at 412 nm was determined using Synergy HT microplate reader (BioTek Instruments, VT). Standard curves of AChE activity were established using plasma exosome standards calibrated by NanoSight analysis (System BioSciences) and used to calculate the exosome quantity.
The levels of exosome protein markers TSG101 and CD63 were analyzed by Western blotting. Samples of exosomes or Donor cell lysates containing 60 μg proteins in RIPA lysis buffer (ThermoFisher Scientific) were mixed with a protease and phosphatase inhibitor cocktail (ThermoFisher Scientific), and the mixture was loaded on 10% Mini-PROTEAN® TGX pre-cast gel (BioRad Laboratories, CA). After electrophoresis, proteins were transferred to a 0.2 μm nitrocellulose membrane (BioRad Laboratories) and probed sequentially with primary antibodies (mouse IgG anti-CD63 and anti-TSG101 from Santa Cruz Biotechnology (Dallas, TX), rabbit IgG anti-beta actin and anti-calnexin from Cell Signaling Technology (Danvers, MA)), and then with fluorescent secondary antibodies (IRDye® 800CW goat anti-mouse and goat anti-rabbit, LI-COR Inc., NE). Fluorescence intensity was quantified using Odyssey CLx system (LI-COR Inc.).
Biological activities of PTX, DOX and PTX-Exo
We measured the cytotoxicity of PTX and DOX on donor cells (Cytotoxicitydonor) and the cytotoxicity of PTX-Exo on Recipient cells (Cytotoxicityrecipient) using the sulforhodamine B (SRB) colorimetric assay [33]. In brief, cells (5000 per 100 μL Growth Medium per well in 96-well plate) were seeded overnight, treated with drugs or PTX-Exo for 48 h, fixed with trichloroacetic acid (10% w/v, 100 μL per well, 30 min at 37°C) after removing the medium, washed gently with tap water three times, air-dried overnight, and stained with SRB (0.1% in 1% acetic acid (v/v), 100 μL per well, 30 min at room temperature) with gentle agitation. After removing the excess SRB by washing four times with 1% acetic acid, the cell-bound SRB was dissolved with Tris base solution (10 mM, 100 μL per well) and the absorbance measured at 510 nm using Synergy HT microplate reader. The concentration-effect relationships were analyzed with a sigmoidal Hill equation using nonlinear least square regression [34] (Prism 7, GraphPad Software, CA). For PTX, the Hill equation was modified with a residual unaffected fraction Re as we previously described [34]. The analysis provided the drug concentration producing 20%, 50% and 80% of the maximal cytotoxicity (EC20, EC50 and EC80, respectively).
Anti-migrationrecipient was measured using the wound healing assay that measures cell migration [35]; the assay used serum-free medium to minimize cell proliferation. Results of pilot studies indicated only one of the four cell lines (LM2) remained viable in serum-free medium after 48 h and PTX-Exo1000nM,24–48h induced shrinkage and fragmentation in Recipient cells. Therefore, the study used LM2 cells as Recipient cells and PTX-Exo5–500nM,24–48h. Briefly, cells were cultured in Growth Medium supplemented with 10% FBS on a tissue culture-coated (vacuum gas-plasma, hydrophilic and negative charge surface) 6-well plate (Corning Incorporated, NY) and allowed to grow to confluence, after which time the medium was replaced with serum-free Growth Medium. The growth surface was scratched with a 200 μL pipet tip to create an open wound area, and cell debris was carefully washed off with pre-warmed DMEM. Afterwards, a suspension of exosomes (containing 100 μg proteins in 2 mL serum-free medium) was added to each well, and microscopic images of a fixed field were obtained before and after scratching (0 h, 24 h and 48 h). The open wound area was measured as fraction of area not covered by cells using TScratch, a stand-alone Matlab application that measures the covered pixels vs. the uncovered pixels of an image [36] with default detection threshold settings.
Quantification of PTX in PTX-Exo and cell lysates
Cells were treated with PTX for 24 h. PTX-Exo was collected from Conditioned Medium as described above. The remaining cells were washed with PBS and trypsinized, collected, and stained with trypan blue. The number of trypan blue-excluding cells were counted and lyzed with RIPA lysis buffer. Drug concentrations in PTX-Exo and cell lysates were measured using high performance liquid chromatography-tandem mass spectrometry (LC-MS/MS). First, PTX in the sample was extracted with 10x volume of methyl t-butyl ether containing 10 ng/mL of the internal standard docetaxel. The organic phase containing paclitaxel was separated, evaporated, and reconstituted with 10 μL of 0.1% aqueous formic acid/methanol (40/60, v/v). A calibration curve was performed using seven calibration standards covering a dynamic range of 1 – 1,000 ng/mL, prepared in the same biological matrix and run in duplicates. The best-fit line was obtained using linear regression with 1/x2 weighting. Quality control standards were prepared in quintuplet at each of three levels (low (3 ng/mL), mid (50 ng/mL), high (800 ng/mL)), and back-calculated against the best-fit line to ensure accuracy and precision of the assay.
For LC-MS/MS analysis, samples (10 μL) were injected onto a Symmetry Shield RP18 column (2.1×50 mm, 3.5 μm; Waters, MA), using a gradient elution scheme of 0.1% aqueous formic acid in methanol, where methanol was increased from 40% to 100% in 7 min before returning to 40% by the end of the 10 min run (flow rate 0.2 mL/min). The column eluent was directed into an electrospray ionization triple quadrupole mass spectrometer (Micromass Quattro Premier XE; Waters) for detection based on multiple reaction monitoring of a structural fragment in the positive ion mode. PTX was monitored by the mass transition from precursor ions to product ions of m/z 854.0➔287.0 (collision energy 16 V), and docetaxel by transition of m/z 806.8➔526.3 (collision energy 9 V). Universal mass spectrometer settings included capillary voltage of 3500 V, cone voltage of 25 V, source temperature 120°C, desolvation temperature 400°C, and desolvation gas flow (N2) of 600 L/hr.
Quantitative pharmacology modeling of exosome-mediated intercellular drug transfer and biological effects: Overview
The experimental results indicated exosomes collected from PTX-treated Donor cells contained high drug concentrations, served as a mechanism of intercellular transfer and conferred biological activities on drug-naïve Recipient cells. We established predictive quantitative pharmacology (QP) models to depict these processes, as follows. First, we modified our previously published PTX cellular pharmacokinetic (PK) models [37–39] to include the sorting and release of exosomes and their re-uptake via receptor-mediated endocytosis. These models were used together with previously published model parameters and experimental results from the current study to obtain values of the exosome-related model parameters. The resulting equations plus parameter values were used to simulate the drug concentrations derived from PTX-Exo, in cells and medium. Next, we established PD models to depict the relationships between total extracellular/intracellular drug concentrations (including the pharmacologically active tubulin-bound moiety) and drug-induced cytotoxicity. Model simulations and data fitting (nonlinear least-squares algorithm) were performed using Matlab Simbiology (Release 2016a, Mathworks, MA).
Development of cellular PK models
We previously established cellular PTX PK models that have since been adopted by multiple investigator groups [34, 38–43]. The current study extended these earlier models to account for exosome-related processes. The model assumptions are (a) unbound drug in medium enters cells by passive diffusion, (b) saturable drug binding to proteins in extracellular fluid and to intracellular tubulin, plus non-saturable drug binding to other intracellular organelles, (c) drug efflux from cells uses a combination of passive diffusion, p-glycoprotein (Pgp)-mediated saturable efflux, and release of drug-containing exosomes, (d) first order sorting of intracellular unbound drug into exosomes through the endosomal transport system, (e) first order release of exosomes from the cell into extracellular space, and (f) internalization of drug-containing exosomes through saturable receptor-mediated endocytosis [14, 44]. The exosome-related processes in (c) through (f) are new, whereas the remaining processes describe the cross-membrane drug transportation and intracellular drug distribution as established previously [37–39]. To simplify the model, we assumed negligible PTX transfer between Cmedium,free and Cmedium,exo.
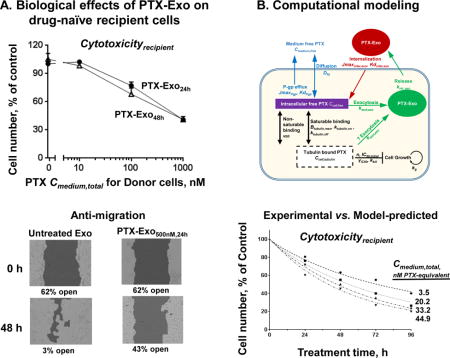
The above processes are summarized in Figure 1. In general, subscripts are used to denote the various drug entities, their locations and transport mechanisms. For drug concentrations C, the first subscript indicates if the drug is located intracellularly or extracellularly (cell vs. medium) and the second subscript indicates if it is bound to proteins or exosomes, e.g., Ccell,tubulin is tubulin-bound drug concentration in cells, and Cmedium,exo and Ccell,exo are the respective drug concentrations in medium and cells derived from exosomes. Note that because Cmedium,exo and Ccell,exo were calculated as amount divided by volume, Cmedium,exo does not equal Ccell,exo due to difference in medium and cell volumes.
Figure 1. Cellular pharmacokinetic/pharmacodynamic models of paclitaxel.
Cmedium,free or Ccell,free is free (unbound) drug concentration in medium or cells, respectively. Ccell,tubulin is the tubulin-bound drug concentration. Dfd is the rate constant of passive diffusion of free drug. JmaxPgp is the maximum Pgp-mediated drug efflux rate. KdPgp is the dissociation constant of drug from Pgp. Cmedium,exo or Ccell,exo is the drug concentration in extracellular or intracellular exosomes, respectively. Jmaxinter,exo is the maximum rate of receptor-mediated internalization of exosomes. Kdinter,exo is the dissociation constant of exosome from receptor for internalization. ksort,exo is the rate constant of drug sorting into exosomes. krel,exo is the rate constant of exosome release into extracellular space. Btubulin,max is the maximum available drug binding sites in tubulin. ktubulin,on or ktubulin,off are the rate constants of drug association and disassociation with tubulin, respectively. NSB is the proportion constant for the linear nonsaturable drug binding in cells. For PD parameters, IC50 is the tubulin-bound drug concentration needed to generate 50% of maximum drug effect, kkni is the maximal rate constant of cell kill. n is Hill exponent.
For transport, Jmax is maximum rate and Kd denotes dissociation constant, and their subscript denotes the transport mechanism, e.g., JmaxPgp is maximum Pgp-mediated efflux rate per cell and KdPgp is dissociation constant of drug from Pgp, whereas Jmaxinter,exo is maximum rate of receptor-mediated internalization of exosomes per cell and Kdinter,exo is dissociation constant of exosome bound to the receptor for internalization. ksort,exo and krel,exo denote the rate constant for sorting and release of exosomes, respectively. For intracellular drug binding to its molecular target tubulin, Btubulin,max is the maximal available binding sites, and ktubulin,on and ktubulin,off are the respective association or disassociation rate constant. NSB is the proportionality constant for the linear nonsaturable drug binding in cells. Dfd is the diffusion rate constant of the free drug (i.e., unbound to macromolecules) through cell membrane. Vcell and Vmedium are volume of a single cell and extracellular medium, respectively. ICN and TCN are respectively the initial and total cell number, and were experimentally counted.
Eq. 1–2 depict Ccell,total and Cmedium,total as sums of various intracellular and extracellular drug entities, respectively.
| (1) |
| (2) |
Eq. 3–5 describe the time-dependent changes of Ccell,free, Ccell,exo, and Ccell,tubulin. Based on the reported value of 0.99 ± 0.04 ligand per alpha/beta tubulin dimer [45, 46], we used a 1:1 PTX-tubulin binding stoichiometry (i.e., binding of Ccell,free to tubulin increases linearly with Btubulin,max). As reflected in these equations, there are three drug efflux mechanisms: passive diffusion (equals Dfd · Ccell,free), and active efflux via Pgp (equals to (JmaxPgp · Ccell,free)/(KdPgp + Ccell,free)) and exosomes (equals krel,exo · Ccell,exo · Vcell).
| (3) |
| (4) |
| (5) |
We reported Btubulin,max increases linearly with time (from an initial value Btubulin,initial) and the corresponding rate constant kBtubulin,max changes with drug concentration [39]; these kinetic processes are captured in Eq. 6 and 7. Eq. 7 was obtained by nonlinear regression of the plot of kBtubulin,max vs. Ccell,free (r2 = 0.998), where the experimental results were obtained from our earlier study [39].
| (6) |
| (7) |
Eq. 8–10 describe changes of Cmedium,free, Cmedium,exo, and Cmedium,bound with time, where Bmedium,max is maximum saturable drug binding sites in medium.
| (8) |
| (9) |
| (10) |
For initial conditions (before treatment), Cmedium,exo equals zero, and Cmedium,free and Cmedium,bound were calculated from Cmedium,total using Eq. 11 (obtained by substituting Eq. 10 into Eq. 2, followed by rearrangement).
| (11) |
Development of PD models
PD models describe cytotoxicity as a function of drug treatment (concentration and time). Eq. 12 depicts the net change in TCN due to (a) cell growth over time at a rate constant kg until confluence, and (b) drug-induced cell kill as function of concentration of tubulin-bound drug. TCNss is maximal cell number at confluence. kkill is maximum cell kill rate constant. Note that Eq. 12 does not account for potential concentration-dependent changes in kkill. IC50 is tubulin-bound drug concentration that generates 50% maximum cell kill. n is the Hill exponent. In view of the time- and dose-dependent PTX cytotoxicity [34] and the development of drug resistance over time in MCF7 cells [47], we used Eq. 13 to depict the time-dependent changes in IC50, where IC50,initial is the IC50 value at time zero and γIC50 is the rate of IC50 change per unit time t.
| (12) |
| (13) |
Model parameterization
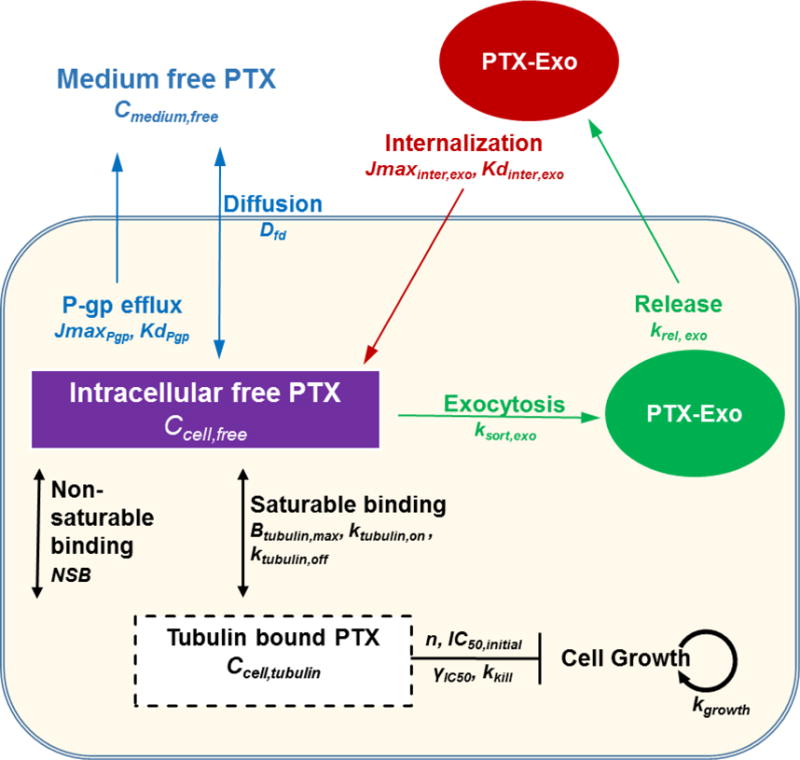
The parameters for exosome-independent processes were obtained as follows. Dfd, and KdPgp were taken from our published results [37, 39]. The calculation of PTX sequestration into exosomes required the Ccell,free-time profiles, which in turn required the rate constants of drug-tubulin interactions (ktubulin,on, ktubulin,off). PTX binding to tubulin depends on their nucleotide contents (GTP or GDP) and can be high affinity (Kd of 15 to 60 nM) or low affinity (Kd of 2 to 3 μM) [46, 48–51]. We used the reported parameters for high affinity binding (ktubulin,on of 2 nM−1s−1, ktubulin,off of 30 s−1, Kd of 15 nM [48]). The current models also required Btubulin,initial and kBtubulin, which were obtained in two-steps: we first modified Eq. 1 to 6 to remove the exosome-related components because drug binding to tubulin is independent of exosomes, and then we fitted the modified equations to our previously obtained experimental results (Ccell,total vs. time plots after treatment with 0.1–1000 nM PTX [39]) to obtain the best-fitting parameters values.
To obtain the rate constants for the exosome-related PK processes (sorting, release, endocytosis), we used the above parameters together with the current models (Eq. 1–9 that incorporated exosome-related processes) to fit four sets of experimental data in PTX-treated Donor MCF7 cells obtained in our previous studies [27, 29], i.e., (a) Ccell,total vs. time plots, (b) time-dependent depletion of Cmedium,total as function of initial Cmedium,total (1 to 1000 nM), (c) Cytotoxicitydonor vs. treatment duration plots, and (d) ratio of Amedium,exo [drug amount in exosomes in culture medium, equals Cmedium,total times Vmedium] to Acell,total [drug amount in Donor cells, equals Ccell,total times Vcell times TCN] at 24 h. The first three data sets were obtained from our earlier studies [34, 38, 39], whereas the last data set was from the current study (see Table 2 in Results). For example, ksort,exo, krel,exo, Jmaxinter,exo, and Kdinter,exo were obtained by simultaneously fitting the plots (a), (b) and (d) (see Figures 5A, 5B and 5D in Results) with Eq. 1–5, Eq. 2 and 8–10, and Eq. 1 and 4, respectively.
Table 2. PTX cellular pharmacokinetic and pharmacodynamic model parameters.
Some parameter values were obtained from the literature and some were obtained from analyzing the experimental results of the current study (indicated as Analysis). Mean values of parameters obtained from Analysis are shown with % CV in parentheses.
| Category | Parameter | Annotation | Value (% CV) |
Source |
|---|---|---|---|---|
|
A. Cellular pharmacokinetic model
| ||||
| Pgp-mediated efflux | JmaxPgp (pmoleh−1·cell−1) | Maximum Pgp efflux rate | 2.8 × 10−6 | [37] |
| KdPgp (nM) | Dissociation constant of drug from Pgp | 13.9 | [37] | |
|
| ||||
| Diffusion | Dfd (μL·h−1·cell−1) | Diffusion rate constant of free PTX across cell membrane | 3.34×10−3 | [39] |
|
| ||||
| Intracellular microtubule binding | Btubulin,initial (μM) | Maximal initial PTX binding sites on tubulin | 59.2 | [39] |
| ktubulin,off (s−1) | Rate constant of PTX dissociation from tubulin | 30 | [48] | |
| ktubulin,on (nM−1·s−1) | Rate constant of PTX association with tubulin | 2 | [48] | |
|
| ||||
| Binding to cell or extracellular fluid | Bmedium,max (μM) | Maximal extracellular drug binding sites | 3.94 | [39] |
| Kdmedium,bound (nM) | Dissociation constant of PTX from extracellular binding sites | 781 | [39] | |
| Vcell (μL) | Volume of single cell | 2.06 × 10−6 | [39] | |
| Vm (μL) | Medium volume | 1000 | Experimental set-up | |
|
| ||||
| Exosome release and internalization | Jmaxinter,exo (pmole·h−1·cell−1) | Maximum rate of endocytosis of extracellular exosomes | 0.038 (23) |
Analysis |
| Kdinter,exo (nM) | Dissociation constant of exosomes from endocytosis receptor | 63 (45) |
Analysis | |
| ksort,exo (h−1) | Rate constant for sorting intracellular PTX into exosome | 31 (67) |
Analysis | |
| krel,exo (h−1) | Rate constant for exosomes release into extracellular fluid | 0.105 (12) |
Analysis | |
|
B. Pharmacodynamic model | ||||
| Drug cytotoxicity | kgrowth (h−1) | Cell growth rate constant | 0.0288 (8) |
Analysis |
| kkill (h−1) | Maximum cell kill rate constant | 0.0202 (11) |
Analysis | |
| IC50,initial (nM) | Tubulin-bound concentration to produce 50% Emax at time zero | 826 (27) |
Analysis | |
| γIC50 (nM·h−1) | Change in IC50 over treatment time | 7.2 (48) |
Analysis | |
| n | Hill exponent of PTX concentration- cytotoxicity curve | 1.67 (4) |
Analysis | |
| Nss | Maximum cell number at confluence | 1.1 × 106 | Experimentally determined | |
| ICN | Initial cell number | 5 × 103 | Experimental set-up | |
Figure 5. Obtaining model parameters.
(A) Donor cell Ccell,total vs. time plots. (B) Time-dependent changes in Cmedium, total at four initial Cmedium,total (1 to 1000 nM). (C) Cytotoxicitydonor vs. treatment concentration and duration plots. (D) Changes of Amedium,exo as a fraction of Acell,total in Donor cells with time. The experimental results in (A), (B) and (C) were from our earlier studies ([34, 39] with permission), and the results in (D) are from the current study (calculated from Table 1; 24 h results only). Fitting Eq. 1–13 to the experiment data points (symbols) yielded the best-fitting curves and model parameter values (see text). Note the different x and y scales (i.e., 24 h in (A), (B) and (D) vs. 96 h in (C); logarithmic in (A) and (B) vs. linear in (C) and (D)).
PD parameters (kkill, n, IC50,initial, γIC50) were obtained using experimental results from our previous study [34] and the current study, by fitting Eq. 1–13 to the above plot (c) (see Figure 5C in Results). kg was calculated from the cell doubling time [52]. TCNss was experimentally counted.
Evaluate model performance
QP models were used to predict the changes in Cytotoxicityrecipient of PTX-Exo and Cytotoxicitydonor of PTX due to exosome-mediated drug efflux. The model-predicted data were then compared with experimental results to evaluate model performance. The deviation between model-predicted results and experimentally observed results, as percentage, was calculated using Eq. 14 where Oi is the observation value and Pi is the predicted value.
| (14) |
Sensitivity analysis
The four parameters on exosome-mediated drug transfer processes (ksort,exo, krel,exo, Jmaxinter,exo, Kdinter,exo) were evaluated for their effects on three cellular PK and PD endpoints (Ccell,total, Ccell,tubulin, Cytotoxicity) in both Donor and Recipient cells. Sensitivity analysis was performed by (a) changing the value (10-fold increase or decrease) of a selected parameter while keeping all other parameters constant in Donor cells, and (b) comparing the differences in the simulated outcomes without or with the parameter value change. These exosome-related effects were further compared to the effects caused by changes in tubulin binding capacity, a parameter we have demonstrated to significantly alter the cellular PTX PK [37, 39]. Simulations of Cytotoxicityrecipient of PTX-Exo used equal cell numbers, i.e., PTX-Exo derived from a selected number of Donor cells were applied to the same number of Recipient cells.
Statistical analysis
Experimental results were analyzed for statistical significance using Student’s t-test (unpaired, two-tailed) or Dunnett’s test (Prism 7). p<0.05 was considered as significant. Data are presented as mean ± SD (standard deviation) or mean ± SEM (standard error of the mean).
Results
Characterization of exosomes
Transmission electron microscopy (Figure 2A) showed the spherical shape of exosomes (from MCF7 cells) obtained after drying and processing. The average particle diameter, based on nanoparticle tracking analysis, was 108 nm (n=100, Figure 2B). Exosomes contained high levels of CD63 (a tetraspanin and marker of exosomes) and TSG101 (a component of ESCRT-I complex or endosomal sorting complexes required for transport), but undetectable levels of skeleton protein β-actin and endoplasmic reticulum protein calnexin, whereas the reverse was found for Donor cell lysates (Figure 2C).
Figure 2. Characterization of cancer cell-derived exosomes.
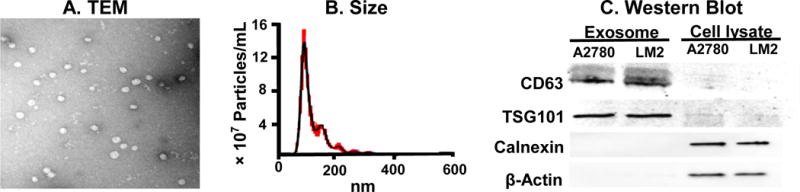
(A) Transmission electron microscopy. Representative image of MCF7 exosomes. (B) Size distribution of MCF7 exosomes by nanoparticle tracking analysis. Black: rolling average. Red: SD of results from three repeated measurements. (C) Western blot results. Exosomes and Donor cell lysates, collected from A2780 cells and LM2 cells, were analyzed for CD63, TSG101, calnexin and β-actin.
Cytotoxicitydonor of PTX and DOX
Figure 3A shows Cytotoxicitydonor of PTX and DOX. PTX yielded incomplete cytotoxicity in all 4 cells; MCF7 cells were the least sensitive with the highest residual unaffected fraction Re. In contrast, DOX yielded complete cytotoxicity in the two cells studied, with ~8-times higher activity in A2780 cells compared to MCF7 cells. Subsequent studies on the effects of PTX and DOX on exosomes used drug concentrations that corresponded to their EC20, EC50, and EC80 values in individual cell lines.
Figure 3. Cytotoxic concentrations of paclitaxel and doxorubicin induced exosome production and/or release.
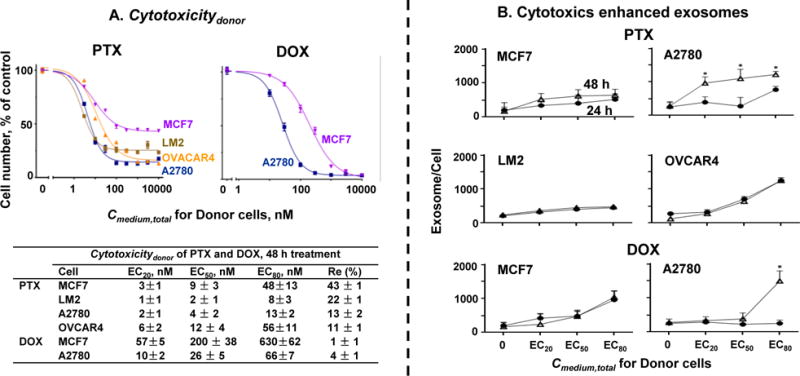
(A) Cytotoxicitydonor of PTX and DOX. MCF7, LM2, A2780, and OVCAR4 cells were treated for 24 or 48 h with PTX or DOX, and the remaining cell numbers were determined using SRB assay. Results were analyzed for the values of drug concentrations producing 20%, 50% and 80% of the maximal cytotoxicity (EC20, EC50 and EC80, respectively). (B) Cytotoxics enhanced exosomes. Cells were treated for 24 h or 48 h with PTX or DOX at Cmedium,total equivalent to their EC20, EC50 and EC80 for 48 h treatment. Number of exosomes in culture medium after drug treatment for 24 h (circles) and 48 h (open triangles) were standardized by cell numbers (mean ± SE, n = 3 experiments, four replicates per experiment). *p<0.01 for 24 h vs. 48 h (unpaired Student’s t-test).
PTX and DOX stimulated exosome production and/or release
Figure 3B shows the levels of exosomes in Conditioned Medium. Without drug treatments, the four cells during exponential growth phase (over 24 to 48 h) yielded comparable exosome levels, ranging from 170 ± 35 to 280 ± 68 exosomes per cell (mean ± SEM). Treatments with cytotoxic concentrations of PTX or DOX generally enhanced the exosome levels, in drug concentration- and time-dependent manners. The data also showed drug- and cell-specific differences. For example, compared to untreated control, treatments with PTX at EC80 for 48 h increased the exosome level by ~3-folds in the two breast cells (LM2 and MCF7) and ~10-folds in the two ovarian cells (A2780 and OVCAR4), and the exosome level in MCF7 and A2780 cells continued to increase with time (50–200% higher after 48 h treatment compared to 24 h) whereas the maximal increase was reached at 24 h with no further increases in LM2 or OVCAR4 cells. Similarly, treatment with DOX at EC80 enhanced the exosome level in MCF7 by 4- to 6-folds after 24 or 48 h, but required longer treatment duration to produce changes in A2780 cells (no change after 24 h and 7-fold increase after 48 h).
Quantification of PTX in PTX-Exo and cell lysate by LC-MS/MS
Table 1 summarizes the LC-MS/MS results of PTX concentrations or amounts in PTX-Exo or cell lysates, after 24 h treatment. In general, the four cancer cells showed comparable PTX concentrations in cell lysates or exosomes (e.g., ~27 pmole in lysates and ~1.5 pmole in exosomes per 106 trypan blue-excluding cells at 100 nM Cmedium,total), and increasing PTX concentrations/amounts with increasing Cmedium,total. However, the ratios of PTX concentration in PTX-Exo and cell lysate indicate (a) substantial cell-specific differences, i.e., up to two-fold difference between cells (e.g., 4.3% in LM2 cells vs. 8.3% in OVCAR4 cells at 100 nM Cmedium,total) and (b) nonlinear uptake of PTX and nonlinear drug sorting into PTX-Exo (e.g., the PTX concentration increases in PTX-Exo and cell lysate were not proportional to either the increase in Cmedium,total (average of ~4-folds vs. 10 folds) or to each other (e.g., average of ~2.5-folds vs. ~4-folds)).
Table 1. Amount of PTX in PTX-Exo or cell lysates.
Cells were treated with 100 or 1000 nM PTX for 24 h. PTX-Exo was collected from Conditioned Medium. Cells were washed and collected with trypsinization, counted and lyzed. Drug concentrations in exosomes and cell lysates were analyzed using LC-MS/MS, and standardized to cell number. The numbers of exosomes per LM2, MCF7, A2780 and OVCAR4 cell were, respectively, 1170, 955, 2236 and 2457 at 100 nM PTX and 1232, 1060, 2513 and 2794 at 1000 nM PTX. Results are mean ± SD (three independent experiments with three replicates per experiment).
| Cell line | PTX, pmole per 106 Donor cells
|
|||||
|---|---|---|---|---|---|---|
| Treated with 100 nM PTX
|
Treated with 1000 nM PTX
|
|||||
| Exosome | Lysate | Exosome-to-Lysate ratio, % | Exosome | Lysate | Exosome-to-Lysate ratio % | |
|
|
|
|||||
| LM2 | 1.13±0.02 | 25.0±0.10 | 4.27±0.22 | 6.18±0.65 | 62.2±0.19 | 9.29 ± 1.08 |
| MCF7 | 1.60±0.04 | 29.3±0.54 | 5.12±0.36 | 8.33±0.16 | 69.7±0.53 | 11.59 ± 0.79 |
| A2780 | 1.44±0.08 | 23.8±0.23 | 6.67±1.12 | 5.38±0.27 | 73.4±3.49 | 7.67 ± 1.64 |
| OVCAR4 | 2.38±0.06 | 28.6±0.47 | 8.28±0.69 | 6.71±0.46 | 71.7±2.72 | 9.65 ± 0.33 |
|
|
|
|||||
| Average | 1.64±0.27 | 26.7±1.34 | 6.08±1.77 | 6.65±0.62 | 69.3±2.46 | 9.55 ± 1.61 |
The amount of PTX in PTX-Exo was between 6–10% of the amount in Donor cell lysate (Table 1). We further calculated the PTX concentration in PTX-Exo from MCF7 cells treated with 100 nM Cmedium,total for 24 h, as [Amountmedium,exo of 1.6 pmole per 106 cells] divided by [Volume of exosomes excreted by 106 cells, calculated using an average exosome radius of 54 nm and release of 103 exosomes per cell (see Table 1)]; the resulting concentration in exosomes equaled 2.4 mM, which is ~60-times the Ccell,total and ~30,000-times the Cmedium,total (experimentally determined to be 39 μM and 83 nM, respectively, after treatment with 100 nM for 24 h [39]).
PTX-Exo exhibited Cytotoxicityrecipient and Anti-migrationrecipient
Figure 4A compares the Cytotoxicityrecipient of PTX to that of PTX-Exo. Each treatment condition had its own control, i.e., no drug for the PTX treatment and exosomes collected from untreated cells (Untreated-Exo) for the PTX-Exo treatment. The amount of PTX-Exo added was calculated based on the drug concentration in PTX-Exo as determined by LC-MS/MS, whereas the amount of Untreated-Exo added was calculated to contain the same protein amount as in PTX-Exo. No cytotoxicity was observed in all control groups, confirming Untreated-Exo had no cytotoxicity (not shown). In contrast, PTX-Exo10–1000nM,24–48h induced significant Cytotoxicityrecipient in all four cells; the extent increased with the drug treatment concentration and duration of Donor cells (e.g., greater cytotoxicity for PTX-Exo48h compared to PTX-Exo24h) and followed the rank order of the chemosensitivity of individual cells (LM2>MCF7>A2780>OVCAR4).
Figure 4. PTX-Exo conferred dose-dependent Cytotoxicityrecipient (A and B) and Anti-migrationrecipient (C and D).
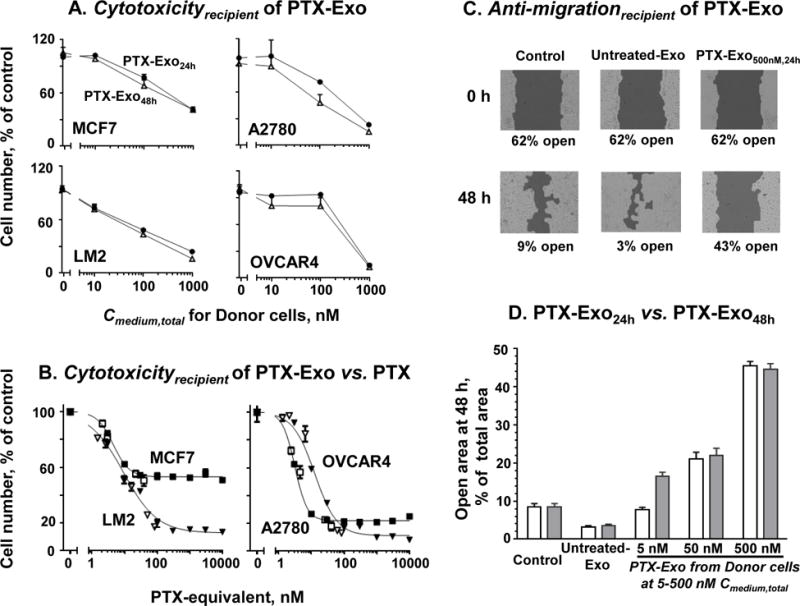
For (A) and (B), drug-naïve cells (MCF7, LM2, A2780, OVCAR4) were treated with PTX or PTX-Exo collected from Conditioned Medium of their corresponding Donor cells. The Recipient cell number was determined using SRB assay. Data are Mean ± SD (n=4 experiments with triplicates per experiment). (A) Cytotoxicityrecipient of PTX-Exo. PTX-Exo24h (circles) and PTX-Exo48h (open triangles), both containing 10 μg proteins, were incubated with Recipient cells for 48 h. (B) Comparison of Cytotoxicityrecipient of PTX-Exo and PTX. Recipient cells were treated with PTX (solid symbols) or PTX-Exo (open symbols) collected from Donor cells after 24 h PTX treatment. The x-axis denotes the PTX-equivalent concentration added to the culture medium, where the amount of the added PTX-Exo was calculated based on the LC-MS/MS results. Note the different scales in (A) and (B). For (C) and (D), LM2 cells were cultured in 6-well plates, without drug treatment (control), or treatment with exosomes collected from untreated Donor cells (Untreated Exo) or PTX-Exo for 48 h. The exosomes added to each well contained 100 μg proteins. (C) Inhibition of cell migration. The initial open areas (i.e., not covered by cells) in untreated control and Untreated-Exo groups were equal (62 ± 2%; mean ± SD, n=6 experiments, duplicates in each experiment). (D) Concentration- and time-dependent Anti-migrationrecipient by PTX-Exo24h (open bars) and PTX-Exo48h (filled bars) harvested from Donor cells treated with 5, 50 or 500 nM PTX. Statistically significant differences between (a) Control and other groups (p<0.01, Dunnett’s test), (b) Untreated-Exo and PTX-Exo groups (p<0.01, Dunnett’s test), (c) among the three PTX-Exo groups at different Cmedium,total (p<0.01, Dunnett’s test), and (d) between PTX-Exo5nM,24h and PTX-Exo5nM,48h (*p<0.01, unpaired Student’s t-test).
We further compared Cytotoxicityrecipient of PTX and PTX-Exo, at equal PTX-equivalent concentrations (Figure 4B); the overlapping concentration-effect relationships of PTX and PTX-Exo in all 4 cells indicate Cytotoxicityrecipient of PTX-Exo was due to its PTX content.
Figure 4C shows Anti-migrationrecipient of PTX-Exo. After 48 h incubation, the untreated control showed a much smaller scratch-induced open-wound gap (from 62% to 9%, equivalent to 85% reduction), indicating substantial cell migration. In comparison, Untreated-Exo slightly but significantly promoted the cell migration as indicated by a yet smaller open-wound gap (>90% reduction, p<0.05 compared to untreated control); this minor effect is consistent with the reported invasion and pre-metastasis functions of tumor-derived exosomes [16, 53]. In contrast, PTX-Exo5–500nM,24–48h significantly inhibited the migration, as indicated by a larger open wound area (e.g., decreased from 62% to 43%, equivalent to 31% reduction, for PTX-Exo500nM,48h). As observed for Cytotoxicityrecipient, Anti-migrationrecipient of PTX-Exo generally increased with PTX treatment concentration and duration of Donor cells (Figure 4D); the differences between Cmedium,total of 5, 50 and 500 nM were significant (p<0.05 compared to untreated control or Untreated-Exo groups). However, prolonging treatment from 24 to 48 h yielded a significant difference only at the lowest concentration and not at the two higher concentrations; this concentration-dependence may be due to the different rates for Cexo,total to equilibrate with Ccell,total.
Quantitative pharmacology models of exosome-mediated effects
Computational models were constructed to capture the above cellular PK and PD findings (Eq. 1–13). Figures 5A–5D show the best-fitting curves for changes in Ccell,total over time at different initial Cmedium,total, depletion of Cmedium,total as function of initial Cmedium,total, Cytotoxicitydonor as function of drug treatment duration and initial Cmedium,total, and changes in % of Amedium,exo as a fraction of Acell,total with treatment time at initial Cmedium,total of 100 and 1000 nM, respectively. Table 2 summarizes the best-fitting PK and PD model parameter values. The coefficients of variations (CV) for most parameters were between <10% to 25%; the three exceptions are the parameters for exosome transport Kdinter,exo and ksort,exo and for resistance development over time γIC50 where the CV was between 45% and 67%; these high CV were in part due to the inability to experimentally measure Ccell,exo.
Comparisons of model parameter values provided the following insights regarding exosome-mediated drug transfer and PD. The value of ksort,exo of ~31 h−1 corresponds to a half-life of <2 min, indicating rapid sorting of Ccell,free into exosomes. Internalization of PTX-Exo into Recipient cells was also rapid, as demonstrated by a relative high Jmaxinter,exo value. In comparison, the release of PTX-Exo into extracellular medium was much slower (krel,exo of 0.105 h−1 per cell, which corresponds to a half-life of ~6–7 h). Hence, release of PTX-Exo from Donor cells is rate-limiting for their appearance in extracellular fluid and Recipient cells. With respect to PD, the γIC50 of 7.2 nM per h indicates a doubling of the initial IC50 of 826 nM in about 115 h, which is consistent with the finding of a separate study showing that continuous PTX treatment led to a 2-fold higher IC50 in human prostate PC3 cells after 120 h (unpublished results).
Evaluation of model performance
Figure 6 compares the QP model-simulated Cytotoxicityrecipient of PTX-Exo with the experimental results, at four initial Cmedum,exo (3.5 to 45 nM) and four treatment durations (24 to 96 h). The good agreement between these results (average deviation of <10% for all data, <15% deviations for individual data points) indicates the QP models in Eq. 1–13 successfully depicted the pharmacological activity of PTX-Exo.
Figure 6. QP model-simulated pharmacodynamics of PTX-Exo: Comparison with experimental results.
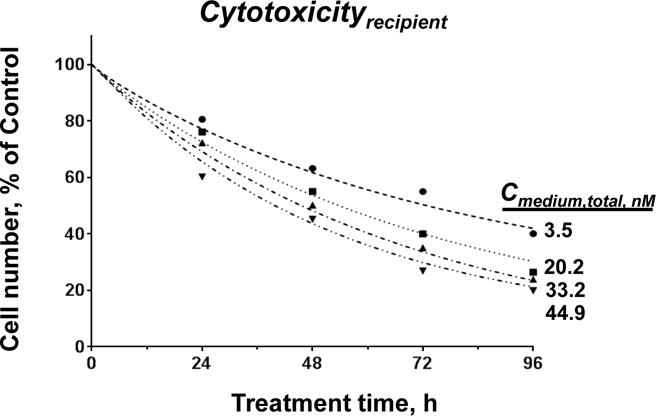
Model-simulated results (curves) vs. experimentally observed results (symbols) on Cytotoxicityrecieint of PTX-Exo as functions of drug treatment concentration and duration. Drug concentrations in culture medium at time zero were calculated as [drug amount in PTX-Exo] divided by extracellular fluid volume (equaled 1000 μL). For experimental data, the remaining cell number was measured using SRB assay (n = 3 experiments, four replicates per experiment). Deviations between simulated and experimental results were 12 ± 7% (mean ± SD).
Sensitivity analysis
The sensitivity analysis results identified the different key determinants of three PK and PD endpoints (Ccell,total, Ccell,tubulin and Cytotoxicity) in Donor and Recipient cells; these endpoints are expressed as area-under-curve from 0 to 24 h (Table 3). The three model parameters (ksort,exo, krel,exo, Jmaxinter,exo) exerted different effects at low and high initial Cmedium,total (1 vs. 1000 nM) and in Donor vs. Recipient cells, as follows.
Table 3. Effect of exosome production and internalization on PTX accumulation and cytotoxicity in cells: Model-simulated results.
Simulations were performed for Donor cells treated with 1 or 1000 nM PTX for 24 h. The resulting values of Ccell,total, Ccell,tubulin and Cytotoxicity (E, rate constant of drug-induced cell death) are represented as area-under-curve from 0 to 24 h. E was calculated as . The original values of model parameters are 31 h−1 for ksort,exo, 0.105 h−1 for krel,exo, 0.038 pmole·h−1·cell−1 for Jmaxinter,exo, and 59.2 μM for Btubulin,initial. With the exception of Btubulin,initial where the low and high values were one-half or 2-times the original values, the low and high values of all other parameters were one-tenth or 10-times the original values. Signs in the table correspond to minimal change of <20% (−), increase or decrease of 20–50% (↑ and ↓), 51–100% (↑↑ and ↓↓), 101–200% (↑↑↑ and ↓↓↓), and >200% (↑↑↑↑ and ↓↓↓↓).
| Parameter | A. Effects on Donor cells | B. Effects on Recipient cells | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| 1 nM | 1000 nM | 1 nM | 1000 nM | ||||||||||||
| Ccell,total | Ccell,tubulin | E | Ccell,total | Ccell,tubulin | E | Ccell,total | Ccell,tubulin | E | Ccell,total | Ccell,tubulin | E | ||||
| Exosome production | ksort,exo | Low | – | – | – | ↑ | – | – | ↓ | ↓ | ↓↓ | ↓ | ↓ | ↓↓ | |
| High | – | – | – | ↓ | – | – | ↑ | ↑ | ↑↑ | ↑↑ | ↑↑ | ↑↑ | |||
|
| |||||||||||||||
| krel,exo | Low | ↑ | ↑↑ | ↑↑ ↑↑ |
↑ | ↑ | – | ↓ | ↓ | ↓↓ | ↓↓ | ↓↓ | ↓↓↓ | ||
| High | ↓ | ↓↓ | ↓↓ ↓↓ |
↓ | ↓ | – | ↑↑ | ↑↑ | ↑↑ ↑ |
↑↑ | ↑↑ | ↑↑↑ ↑ |
|||
|
| |||||||||||||||
| Exosome internalization | Jmaxinter,exo | Low | – | ↓ | ↓ | – | – | – | ↑ | ↑ | ↑↑ ↑ |
↑↑ | ↑↑ | ↑↑↑ ↑ |
|
| High | – | ↑ | ↑ | – | – | – | ↓ | ↓ | ↓ | ↓ | ↓ | ↓↓ | |||
|
| |||||||||||||||
| Tubulin binding capacity | Btubulin,initial | Low | ↓ | ↓↓ | ↓↓ ↓ |
↓↓ | ↓↓↓ | – | ↑ | ↑ | ↑ | ↑ | ↑↑ | ↑↑ | |
| High | ↑ | ↑↑ | ↑↑ ↑ |
↑↑ | ↑↑↑ | – | ↓ | ↓ | ↓↓ | ↓ | ↓ | ↓↓ | |||
For Donor cells treated with 1 nM Cmedium,total, a 10-fold increase or decrease in krel,exo produced small-to-moderate changes in Ccell,total (20–50%) and Ccell,tubulin (51–100%), but significantly greater changes in Cytotoxicitydonor (241%), whereas similar changes in ksort,exo or Jmaxinter,exo yielded minor or no changes in all three endpoints (<20%). At 1000 nM Cmedium,total or when drug binding to tubulin would be saturated and consequently maximal cytotoxicity would have been reached, 10-fold changes in ksort,exo or krel,exo produced 20–50% changes in Ccell,total and/or Ccell,tubulin, whereas Jmaxinter,exo had little or no effects, and none of the three parameters affected Cytotoxicitydonor. These findings indicate (a) exosome release played a greater role on Ccell,tubulin and Ccell,total compared to exosome sorting or re-uptake, especially at low Cmedium,total of 1 nM, and (b) exosome release played a major role in Cytotoxicitydonor at low Cmedium,total but had no effect at high Cmedium,total. These concentration-dependent outcomes indicate complex interplay between exosome-mediated processes and other competing linear and nonlinear processes that also determine Ccell,tubulin and hence Cytotoxicitydonor.
Recipient cells showed changes in the opposite direction with substantial quantitative differences. Because <10% Ccell,total in Donor cells was released in PTX-Exo, which was the drug source for Recipient cells, Ccell,total in Recipient cells was much lower compared to Donor cells. In general, changes in exosome-related model parameters had greater effects on Cytotoxicityrecipient than on Cytotoxicitydonor, especially at the high Cmedium,total of 1000 nM. Changes in Jmaxinter,exo also affected Ccell,total and Ccell,tubulin in Recipient cells more than in Donor cells.
We further compared the effect of exocytosis of PTX-Exo to the effect of a known paclitaxel resistance mechanism, i.e., tubulin alteration resulting in reduced drug binding as reflected by a lower Btubulin,initial [39, 54]. The simulation results indicate a 10-fold increase in krel,exo yielded effects that are comparable to a 2-fold decrease in Btubulin,initial on all three endpoints in Donor cells. Changes in Btubulin,initial had less impact on Recipient cells compared to Donor cells, presumably due to nonlinear exocytosis and endocytosis of PTX-Exo.
Comparison of drug efflux rates by different mechanisms
We used simulations to compare the drug efflux rates via passive diffusion and the two active efflux by Pgp and exosomes; the simulations were for cells treated with PTX Cmedium,total of 0.1–1000 nM for 24 h or after Ccell,total has reached a plateau level. The results, shown in Figure 7, indicate passive diffusion was the dominant efflux mechanism at all Cmedium,total, accounting for at least 92% of total efflux. At Cmedium,total exceeding 30 nM or when PTX binding to tubulin was saturated, Ccell,free increased nonlinearly with Cmedium,total and resulted in greater efflux by all three mechanisms such that efflux by passive diffusion increased from ~12-times to 44-times the active efflux at 1000 nM. The contributions of two active efflux mechanisms changed with Cmedium,total due to saturation of Pgp-efflux, i.e., Pgp-efflux initially exceeded exosome-efflux (e.g., 235%–88% higher at up to 100 nM Cmedium,total) but became less efficient at higher Cmedium,total (e.g., one-fourth the exosome-efflux at 1000 nM).
Figure 7. Contribution of drug efflux mechanisms.
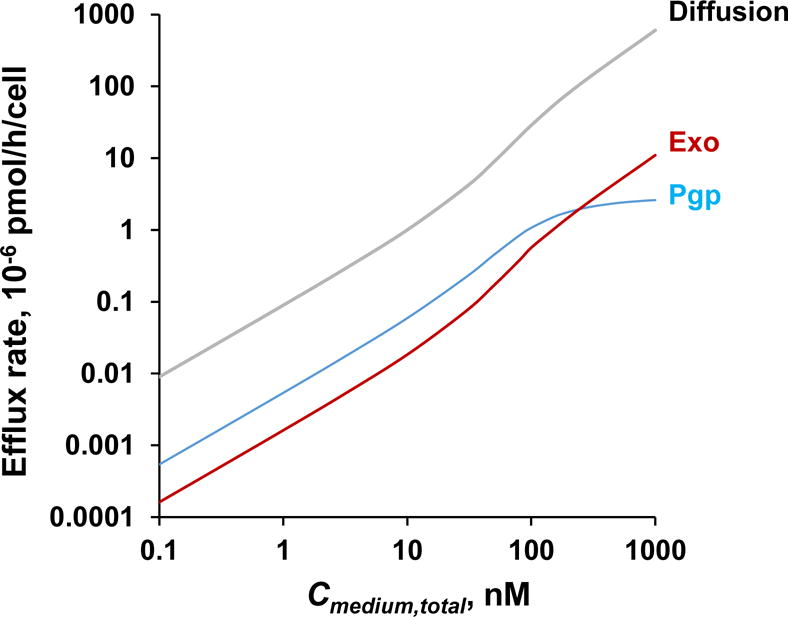
Drug efflux rates by passive diffusion and two active efflux mechanisms (Pgp and exosomes) were simulated using Eq. 3–4 for cells treated with 0.1–1000 nM PTX for 24 h.
Discussion
The present study indicated several new findings regarding the potential roles for exosomes in pharmacological effects of cytotoxics in solid tumors.
We observed that cytotoxics such as DOX and PTX stimulated the production and/or release of exosomes containing high drug levels. The sorting of intracellular drug content into exosomes and their release allows the cell to reduce the intracellular drug concentration (e.g., 15–50% reduction of Ccell,tubulin in the current study). Hence, drug-stimulated exosome production/release may reduce drug activity, rendering this effect a potential new chemoresistance mechanism. This is supported by our QP model-simulated results indicating that in PTX treatments, a 10-fold increase in krel,exo was as effective in reducing Cytotoxicitydonor as a 2-fold reduction in drug binding to tubulin, a known mechanism of PTX resistance [54]. Note the current study was conducted using MCF7 cells which have relatively low Pgp level; under this setting the exosome-efflux accounted for up to 80% of total active drug efflux at initial Cmedium,total of 1000 nM, whereas Pgp-efflux accounted for the remaining 20%. It is conceivable that drug elimination via exosomes may become less pronounced in cells with higher Pgp levels. Studies to investigate the relative contribution of exosome- and Pgp-mediated drug depletion in cells with different Pgp expression are ongoing in our laboratory.
Second, we calculated the drug concentration in PTX-Exo per unit volume (i.e., 2.4 mM in PTX-Exo from MCF7 cells treated with 100 nM Cmedium,total) was 60-times the Ccell,total and 30,000-times the Cmedium,total, indicating PTX-Exo represents an important drug depot. This high PTX level in exosomes is not likely due to pH trapping in acidic exosomes as reported for weakly basic drugs such as DOX and cisplatin [55–57], since the pKa of paclitaxel is 11.5 and is unionized at physiological pH [58]. A possible cause is exosomes contain tubulin [59, 60], which, based on the extensive PTX binding to tubulin and the following calculation, would readily account for the high drug concentration in PTX-Exo. Based on our previously reported Ccell,total of 39 μM at 100 nM Cmedium,total [39] and cell volume of 2 μL per 106 cells [39], and since nearly all Ccell,total is due to macromolecule-bound drug, we calculated the maximal bound drug would be 78 pmole PTX per 106 cells. With tubulin being the primary binding site and because tubulins constitute ~1% of total proteins in neuroblastoma and HeLa cells (3 vs. 300 pg per cell [61]), the amount of tubulin-bound PTX would be 26 μmol per gram tubulin (equals 78 pmole divided by 3 × 106 pg). Using the experimentally determined protein amount in PTX-Exo (1.3 μg per 106 MCF7 cells) and assuming the same 1% protein-to-tubulin ratio as in cells, the tubulin amount in PTX-Exo would be 13 ng per 106 MCF7 cells, corresponding to a maximal tubulin-bound drug concentration of ~3.4 mM in PTX-Exo. This calculation shows that preferential sorting of tubulin and tubulin-bound PTX would lead to the high drug concentration in PTX-Exo observed in the present case. We further evaluated whether inclusion of this process (i.e., sorting of tubulin-bound drug into exosomes) in the QP model would significantly alter the PK/PD in Donor cells; the simulations indicated the model modification yielded negligible changes in the Ccell,total-time profiles (~5% compared to without this additional sorting step).
Third, our results indicate exosome is a mechanism of intercellular drug transfer. Unlike diffusion which is driven by the concentration gradient, exosomes, which use receptor-mediated endocytosis for internalization [14], enable the drug sequestered in exosomes to enter cells irrespective of the concentration gradient. An earlier study shows that exosomes isolated from prostate cancer cells and loaded with PTX extracellularly enter cells via endocytosis [62]. Another notable property is that exosomes, as nanoparticles, are less readily removed from tumor interstitium compared to free drug and thereby provide sustained drug exposure in tumors. This property may have therapeutic importance as our experimental results indicate substantial Cytotoxicityrecipient by PTX-Exo.
Fourth, our study used PTX-Exo collected from epithelial cancer cells. Their pharmacological effects, together with an earlier report that PTX-treated dendritic cells yielded exosomes capable of inducing cytotoxicity in mdr1-transfected canine kidney cells [63], suggest release of pharmacologically active PTX-Exo is a general property that occurs in different cell types/lineages.
Fifth, the finding that exosome release, rather than sorting or internalization via receptor-mediated endocytosis, is the rate-limiting step of exosome appearance in Recipient cells suggests that its perturbation, e.g., by agents that block exosome release, will be a critical determinant of intercellular drug transfer.
Finally, the current study established the utility of QP models to delineate the complex interplay between exosome-related processes and other competing intracellular and extracellular processes, and the pharmacological consequences of these various processes. The current, first-generation model does not account for cell doubling leading to dilution of drug concentration, concentration-dependent cell kill mechanisms (e.g., apoptosis, necrosis), potential cytostasis (which would decrease the kgrowth), nor PTX transfer between Cmedium,free and Cmedium,exo. Such dynamic changes may be considered in future model development. Additional areas that warrant further studies include spatial distribution and residence of exosomes within tumor interstitium, their transport into systemic circulation, and factors that alter their production and release.
Conclusion
The present study demonstrated exosomes as a mechanism to reduce intracellular drug concentration as well as a mechanism of intercellular drug transfer, with significant pharmacological consequences. We further provided the first QP models that (a) captured the PK of cellular PTX processing and drug release through exosomes, and (b) successfully described the effects of exosome exocytosis on PD in Donor cells and drug-naïve Recipient cells. These findings and computational tools may be used to interrogate exosomes as a resistance mechanism and as a means to deliver drugs to the hard-to-reach regions in solid tumors, and to predict the contributions of these opposite effects of exosomes to therapeutic outcomes.
Acknowledgments
Supported in part by research grants R01CA158300 (MGW, JLA), R01CA163015 (MGW, JLA), R01GM100487 (JLA, MGW) and R01EB015253 (JLA, MGW), from National Cancer Institute, National Institute of General Medical Sciences, and National Institute of Biomedical Imaging and Bioengineering, NIH, DHHS, and by RSG-16-006-01-CCE (SW) from American Cancer Society. Authors grateful acknowledge the assistance of Imaging Core Facility at Oklahoma Medical Research Foundation.
Abbreviations
- AChE
acetylcholinesterase
- Conditioned Medium
culture medium collected after incubating cells under preselected conditions
- CV
coefficient of variation
- Cytotoxicitydonor
cytotoxicity in Donor cells
- Cytotoxicityrecipient
cytotoxicity in Recipient cells
- DMSO
dimethyl sulfoxide
- Donor cells
cells that provide drug-containing exosomes
- DOX
doxorubicin
- Drug-Exoconc,time
exosomes collected from Donor cells treated with a drug at a preselected concentration for a preselected duration
- Emax
maximum effect
- ECn
drug concentration producing n% of Emax
- ESCRT
endosomal sorting complexes required for transport
- Growth Medium
cell culture medium supplemented with 10% fetal bovine serum (either unaltered or exosome-depleted) and antibiotics
- IC50
tubulin-bound paclitaxel concentration that generates 50% maximum cell kill
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- PBS
phosphate-buffered saline
- PD
pharmacodynamics
- Pgp
P-glycoprotein
- PK
pharmacokinetics
- PTX
paclitaxel
- QP
quantitative pharmacology
- Recipient cells
drug-naïve cells receiving treatment with drug-containing exosomes
- SD
standard deviations
- SEM
standard error of the mean
- SRB
sulforhodamine B
For abbreviations used in equations
- A
denotes drug amount
- B
denotes drug binding sites
- C
denotes concentration
- D
denotes diffusion
- J
denotes flux
- Kd
denotes dissociation constant
- k
denotes rate constant
- NSB
denotes non-saturable binding
- t
-
denotes time
subscripts are used to denote location (extracellular, intracellular, exosomes), drug moieties with respect to binding status (free, bound, total) to macromolecules/organelles (tubulin, exosomes)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Au JL, Yeung BZ, Wientjes MG, Lu Z, Wientjes MG. Delivery of cancer therapeutics to extracellular and intracellular targets: Determinants, barriers, challenges and opportunities. Adv Drug Deliv Rev. 2016;97:280–301. doi: 10.1016/j.addr.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359–1370. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 3.Fisher DT, Muhitch JB, Kim M, Doyen KC, Bogner PN, Evans SS, Skitzki JJ. Intraoperative intravital microscopy permits the study of human tumour vessels. Nat Commun. 2016;7:10684. doi: 10.1038/ncomms10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM. Exosome mediated communication within the tumor microenvironment. J Control Release. 2015;219:278–294. doi: 10.1016/j.jconrel.2015.06.029. [DOI] [PubMed] [Google Scholar]
- 6.Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 7.Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010;78:838–848. doi: 10.1038/ki.2010.278. [DOI] [PubMed] [Google Scholar]
- 8.Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, Laszlo V, Pallinger E, Pap E, Kittel A, Nagy G, Falus A, Buzas EI. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68:2667–2688. doi: 10.1007/s00018-011-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, Wu Y, Qin J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PloS one. 2015;10:e0125625. doi: 10.1371/journal.pone.0125625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 11.Woodman PG. Biogenesis of the sorting endosome: the role of Rab5. Traffic. 2000;1:695–701. doi: 10.1034/j.1600-0854.2000.010902.x. [DOI] [PubMed] [Google Scholar]
- 12.Savina A, Vidal M, Colombo MI. The exosome pathway in K562 cells is regulated by Rab11. J Cell Sci. 2002;115:2505–2515. doi: 10.1242/jcs.115.12.2505. [DOI] [PubMed] [Google Scholar]
- 13.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Bio Chem. 2010;285:17442–17452. doi: 10.1074/jbc.M110.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, Belting M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A. 2013;110:17380–17385. doi: 10.1073/pnas.1304266110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Logozzi M, De Milito A, Lugini L, Borghi M, Calabro L, Spada M, Perdicchio M, Marino ML, Federici C, Iessi E, Brambilla D, Venturi G, Lozupone F, Santinami M, Huber V, Maio M, Rivoltini L, Fais S. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS One. 2009;4:e5219. doi: 10.1371/journal.pone.0005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, Xiang J, Zhang T, Theilen TM, Garcia-Santos G, Williams C, Ararso Y, Huang Y, Rodrigues G, Shen TL, Labori KJ, Lothe IM, Kure EH, Hernandez J, Doussot A, Ebbesen SH, Grandgenett PM, Hollingsworth MA, Jain M, Mallya K, Batra SK, Jarnagin WR, Schwartz RE, Matei I, Peinado H, Stanger BZ, Bromberg J, Lyden D. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–826. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, Lucci A, Ivan C, Calin GA, Kalluri R. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26:707–721. doi: 10.1016/j.ccell.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, Chen W, Liu F, Sun W, Li XF, Wang X, Wang Y, Xu ZY, Gao L, Yang Q, Xu B, Li YM, Fang ZY, Xu ZP, Bao Y, Wu DS, Miao X, Sun HY, Sun YH, Wang HY, Wang LH. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell. 2016;29:653–668. doi: 10.1016/j.ccell.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Stone L. Kidney cancer: Exosome transmission of sunitinib resistance. Nat Rev Urol. 2016;13:297. doi: 10.1038/nrurol.2016.88. [DOI] [PubMed] [Google Scholar]
- 20.Muller L, Mitsuhashi M, Simms P, Gooding WE, Whiteside TL. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci Rep. 2016;6:20254. doi: 10.1038/srep20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, Coscia C, Iessi E, Logozzi M, Molinari A, Colone M, Tatti M, Sargiacomo M, Fais S. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Bio Chem. 2009;284:34211–34222. doi: 10.1074/jbc.M109.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savina A, Furlan M, Vidal M, Colombo MI. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Bio Chem. 2003;278:20083–20090. doi: 10.1074/jbc.M301642200. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y, Chen Y, Zhang F, Zhao Q, Zhong H. Increased anti-tumour activity by exosomes derived from doxorubicin-treated tumour cells via heat stress. International journal of hyperthermia : the official journal of European Society for Hyperthermic Oncology, North American Hyperthermia Group. 2015;31:498–506. doi: 10.3109/02656736.2015.1036384. [DOI] [PubMed] [Google Scholar]
- 25.Lv LH, Wan YL, Lin Y, Zhang W, Yang M, Li GL, Lin HM, Shang CZ, Chen YJ, Min J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J Bio Chem. 2012;287:15874–15885. doi: 10.1074/jbc.M112.340588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walter S, Buchner J. Molecular chaperones–cellular machines for protein folding. Angew Chem Int Ed Engl. 2002;41:1098–1113. doi: 10.1002/1521-3773(20020402)41:7<1098::aid-anie1098>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 27.Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, Thonel A De, Hamman G, Multhoff A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rebe C, Ghiringhelli F. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. 2010;120:457–471. doi: 10.1172/JCI40483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li XQ, Liu JT, Fan LL, Liu Y, Cheng L, Wang F, Yu HQ, Gao J, Wei W, Wang H, Sun GP. Exosomes derived from gefitinib-treated EGFR-mutant lung cancer cells alter cisplatin sensitivity via up-regulating autophagy. Oncotarget. 2016;7:24585–24595. doi: 10.18632/oncotarget.8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.in, National Cancer Institute.
- 30.Hu G, Chong RA, Yang Q, Wei Y, Blanco MA, Li F, Reiss M, Au JL, Haffy BG, Kang Y. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell. 2009;15:9–20. doi: 10.1016/j.ccr.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 doi: 10.1002/0471143030.cb0322s30. Chapter 3. Unit 3 22. [DOI] [PubMed] [Google Scholar]
- 32.Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 33.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 34.Au JL, Li D, Gan Y, Gao X, Johnson AL, Johnston J, Millenbaugh NJ, Jang SH, Kuh HJ, Chen CT, Wientjes MG. Pharmacodynamics of immediate and delayed effects of paclitaxel: role of slow apoptosis and intracellular drug retention. Cancer Res. 1998;58:2141–2148. [PubMed] [Google Scholar]
- 35.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 36.Geback T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotech. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- 37.Jang SH, Wientjes MG, Au JL. Interdependent effect of P-glycoprotein-mediated drug effux and intracellular drug binding on intracellular paclitaxel pharmacokinetics: application of computational modeling. J Pharmacol Exp Ther. 2003;304:773–780. doi: 10.1124/jpet.102.044172. [DOI] [PubMed] [Google Scholar]
- 38.Jang SH, Wientjes MG, Au JL. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J Pharmacol Exp Ther. 2001;298:1236–1242. [PubMed] [Google Scholar]
- 39.Kuh HJ, Jang SH, Wientjes MG, Au JL. Computational model of intracellular pharmacokinetics of paclitaxel. J Pharmacol Exp Ther. 2000;293:761–770. [PubMed] [Google Scholar]
- 40.Venkatasubramanian R, Arenas RB, Henson MA, Forbes NS. Mechanistic modelling of dynamic MRI data predicts that tumour heterogeneity decreases therapeutic response. British journal of cancer. 2010;103:486–497. doi: 10.1038/sj.bjc.6605773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams DP, Shipley R, Ellis MJ, Webb S, Ward J, Gardner I, Creton S. Novel in vitro and mathematical models for the prediction of chemical toxicity. Toxicology research. 2013;2:40–59. doi: 10.1039/c2tx20031g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min KA, Zhang X, Yu JY, Rosania GR. Computational approaches to analyse and predict small molecule transport and distribution at cellular and subcellular levels. Biopharm & Drug Dispo. 2014;35:15–32. doi: 10.1002/bdd.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C, Krishnan, Xu XY. Towards an integrated systems-based modelling framework for drug transport and its effect on tumour cells. J Bio Eng. 2014;8:3. doi: 10.1186/1754-1611-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang R, Ding Q, Yaqoob U, de Assuncao TM, Verma VK, Hirsova P, Cao S, Mukhopadhyay D, Huebert RC, Shah VH. Exosome Adherence and Internalization by Hepatic Stellate Cells Triggers Sphingosine 1-Phosphate-dependent Migration. J Bio Chem. 2015;290:30684–30696. doi: 10.1074/jbc.M115.671735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diaz JF, Andreu JM. Assembly of purified GDP-tubulin into microtubules induced by taxol and taxotere: reversibility, ligand stoichiometry, and competition. Biochemistry. 1993;32:2747–2755. doi: 10.1021/bi00062a003. [DOI] [PubMed] [Google Scholar]
- 46.Ross JL, Santangelo CD, Makrides V, Fygenson DK. Tau induces cooperative Taxol binding to microtubules. Proc Natl Acad Sci U S A. 2004;101:12910–12915. doi: 10.1073/pnas.0402928101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ajabnoor GM, Crook T, Coley HM. Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells. Cell Death Dis. 2012;3:e260. doi: 10.1038/cddis.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caplow M, Shanks J, Ruhlen R. How taxol modulates microtubule disassembly. J Bio Chem. 1994;269:23399–23402. [PubMed] [Google Scholar]
- 49.Diaz JF, Strobe R, Engelborghs Y, Souto AA, Andreu JM. Molecular recognition of taxol by microtubules. Kinetics and thermodynamics of binding of fluorescent taxol derivatives to an exposed site. J Bio Chem. 2000;275:26265–26276. doi: 10.1074/jbc.M003120200. [DOI] [PubMed] [Google Scholar]
- 50.Li Y, Edsall R, Jr, Jagtap PG, Kingston DG, Bane S. Equilibrium studies of a fluorescent paclitaxel derivative binding to microtubules. Biochem. 2000;39:616–623. doi: 10.1021/bi992044u. [DOI] [PubMed] [Google Scholar]
- 51.Diaz JF, Barasoain I, Andreu JM. Fast kinetics of Taxol binding to microtubules. Effects of solution variables and microtubule-associated proteins. The Journal of biological chemistry. 2003;278:8407–8419. doi: 10.1074/jbc.M211163200. [DOI] [PubMed] [Google Scholar]
- 52.Sutherland RL, Hall RE, Taylor IW. Cell proliferation kinetics of MCF-7 human mammary carcinoma cells in culture and effects of tamoxifen on exponentially growing and plateau-phase cells. Cancer Res. 1983;43:3998–4006. [PubMed] [Google Scholar]
- 53.Atay S, Banskota S, Crow J, Sethi G, Rink L, Godwin AK. Oncogenic KIT-containing exosomes increase gastrointestinal stromal tumor cell invasion. Proc Natl Acad Sci U S A. 2014;111:711–716. doi: 10.1073/pnas.1310501111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10:194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 55.Raghunand N, Martinez-Zaguilan R, Wright SH, Gillies RJ. pH and drug resistance. II. Turnover of acidic vesicles and resistance to weakly basic chemotherapeutic drugs. Biochem Pharmacol. 1999;57:1047–1058. doi: 10.1016/s0006-2952(99)00021-0. [DOI] [PubMed] [Google Scholar]
- 56.Avnet S, Lemma S, Cortini M, Pellegrini P, Perut F, Zini N, Kusuzaki K, Chano T, Grisendi G, Dominici M, De Milito A, Baldini N. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget. 2016;7:63408–63423. doi: 10.18632/oncotarget.11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Federici C, Petrucci F, Caimi S, Cesolini A, Logozzi M, Borghi M, D’Ilio S, Lugini L, Violante N, Azzarito T, Majorani C, Brambilla D, Fais S. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS One. 2014;9:e88193. doi: 10.1371/journal.pone.0088193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shah AK, Wyandt CM, Stodghill SP. Physico chemical characterization of a novel anticancer agent and its comparison to Taxol((R)) Drug Dev Ind Pharm. 2013;39:89–101. doi: 10.3109/03639045.2012.659187. [DOI] [PubMed] [Google Scholar]
- 59.Hegmans JP, Bard MP, Hemmes A, Luider TM, Kleijmeer MJ, Prins JB, Zitvogel L, Burgers SA, Hoogsteden HC, Lambrecht BN. Proteomic analysis of exosomes secreted by human mesothelioma cells. American J Patho. 2004;164:1807–1815. doi: 10.1016/S0002-9440(10)63739-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ribatti D, Marimpietri D, Petretto A, Raffaghello L, Pezzolo A, Gagliani C, Tacchetti C, Mauri P, Melioli G, Pistoia V. Proteome Profiling of Neuroblastoma-Derived Exosomes Reveal the Expression of Proteins Potentially Involved in Tumor Progression. PLoS ONE. 2013;8:e75054. doi: 10.1371/journal.pone.0075054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van de Water L, 3rd, Olmsted JB. The quantitation of tubulin in neuroblastoma cells by radioimmunoassay. J Bio Chem. 1980;255:10744–10751. [PubMed] [Google Scholar]
- 62.Saari H, Lazaro-Ibanez E, Viitala T, Vuorimaa-Laukkanen E, Siljander P, Yliperttula M. Microvesicle-and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J Control Release. 2015;220:727–737. doi: 10.1016/j.jconrel.2015.09.031. [DOI] [PubMed] [Google Scholar]
- 63.Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, Inskoe E, Piroyan A, Sokolsky M, Okolie O, Hingtgen SD, Kabanov AV, Batrakova EV. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine. 2016;12:655–664. doi: 10.1016/j.nano.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]