

Abstract
We have witnessed major successes in the development of effective immunomodulatory therapies capable of reducing adaptive immune-mediated myelin damage in MS over the last 30 years. However, until it is possible to prevent MS or initiate treatment before it has already caused lesions there is a need to repair myelin damage to prevent further axonal loss. The past decade has brought remarkable advances in our understanding of oligodendrocyte biology and the related search for remyelinating therapies in humans. In this review, we first outline the basic biology of central nervous system myelin and remyelination, including a discussion of the major identified pathways and targets that might help yield CNS remyelinating drugs. In conjunction, we provide an overview of techniques that have helped identify compounds capable of promoting oligodendrocyte precursor cell differentiation and myelination. This includes the methods for both initial in vitro screening and subsequent in vivo confirmation of the target. We then review methods proposed to quantify human remyelination in vivo, including visual evoked potentials and putative imaging modalities. As the remyelination era approaches, with the announcement of the first positive trial in remyelination, we are now tasked with answering new questions regarding patient-specific factors (e.g., age) that may influence the extent and optimal therapeutic window for remyelination.
Electronic supplementary material
The online version of this article (10.1007/s13311-017-0577-0) contains supplementary material, which is available to authorized users.
Keywords: Remyelination, Clemastine, LINGO1, Neurodegeneration, Muscarinic, Multiple sclerosis
Introduction: Rationale for Remyelination
Great strides have been made in the past generation to develop drugs that can attenuate and hopefully eliminate new immune-mediated damage in multiple sclerosis (MS) particularly limiting the aberrant self-targeted peripherally generated immune response that characterizes the disease. These new treatments are resulting in delayed progression of disease, and the newer MS therapies may even represent “the end of the beginning”. We now expect significant and robust constraint on adaptive immune-mediated injury and marked reduction of new inflammatory demyelinating MS lesions in relapsing disease [1] and have even seen the first evidence of a dent in the course of primary progressive MS [2]. Many new treatments promise to effectively stop the development of new lesions, but until it is possible to predict and eventually prevent MS, there is a need to promote repair of existing lesions in people already living with MS.
One major goal for repair is the remyelination of demyelinated axons. Myelination is a natural consequence of oligodendrocyte differentiation. Remyelination therefore involves promoting the endogenous pool of oligodendrocyte precursor cells (OPCs) in the brain to differentiate into mature oligodendrocytes, and thereby wrap new myelin around axons. The number of potential therapies capable of possible remyelination in the translational pipeline has expanded significantly in the past 5 years, and the first successful phase II clinical trial for remyelination meeting its a priori-defined clinical endpoints in an intention-to-treat analysis was recently announced [3]. In this article, we review the basic mechanisms for remyelination, as well as the advances that led to the identification of promising therapies in various current stages of translational development. Then, we move towards a more clinical perspective: when during the clinical course of MS should remyelination be measured, and how should we measure its effectiveness?
Remyelination: Overview of Basic Mechanisms
Myelination appears to have evolved in jawed vertebrates to improve the speed and efficiency of conduction of action potentials over long axonal distances by using saltatory, rather than membrane, conduction. The existence of insulating myelin around most of the axonal length means that membrane potential-sensing ion channels can be aggregated at nodes where ion exchange is concentrated and focused. This dramatically speeds axon potential transmission by up to 2 orders of magnitude (holding axonal diameter constant), and is, in turn energetically favorable for the neuron. Myelin also serves other important functions including likely providing physical and metabolic support for axons [4, 5], and may play a role in tuning and adjusting the exquisite timing required for the coordinated integration of action potentials coming in from multiple areas of the central nervous system (CNS) [6].
Myelination of the CNS during development, i.e., oligodendrogenesis, requires the following steps: 1) OPCs arise from differentiation of immature progenitors (neural stem cells); 2) OPCs then proliferate and tile themselves, i.e., distribute themselves, in a grid-like manner occupying nonoverlapping domains (through contact-mediated inhibition of growth) across the CNS [7]; then 3) OPCs differentiate into myelin-forming oligodendrocytes, under regulation of genes and transcription factors (e.g., Olig1, Olig2, Sox10, NKx2.2 and Myr; see, e.g., [8] for a review); and finally 4) oligodendrocytes wrap myelin around axons. A number of pathways related to OPC differentiation have been identified, and the fact that many of these are inhibitory, including LINGO-1 [9, 10], polysialylated neural cell adhesion molecule (PSA-NCAM) [11, 12], Notch signaling [13–16], hyaluronan [17], Nogo-A [18], and the Wnt/β-catenin pathway, suggest that endogenous inhibitors of developmental myelination ensure that the myelination is (at least partially) a regulated process [8]. However, the retinoic acid receptor RXR-γ pathway facilitates oligodendroglial development and myelination [19], and the exact cues required for the initiation of myelination remain obscure.
A traditional and historically canonical view in the field had been that axonal signaling drives myelination. Many of the signaling pathways initially identified were axonally derived (LINGO-1 [9, 10], PSA-NCAM [11], Notch signaling [13–16], retinoic acid receptor retinoid X receptor (RXR-γ) pathway [19]), supporting the perception that axons regulate myelination. More recently, this perception that myelination depends on axon-derived inductive cues has been challenged by evidence that oligodendrocytes can spontaneously myelinate not only chemically cross-linked dead axons [20], but also other inert structures that are designed to have the mechanical properties of axons, such as polystyrene nanofibers above a threshold diameter [21].
After injury, remyelination is necessary to restore metabolic support and fast conduction of nerve impulses, and to protect axons from degradation. Figure 1 provides an overview of the remyelination process. According to the recapitulation hypothesis of myelin regeneration, the mechanisms of myelin repair recapitulate those of primary developmental myelination [22]. Therefore, for the remyelination process to occur it is necessary that either the endogenous pool of adult endogenous OPCs are available at the site of damage or they must migrate to the lesion site. OPCs originate from neural stem cells in distinct locations in a highly regulated and orchestrated process during development and from specific areas such as the subventricular zone in the adult organism. OPCs must proliferate and regulate their spacing via a self-repulsive property that creates a relatively uniform tiling of NG2+ OPCs throughout the adult CNS. It has been clearly established that OPCs are found even within MS lesions on histopathology [23]. Following differentiation oligodendrocytes are capable of extending processes towards demyelinated/injured axons, making contact with them, and then wrapping concentric new layers of myelin around them, which is then compacted [24]. Remyelination has been demonstrated to occur in the adult mammalian brain [25], and may involve either 1) replacement of existing quiescent oligodendrocytes; or 2) addition of additional myelin internodes.
Fig. 1.
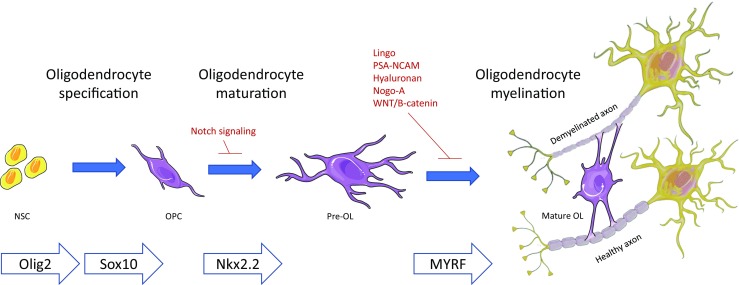
Remyelination. Oligodendrocyte precursor cells (OPCs) undergo maturation to oligodendrocytes, which then wrap myelin around demyelinated axons. At each step, the remyelination process may be promoted or inhibited by intrinsic and extrinsic signals or by environmental cues (adapted from [40] and [107]). NSC = neural stem cell; OPC = Oligodendrocyte precursor cell; OL = Oligodendrocyte; PSA-NCAM = polysialylated neural cell adhesion molecule; MYRF = Myelin Regulatory Factor
In a young healthy animal, chemical demyelination of the CNS is followed by robust and predictable remyelination as a testament to the viability and availability of OPCs. However, the completeness and efficiency of the remyelinating process may be influenced by a number of endogenous factors, which may include species-specific factors (e.g., more complete in the zebrafish than in mammals [26]), neuroanatomical location (e.g., may be faster in the murine corpus callosum than in the cortical gray matter [27]; potentially more extensive in subcortical lesions than periventricular lesions and near absent in cerebellar lesions in humans [28]), and aging. Aging is associated with a number of impairments of remyelination, including decreased OPC recruitment and differentiation [23, 29], and shorter internodes [26, 30, 31]. These age-associated changes likely arise from changes in environmental signaling [32] and in epigenetic regulation [33, 34]. The effect of increasing age on impaired remyelination is further supported by reversal of age-associated remyelination deficits with telomerase reactivation [35], and with enhancement of remyelination after heterochronic parabiosis, in which unidentified circulating factors may, respectively, inhibit or enhance remyelination, including blood-derived monocytes from a younger mouse recruited to demyelinating lesions, by increasing M2 microglia and macrophages in these lesions [36, 37]. In humans, OPCs may represent a smaller proportion of the total glial population [38] than in other mammals such as rodents, where they make up approximately 8% to 9% of the total cell population in white matter [39].
In MS, in the lesions caused by demyelinating injury, remyelination appears to be impaired, particularly with advancing age and disease duration [24, 28]. However, much remains to be understood about the pattern and pace of remyelination over the course of the disease, and the reasons for the impaired signaling that leads to failure of remyelination (reviewed in [40]). The following three steps have been suggested to be major contributors to this failure: 1) failure of recruitment of OPCs to demyelinated lesions because of aberrant cues (from netrins [41], semaphorins [42], or guidance chemokines, e.g., CXCL1 [43]); 2) failure of OPC differentiation because of ineffective clearance of myelin debris by microglia or monocyte-derived macrophages, and expression of endothelin-1 by reactive astrocytes in demyelinated lesions, which promotes Notch activation in OPCs, and their consequent failure to differentiate and mature [44]; 3) and ultimately, failure of OPC maturation because of inadequacy of myelin-specific gene expression and the consequent increases in myelin wrapping. Additional factors include re-expression on the surface of demyelinated axons of PSA-NCAM. This glycoprotein negatively regulates myelin, preventing myelin-producing cells from attaching to the axon and blocking OPC differentiation [11, 45]. Hyaluronan has also been reported to block OPC maturation and myelin repair: present in MS lesions, it is degraded by hyaluronidases into oligomers that block OPC maturation and myelin repair through Toll-like receptor 2–MyD88 (Toll-like receptor-2 adaptor molecule) signaling. [46]
Astroglial communications, in particular, appear to be impaired in MS, through mechanisms that include disruption of the juxtaparanodal proteins (which neighbor the nodes of Ranvier) [47], disruption of oligodendrocyte gap junctions [48], and dysregulated inhibitory signaling.
Pharmacological Approach to Remyelination: Which Drugs and How to Test Them?
Types of Approaches
A number of approaches have been advanced to overcome the signaling barriers to effective remyelination of endogenous OPCs in MS. Pharmacological approaches represent the focus of the current review, but non-medicinal approaches, as well as approaches aimed at cell reprogramming, have also been developed. Rodent models suggest a pro-remyelinating effect of for activity including natural endogenous processes, such as motor learning leading to increased OPC differentiation [49], as well as artificial approaches such as electroacupuncture. For example, electroacupuncture has been reported to enhance the removal of myelin debris by activated microglia after cuprizone-induced demyelination [50]. Activation of the molecular program of oligodendrogenesis for remyelination through reprogramming of stem cells is also being explored. This has already been reported in the peripheral nervous system through induction of human bone marrow-derived Schwann cells, [51], although differences between the transcriptional programs of remyelination and oligodendrogenesis likely exist [52].
High-Throughput Screening for Compounds
The search for pharmacological compounds for remyelination has been bolstered not only by an expanding understanding of the signaling pathways involved in remyelination, but also by a series of innovative screening techniques that have substantially increased the number of candidate compounds. We describe 3 of the recent independent techniques that have been developed to screen for drugs that promote OPC differentiation and myelin wrapping. 1) The first approach deployed larval zebrafish, quantifying the in vivo effect of compounds from a re-profiling library on the number of dorsally migrated spinal cord olig2 + cells (OPC proliferation) and levels of larval myelin basic protein transcripts (myelination level). Both novel compounds (src family kinase inhibitor PP2, a biogenic amine, and a thioxanthene), as well as previously described ones [peroxisome proliferator-activated receptor (PPAR) agonist, steroid hormones and src family kinase inhibitors] were identified by this screen [53]. 2) A second, in vitro, approach leveraged the ability of polystyrene nanofibers above a threshold diameter to mimic an effective mechanically based cue to induce OPCs to proliferate and differentiate. From this observation, an alternative but related approach known as the binary indicant for myelination using micropillar arrays (BIMA) high-throughput assay was developed, allowing quantification of the extent and length of myelin wrapping in response to compounds from a library of compounds. This technique was used to rapidly screen 1000 Food and Drug Administration (FDA)-approved bioactive molecules. Major clusters identified through this approach included antimuscarinic compounds, which enhanced oligodendrocyte differentiation and remyelination [54], as well as selective estrogen receptor modulators (unpublished, personal communication). 3) A third approach involves screening a library of bioactive small molecules on rodent pluripotent epiblast stem cell-derived OPCs. This yielded miconazole and clobetasol as possible remyelinating agents, and included a description of further functional validation [55].
Quantifying Remyelination in vivo in Humans
In the absence of tissue specimens, several approaches have been proposed to measure the efficacy of candidate compounds on human remyelination in vivo. As clinical trials for remyelination begin to show positive results, there is a need to identify and/or develop more sensitive and specific measures of remyelination, given the multiple potential mechanisms other than remyelination through which clinical improvement may be achieved (decreased inflammation, increased synaptic plasticity, neuronal survival, or other compensatory strategies).
Magnetic Resonance Imaging
Using conventional magnetic resonance imaging (MRI), an early approach suggested that we might approximate the extent of remyelination by calculating the ratio between the size or number of T2 hyperintense lesions and their accompanying T1 hypointensity. A number of advanced MRI techniques—such as magnetization transfer imaging [56, 57], including magnetization transfer ratio (MTR) [58], diffusion-weighted imaging, and T2 relaxometry—generate quantitative measures [59–61], and have been postulated to reflect important radiological properties of myelin status (see [62] for a review). Altered ratios of choline, N-acetylaspartate, myoinositol, and glutamate detected by MR spectroscopy may also play a role in detecting patterns of demyelination and remyelination in vivo [63]. Further, MRI of the spinal cord represents an increasingly attractive surrogate marker for multicenter clinical trials of progression, and advanced cervical cord MRI measures, including spinal cord magnetization transfer, may hold promise as outcome measures in remyelination trials [62, 64].
However, these approaches have only been assessed with clinical demyelination and partial remyelination in animal models, and their specificity for myelin remains unclear as their utility in measuring remyelination has never been demonstrated. Some of these methods have been referred to using terms such as “myelin water fraction”, but the method measures pools of large macromolecules—not myelin specifically. All existing myelin imaging techniques may reflect not only myelin content, but also inflammatory infiltrates, edema/water, as well as axons. [62, 64].
Positron Emission Tomography
Positron emission tomography using the intravenously injected radiolabeled amyloid tracer Pittsburgh compound B (PiB), i.e., [11C]PIB, has been investigated as a potential tool for imaging CNS myelin [65]. An exploratory study of longitudinal (1–4 months) changes in [11C]PiB in 20 individuals with MS [66] raised a number of important questions about the spatial and temporal associations between myelin repair and axonal damage. First, changes in myelin content were noted primarily in the peripheral lesional area, presumably the area where both new demyelination and active remyelination occur. Second, a high between-patient variability was found for all the indices of dynamic myelin content change (particularly the index of dynamic remyelination), suggesting that patient-specific features (e.g. age or disease stage) may determine the balance between demyelination and remyelination. Third, a strong correlation between remyelination and clinical disability was noted, raising the possibility of a fast and severe process of axonal degeneration occurring immediately after acute demyelination in some patients with a worse prognosis, which precedes any remyelination and results in fewer remyelinating voxels. While requiring ongoing validation and expansion, these observations raise the possibility that it will eventually be possible to ascertain patient-specific threshold levels of, and temporal windows for, remyelination after demyelinating events [66].
Visual Evoked Potential
This electric potential recorded from the visual cortex in response to a repeating visual stimulus evaluates the time needed for a signal to travel from the retina to the brain’s visual cortex, while averaging out background cortical potentials. Delay in the P100 latency component of visual evoked potentials (VEPs) is common in MS [67]. Our preliminary findings suggest that VEP is an even more sensitive indicator of de- and remyelination than clinical scores in both cuprizone and experimental autoimmune encephalomyelitis (EAE) models. VEP latency was the most sensitive predictor of remyelination in the clemastine phase II clinical trial [3]. Results strongly suggest that the observed electrophysiologic effects are evidence of remyelination [68], and establish VEPs as a robust, reliable method of evaluating remyelination at a clinical level in humans [3, 67].
Overview of Lead Remyelinating Compounds
As discovery of remyelinating pathways and development of methods for high-throughput screening continue to expand, the number of candidate remyelinating compounds continues to increase. For the current review, we focus on the most clinically promising, as well as the most clinically advanced, compounds that have undergone testing in humans. We then briefly review other compounds in the translational pipeline. These are summarized in Table 1.
Table 1.
Published preclinical and clinical trials results related to remyelination.
| Compound | Preclinical data | Phase I | Phase II | Phase III |
|---|---|---|---|---|
| Clemastine (antimuscarinic) | Y | Y | 150-day, placebo-controlled, crossover trial Reduced latency of VEPs in eyes affected by chronic optic neuritis, relative to placebo |
|
| GSK239512 (antimuscarinic) | Y | Y | Randomized, parallel-group, placebo-controlled, double-blind, multicenter trial Did not meet primary or secondary endpoints; did increase lesional MTR relative to placebo |
|
| Benztropine (antimuscarinic) | Y | Y | ||
| Opicinumab (anti-LINGO1) | Y | Y | 4 phase II trials NCT01721161: reported no significant difference in VEP latency | |
| Domperidone | Y | Y | Ongoing | |
| rHIgM22 | Y | Not published | ||
| Quetiapine | Y | Y | ||
| Anti-SEMA4D (VX15/2503) | Y | Not published | ||
| Thyroid hormone | Y | |||
| SERMs | Y | |||
| RXR-γ | Y | Y | ||
| VDR | Y | |||
| Clobetasol | Y | |||
| Kappa opioid | Y | |||
| Fingolimod | +/- | +/- | Phase II and III clinical trials have been completed, but there was no assessment of remyelinating activity. | |
| Biotin | Y |
Y = yes; VEP = visual evoked potential; MTR = magnetization transfer ratio; LINGO1 = leucine-rich repeat and immunoglobulin-like domain-containing protein 1; SEMA4D = semaphorin 4D; SERM = selective estrogen receptor modulator; RXR = retinoid X receptor; VDR = vitamin D receptor
Compounds With Reported Efficacy Data From Clinical Trials
Antimuscarinic compounds
Clemastine
Clemastine is an older-generation, widely available antihistamine (H1 receptor antagonist) and M1/M3 muscarinic receptor reverse antagonist.
As noted above, clemastine emerged from screening in BIMA, a high-throughput in vitro screening system for compounds that promote OPC proliferation or oligodendrocyte differentiation [69]. Muscarinic compounds have also been identified using another system, primary rat optic nerve-derived progenitor cells [70]. It is the most robustly validated compound with remyelinating capacity identified to date with demonstration of effect documented in multiple independent laboratories and using a wide variety of model systems. Clemastine has subsequently been found to promote remyelination in the cuprizone model of demyelinating injury [69], as well as to enhance oligodendrocyte differentiation and myelination in the prefrontal cortex after social isolation in adult mice, through epigenetic regulation [71]. In further analyses, the remyelinating effects of clemastine were found to be mediated specifically by the M1 muscarinic receptor: oligodendroglial-specific genetic ablation of M1 was found to lead to accelerated remyelination in EAE absent the administration of clemastine, preventing axonal loss and improving functional recovery [68].
A successful phase II, 150-day, double-blind, placebo-controlled, crossover trial has been completed (NCT02040298). In this trial, 50 patients with relapsing–remitting MS with chronic longstanding optic neuropathy were randomized to receive either clemastine (5.3 mg PO twice daily) for 90 days, followed by placebo for 60 days, or vice versa. The trial exploited the capacity to assess participants who had adequate axonal substrate to remyelinate by screening potential participants using optical coherence tomography. All participants underwent VEPs at baseline, month 1, drug transition point (month 3), and end of trial (day 150). Clemastine was found to reduce P100 latency by 3.2 ms/eye if the trial results were analyzed as a “delayed treatment trial”, or 1.7 ms/eye when considered as a “crossover trial” [3].
Other Drugs Targeting Neurotransmitter Receptors
GSK239512
The remyelinating properties of this second antihistamine, a selective, orally bioavailable, and CNS-penetrant H3 receptor antagonist/inverse agonist originally developed to treat schizophrenia and Alzheimer’s disease, were investigated based on proprietary screens separate from those discussed above. GSK239512 was well tolerated and with good CNS penetrance (> 90% H3 receptor occupancy in the brain, at trough plasma concentrations) [72–74].
The efficacy of GSK239512 was tested in a small phase II, randomized, parallel-group, placebo-controlled, double-blind international, multicenter study of adults with relapsing–remitting MS (NCT01772199). This trial failed to meet its primary endpoint or any of its major secondary endpoints. However, in a post-hoc analysis on MTR to assess in vivo myelin content, oral GSK239512 daily as an add-on to the injectable disease-modifying treatments showed possible small mean improvements in lesional MTR relative to placebo [75].
Benztropine
Benztropine is an FDA-approved treatment for Parkinson’s disease, owing to its anticholinergic effects. However, it also works as an antihistamine and as a dopamine reuptake inhibitor, and in preclinical studies was found to induce OPC differentiation by directly antagonizing M1/M3 receptors, decrease clinical severity in EAE, and enhance remyelination after cuprizone-induced toxic demyelination [70]. To our knowledge, this medication has not yet been tested in clinical trials for remyelination.
LINGO1
The leucine-rich repeat and immunoglobin-like domain-containing protein 1 (LINGO1) is expressed on OPCs and neurons and may negatively regulate myelination. Initial mechanistic hypotheses centered on its role as part of a ternary receptor complex. This complex includes LINGO1, Nogo-66 receptor (ligand-binding subunit), and tumor necrosis factor receptor superfamily members p75NTR or Troy/Taj (signal-transducing subunit). To negatively regulate myelination in the CNS, the complex inhibits the RhoA signaling pathway engaged by the p75NTR or Troy components of the Nogo-66 receptor/LINGO1/p75NTR–Troy signaling complex, in response to the presence of myelin oligodendrocyte glycoprotein, myelin-associated glycoprotein, or Nogo-66. [76] LINGO1 has also been reported to work through other mechanisms, including by negatively regulating signaling by the neuregulin receptor subunit ErbB2, or signaling by receptor tyrosine kinases (ErbB1, ErbB2, ErbB4, TrkA, TrkB, and TrkC).
Preclinical studies from a single laboratory suggested LINGO1 as a potential target included the observation that in LINGO1−/− mice, OPC differentiation and OL maturation are enhanced, leading to precocious and enhanced myelination. [10, 77, 78]. Further, short hairpin RNA knockdown or functional inhibition of LINGO1 in oligodendrocytes in vitro enhanced oligodendrocyte maturation [79]. These observations led to the development of a functionally inhibitory monoclonal antibody to LINGO1 (mAb3B5), which promoted remyelination and recovery of axonal function in the spinal cord after focal lysolecithin-induced toxic demyelination, as well as after EAE [80].
A LINGO1 function-blocking monoclonal antibody, opicinumab (BIIB033; Biogen, Cambridge, MA, USA), was developed for humans. Opicinumab passed a phase I safety trial in subjects with MS (NCT01244139) [81]. The protein was evaluated in 4 phase II randomized, double-blind, placebo-controlled clinical trials (NCT02657915, NCT01864148, NCT01721161, NCT01721161). A final publication is available for the acute optic neuritis trial (NCT01721161, RENEW study), with the primary intent-to-treat analysis revealing no significant difference in remyelination at week 24, as measured by optic nerve conduction latency of the affected eye versus the unaffected eye at baseline, using full-field VEP. However, there was a signal in the per-protocol analysis, suggesting that further evaluation may be warranted [82]. A non-specific effect related to the use of immunoglobulin also cannot be ruled out based on the high dose (100 mg/kg) and the use of placebo as a control. An assessment of the drug in chronic demyelination yielded negative results for the primary endpoint, but the manufacturer suggested that in post-hoc analyses there was a possible effect in some subgroups. Preliminary results from the SYNERGY trial (NCT01864148) testing opicinumab over a range of doses as an add-on to interferon therapy, reported no significant increase in the percentage of patients who improved in a range of MS disability scores (the EDSS, Timed 25-Foot Walk, Paced Auditory Serial Addition Test, and 9-Hole Peg Test) over 3 months, relative to placebo. Some more significant improvements were noted in individuals receiving middle-range doses of opicinumab. Final peer-reviewed data are awaited.
Overall, limitations of this therapeutic approach may include 1) downstream effects due to cross-talk between signaling pathways; 2) poor expression of LINGO1 at the cell surface relative to intracellular vehicles of the endocytic pathways; 3) the low CNS penetrance of systemically injected opicinumab. Additionally, limitations in the SYNERGY trial included 4) selection of lower-efficacy disease-modifying treatments to which opicinumab was “added on”, which may have only partially reduced inflammatory activity in the brain prior to assessing remyelination, as well as 5) selection of functional outcomes that may reflect a variety of neuropathological processes and show low specificity for remyelination [83, 84].
Compounds Without Published Efficacy Data From Clinical Trials
rHIgM22
CNS-reactive, remyelination-promoting IgMs are germline gene-encoded autoantibodies that are part of the natural immunoglobulin repertoire in humans; and target cell-surface antigens of oligodendrocytes and myelin. The recombinant human remyelination-promoting monoclonal IgM antibody was developed through the observation that serum-derived IgM22 bind to human oligodendrocyte surface antigens and exerts possible remyelinating effects [85].
rHIgM22 has been reported to induce OPC proliferation; however, the relationship of this proliferation to OPC differentiation remains unclear. The antibody has also been reported to protect oligodendrocytes from proapoptotic signaling, possibly through the platelet-derived growth factor/platelet-derived growth factor-α receptor signaling pathway [85–87].
Two phase I studies have been developed (Acorda Therapeutics, Ardsley, NY, USA), 1 completed in January 2015 (NCT01803867) and 1 ongoing (NCT02398461), but complete descriptions of these trial results have not been published.
Quetiapine
This nonselective G protein-coupled receptor antagonist with a number of remyelinating properties in preclinical models [88]. It is currently undergoing phase I clinical testing in MS (NCT02087631). However, its clinical profile (sedation) may be limiting.
Domperidone
This D2/D3 dopamine receptor antagonist is the subject of 2 phase II clinical trials in MS (NCT02493049; NCT02308137), based on its effects on the prolactin receptor signaling pathway.
Antisemaphorin 4D (VX15/2503)
Antisemaphorin D is a monoclonal antibody to semaphorin 4D, which inhibits remyelination through a number of mechanisms, including OPC survival and differentiation [89]. Results from the phase I clinical trial (NCT01764737) are awaited.
Nuclear Hormone Agonists
Thyroid hormone plays a well-recognized role in regulating oligodendrocyte differentiation and myelination during development [83], and liothyronine sodium is currently undergoing phase I clinical trial to evaluate its effect on VEPs relative to placebo (NCT02760056). However, owing to the widespread effects of thyroid hormone, there has been a search for more specific agents capable of promoting remyelination. One such agent, GC-1, is a thyromimetic with selective thyroid receptor β action, and with a potentially limited side effect profile. GC-1 has been reported to enhance oligodendrocyte differentiation in vitro and during developmental myelination in vivo [84].
Compounds With Promising Preclinical Data That to Our Knowledge (clinicaltrials.gov search 10 April 2017) Have Not Yet Been Tested in Clinical Trials for Remyelination
Nuclear Hormone Receptor Agonists
Selective Estrogen Receptor Modulators
Selective estrogen receptor modulators have been identified using high-throughput screening system as molecules that promote oligodendrocyte differentiation, as well as protect their viability and differentiation under inflammatory conditions [90, 91].
RXR-γ
RXR-γ is highly expressed in MS lesions, both acute and remyelinating [92]. Mice that lack RXR-γ show OPC proliferation but delayed differentiation; the RXR agonist 9-cis-retinoic acid promotes OPC differentiation and remyelination [19]. Bexarotene is a licensed RXR agonist used as an antineoplastic, and has been investigated for its effects on brain amyloid in Alzheimer’s disease [93]. Interestingly, the vitamin D receptor has recently been reported to serve as one of the nuclear receptors of RXR-γ in OPCs and oligodendrocytes [94]; in fact, promotion of VDR signaling through vitamin D has been suggested as an additional mechanism through which to promote myelin repair [94].
PPAR-γ
Agonism of the nuclear receptor PPAR-γ has been reported to protect mitochondrial function and thereby OPC differentiation [95].
Clobetasol
Clobetasol is a glucocorticoid used for a number of inflammatory dermatoses and psoriasis, and off-label for alopecia. It emerged from the pluripotent epiblast stem cell-derived OPCs screen for remyelinating compounds, and subsequently underwent functional validation [55]. It also appeared to increase the number of mature oligodendrocytes in a mouse model of neuromyelitis optica [96].
Kappa Opioid
The kappa opioid receptor (KOR) forms part of the endogenous opioid system along with the mu-opioid receptor (MOR), the delta opioid receptor (DOR), and their ligands. In the screen of a G protein-coupled receptor small-molecule library using BIMA, a cluster of KOR compounds was identified that promoted oligodendrocyte differentiation and myelination in purified oligodendroglial co-culture. The compound (±)U-50488, the most effective in promoting differentiation, accelerated the kinetics of remyelination in vivo after focal lysolethicin-induced toxic demyelination, and enhanced the differentiation of human-induced pluripotent stem cell-derived OPCs into mature oligodendrocytes. Deletion of KOR abolished these effects [97]. Further, deletion of KOR has been found to exacerbate EAE, with the converse observed with KOR activation; these effects appear to be mediated not by immune cell differentiation and function, but confirm KOR effects on oligodendrocyte differentiation and myelination in vitro and in vivo [98].
Fingolimod
Fingolimod is an FDA-approved disease-modifying therapy for MS whose primary disease-modifying mechanism relies on lymphocyte sequestration within lymph nodes. At nanomolar concentrations, it has been reported to promote the survival of neural stem cells, which the authors suggested may enhance the ultimate development of mature oligodendrocytes in vivo and subsequent remyelination [99]. However, given the need for OPC differentiation the exact mechanisms for this reported effect remain unclear and inadequately described.
Biotin
Interest in biotin as a potential reparative agent arose from observations, in an open-label pilot phase II trial of high-dose biotin (MD1003, 100–300 mg/day), that some patients with primary or secondary progressive MS experienced clinical improvement. While in some this improvement was in walking distance and walking speed, the investigators also noted 1) improved visual acuity and normalization of VEPs; and 2) normalization of the choline/creatine ratio on magnetic resonance spectroscopy) [100]. However, given the open-label nature of the study, these reports must be viewed with caution. Publication of results from a follow-up double-blind, placebo-controlled, multisite study is expected, but in conference proceedings a delayed clinical improvement was announced, which was attributed to a remyelination effect (rather than a more immediate effect on neurotransmission). The postulated mechanism for remyelination is that biotin is an essential cofactor for ACC1 and ACC2, two isoforms of acetyl-CoA carboxylase, expressed in the CNS by oligodendrocytes, and involved in regulation of the rate of fatty-acid synthesis (building block of myelin) [101]. However there is no direct experimental evidence from any animal model or in vitro data that suggests that biotin has a remyelinating effect or that it impacts oligodendrocyte biology.
Towards Remyelination in Clinical Practice: When and in Whom?
As the era of effective remyelinating therapies appears on the horizon, there is a need to define the demographic and clinical characteristics of the patients who might benefit from them, as well as the optimal time window for treatment: when during the course of disease and for how long. It is unclear, for instance, if remyelinating effects are more robust at the time of acute injury and how long after lesion formation remyelinating potential is lost. The overarching goal should be to enhance endogenous capacity for OPC differentiation and myelination before neurons enter neurodegenerative pathways [102]. Reassuringly, data from EAE suggest that the early myelin formed after EAE induction appears to be stable, possibly because immature myelin does not express myelin oligodendrocyte glycoprotein, and it supports axonal integrity and neuronal function [68].
As suggested by the paper assessing myelin formation after EAE and treatment with clemastine, as well as the initial longitudinal positron emission tomography data described above, remyelination is likely coincident or immediately follows demyelination [66], and is perhaps associated with inflammatory activity [103]. However, the positive results from the phase II trial of clemastine for chronic optic neuritis also suggest that even long after inflammation, ongoing remyelination may be achievable. These results also potentially expand the optimal duration of treatment, from a time-limited window after acute clinical events or new inflammatory lesions to a more chronic timeframe. It is unclear if remyelinating therapies will need to be given chronically to help overcome or optimally limit injury.
Finally, a high between-patient variability suggests that age, disease duration, as noted above, or other patient-specific factors may influence a person’s capacity for dynamic remyelination [66]. For example, it has been suggested that propensity for remyelination might be responsive to gonadal hormones, which could explain why women with MS, who experience a relatively more benign course during their reproductive years, tend to “catch up” to men with ovarian aging [104, 105].
Summary
Many questions about remyelination in MS remain to be solved, the most important of which concerns how well remyelination in humans can be predicted based on preclinical models [106].
In the coming years, we anticipate significant advances in the ability to promote, measure, and predict remyelination in individuals with MS at various stages in their disease course. We have recently seen the first evidence that remyelination is a tractable and assailable clinical problem, and we are now at the precipice of an important new era in MS therapeutics.
Electronic supplementary material
(PDF 1224 kb)
(PDF 1224 kb)
Acknowledgments
We thank Michael Devereux for his assistance with figure preparation and Dr. Jonah Chan for his scientific collaboration and critical review of the manuscript.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Contributor Information
Riley M. Bove, Email: riley.bove@ucsf.edu
Ari J Green, Email: agreen@ucsf.edu.
References
- 1.Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376:221–234. doi: 10.1056/NEJMoa1601277. [DOI] [PubMed] [Google Scholar]
- 2.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;276:209–220. doi: 10.1056/NEJMoa1606468. [DOI] [PubMed] [Google Scholar]
- 3.Green AJ, Gelfand JM, Cree BA, et al. A randomized-controlled double-blinded trial of clemastine fumarate as a remyelinating therapy for chronic optic neuropathy in multiple sclerosis. Lancet. in press. [DOI] [PubMed]
- 4.Hirrlinger J, Nave KA. Adapting brain metabolism to myelination and long-range signal transduction. Glia. 2014;62(11):1749–1761. doi: 10.1002/glia.22737. [DOI] [PubMed] [Google Scholar]
- 5.Funfschilling U, Supplie LM, Mahad D, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485(7399):517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomassy GS, Berger DR, Chen HH, et al. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science. 2014;344(6181):319–324. doi: 10.1126/science.1249766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci. 2013;16(6):668–676. doi: 10.1038/nn.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- 9.Mi S, Hu B, Hahm K, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13(10):1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- 10.Mi S, Miller RH, Lee X, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8(6):745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- 11.Charles P, Hernandez MP, Stankoff B, et al. Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc Natl Acad Sci U S A. 2000;97(13):7585–7590. doi: 10.1073/pnas.100076197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fancy SP, Baranzini SE, Zhao C, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23(13):1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Sdrulla AD. diSibio G, et al. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 1998;21(1):63–75. doi: 10.1016/S0896-6273(00)80515-2. [DOI] [PubMed] [Google Scholar]
- 14.Genoud S, Lappe-Siefke C, Goebbels S, et al. Notch1 control of oligodendrocyte differentiation in the spinal cord. J Cell Biol. 2002;158(4):709–718. doi: 10.1083/jcb.200202002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watkins TA, Emery B, Mulinyawe S, Barres BA. Distinct stages of myelination regulated by gamma-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron. 2008;60(4):555–569. doi: 10.1016/j.neuron.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Argaw AT, Gurfein BT, et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc Natl Acad Sci U S A. 2009;106(45):19162–19167. doi: 10.1073/pnas.0902834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Back SA, Tuohy TM, Chen H, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11(9):966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- 18.Chong SY, Rosenberg SS, Fancy SP, et al. Neurite outgrowth inhibitor Nogo-A establishes spatial segregation and extent of oligodendrocyte myelination. Proc Natl Acad Sci U S A. 2012;109(4):1299–1304. doi: 10.1073/pnas.1113540109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang JK, Jarjour AA, Nait Oumesmar B, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci. 2011;14(1):45–53. doi: 10.1038/nn.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenberg SS, Kelland EE, Tokar E, De la Torre AR, Chan JR. The geometric and spatial constraints of the microenvironment induce oligodendrocyte differentiation. Proc Natl Acad Sci U S A. 2008;105(38):14662–14667. doi: 10.1073/pnas.0805640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee S, Leach MK, Redmond SA, et al. A culture system to study oligodendrocyte myelination processes using engineered nanofibers. Nat Methods. 2012;9(9):917–922. doi: 10.1038/nmeth.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franklin RJ, Hinks GL. Understanding CNS remyelination: clues from developmental and regeneration biology. J Neurosci Res. 1999;58(2):207–213. doi: 10.1002/(SICI)1097-4547(19991015)58:2<207::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 23.Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Bruck W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131(Pt 7):1749–1758. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- 24.Miron VE, Kuhlmann T, Antel JP. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim Biophys Acta. 2011;1812(2):184–193. doi: 10.1016/j.bbadis.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Dimou L, Simon C, Kirchhoff F, Takebayashi H, Gotz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. 2008;28(41):10434–10442. doi: 10.1523/JNEUROSCI.2831-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gledhill RF, Harrison BM, McDonald WI. Pattern of remyelination in the CNS. Nature. 1973;244(5416):443–444. doi: 10.1038/244443a0. [DOI] [PubMed] [Google Scholar]
- 27.Clarke LE, Young KM, Hamilton NB, Li H, Richardson WD, Attwell D. Properties and fate of oligodendrocyte progenitor cells in the corpus callosum, motor cortex, and piriform cortex of the mouse. J Neurosci. 2012;32(24):8173–8185. doi: 10.1523/JNEUROSCI.0928-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldschmidt T, Antel J, Konig FB, Bruck W, Kuhlmann T. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology. 2009;72(22):1914–1921. doi: 10.1212/WNL.0b013e3181a8260a. [DOI] [PubMed] [Google Scholar]
- 29.Sim FJ, Zhao C, Penderis J, Franklin RJ. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 2002;22(7):2451–2459. doi: 10.1523/JNEUROSCI.22-07-02451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kenakin TP. Synoptic pharmacology: detecting and assessing the pharmacological significance of ligands for orphan receptors. Pharmacol Res. 2016;114:284–290. doi: 10.1016/j.phrs.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 31.Munzel EJ, Becker CG, Becker T, Williams A. Zebrafish regenerate full thickness optic nerve myelin after demyelination, but this fails with increasing age. Acta Neuropathol Commun. 2014;2:77. doi: 10.1186/s40478-014-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinks GL, Franklin RJ. Delayed changes in growth factor gene expression during slow remyelination in the CNS of aged rats. Mol Cell Neurosci. 2000;16(5):542–556. doi: 10.1006/mcne.2000.0897. [DOI] [PubMed] [Google Scholar]
- 33.Shen S, Sandoval J, Swiss VA, et al. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008;11(9):1024–1034. doi: 10.1038/nn.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang DG, Tokumoto YM, Raff MC. Long-term culture of purified postnatal oligodendrocyte precursor cells. Evidence for an intrinsic maturation program that plays out over months. J Cell Biol. 2000;148(5):971–984. doi: 10.1083/jcb.148.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaskelioff M, Muller FL, Paik JH, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469(7328):102–106. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruckh JM, Zhao JW, Shadrach JL, et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10(1):96–103. doi: 10.1016/j.stem.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miron VE, Boyd A, Zhao JW, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16(9):1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeung MS, Zdunek S, Bergmann O, et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell. 2014;159(4):766–774. doi: 10.1016/j.cell.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 39.Levine JM, Reynolds R, Fawcett JW. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001;24(1):39–47. doi: 10.1016/S0166-2236(00)01691-X. [DOI] [PubMed] [Google Scholar]
- 40.Motavaf M, Sadeghizadeh M, Javan M. Attempts to overcome remyelination failure: toward opening new therapeutic avenues for multiple sclerosis. Cell Mol Neurobiol. 2017. [DOI] [PMC free article] [PubMed]
- 41.Tepavcevic V, Kerninon C, Aigrot MS, et al. Early netrin-1 expression impairs central nervous system remyelination. Ann Neurol. 2014;76(2):252–268. doi: 10.1002/ana.24201. [DOI] [PubMed] [Google Scholar]
- 42.Syed YA, Hand E, Mobius W, et al. Inhibition of CNS remyelination by the presence of semaphorin 3A. J Neurosci. 2011;31(10):3719–3728. doi: 10.1523/JNEUROSCI.4930-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel JR, Williams JL, Muccigrosso MM, et al. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012;124(6):847–860. doi: 10.1007/s00401-012-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hammond TR, Gadea A, Dupree J, et al. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron. 2014;81(3):588–602. doi: 10.1016/j.neuron.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charles P, Reynolds R, Seilhean D, et al. Re-expression of PSA-NCAM by demyelinated axons: an inhibitor of remyelination in multiple sclerosis? Brain. 2002;125(Pt 9):1972–1979. doi: 10.1093/brain/awf216. [DOI] [PubMed] [Google Scholar]
- 46.Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A. 2010;107(25):11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kastriti ME, Sargiannidou I, Kleopa KA, Karagogeos D. Differential modulation of the juxtaparanodal complex in multiple sclerosis. Mol Cell Neurosci. 2015;67:93–103. doi: 10.1016/j.mcn.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 48.Markoullis K, Sargiannidou I, Schiza N, et al. Gap junction pathology in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol. 2012;123(6):873–886. doi: 10.1007/s00401-012-0978-4. [DOI] [PubMed] [Google Scholar]
- 49.McKenzie IA, Ohayon D, Li H, et al. Motor skill learning requires active central myelination. Science. 2014;346(6207):318–322. doi: 10.1126/science.1254960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu K, Sun J, Kang Z, Zou Z, Wu G, Wang J. Electroacupuncture promotes remyelination after cuprizone treatment by enhancing myelin debris clearance. Front Neurosci. 2016;10:613. doi: 10.3389/fnins.2016.00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai S, Han L, Ao Q, Chan YS, Shum DK. Human induced pluripotent cell-derived sensory neurons for fate commitment of bone marrow-derived Schwann cells: implications for remyelination therapy. Stem Cells Transl Med. 2017;6(2):369–381. doi: 10.5966/sctm.2015-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hollingsworth E, Khouri J, Imitola J. Endogenous repair and development inspired therapy of neurodegeneration in progressive multiple sclerosis. Expert Rev Neurother. 2017;17:611–629. doi: 10.1080/14737175.2017.1287564. [DOI] [PubMed] [Google Scholar]
- 53.Buckley CE, Marguerie A, Roach AG, et al. Drug reprofiling using zebrafish identifies novel compounds with potential pro-myelination effects. Neuropharmacology. 2010;59(3):149–159. doi: 10.1016/j.neuropharm.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 54.Mei F, Fancy SP, Shen YA, et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat Med. 2014;20(8):954–960. doi: 10.1038/nm.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Najm FJ, Madhavan M, Zaremba A, et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature. 2015;522(7555):216–220. doi: 10.1038/nature14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmierer K, Wheeler-Kingshott CA, Tozer DJ, et al. Quantitative magnetic resonance of postmortem multiple sclerosis brain before and after fixation. Magn Reson Med. 2008;59(2):268–277. doi: 10.1002/mrm.21487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen JT, Collins DL, Freedman MS, et al. Local magnetization transfer ratio signal inhomogeneity is related to subsequent change in MTR in lesions and normal-appearing white-matter of multiple sclerosis patients. Neuroimage. 2005;25(4):1272–1278. doi: 10.1016/j.neuroimage.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 58.Chen JT, Kuhlmann T, Jansen GH, et al. Voxel-based analysis of the evolution of magnetization transfer ratio to quantify remyelination and demyelination with histopathological validation in a multiple sclerosis lesion. Neuroimage. 2007;36(4):1152–1158. doi: 10.1016/j.neuroimage.2007.03.073. [DOI] [PubMed] [Google Scholar]
- 59.Filippi M, Rocca MA, Barkhof F, et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol. 2012;11(4):349–360. doi: 10.1016/S1474-4422(12)70003-0. [DOI] [PubMed] [Google Scholar]
- 60.Hickman SJ, Toosy AT, Jones SJ, et al. Serial magnetization transfer imaging in acute optic neuritis. Brain. 2004;127(Pt 3):692–700. doi: 10.1093/brain/awh076. [DOI] [PubMed] [Google Scholar]
- 61.Schmierer K, Parkes HG, So PW. Direct visualization of remyelination in multiple sclerosis using T2-weighted high-field MRI. Neurology. 2009;72(5):472. doi: 10.1212/01.wnl.0000341878.80395.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahajan KR, Ontaneda D. The role of advanced magnetic resonance imaging techniques in multiple sclerosis clinical trials. Neurotherapeutics 2017. [DOI] [PMC free article] [PubMed]
- 63.Orije J, Kara F, Guglielmetti C, et al. Longitudinal monitoring of metabolic alterations in cuprizone mouse model of multiple sclerosis using 1H-magnetic resonance spectroscopy. Neuroimage. 2015;114:128–135. doi: 10.1016/j.neuroimage.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 64.Martin AR, Aleksanderek I, Cohen-Adad J, et al. Translating state-of-the-art spinal cord MRI techniques to clinical use: a systematic review of clinical studies utilizing DTI, MT, MWF, MRS, and fMRI. Neuroimage Clin. 2016;10:192–238. doi: 10.1016/j.nicl.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stankoff B, Freeman L, Aigrot MS, et al. Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-(1)(1)C]-2-(4'-methylaminophenyl)- 6-hydroxybenzothiazole. Ann Neurol. 2011;69(4):673–680. doi: 10.1002/ana.22320. [DOI] [PubMed] [Google Scholar]
- 66.Bodini B, Veronese M, Garcia-Lorenzo D, et al. Dynamic imaging of individual remyelination profiles in multiple sclerosis. Ann Neurol 2016. [DOI] [PMC free article] [PubMed]
- 67.Niklas A, Sebraoui H, Hess E, Wagner A, Then BF. Outcome measures for trials of remyelinating agents in multiple sclerosis: retrospective longitudinal analysis of visual evoked potential latency. Mult Scler. 2009;15(1):68–74. doi: 10.1177/1352458508095731. [DOI] [PubMed] [Google Scholar]
- 68.Mei F, Lehmann-Horn K, Shen YA, et al. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 2016;5. [DOI] [PMC free article] [PubMed]
- 69.Li Z, He Y, Fan S, Sun B. Clemastine rescues behavioral changes and enhances remyelination in the cuprizone mouse model of demyelination. Neurosci Bull. 2015;31(5):617–625. doi: 10.1007/s12264-015-1555-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deshmukh VA, Tardif V, Lyssiotis CA, et al. A regenerative approach to the treatment of multiple sclerosis. Nature. 2013;502(7471):327–332. doi: 10.1038/nature12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu J, Dupree JL, Gacias M, et al. Clemastine enhances myelination in the prefrontal cortex and rescues behavioral changes in socially isolated mice. J Neurosci. 2016;36(3):957–962. doi: 10.1523/JNEUROSCI.3608-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grove RA, Harrington CM, Mahler A, et al. A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer's disease. Curr Alzheimer Res. 2014;11(1):47–58. doi: 10.2174/1567205010666131212110148. [DOI] [PubMed] [Google Scholar]
- 73.Nathan PJ, Boardley R, Scott N, et al. The safety, tolerability, pharmacokinetics and cognitive effects of GSK239512, a selective histamine H(3) receptor antagonist in patients with mild to moderate Alzheimer's disease: a preliminary investigation. Curr Alzheimer Res. 2013;10(3):240–251. doi: 10.2174/1567205011310030003. [DOI] [PubMed] [Google Scholar]
- 74.Ashworth S, Berges A, Rabiner EA, et al. Unexpectedly high affinity of a novel histamine H(3) receptor antagonist, GSK239512, in vivo in human brain, determined using PET. Br J Pharmacol. 2014;171(5):1241–1249. doi: 10.1111/bph.12505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schwartzbach CJ, Grove RA, Brown R, Tompson D, Then Bergh F, Arnold DL. Lesion remyelinating activity of GSK239512 versus placebo in patients with relapsing-remitting multiple sclerosis: a randomised, single-blind, phase II study. J Neurol. 2017;264(2):304–315. doi: 10.1007/s00415-016-8341-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mi S, Lee X, Shao Z, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7(3):221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- 77.Mi S, Miller RH, Tang W, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. 2009;65(3):304–315. doi: 10.1002/ana.21581. [DOI] [PubMed] [Google Scholar]
- 78.Sun JJ, Ren QG, Xu L, Zhang ZJ. LINGO-1 antibody ameliorates myelin impairment and spatial memory deficits in experimental autoimmune encephalomyelitis mice. Sci Rep. 2015;5:14235. doi: 10.1038/srep14235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang J, Ye Z, Zheng S, et al. Lingo-1 shRNA and Notch signaling inhibitor DAPT promote differentiation of neural stem/progenitor cells into neurons. Brain Res. 1634;2016:34–44. doi: 10.1016/j.brainres.2015.11.029. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Zhang YP, Pepinsky B, et al. Inhibition of LINGO-1 promotes functional recovery after experimental spinal cord demyelination. Exp Neurol. 2015;266:68–73. doi: 10.1016/j.expneurol.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 81.Tran JQ, Rana J, Barkhof F, et al. Randomized phase I trials of the safety/tolerability of anti-LINGO-1 monoclonal antibody BIIB033. Neurol Neuroimmunol Neuroinflamm. 2014;1(2):e18. doi: 10.1212/NXI.0000000000000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cadavid D, Balcer L, Galetta S, et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2017;16(3):189–199. doi: 10.1016/S1474-4422(16)30377-5. [DOI] [PubMed] [Google Scholar]
- 83.Rodriguez-Pena A. Oligodendrocyte development and thyroid hormone. J Neurobiol. 1999;40(4):497–512. doi: 10.1002/(SICI)1097-4695(19990915)40:4<497::AID-NEU7>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 84.Baxi EG, Schott JT, Fairchild AN, et al. A selective thyroid hormone beta receptor agonist enhances human and rodent oligodendrocyte differentiation. Glia. 2014;62(9):1513–1529. doi: 10.1002/glia.22697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Warrington AE, Asakura K, Bieber AJ, et al. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci U S A. 2000;97(12):6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Watzlawik J, Holicky E, Edberg DD, et al. Human remyelination promoting antibody inhibits apoptotic signaling and differentiation through Lyn kinase in primary rat oligodendrocytes. Glia. 2010;58(15):1782–1793. doi: 10.1002/glia.21048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bieber AJ, Warrington A, Asakura K, et al. Human antibodies accelerate the rate of remyelination following lysolecithin-induced demyelination in mice. Glia. 2002;37(3):241–249. doi: 10.1002/glia.10033. [DOI] [PubMed] [Google Scholar]
- 88.Zhornitsky S, Wee Yong V, Koch MW, et al. Quetiapine fumarate for the treatment of multiple sclerosis: focus on myelin repair. CNS Neurosci Ther. 2013;19(10):737–744. doi: 10.1111/cns.12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smith ES, Jonason A, Reilly C, et al. SEMA4D compromises blood-brain barrier, activates microglia, and inhibits remyelination in neurodegenerative disease. Neurobiol Dis. 2015;73:254–268. doi: 10.1016/j.nbd.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 90.Lariosa-Willingham KD, Rosler ES, Tung JS, Dugas JC, Collins TL, Leonoudakis D. Development of a high throughput drug screening assay to identify compounds that protect oligodendrocyte viability and differentiation under inflammatory conditions. BMC Res Notes. 2016;9(1):444. doi: 10.1186/s13104-016-2220-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lariosa-Willingham KD, Rosler ES, Tung JS, Dugas JC, Collins TL, Leonoudakis D. A high throughput drug screening assay to identify compounds that promote oligodendrocyte differentiation using acutely dissociated and purified oligodendrocyte precursor cells. BMC Res Notes. 2016;9(1):419. doi: 10.1186/s13104-016-2220-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Diab A, Hussain RZ, Lovett-Racke AE, Chavis JA, Drew PD, Racke MK. Ligands for the peroxisome proliferator-activated receptor-gamma and the retinoid X receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148(1-2):116–126. doi: 10.1016/j.jneuroim.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 93.Cummings JL, Zhong K, Kinney JW, et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer's disease. Alzheimers Res Ther. 2016;8:4. doi: 10.1186/s13195-016-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de la Fuente AG, Errea O, van Wijngaarden P, et al. Vitamin D receptor-retinoid X receptor heterodimer signaling regulates oligodendrocyte progenitor cell differentiation. J Cell Biol. 2015;211(5):975–985. doi: 10.1083/jcb.201505119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.De Nuccio C, Bernardo A, Cruciani C, De Simone R, Visentin S, Minghetti L. Peroxisome proliferator activated receptor-gamma agonists protect oligodendrocyte progenitors against tumor necrosis factor-alpha-induced damage: effects on mitochondrial functions and differentiation. Exp Neurol. 2015;271:506–514. doi: 10.1016/j.expneurol.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 96.Yao X, Su T, Verkman AS. Clobetasol promotes remyelination in a mouse model of neuromyelitis optica. Acta Neuropathol Commun. 2016;4(1):42. doi: 10.1186/s40478-016-0309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mei F, Mayoral SR, Nobuta H, et al. Identification of the kappa-opioid receptor as a therapeutic target for oligodendrocyte remyelination. J Neurosci. 2016;36(30):7925–7935. doi: 10.1523/JNEUROSCI.1493-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Du C, Duan Y, Wei W, et al. Kappa opioid receptor activation alleviates experimental autoimmune encephalomyelitis and promotes oligodendrocyte-mediated remyelination. Nat Commun. 2016;7:11120. doi: 10.1038/ncomms11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Y, Li X, Ciric B, et al. Effect of fingolimod on neural stem cells: a novel mechanism and broadened application for neural repair. Mol Ther. 2017;25(2):401–415. doi: 10.1016/j.ymthe.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sedel F, Papeix C, Bellanger A, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159–169. doi: 10.1016/j.msard.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 101.Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2016;110(Pt B):644–653. doi: 10.1016/j.neuropharm.2015.08.028. [DOI] [PubMed] [Google Scholar]
- 102.Franklin RJ, ffrench-Constant C, Edgar JM, Smith KJ. Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol. 2012;8(11):624–634. doi: 10.1038/nrneurol.2012.200. [DOI] [PubMed] [Google Scholar]
- 103.Chan J, Ban EJ, Chun KH, et al. Methylprednisolone induces reversible clinical and pathological remission and loss of lymphocyte reactivity to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis. Autoimmunity. 2008;41(5):405–413. doi: 10.1080/08916930802011258. [DOI] [PubMed] [Google Scholar]
- 104.Bove RM, Healy B, Augustine A, Musallam A, Gholipour T, Chitnis T. Effect of gender on late-onset multiple sclerosis. Mult Scler. 2012;18(10):1472–1479. doi: 10.1177/1352458512438236. [DOI] [PubMed] [Google Scholar]
- 105.Bove R, Healy BC, Musallam A, Glanz BI, De Jager PL, Chitnis T. Exploration of changes in disability after menopause in a longitudinal multiple sclerosis cohort. Mult Scler. 2016;22(7):935–943. doi: 10.1177/1352458515606211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bothwell M. Mechanisms and medicines for remyelination. Annu Rev Med. 2017;68:431–443. doi: 10.1146/annurev-med-050715-104400. [DOI] [PubMed] [Google Scholar]
- 107.Luessi F, Kuhlmann T, Zipp F. Remyelinating strategies in multiple sclerosis. Expert Rev Neurother. 2014;14(11):1315–1334. doi: 10.1586/14737175.2014.969241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1224 kb)
(PDF 1224 kb)