

Abstract
Objective(s):
Stroke may cause severe neuronal damage. The sesamin have been demonstrated to possess neuroprotection by its antioxidant and anti-inflammatory properties. One sesamin derivative was artificially composited, 1, 2-bis [(3-methoxyphenyl) methyl] ethane-1, 2-dicaroxylic acid (MMEDA) had been developed to study its antioxidative activity and neuroprotection.
Materials and Methods:
The infaction of Sprague Dawley (SD) rats and hypoxia models of BV-2 microglia or PC12 cells were investigated for in vivo and in vitro test respectively. Lipid peroxidation and reactive oxygen species (ROS), prostaglandin E2 (PGE2) and related signaling pathways from hypoxic cells were analyzed by ELISA or Western blot assay, respectively.
Results:
MMEDA showed a protective effect when given 90 min after the focal cerebral ischemia. The neuroprotection of MMEDA was further confirmed by attenuating ROS and PGE2 release from hypoxic BV-2 or PC12 cells. MMEDA significantly reduced hypoxia-induced JNK and caspase-3 (survival and apoptotic pathways) in PC12 cells.
Conclusion:
The neuroprotective effect of MMEDA on ischemia/hypoxia models was involved with its antioxidative activity and anti-inflammatory effects. These results suggest that MMEDA exert effective neuroprotection against ischemia/hypoxia injury.
Keywords: Anti-inflammatory, Cerebral ischemia, Hypoxia, Neuroprotection, Reactive oxygen species Sesamin derivative
Introduction
Stroke occurs in the blood supply to brain is blocked and this reduced the transport of oxygen and nutrients, which can cause neurons to damage. Cerebral ischemia resulted from low oxygen and glucose supply evidently decreases the formation of ATP (1-3). Brain ischemia injury is well known that it increases reactive oxygen species (ROS) generation and inflammatory reactions (1). The reports have shown that ischemia/reperfusion of brain always cause cell damage by oxidative stress (2, 3). Previously we reported that the sesamin protected brain and neuron from cerebral ischemia and hypoxic injuries (4-6).
The cerebral ischemia or hypoxia-induced ROS and cytokine could be eliminated by the antioxidants (7). The PC12 cells of rat pheochromacytoma and the BV-2 cells of murine microglia were well known as neuronal stress models (4, 5). Especially, the survival pathways including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) signaling pathways would be activated by subjecting with ROS in PC12 or BV-2 cells (4, 5). The cerebral ischemia induces apoptosis in the brain is identified by activation of caspase-3 (8). Therefore, a compound, 1, 2-bis [(3-methoxyphenyl) methyl] ethane-1, 2-dicaroxylic acid (MMEDA), was investigated for its neuroprotective effect in the present study. Therefore, the possible mechanism of MMEDA was examined with ischemic brain and cell hypoxia models for its antioxidant, anti-inflammatory properties, and the cell signal of hypoxia-induced survival and apoptotic pathways.
Materials and Methods
Reagents
Dimethylsulfoxide was obtained from Sigma-Aldrich Chemical (St Louis, MO, USA). Porcine polar brain lipid (PBL) was purchased from Avanti Polar Lipids Inc (Alabaster, AL, USA). 2’,7’-Dichlorodihydrofluorescein diacetate (H2DCF-DA) was obtained from Molecular Probe (Eugene, Oregon, USA). Fetal bovine serum (FBS) was obtained from Gibco Invitrogen (Grand Island, NY, USA). Dulbecco’s Modified Eagle’s medium (DMEM) were purchased from GIBCO (Grand Island, NY, USA). Anti-phospho-p38, ERK, JNK, and β-actin antibodies were purchased from Abcam (Cambridge, UK). Anti-Akt1 (pSer473) antibody was purchased from Calbiochem (Darmstadt, Germany). 1, 2-bis [(3-methoxyphenyl) methyl] ethane-1, 2-dicaroxylic acid (MMEDA) was donated by Joben Bio-Medical Co (Kaohsiung, Taiwan).
Animal study
Twenty Sprague Dawley (SD) rats (male, 200~250 g) were obtained from BioLASCO (Taipei, Taiwan) and divided into the control (normal saline), and MMEDA groups randomly. The experiment was approved by Institutional Animal Care and Use Committee, Taichung Veterans General Hospital (IACUC LA-97490). Rats were injected intraperitoneally (IP) with MMEDA (10 mg/kg) 90 min after MACO experiment. Each SD rat was anesthetized with chlorohydrate (400 mg/kg) IP and its body temperature was maintained at 37 °C with a heating pad (CMA/150). A midline neck incision was made and the right carotid artery was exposed and separated from the vago-sympathetic trunk. The right carotid artery was loosely encircled with a 4-O suture for later occlusion. The SD rat’s head was placed in a stereotaxic frame (David Kopf, CA, USA) with the nose bar positioned 4.0 mm below the horizontal line. Following a midline incision, the skull was partially removed to expose the right middle cerebral artery. The middle cerebral artery was loosely encircled with an 8-O suture for later occlusion. A focal cerebral ischemia was induced by occlusion of the right common carotid artery and the right cerebral artery (MCAO) for 60 min, followed by reperfusion. A laser probe (0.8 mm in diameter) of a Laser Doppler Blood Flow monitor (MBF 3D, Moor Instruments, Axminster, UK) was positioned onto the cortex with its tip close to the middle of cerebral artery. Cerebral blood flow dropped to less than 5% of basal after the occlusion of the MCAO. Cerebral blood flow reached its minimal levels within 5 min after the start of the occlusion and was confirmed to remain at this level throughout the monitoring period to ensure the validity of the stroke model. Twenty-four hours after cerebral ischemia, each SD rat was anesthetized and perfused transcardially with isotonic heparinized saline and 2,3,5-triphenyltetrazolium chloride (TTC). The brain was then removed and sliced into five 2-mm-thick coronal sections for TTC staining.
Cell culture
The BV-2 cell line was maintained in DMEM supplemented with 10% (v/v) FBS in a humidified incubator under 5% CO2 at 37 °C. The PC12 cell line was maintained in DMEM supplemented with 10% FBS, 5% horse serum, at 37 °C under 5% CO2. Confluent cultures were passaged by trypsinization. In all experiments, the cells were treated with 1, 10, 50 µM of MMEDA immediately before hypoxia. MMEDA was dissolved in 95% alcohol. The final concentration of alcohol added to cells never exceeded 0.1% (v/v).
Hypoxia
In experiment, culture media were replaced with glucose-free DMEM, then, gassed with 85% N2, 10% H2, and 5% CO2 for various time periods in the absence or presence of various doses of MMEDA.
MTT assay
Cell viability was measured using blue formazan that was metabolized from colorless 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) by mitochondrial dehydrogenases, which are active only in live cells. PC12 or BV2 cells were pre-incubated in 24-well plates at a density of 5×105 cells per well for 24 hr. Cells incubated with various concentrations of MMEDA (1, 10, 50 µM) were under hypoxia for 30 min, and incubated in 0.5 mg/ml MTT at 37 °C. One hour later, 200 μl of solubilization solution were added to each well and absorption values read at 540 nm on microtiter plate reader (spectraMAX 340, Molecular Devices, Sunnyvale, CA, USA). Data were expressed as the mean percent of viable cells vs. control.
LDH assay
Cytotoxicity was determined by measuring the release of LDH. PC12 or BV-2 cells treated with various concentrations of MMEDA were stressed with hypoxia for one hour and the supernatant was then assayed for LDH activity. An absorbance was read at 490/630 nm using a spectraMAX 340 microtiter plate reader. Data were expressed as the mean percent of viable cells vs. the control.
Generation of reactive oxygen species
Intracellular accumulation of ROS was determined using H2DCF-DA, which is a non-fluorescent compound that accumulates in cells following deacetylation. H2DCF then reacts with ROS to form fluorescent dichlorofluorescein (DCF). PC12 cells were plated in 96-well plates and grown for 24 hr before addition of DMEM plus 10 μM H2DCF-DA, incubated for 60 min at 37 °C, and treated with various concentrations of MMEDA for hypoxia 30 min. Cells were then washed twice at room temperature with Hank’s balanced salt solution (HBSS without phenol red). Cellular fluorescence was monitored on a Fluoroskan Ascent fluorometer (Labsystems Oy, Helsinki, Finland) using an excitation wavelength of 485 nm and emission wavelength of 538 nm.
Measurement of PGE2 assay
PGE2 was measured using ELISA kits (R&D, Minneapolis, MN, USA). The absorbance at 450 nm was determined using a microplate reader (spectraMAX 340).
Western blot
Samples containing 25 μg of protein were separated on 12.5% (w/v) sodium dodecyl sulfate-polyacrylamide gels, and transferred to immobilon polyvinylidenedifluoride membranes (Millipore, Bed-ford, USA). The membranes were incubated for 2 hr with 5% (w/v) dry skim milk in TBST buffer to block non-specific binding, then ERK, p38 JNK, caspase-3, β-actin proteins for neuron cells were detected by a chemiluminescence detection system according to the manufacturer’s instructions (ECL, Amersham, Berkshire, UK).
Lipid peroxidation
Lipid peroxidation is quantified by measuring malondialdehyde (MDA) of PC12 cells and brain tissue of SD rats by lipid peroxidation (LPO) assay kit (Cayman). This kit works on the principle of condensation of one molecule of either MDA or 4-hydroxyalkenals with two molecules of N-methyl-2-phenylindole to yield a stable chromophore. MDA levels were assayed by measuring the amount expressed in 5×105 cells of PC12 and SD brain tissue, and the absorbance at 500 nm was determined using a microplate reader (spectraMAX 340).
Statistical analysis
Data were expressed as the mean ± SEM. In animal study, TTC data were analyzed by analysis of variance (ANOVA) with Student’s t-tests. A P value less than 0.05 was considered to be statistically significant. For in vitro study with single variable comparisons, Student’s t-test was used. For multiple variable comparisons, data were analyzed by one-way ANOVA followed by Scheffe’s test.
Results
The effect of MMEDA on the cerebral ischemia
SD rats treated IP with MMEDA (10 mg/kg) 90 min after MCAO-induced ischemia reduced 41% of the infarct size as compared to the cerebral ischemia group (from 582.46±13.67 mm to 343.07±15.79 mm, P<0.05; Figure 2A).
Figure 2.
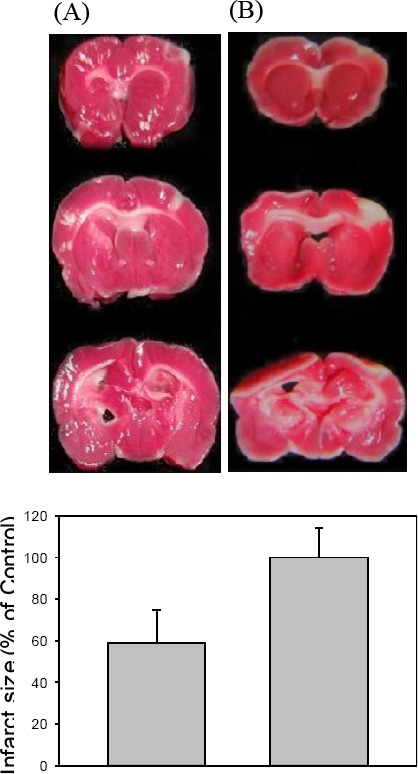
In vivo effect of MMEDA on the cerebral ischemia. SD rats treated with MMEDA IP 90 min after MACO had reduced 41 infarct sizes of brains (A). Data are expressed as mean±SEM.*: P<0.05 as compared to the non-treated ischemic control (A, n = 10). Data are expressed as mean±SEM. *: P<0.05 as compared to the hypoxia control (B).
Figure 1.
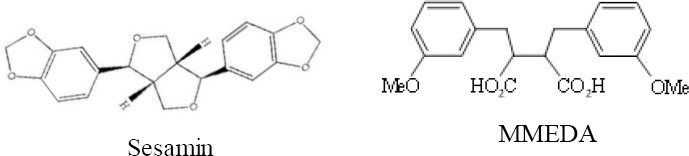
The chemical structure of sesamin and MMEDA
The protection of MMEDA against hypoxia
The neuroprotection of MMEDA were investigated by hypoxia model. Hypoxia can induce damage neuronal cells, therefore the cell viability was measured using MTT assay and the cytosol LDH released was measured using LDH ELISA assay from PC12 or BV-2 cells. In Figure 3B, the cell viability of PC12 cells subjected hypoxia for 30 min was increased by the presence of MMEDA (1, 10, 50 μM).
Figure 3.
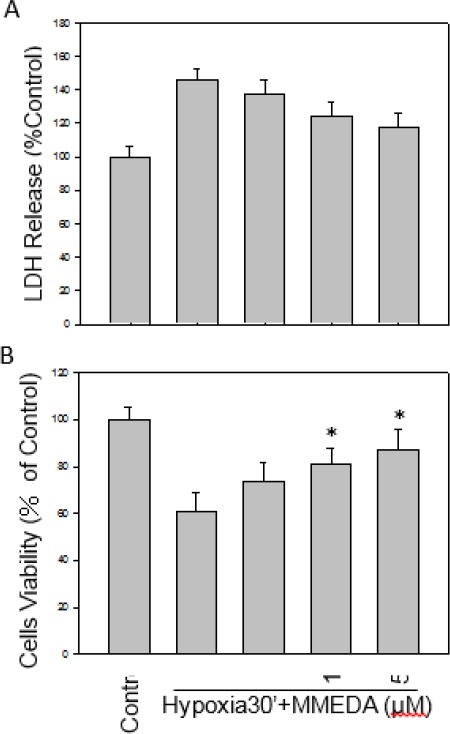
Effect of MMEDA on cell viability and cytotoxicity of PC12 cells under hypoxia. The cells were treated with hypoxia alone or in the presence of various concentrations (1, 10, 50 µM) of MMEDA for 30 min. Cell viability of PC12 (A) was increased and the LDH release, reduced (B) from hypoxia dose-dependently by MMEDA. Data are expressed as mean ± SEM of three independent experiments in triplicate. *: P<0.05 as compared to the hypoxia control
Hypoxia-induced LDH released was also decreased by MMEDA (P<0.05, Figure 3A). Similarly, BV-2 cells were protected by MMEDA under hypoxia (data not shown).
ROS scavenging
The neuronal cells induce free radicals release by hypoxia stress. Subjecting with hypoxia 30 mins, ROS (as DCF signal) was increased 60% in BV-2 cells in compared with the control cells. MMEDA protected cells against hypoxia-induced cell toxicity by scavenging the ROS accumulation level, was decreased 23 and 26% in 10, 50 µM of MMEDA (Figure 4A). The increased MDA level was suppressed by MMEDA in hypoxia-stressed BV-2 cells. MDA level of the MMEDA group was decreased 17 % (from 154± 8 μM to 128±7 μM) as compared to the hypoxic group (Figure 4B, P<0.05).
Figure 4.
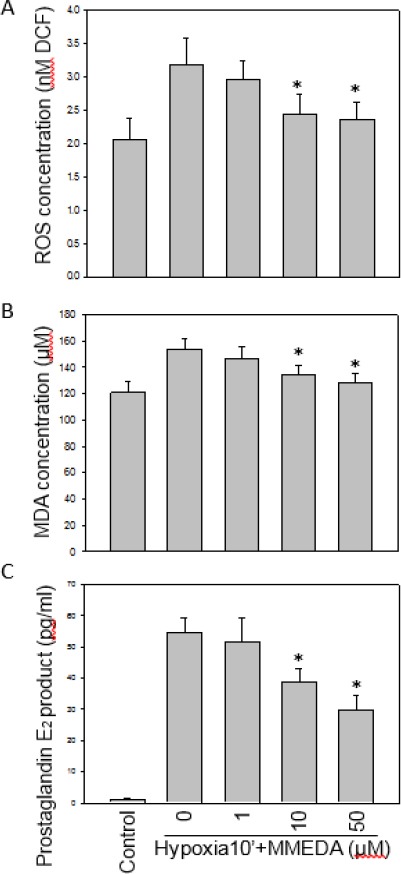
Effects of MMEDA on hypoxia-induced ROS generation, MDA and PGE2 productions. BV-2 cells were treated with 1, 10, 50 µM of MMEDA for 30 min. MMEDA reduced the generation of ROS under hypoxia in BV-2 cells (A). MDA of PC12 cells were induced by 30-min hypoxia and reduced by MMEDA (B). BV-2 cells were treated with 1, 10, 50 µM of MMEDA under hypoxia 30 min. MMEDA of 10 and 50 µM, concentration-dependently reduced hypoxia-induced PGE2 production from BV-2 cells (C). Data are expressed as mean±SEM of 3 independent experiments in triplicate. *: P<0.05 as compared to the hypoxia control
MMEDA inhibited PGE2
We further examined the anti-inflammatory property of MMEDA on hypoxia-induced PGE2 production. BV-2 cells were pretreated with 1, 10, 50 µM of MMEDA, and then exposed to hypoxia for 30 min. The MMEDA (10 and 50 µM) dose-dependently decreased PGE2 release 29 and 45% level of BV-2 cells (P<0.05, Figure 4C).
MMEDA inhibited hypoxia-induced JNK MAPK, caspase-3 activation and JNK mRNA expression
The effects of MMEDA (1, 10, 50 μM) on hypoxia-induced signaling pathways were evaluated by Western blot assay (Figure 5A). There is no effect of the phospho-ERK and P38 protein expression subjected to 10 min hypoxia in PC12 cells. The 50 μM MMEDA reduced expression of the phospho-JNK (63%), and caspase-3 (70%), respectively for the 10 min hypoxia-induced PC12 cells (*P<0.05; Figure 5A). PC12 cells were treated with MMEDA and subjected to hypoxia for 10 min. Levels of JNK, and β-actin mRNA was assayed by RT-PCR (5B). MMEDA decreased JNK mRNA as compared with that of the hypoxia and the 50 μM MMEDA reduced 63% expression of the JNK mRNA (*P<0.05; Figure 5B).
Figure 5.
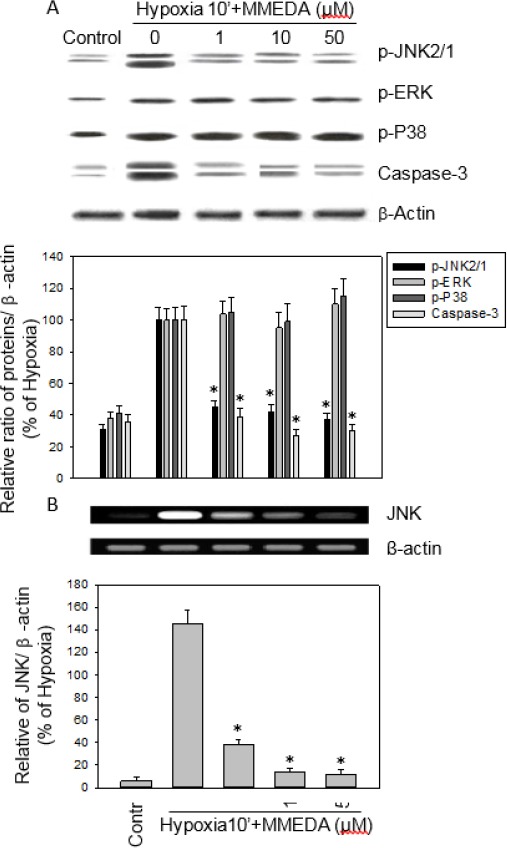
MMEDA inhibited hypoxia-induced phospho-JNK MAP kinase and JNK mRNA expression. The effect of MMEDA (1, 10, 50 μM) on hypoxia-activated cell signaling pathways was determined by Western blotting under hypoxia stress in PC12 cells for 10 min. MMEDA effectively reduced the activation of JNK MAP kinase and caspase-3 in PC12 cells (A). PC12 cells were treated with MMEDA and subjected to hypoxia for 10 min. Levels of JNK, and β-actin mRNA were assayed by RT-PCR (B). Values represent the mean from three independent experiments. MMEDA decreased JNK mRNA as compared with that of the hypoxia. Data are expressed as mean±SEM of three independent experiments. *: P<0.05 as compared to hypoxia control
Discussion
This research showed that the MMEDA (10, 50 μM) significantly protected SD rats from the focal cerebral ischemia damage. And, the MMEDA protected the BV-2 or PC12 cells from hypoxia-induced cell cytotoxicity, ROS generation and MDA levels.
The free radical ROS production in the brain contributed to the degenerative processes of neuron (9, 10). Reports show that the dietary-derived antioxidants inhibit the hypoxia-exposed inflammation response existence neuroprotective potential (11, 12). In our previous studies sesamin and its related structure were supported the protective effect on the hypoxia-stressed inflammatory and oxidative stress (5, 13), There is a similar effect of MMEDA, a sesamin novel derivative. Protective effect of MMEDA on hypoxia-stressed MDA might be through the activation of antioxidant signaling pathway such as Nrf2/ARE (14).
Western blot showed that hypoxia of 10 min could significantly activate the phospho-MAPKs, and caspase-3 expression in PC12 cells. Inhibition of apoptotic pathways by decreasing the phospho-JNK MAPK and caspase-3 expression could be expected to be beneficial in damage involving microglia activation and inflammation. The specific inhibitors of JNK MAPK would provide the reducing of inflammation and neuroprotective effects (4, 15, 16). Studies reported that the various antioxidant compounds inhibit JNK MAPK signal expression in microglia represent potential anti-inflammatory effects and neuroprotection (5, 17, 18). In addition, antioxidants inhibit JNK MAPK signal expression in neuron and cardiomyocyte cells represent potential protective effects from hypoxic injury (5, 19-21). We know that the sesamin regulate microglial activities by inhibition of the intracerebral hemorrhage-induced ERK cell signal pathway of MAPK and protect neurons from hypoxia damage by inhibition of hypoxia-induced ERK, JNK, p38 MAPK (5, 22). The Western blot show MMEDA also inhibited phospho-JNK MAPK expression in PC12 cells subjected to hypoxia significantly (Figure 5). Studies reported that hypoxia activates MAPK and apoptosis factor caspase-3 cell signal expression in vitro and in vivo (4, 8). Therefore, we evaluated the effect of MMEDA on the survival and apoptotic signaling pathways including phospho-MAP kinases (JNK, ERK, p38), and Caspase-3 with hypoxiastressed PC12 cells. Western blot analysis revealed that MMEDA (10 and 50 μM) significantly reduced phospho-JNK MAPK, and caspase-3 expression in PC12 cells as compared to hypoxia controls (Figure 5A, 5B). The results suggested that the MMEDA repaired the cell viability under hypoxic stress (Figure 3) through survival and apoptotic signaling pathways in PC12 cells. This consistent with a research that the Tat-glyoxalase protein protects neuronal cells from H2O2-induced neuron damage, and DNA fragmentation by through with inhibition of caspase-3 and MAP kinase expression, and then ameliorate ischemic brain injury (23).
Previous studies shown that neuroprotection of antioxidants are due to increasing the antioxidative enzymes, or scavenging of ROS, and preventing calcium release (5). Recently, a study shows that sesamin and metabolites activate heme oxygenase-1 (HO-1) by increasing cell signal of Nrf2/ARE protein expression, and reduce oxidative stress (14). According to MMEDA was able to inhibit MDA generation and 23~26% ROS, and 29~45% of PGE2 production of hypoxia stress. The free radical ROS could induce cell damage by upregulating MAPK expression (24). The MMEDA might downstream inflammatory proteins by way of the suppression of ROS generation which would reduce the activation of PGE2 in BV-2 cells subjected with hypoxia. The caspase-3 is the key point apoptosis factor for cells (23). Pretreating with MMEDA (100 μM) alone was not toxic to PC12/BV-2 cells (data not shown) and MMEDA at the concentrations of 10 and 50 μM inhibited the inflammation response, however, there is no effect of 1 μM MMEDA on the PGE-2 response in hypoxia-stressed BV-2 cells (Figure 4C). The MMEDA signi ficantly restored the infarct size (about 41 %) of the focal cerebral ischemic brain as compared to the control group SD rats. Although the precise mechanism of MMEDA neuroprotection is not clear, the present in vitro and in vivo results suggest that its protection might be involved with the suppression of ROS and inflammation during cerebral ischemia.
Conclusion
The present study shows that MMEDA restored the brain subjected to the focal cerebral ischemia in vivo and, protected the BV-2 or PC12 cells from hypoxia-induced cell cytotoxicity in vitro. It also neutralized ROS generation and reduced lipid peroxidation in the BV-2 cells subjected to hypoxia. The protective mechanisms of MMEDA might be involved with the downregulation of phospho-JNK MAPK, and caspase-3 signal pathway expression. Therefore, the MMEDA should have a neuroprotective effect against ischemia stress.
Acknowledgment
The results described in this paper were part of student thesis. This study was supported in part by grant from Taichung Veterans General Hospital, Taiwan. We are grateful of manuscript proofreading support of prof K-C Jeng.
Conflict of interest
The authors declare that they have no competing interests.
References
- 1.Ishida I, Kubo H, Suzuki S, Suzuki T, Akashi S, Inoue K, et al. Hypoxia diminishes toll-like receptor 4 expression through reactive oxygen species generated by mitochondria in endothelial cells. J Immunol. 2002;15:2069–2075. doi: 10.4049/jimmunol.169.4.2069. [DOI] [PubMed] [Google Scholar]
- 2.Koga Y, Fujita M, Tsuruta R, Koda Y, Nakahara T, Yagi T, et al. Urinary trypsin inhibitor suppresses excessive superoxide anion radical generation in blood, oxidative stress, early inflammation, and endothelial injury in forebrain ischemia/reperfusion rats. Neurol Res. 2010;32:925–932. doi: 10.1179/016164110X12645013515133. [DOI] [PubMed] [Google Scholar]
- 3.Tsuruta R, Fujita M, Ono T, Koda Y, Koga Y, Yamamoto T, et al. Hyperglycemia enhances excessive superoxide anion radical generation, oxidative stress, early inflammation, and endothelial injury in forebrain ischemia/reperfusion rats. Brain Res. 2010;1309:155–163. doi: 10.1016/j.brainres.2009.10.065. [DOI] [PubMed] [Google Scholar]
- 4.Hou CW, Wu CC, Yang CH, Jeng KC. Protective effects of sesamin and sesamolin on murine BV-2 microglia cell line under hypoxia. Neuroscience Letters. 2004;367:10–13. doi: 10.1016/j.neulet.2004.05.073. [DOI] [PubMed] [Google Scholar]
- 5.Hou CW, Huang HM, Tzen JT, Jeng KC. Protective effects of sesamin and sesamolin on hypoxic neuronal and PC12 cells. J Neurosci Res. 2003;74:123–133. doi: 10.1002/jnr.10749. [DOI] [PubMed] [Google Scholar]
- 6.Khan MM, Ishrat T, Ahmad A, Hoda MN, Khan MB, Khuwaja G, et al. Sesamin attenuates behavioral, biochemical and histological alterations induced by reversible middle cerebral artery occlusion in the rats. Chem Biol Interact. 2010;183:255–263. doi: 10.1016/j.cbi.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 7.Bush ML, Miyashiro JS, Ingram VM. Activation of a neurofilament kinase, a tau kinase, and a tau phosphatase by decreased ATP levels in nerve growth factor-differentiated PC-12 cells. Proc Natl Acad Sci USA. 1995;92:1861–1865. doi: 10.1073/pnas.92.6.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, et al. Sonic hedgehog--regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron. 2000;25:317–329. doi: 10.1016/s0896-6273(00)80897-1. [DOI] [PubMed] [Google Scholar]
- 9.Chao CC, Hu S, Tsang M, Weatherbee J, Molitor TW, Anderson WR, et al. Effects of transforming growth factor-beta on murine astrocyte glutamine synthetase activity. Implications in neuronal injury. J Clin Invest. 1992;90:1786–1793. doi: 10.1172/JCI116053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun AY, Cheng JS. Neuroprotective effects of poly (ADP-ribose) polymerase inhibitors in transient focal cerebral ischemia of rats. Zhongguo Yao Li Xue Bao. 1998;19:104–108. [PubMed] [Google Scholar]
- 11.Hou RCW, Chen HL, Tzen JTC, Jeng KC. Effect of sesame antioxidants on LPS-induced NO production by BV2 microglial cells. Neuroreport. 2003;14:1815–1819. doi: 10.1097/00001756-200310060-00011. [DOI] [PubMed] [Google Scholar]
- 12.Hou RCW, Chen YS, Chen CH, Chen YH, Jeng JC. Protective effect of 1,2,4-benzenetriol on LPS-induced NO production by BV2 microglial cells. J Biomed Sci. 2006;13:89–99. doi: 10.1007/s11373-005-9039-5. [DOI] [PubMed] [Google Scholar]
- 13.Hamada N, Tanaka A, Fujita Y, Itoh T, Ono Y, Kitagawa Y, et al. Involvement of heme oxygenase-1 induction via Nrf2/ARE activation in protectionagainst H2O2-induced PC12 cell death by a metabolite of sesamin contained in sesame seeds. Bioorg Med Chem. 2011;19:1959–1965. doi: 10.1016/j.bmc.2011.01.059. [DOI] [PubMed] [Google Scholar]
- 14.Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, et al. a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther. 2001;296:312–321. [PubMed] [Google Scholar]
- 15.Kaminska B, Gozdz A, Zawadzka M, Ellert-Miklaszewska A, Lipko M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat Rec(Hoboken) 2009;292:1902–1913. doi: 10.1002/ar.21047. [DOI] [PubMed] [Google Scholar]
- 16.Zeng KW, Fu H, Liu GX, Wang XM. Icariin attenuates lipopolysaccharide-induced microglial activation and resultant death of neurons by inhibiting TAK1/IKK/NF-kappaB and JNK/p38 MAPK pathways. Int Immunopharmacol. 2010;10:668–678. doi: 10.1016/j.intimp.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Tabakman R, Jiang H, Levine RA, Kohen R, Lazarovici P. Apoptotic characteristics of cell death and the neuroprotective effect of homocarnosine on pheochromocytoma PC12 cells exposed to ischemia. J Neurosci Res. 2004;75:499–507. doi: 10.1002/jnr.20008. [DOI] [PubMed] [Google Scholar]
- 18.Ha SK, Moon E, Kim SY. Chrysin suppresses LPS-stimulated proinflammatory responses by blocking NF-κB and JNK activations in microglia cells. Neurosci Lett. 2010;485:143–147. doi: 10.1016/j.neulet.2010.08.064. [DOI] [PubMed] [Google Scholar]
- 19.Liou SF, Hsu JH, Liang JC, Ke HJ, Chen IJ, Wu JR, et al. San-Huang-Xie-Xin-Tang protects cardiomyocytes against hypoxia/reoxygenation injury via inhibition of oxidative stress-induced apoptosis. J Nat Med. 2012;66:311–320. doi: 10.1007/s11418-011-0592-0. [DOI] [PubMed] [Google Scholar]
- 20.Chiu PY, Chen N, Leong PK, Leung HY, Ko KM. Schisandrin B elicits a glutathione antioxidant response and protects against apoptosis via the redox-sensitive ERK/Nrf2 pathway in H9c2 cells. Mol Cell Biochem. 2011;350:237–250. doi: 10.1007/s11010-010-0703-3. [DOI] [PubMed] [Google Scholar]
- 21.Jamarkattel-Pandit N, Pandit NR, Kim MY, Park SH, Kim KS, Choi H, et al. Neuroprotective effect of defatted sesame seeds extract against in vitro and in vivo ischemicneuronal damage. Planta Med. 2010;76:20–26. doi: 10.1055/s-0029-1185903. [DOI] [PubMed] [Google Scholar]
- 22.Fujimura N, Sumita S, Narimatsu E. Alteration in diaphragmatic contractility during septic peritonitis in rats: effect of polyethylene glycol-absorbed superoxide dismutase. Crit Care Med. 2000;28:2406–2414. doi: 10.1097/00003246-200007000-00036. [DOI] [PubMed] [Google Scholar]
- 23.Shin MJ, Kim DW, Lee YP, Ahn EH, Jo HS, Kim DS, et al. Tat-glyoxalase protein inhibits against ischemic neuronal cell damage and ameliorates ischemic injury. Free Radic Biol Med. 2013;67C:195–210. doi: 10.1016/j.freeradbiomed.2013.10.815. [DOI] [PubMed] [Google Scholar]
- 24.Haddad JJ, Land SC. Redox/ROS regulation of lipopolysaccharide-induced mitogen-activated protein kinase (MAPK) activation and MAPK-mediated TNF-alpha biosynthesis. Br J Pharmacol. 2002;135:520–536. doi: 10.1038/sj.bjp.0704467. [DOI] [PMC free article] [PubMed] [Google Scholar]