

Abstract
Background
Coronary artery calcified atherosclerotic plaque (CAC) predicts cardiovascular disease (CVD). Despite exposure to more severe conventional CVD risk factors, African Americans (AAs) are less likely to develop CAC, and when they do, have markedly lower levels than European Americans. Genetic factors likely contribute to the observed ethnic differences. To identify genes associated with CAC in AAs with type 2 diabetes (T2D), a genome-wide association study (GWAS) was performed using the Illumina 5 M chip in 691 African American-Diabetes Heart Study participants (AA-DHS), with replication in 205 Jackson Heart Study (JHS) participants with T2D. Genetic association tests were performed on the genotyped and 1000 Genomes-imputed markers separately for each study, and combined in a meta-analysis.
Results
Single nucleotide polymorphisms (SNPs), rs11353135 (2q22.1), rs16879003 (6p22.3), rs5014012, rs58071836 and rs10244825 (all on chromosome 7), rs10918777 (9q31.2), rs13331874 (16p13.3) and rs4459623 (18q12.1) were associated with presence and/or quantity of CAC in the AA-DHS and JHS, with meta-analysis p-values ≤8.0 × 10−7. The strongest result in AA-DHS alone was rs6491315 in the 13q32.1 region (parameter estimate (SE) = −1.14 (0.20); p-value = 9.1 × 10−9). This GWAS peak replicated a previously reported AA-DHS CAC admixture signal (rs7492028, LOD score 2.8).
Conclusions
Genetic association between SNPs on chromosomes 2, 6, 7, 9, 16 and 18 and CAC were detected in AAs with T2D from AA-DHS and replicated in the JHS. These data support a role for genetic variation on these chromosomes as contributors to CAC in AAs with T2D, as well as to variation in CAC between populations of African and European ancestry.
Electronic supplementary material
The online version of this article (10.1186/s12863-017-0572-9) contains supplementary material, which is available to authorized users.
Keywords: African Americans, Coronary artery calcified atherosclerotic plaque, Genome-wide association study, Cardiovascular disease, Type 2 diabetes, Genetics
Background
Atherosclerotic coronary artery disease remains a leading cause of death in Western societies. In 2011, cardiovascular disease (CVD) accounted for 31.3% of all-cause mortality in the US [1]. Non-invasive computed tomography (CT)-based measures of coronary artery calcified atherosclerotic plaque (CAC) are useful for risk stratification. Higher levels of CAC are associated with CVD risk and death and may refine existing algorithms used to predict CVD events [2, 3].
Individuals with type 2 diabetes (T2D) have an elevated burden of subclinical coronary atherosclerosis. The severity of CAC and suboptimal glycemic control are strong risk factors for CAC progression [4]. However, marked variation in levels of CAC and calcified atherosclerotic plaque (CP) in other vascular beds, including the carotid arteries and aorta, are observed between African and European ancestral groups [5–7]. Despite exposure to more severe conventional CVD risk factors, African Americans (AAs) have the same or lower CP levels than European Americans (EAs) [8, 9]. These risk factors include more frequent and more severe hypertension, higher LDL-cholesterol levels, and excess albuminuria [10–12]. Similar population ancestry-based CAC differences are observed in T2D individuals, with CAC levels generally increased relative to those lacking diabetes. Despite higher blood sugar, AAs with T2D have markedly lower CAC levels than similarly affected EAs [13–15].
In addition to differences in environmental exposure, abundant evidence supports inherited contributions to ethnic-specific T2D rates and CP susceptibility. A genetic basis for CAC differences based on ancestry is supported by results of the Multi-Ethnic Study of Atherosclerosis (MESA) and the African American-Diabetes Heart Study (AA-DHS) [13, 14]. Higher proportions of European ancestry were observed in AAs with higher CAC levels in both studies. AAs are an admixed population with approximately 80% African and 20% European ancestry [16]. Mapping by admixture linkage disequilibrium (MALD or admixture mapping) was performed in the AA-DHS [14]. Eleven genomic regions were suggestively or significantly linked with CAC in the AA-DHS MALD study; all demonstrated excess European ancestry in CAC-linked regions in support of European-derived risk. The present analysis reports results of the first genome-wide association study (GWAS) for CAC in AAs with T2D, the second GWAS for CAC in AAs to date.
Methods
Subjects
The present report contains GWAS results in 691 AAs with T2D from AA-DHS. Replication analyses were performed in AAs with T2D (N = 205) from the Jackson Heart Study (JHS). Diabetes was defined as fasting blood glucose (FBG) ≥126 mg/dL or a random glucose ≥200 mg/dL, history of physician diagnosis of diabetes, or use of insulin or an oral hypoglycemic agent.
African American-Diabetes Heart Study
As reported, AA-DHS consisted of AAs with T2D recruited from two Wake Forest School of Medicine (WFSM) studies: the family-based Diabetes Heart Study (DHS) and unrelated individuals in the AA-DHS. DHS is a cross-sectional study of EA and AA families with siblings concordant for T2D. AA-DHS started after DHS and enrolled unrelated AAs. AA-DHS objectives are to improve understanding of ethnic differences in CAC and CP in populations of African and European ancestry. T2D in AAs was diagnosed after the age of 30 years in the absence of diabetic ketoacidosis. Individuals who underwent prior coronary artery bypass surgery or coronary artery angioplasty and/or stent placement were not included in the analyses, because CAC scores could have been impacted by the procedures. Those with prior myocardial infarction (MI) or stroke were included. The final analysis included 691 unrelated AAs and AA sibpairs concordant for T2D obtained by selecting all AA-DHS participants and DHS participants that passed the quality control checks described in the genotyping section (below). The study was approved by the WFSM Institutional Review Board and all participants provided written informed consent.
Jackson Heart Study
The Jackson Heart Study (JHS) is a prospective population-based cohort study initiated in 2000 in the Jackson, Mississippi tri-county area. The primary objective of this study was to understand the determinants of the high prevalence of common complex diseases including CVD, T2D, obesity, chronic kidney disease, and stroke in AAs [17]. The subset of JHS participants with diabetes was between 35 and 84 years old.
Vascular imaging
CAC was quantified by non-contrast, ECG gated, cardiac computed tomography (CT) in both studies using established methods [18]. In AA-DHS, from the start of the study in 1999, four generations of CT scanners all with ECG gating were utilized (CTi, QXi, 16Pro and VCT, GE Healthcare, Waukesha, WI). Image spatial resolution, reconstruction kernel and image technique were held constant. CAC was measured on a workstation and reported as the Agatston Score using a 90 Hounsfield Unit (HU) threshold (SmartScore GE Healthcare). Additional scoring parameters included a 130 HU threshold and 2 adjacent pixels used to define the maximum calcified lesion size; the program accounted for slice thickness. In JHS, calcified plaque was measured on a 16-channel CT system (16 Pro; GE Healthcare). The CAC score was reported as an Agatston score using a 130 HU threshold (TeraRecon Aquarius Workstation, Foster City, CA). Image analysis and quality control for both studies were performed at the same central reading center. The minimum lesion size in AA-DHS was 0.52 mm2 and the minimum lesion size in the Jackson Heart Study was 1 mm2 [19, 20].
The robustness of the CAC score as an imaging biomarker across generations of CT scanners has been previously documented [5, 19]. The calcium mass and traditional Agatston scores are two scales for measuring calcified plaque. The mass score is calibrated on a per pixel basis to an external calibration phantom (Image Analysis, Columbia, KY) which reduces noise related to the method of quantification. The Agatston score uses a threshold of 130 HU and calcified plaque below this cut-point even when present are grouped with the zero scores. This creates a group of false negatives by the design of the scoring system. The calcium mass uses a threshold of 90 and minimizes the probability of a false negative result (increased sensitivity) with a modest reduction in specificity. In AA-DHS, the Pearson and Spearman correlations between the mass score and Agatston scores were 0.97 and 0.95, respectively. However, the interquartile ranges were 397 for the mass score and 180 Agatston score. Therefore, the 90 HU CAC measure is better suited than 130 HU CAC to detect low CP levels in vascular beds. Thus, the 90 HU measure coupled with the MASS scoring algorithm we employed are more sensitive for detecting calcified plaques and reduce noise in the scoring methods. This approach improved power for the CAC study outcome. The 90 HU measure was only available in AA-DHS. Therefore, we opted for a discovery approach which was conducted in AA-DHS using the 90 HU CAC measure as the outcome, followed by replication with the 130 HU CAC in JHS. In addition, two meta-analyses were performed. The main meta-analysis used AA-DHS 90 HU CAC and 130 HU Jackson Heart Study CAC measures; the inverse variance meta-analysis was performed separately in the two studies using standardized coefficients. This approach controls for minor differences in test composition between studies (i.e., 90 HU vs 130 HU). For completeness, we also performed a second meta-analysis using the 130 HU CAC score in both study samples. Results of both analyses are reported in Table 3 and Additional file 1: Table S4 for 90 HU in AA-DHS vs. 130 HU in JHS and 130 HU in both studies, respectively.
Table 3.
Summary of SNPs with meta-analysis p-values ≤8.0 × 10−7
| SNP | CHR | Position | Nearest Gene | Alleles | Outcome | MOI | AA-DHSa | JHSb | Meta-Analysis | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | SE | P-value | Estimate | SE | P-value | DE | P-value | |||||||
| rs113533135 | 2 | 141,026,436 | LRP1B | T/C | CAC > =10 vs. CAC < 10 | Dominant | 0.19 | 0.05 | 1.7 × 10−4 | 0.30 | 0.08 | 3.1 × 10−4 | – | 3.3 × 10−7 |
| rs6754498 | 2 | 186,817,848 | A/G | Log(CAC + 1) | Dominant | 0.83 | 0.22 | 1.9 × 10−4 | 1.12 | 0.32 | 5.2 × 10−4 | ++ | 5.5 × 10−7 | |
| rs9973676 | 2 | 231,818,806 | GPR55 | A/G | CAC > =10 vs. CAC < 10 | Dominant | −0.20 | 0.05 | 5.8 × 10−5 | −0.22 | 0.07 | 3.1 × 10−3 | – | 6.4 × 10−7 |
| rs6898559 | 5 | 125,892,929 | ALDH7A1 | A/G | CAC > =10 vs. CAC < 10 | Additive | 0.09 | 0.03 | 4.1 × 10−4 | 0.18 | 0.04 | 4.3 × 10−5 | – | 5.0 × 10−7 |
| rs16879003 | 6 | 16,745,239 | ATXN1 | T/C | CAC > =10 vs. CAC < 10 | Additive | −0.19 | 0.04 | 7.0 × 10−6 | −0.20 | 0.07 | 4.7 × 10−3 | ++ | 1.1 × 10−7 |
| rs113805659 | 7 | 78,292,791 | MAGI2 | C/G | CAC > =10 vs. CAC < 10 | Additive | −0.19 | 0.04 | 5.5 × 10−6 | −0.21 | 0.08 | 7.5 × 10−3 | – | 1.4 × 10−7 |
| rs117854110 | 7 | 105,243,449 | T/C | Log(CAC + 1) | Dominant | −2.74 | 0.66 | 4.2 × 10−5 | −2.72 | 0.91 | 2.9 × 10−3 | – | 4.5 × 10−7 | |
| rs58071836 | 7 | 118,273,539 | T/C | CAC > =10 vs. CAC < 10 | Additive | −0.13 | 0.04 | 1.7 × 10−4 | −0.21 | 0.06 | 2.2 × 10−4 | ++ | 2.8 × 10−7 | |
| rs10978777* | 9 | 110,030,590 | C/G | Log(CAC + 1) | Additive | −0.63 | 0.16 | 1.3 × 10−4 | −0.95 | 0.24 | 8.7 × 10−5 | – | 1.2 × 10−7 | |
| rs75916004 | 11 | 101,896,887 | A/G | CAC > =10 vs. CAC < 10 | Dominant | 0.22 | 0.04 | 1.2 × 10−6 | 0.21 | 0.09 | 1.5 × 10−2 | – | 5.9 × 10−8 | |
| rs77934287 | 12 | 23,224,253 | AK094733 | A/G | CAC > =10 vs. CAC < 10 | Recessive | −0.38 | 0.10 | 3.0 × 10−4 | −0.59 | 0.16 | 1.8 × 10−4 | – | 4.8 × 10−7 |
| rs448792 | 13 | 32,677,424 | FRY | T/C | CAC > =10 vs. CAC < 10 | Dominant | 0.21 | 0.05 | 2.3 × 10−5 | 0.21 | 0.08 | 8.4 × 10−3 | ++ | 6.1 × 10−7 |
| rs77757620 | 15 | 42,953,597 | STARD9 | T/C | CAC > =10 vs. CAC < 10 | Additive | 0.15 | 0.04 | 5.0 × 10−5 | 0.18 | 0.06 | 2.3 × 10−3 | ++ | 4.3 × 10−7 |
| rs13331874 | 16 | 581,733 | SOLH | A/G | CAC > =10 vs. CAC < 10 | Recessive | −0.28 | 0.06 | 1.6 × 10−6 | −0.29 | 0.11 | 1.2 × 10−2 | ++ | 6.3 × 10−8 |
| rs4459623 | 18 | 28,798,433 | DSC1 | C/G | CAC > =10 vs. CAC < 10 | Recessive | −0.32 | 0.09 | 4.1 × 10−4 | −0.55 | 0.13 | 4.3 × 10−5 | ++ | 5.3 × 10−7 |
CHR chromosome, MOI Mode of Inheritance, DE Direction of effect (+: positive association, −: negative association)
*Upstream to ABCB1
aResults observed with the 90 HU CAC
bResults observed with the 130 HU CAC
Genotyping
DNA in AA-DHS participants was extracted from peripheral blood using the PureGene system (Gentra Systems, Minneapolis, MN). The AA-DHS GWAS utilized the Illumina 5 M chip; the JHS GWAS genotyping was performed with the Affymetrix 6.0 chip. Genotypic data in both studies were imputed to 1000 Genomes (phase 1 version 3, cosmopolitan panel) [21].
Quality control
Quality control (QC) checks were performed before conducting the GWAS. In AA-DHS, these checks led to the exclusion of twelve individuals from the analyses: 6 had call rates <90%, 2 had discordant self-reported and genetically determined sex, 1 had a heterozygosity score outside of the mean ± 4 times the standard error interval, 2 had the same sample identifiers and 1 had 100% European ancestry. Genome-wide association analyses were performed on 691 individuals that met the QC inclusion criteria. We used a classification scheme to rank SNPs and prioritize association results. This scheme was based on the estimated minor allele frequency (MAF), the Hardy-Weinberg equilibrium (HWE) p-value, and the call rate. All SNPs were analyzed; however, results are reported for common variants with a HWE p-value ≤10−4 and a call rate ≥ 95%. SNPs with these qualifications also served as the basis for our imputation efforts. SNPs with a MAF between 1 and 5% that met the HWE p-value and call rate thresholds were also included in imputation. Similar QC procedures were applied in JHS, which led to the exclusion of 2 subjects. The final analysis included 205 JHS study participants with T2D and available CAC measurements.
Local and global admixture
Local ancestry estimation was performed using LAMP-ANC and HAPMIX [22, 23]. A linkage disequilibrium (LD) pruning algorithm was applied with an R-squared threshold of 0.8 to select a subset of SNPs among those that met the above QC criteria. Observed data at these SNPs were then combined with HapMap phase 3 genotypes obtained from Yoruban and CEPH samples; the HapMap samples were used as anchoring populations and were not included in the analysis. The estimation process was repeated twice in AA-DHS, once with LAMP-ANC and once with HAPMIX. Results were comparable; the distribution of Spearman correlation estimates ranged between 0.88 and 0.97. Local admixture estimation in JHS was performed with LAMP-ANC. The global ancestry proportion estimates were obtained by averaging the local ancestry estimates across the genome. These global estimates were used as covariates in the association models and are reported in Tables 1 and 2.
Table 1.
Demographic characteristics of AA-DHS participants, by presence/absence of CAC
| Variables | CAC 130 HU <10 (N = 346) | CAC 130 HU ≥10 (N = 345) | ALL (N = 691) | P-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Median | Mean | SD | Median | Mean | SD | Median | ||
| Age (years) | 52.8 | 8.7 | 52.0 | 59.8 | 9.0 | 60.0 | 56.3 | 9.6 | 56.0 | <0.0001 |
| Female (%) | 66.5 | 53.6 | 60.1 | 0.0006 | ||||||
| African Ancestry proportion (%) | 76.4 | 14.9 | 78.9 | 73.8 | 16.0 | 76.4 | 75.1 | 15.5 | 78.0 | 0.05 |
| Diabetes duration (years) | 8.4 | 6.1 | 7.0 | 12.2 | 9.3 | 10.0 | 10.3 | 8.1 | 8.0 | <0.0001 |
| HbA1c (%) | 8.3 | 2.3 | 7.8 | 8.1 | 1.9 | 7.6 | 8.2 | 2.1 | 7.7 | 0.94 |
| C-reactive protein (mg/dl) | 1.0 | 1.5 | 0.5 | 0.9 | 1.3 | 0.4 | 0.9 | 1.4 | 0.5 | 0.27 |
| Glucose (mg/dl) | 154.7 | 68.8 | 138.0 | 148.9 | 67.1 | 132.0 | 151.7 | 68.0 | 136.0 | 0.19 |
| Low density lipoprotein cholesterol (mg/dl) | 108.5 | 37.4 | 106.0 | 107.1 | 36.6 | 103.0 | 107.8 | 36.9 | 105.0 | 0.46 |
| High density lipoprotein cholesterol (mg/dl) | 48.2 | 14.4 | 46.0 | 47.8 | 14.4 | 45.0 | 48.0 | 14.4 | 46.0 | 0.51 |
| Triglycerides (mg/dl) | 128.2 | 112.0 | 104.0 | 128.2 | 130.4 | 100.0 | 128.2 | 121.5 | 102.0 | 0.66 |
| Body Mass Index (kg/m2) | 35.9 | 8.6 | 34.6 | 34.4 | 8.2 | 32.9 | 35.1 | 8.4 | 33.7 | 0.02 |
| CAC 90 (Hounsfield unit) | 8.8 | 16.1 | 1.5 | 1212.0 | 1810.9 | 442.0 | 609.4 | 1413.3 | 44.0 | NA |
| CAC 130 (Hounsfield unit) | 0.8 | 1.8 | 0.0 | 445.1 | 646.1 | 190.5 | 222.6 | 507.5 | 9.5 | NA |
| ACE inhibitor use (%) | 46.8 | 51.3 | 49.1 | 0.25 | ||||||
| Current smoker (%) | 20.3 | 25.2 | 22.8 | 0.007 | ||||||
| Past smoker (%) | 31.7 | 40.2 | 36.0 | 0.001 | ||||||
| Hypertension (%) | 78.0 | 89.1 | 83.6 | 0.0001 | ||||||
| Lipid-lowering medication (%) | 39.5 | 53.4 | 46.4 | 0.0004 | ||||||
Table 2.
Demographic characteristics of Jackson Heart Study participants with diabetes, by presence/absence of CAC
| Variable | CAC 130 HU <10 (N = 68) | CAC 130 HU ≥10 (N = 137) | Full Sample (N = 205) | P-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Median | Mean | SD | Median | Mean | SD | Median | ||
| Age (years) | 54.3 | 10.1 | 53.0 | 60.7 | 9.3 | 60.0 | 58.4 | 10.1 | 59.0 | <0.0001 |
| Female (%) | 73.5 | 63.7 | 67.2 | 0.10 | ||||||
| African Ancestry proportion (%) | 79.9 | 6.5 | 80.7 | 78.1 | 8.4 | 79.5 | 78.8 | 7.8 | 80.0 | 0.21 |
| Diabetes duration (years) | 8.2 | 7.8 | 6.0 | 11.4 | 9.7 | 10.0 | 10.3 | 9.2 | 8.0 | 0.01 |
| HbA1c (%) | 7.7 | 2.0 | 7.2 | 7.9 | 1.8 | 7.6 | 7.8 | 1.9 | 7.4 | 0.06 |
| C-reactive protein (mg/dl) | 0.6 | 0.8 | 0.4 | 0.7 | 1.0 | 0.4 | 0.7 | 0.9 | 0.4 | 0.79 |
| Glucose (mg/dl) | 147.4 | 62.4 | 133.0 | 145.1 | 53.0 | 130.0 | 146.0 | 56.5 | 132.5 | 0.90 |
| Low density lipoprotein cholesterol (mg/dl) | 115.9 | 33.1 | 116.0 | 122.2 | 39.5 | 120.0 | 119.9 | 37.4 | 117.0 | 0.12 |
| High density lipoprotein cholesterol (mg/dl) | 50.3 | 13.0 | 47.0 | 48.3 | 13.9 | 45.0 | 49.0 | 13.6 | 46.0 | 0.12 |
| Triglycerides (mg/dl) | 118.5 | 63.1 | 100.0 | 142.0 | 100.3 | 113.0 | 133.6 | 89.5 | 108.0 | 0.12 |
| Body Mass Index (kg/m2) | 34.2 | 7.0 | 33.6 | 34.0 | 6.5 | 33.0 | 34.1 | 6.7 | 33.1 | 0.94 |
| CAC 130 HU (Hounsfield unit) | 1.2 | 2.4 | 0.0 | 548.4 | 866.1 | 206.4 | 354.8 | 743.3 | 60.0 | NA |
| ACE inhibitor use (%) | 78.0 | 79.8 | 79.2 | 0.73 | ||||||
| Current smoker (%) | 11.2 | 11.2 | 11.2 | 0.99 | ||||||
| Past smoker (%) | 20.4 | 30.7 | 27.1 | 0.06 | ||||||
| Hypertension (%) | 82.5 | 86.6 | 85.1 | 0.36 | ||||||
| Lipid-lowering medication (%) | 18.6 | 33.5 | 28.4 | 0.01 | ||||||
Imputation
Imputation was performed using IMPUTE2 with phased haplotypic data obtained from Shapeit2 [24]. The imputation effort used all SNPs that passed the QC filters. Imputation was based on 3,436,913 and 733,318 autosomal SNPs in AA-DHS and JHS, respectively. The multi-ethnic 1000 Genomes Phase I integrated variant set release (v3) was used as the reference panel [21]. Imputation was performed separately for each study. Statistical analyses were performed on imputed SNPs that had certainty score above 90%, info score above 50% and MAF greater than 1%.
Statistical analysis
Analyses were run using Log(CAC + 1) and CAC dichotomized (presence (CAC ≥ 10) vs. absence (CAC < 10)). The value of 1 added to the observed CAC score allowed for the inclusion all subjects, even those with a CAC score of zero. This approach is justified based on the assumption that factors governing presence of CAC may differ from those influencing amount of CAC once calcification is initiated [25]. Age, gender, global African ancestry proportion, diabetes duration, hemoglobin A1c, body mass index, smoking status, and use of lipid-lowering medication were included as covariates in the model to test for association between each SNP and CAC. Analyses were run separately in each study using the same outcome definitions based on the 90 HU CAC in AA-DHS and the 130 HU score in JHS. For the continuous outcome, linear mixed models were fitted using Genome-wide Efficient Mixed Model Analysis (GEMMA) software [26]. Generalized estimating equations were implemented to test for associations with the binary outcome. All analyses adjusted for familial relationships estimated using the Relatedness Estimation in Admixed Populations (REAP) software [27]. SNPs were tested for association using the likelihood ratio test for the overall two degrees of freedom mode of inheritance model. If the overall test of association is significant, then the three a priori genetic models (dominant, additive, and recessive) were explored; the model with the best fit for each SNP was used. Correction for this maximization was applied to account for the correlation between tests and to maintain the type 1 error rate [28–30]. This approach is consistent with Fisher’s protected least significant difference multiple comparison procedure. Sample size weighted meta-analysis was performed to compare and combine results observed from each study. Penalized regression with the L 1 norm (LASSO) was used to identify the SNP with the strongest effect size when LD caused several SNPs to display strong association with the outcome. A cross validation approach was used to determine the shrinkage parameter for each region. SNP selection was performed only in the AA-DHS subset, the larger of the two studies, to limit confounding effects. Joint tests of association between local ancestry and genotypes with CAC were also were performed. The model used for testing for association between local ancestry and CAC was similar to the one described for the genetic association tests, with local ancestry replacing the observed or imputed genotypes. If T L and T G denote the test statistics associated with the local ancestry and genotypic association with CAC, the joint test of association with local ancestry and genotype at each marker was calculated as , which follows a Chi-square distribution with 2 degrees of freedom [31]. An alternative test based on the maximum of T G and T L was also computed, assuming these tests follow a bivariate normal distribution with a non-zero correlation. The empirical correlation was computed using the variance-covariance matrix for 2 correlated score tests [32]. Results from these tests are shown in Additional file 2: Table S3.
Significant effects and correction for multiple testing
The analysis involved more than 13 million directly genotyped and imputed SNPs. A strict Bonferroni correction would place the significance threshold at 1.9 × 10−9 for a two-sided test, a highly conservative threshold. The sample sizes required to adequately power genetic association studies in AAs at this significance threshold are not feasible for a single study (or even two). These limitations are more pronounced when the focus is on atherosclerosis in the subset of this population with T2D. We prioritized meta-analysis association results that reached an adjusted p-value ≤8.0 × 10−7, with a minimum adjusted p-value ≤8.0 × 10−3 observed in each study and prior evidence of association of the gene with CAC, T2D and other factors involved in the atherosclerosis process [33, 34]. This approach combines statistical plausibility, with nominal statistical significance from both studies, and documented prior evidence that these genomic regions have been implicated in T2D and one or more processes leading to atherosclerosis. The adjusted p-value (or best p-value) is the p-value associated with the minimum of the three test statistics obtained with the additive, dominant and recessive mode of inheritance models.
Results
Table 1 displays demographic and clinical data in 691 AA-DHS participants with T2D; data in JHS participants are displayed in Table 2. Although 90 HU CAC scores were analyzed in the AA-DHS GWAS, demographic data in both tables uses the 130 HU CAC score to permit comparability. AA-DHS study participants were on average two years younger than those in JHS (56.3 ± 9.6 years vs. 58.4 ± 10.1 years; p-value = 6.7 × 10−3); had slightly higher HbA1c (8.2 ± 2.1% vs. 7.8 ± 1.9%, p-value = 0.02); lower proportion of African ancestry (75.1 ± 15.5% vs. 78.8 ± 7.8%, p-value = 0.001), and lower low density lipoprotein cholesterol (LDL) levels (107.8 ± 36.9 mg/dl vs. 119.9 ± 37.4 mg/dl, p-value = 4.8 × 10−5). At the 130 HU threshold, JHS participant were more likely to have CAC ≥ 10, compared to AA-DHS participants (49.9% in AA-DHS vs. 66.8% in JHS, p-value = 8.6 × 10−6). However, when CAC was present, AA-DHS and JHS had comparable levels (445.1 ± 646.1 vs. 548.4 ± 866.1, p-value = 0.11). In addition, participants in AA-DHS were less likely to be on angiotensin converting enzyme (ACE) inhibitors than JHS (49.1% vs. 79.2%, p-value = 3.2 × 10−35), but more likely to be current or former smokers (58.8% vs. 38.3%, p-value = 7.8 × 10−26) and on lipid lowering medications (46.4% vs. 28.4%, p-value = 6.1 × 10−17).
Within AA-DHS, individuals with 130 HU CAC ≥10 were on average 6 years older (p-value <0.0001), had longer diabetes durations (12.2 ± 9.3 years vs. 8.4 ± 6.1, p-value = 5.3 × 10−5), and were more likely to be on lipid lowering medications (53.4% vs. 39.5%, p-value = 2.2 × 10−4). Similar patterns were observed in JHS; study participants with CAC ≥10 were more likely to be older (average age difference 5.4 years, p-value < 0.0001), have longer diabetes durations (average difference 3.2 years, p-value = 0.01) and more likely to receive lipid lowering medications (33.5% vs. 18.6%, p-value = 0.01). The 90 HU CAC measure was more sensitive than the 130 HU CAC score at the lower end of the calcified plaque (CP) distribution; 37.7% of AA-DHS had 90 HU CAC <10 vs. 51.1% with 130 HU CAC, which supports the use of 90 HU CAC measure as the outcome in the GWAS, especially when focusing on presence of CAC. The kappa statistic measuring the agreement between the 2 measures was 0.75 with the cutoff value of 10. However, the spearman rank correlation between 90 HU CAC and 130 HU CAC was over 99% when the CP level was above 10 with both thresholds.
Complete results of the sample size weighted meta-analysis GWAS for CAC are displayed in Figs. 1 and 2 for the continuous and binary analyses, respectively. SNPs that reach a meta-analysis p-value less than 10−4 are shown in Additional file 3: Table S1. Table 3 displays the top meta-analysis GWAS results for 90 HU CAC in AA-DHS and 130 HU CAC in JHS, limited to those with meta-analysis p-values ≤8.0 × 10−7. SNPs that were associated with both presence and amount of CAC tended to have stronger association effects with the amount of CAC modeled as Log(CAC + 1). In this case, only the association with the continuous outcome is reported. Association effects shown with presence of CAC were only observed with this outcome. We focus on six genomic regions that meet our prioritization rule. Top results for the meta-analysis of the 130 HU CAC in both AA-DHS and JHS are shown in Additional file 1: Table S4. We note that rs113805659 was the only SNP that met the statistical significance threshold in both analyses. Regional association plots for these regions are shown in Figs. 3 and 4.
Fig. 1.
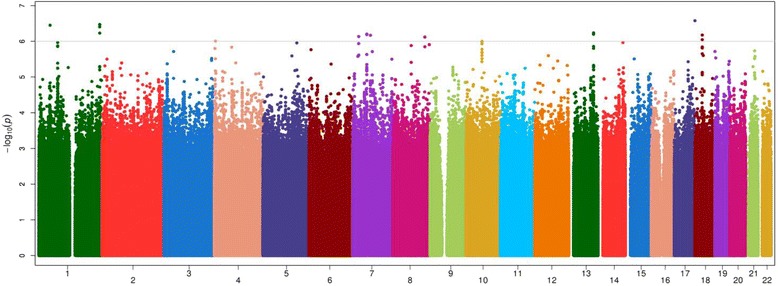
Manhattan Plot of the meta-analysis of Log(CAC + 1) between AA-DHS and JHS CAC was measured at the 90 HU threshold in AADHS and the 130 HU threshold in JHS
Fig. 2.
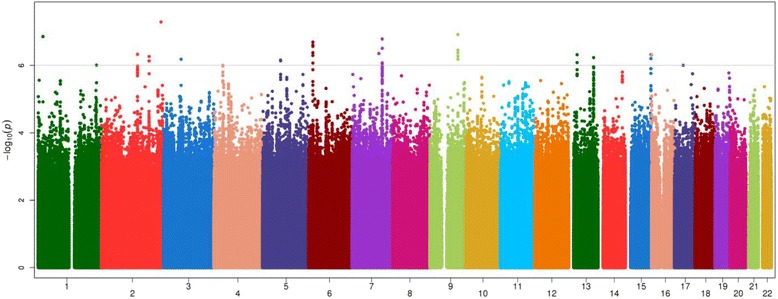
Manhattan Plot of the meta-analysis of Presence of CAC between AA-DHS and JHS CAC was measured at the 90 HU threshold in AADHS and the 130 HU threshold in JHS
Fig. 3.
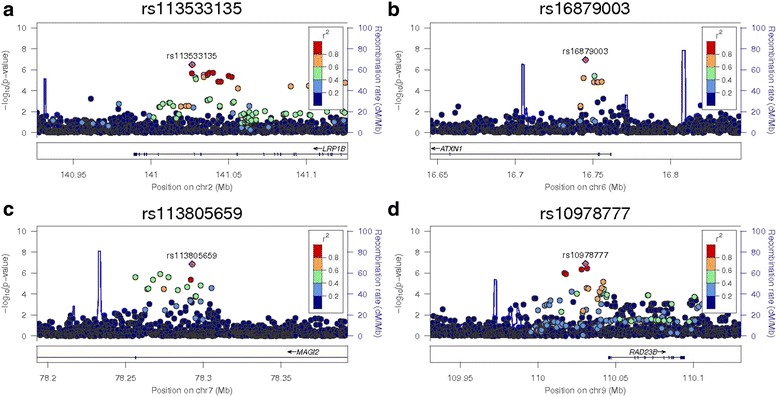
Regional association plots for the ‘sentinel’ SNP in LRP1B, ATXN1, MAGI2, 9q31.2 –log10 (p-values) are shown for all SNPs in each region with the color of circles indicating the degree of LD with the most associated SNP in the region. These regions are: a rs113533135 in LRP1B, b rs16879003 in in ATXN1, c rs113805659 in MAGI2; and d rs10978777 on 9q31.2. Observed p-values came from the meta-analysis for all SNPs
Fig. 4.
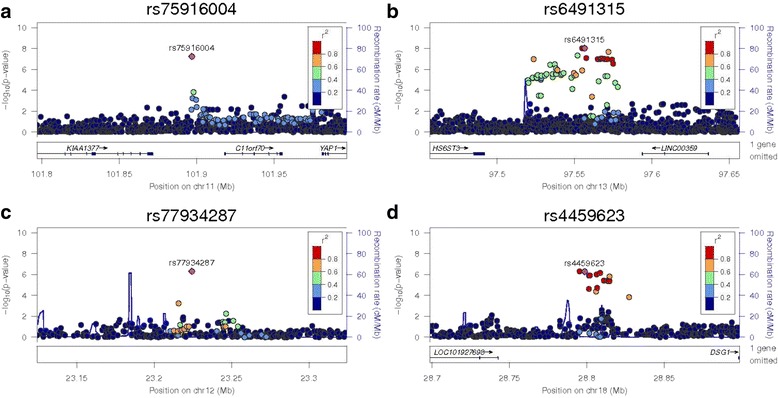
Regional association plots for the ‘sentinel’ SNP in 11q22.1, AK094733, HS6ST3 and DSC1. –log10 (p-values) are shown for all SNPs in each region with the color of circles indicating the degree of LD with the most associated SNP in the region. These regions are: a rs75916004 on 11q22.1, b rs77934287 in chromosome 12, c rs6491315 in HS6ST3 and d rs4459623 in DSC1. Observed p-values came from the meta-analysis for all SNPs, except rs6491315 in HS6ST3. This is region with the strongest result in AA-DHS; however, this signal was not replicated in JHS
The strongest association result was observed in the ataxin-1 (ATXN1) gene located on 6p22.3. Penalized regression suggests that rs16879003 had the strongest estimated effect size under the additive mode of inheritance (MOI) with parameter estimate and (standard error) -0.19(0.04) in AA-DHS and −0.20(0.07) in JHS for a meta-analysis p-value of 1.1 × 10−7. Variants in the 6p22.3 region have been associated with MI and coronary heart disease (CHD) [35].
Association was also detected within LRP1B, a LDL receptor gene, located on 2q22.1, a genomic region previously implicated in CHD and heart failure [36, 37]. Under the dominant MOI, the parameter estimates for the association tests between rs113533135 and presence of CAC were 0.19(0.05) in AA-DHS and 0.30(0.08) in JHS for a meta-analysis p-value of 3.3 × 10−7.
Several SNPs in the membrane-associated guanylate kinase inverted 2 gene (MAGI2) were strongly associated with the binary outcome of presence (versus absence) of CAC. This gene is located on chromosome 7q21.11. Conditional analysis revealed that rs113805659 had the strongest effect; under an additive mode of inheritance model that counts the number of minor alleles, the parameter estimates (standard errors) were −0.19 (0.04) in AA-DHS and −0.21 (0.08) in JHS with p-values of 5.5 × 10−6 and 7.5 × 10−3, respectively. The association effect was in the same direction in both studies, leading to a meta-analysis p-value of 1.4 × 10−7. Variants in MAGI2 have been associated with the disposition index, a measure of the relationship between insulin secretion and insulin sensitivity, cell-cell and cell-matrix adhesion in Hispanics and AAs with T2D, and aortic CP in the Framingham study [38–40].
Several SNPs located in the 7q31.31 region showed significance with presence of CAC. After conditional analyses, rs58071836 had the strongest effect under an additive MOI -0.13(0.04) in AA-DHS and −0.22(0.06) in JHS for a meta-analysis p-value of 1.8 × 10−7. This chromosomal region harbors WNT16, a gene implicated in vascular calcification and bone mineral density (BMD) [41–43].
Similar strong association results were observed on chromosome 9 with rs10918777 located on 9q31.2, under the additive model (−0.63 (0.16), p-value = 1.3 × 10−4 in AA-DHS and −0.95 (0.24), p-value = 8.7 × 10−5 in JHS and meta-analysis p-value = 1.2 × 10−7), and on chromosome 12 with rs77934287, under the recessive model (−0.38 (0.10), p-value = 3.0 × 10−4 in AA-DHS and −0.59 (0.16), p-value = 1.8 × 10−4 in JHS and meta-analysis p-value = 4.8 × 10−7).
Nearly 20 different SNPs were found to be associated with the presence of CAC in the 18q12.1 region. These SNPs were all significant under the recessive model in both datasets. Conditional analyses showed that the number of associated SNPs is primarily due to strong LD in the region. The sentinel SNP (rs4459623) had an effect size of (−0.32 (0.09), p = 4.1 × 10−4) in AA-DHS and (−0.55 (0.13), p = 4.3 × 10−5) in JHS, for a meta-analysis p-value 5.3 × 10−7. These SNPs are in an intergenic region 25 kb distal to the desmocollin 1 gene (DSC1) and 99 kb proximal to desmoglein 1 gene (DSG1) [44, 45].
Our previous admixture mapping effort identified several regions with strong evidence for linkage with local ancestry [14]. All of these regions showed that excess European ancestry is associated with the risk and progression of CAC. However, these regions were identified using a 90 HU threshold Agatston Score, a measure that is not available in JHS. We were able to replicate these results in the AA-DHS analysis, but not in JHS. For example, the admixture mapping signal reported on chromosome 13 near 13q32.1 (LOD = 2.8) replicated with several SNPs showing statistical significance at the 10−8 threshold in AA-DHS alone. In the fully-adjusted model, rs6491315 had an effect size of (−1.14 (0.2), p = 9.1 × 10−9); however, the effect was non-significant in JHS (0.11 (0.36), p-value = 0.75), yielding a meta-analysis p-value 5.8 × 10−7. Joint models that combine local ancestry with the observed genotypic data in AA-DHS are shown in Additional file 4: Table S2 for completeness.
Discussion
This report contains one of the few GWAS for CAC in the understudied AA population with T2D; it includes 896 subjects from two of the largest studies in this population. Several SNPs had a meta-analysis p-value less than 8.0× 10−7, albeit none reached the strict Bonferroni corrected threshold. Two SNPs, rs75916004 located on chromosome 11 in the 11p15.4 region and rs13331874 located on 16p13.3 were close, with meta-analysis p-values of 5.9 × 10−8 and 6.3 × 10−8, respectively. However, their p-values were greater than 8 × 10−3 in JHS. Genome-wide significant association was detected between CAC and multiple SNPs located on chromosome 13 near the HS6ST3 gene in AA-DHS. Several other genomic regions displayed suggestive evidence of association.
The chromosome 11 and 13 GWAS peaks overlay suggestive MALD peaks for CAC in AA-DHS participants. This result suggests that a portion of the ancestry-specific risk for development of CAC lies in this genomic region. Emerging data support a genetic basis for the markedly lower levels of CAC in AAs, relative to EAs. In support of clinical relevance, reduced rates of MI are present in AAs with and without T2D, provided equal access to healthcare as EAs [46–48]. We also note that no SNPs in the 9p21 and 6p24 regions that have been reported in previous GWAS of European ancestry populations met the statistical significance criteria defined for the genetic association tests. The admixture mapping results shown in Additional file 4: Table S2 still suggest that excess European ancestry in these regions contributes to the likelihood and severity of CAC; however, the association signals were weaker.
These results observed in AAs with T2D contrast with those in a recent GWAS meta-analysis for CAC in AAs where the prevalence of T2D varied between 10.3 and 33.2% [49]. That report identified significant heritability for CAC in AAs, but failed to detect genome-wide evidence of association. Gomez et al. identified one statistically significant admixture mapping peaks on chromosome 12 and three suggestive peaks on chromosomes 6, 9, and 15 [50]. These regions did not overlap with the admixture mapping results reported in Divers et al. [14] and those described in Table 3 below. Similar to Wojczynski et al. [49], only a fraction of their sample (~28%) had T2D, which may partly explain the discordant results.
We also note that some of the admixture results observed in AA-DHS did not replicate in JHS (See Additional file 2: Table S3), and we can only speculate on why that would be the case. Possible reasons include the smaller sample size (691 vs. 205), the greater sensitivity of the 90 HU CAC measure that was only available in AA-DHS, and that hyperglycemia and T2D likely provide a modifying environmental stimulus that increases the development of CAC more prominently in individuals who are genetically susceptible to atherosclerosis. All published reports that measured CAC and other sites of CP reveal markedly higher levels in subjects with T2D, relative to those without T2D [51, 52]. Therefore, it is probable that the diabetic milieu predisposed to coronary artery atherosclerosis in our populations, even among participants at lower risk for CAC due to their higher percentage of recent African ancestry. Absence of similar environmental triggers in the Wojczynski et al. AA GWAS and Gomez et al. admixture mapping meta-analyses with lower levels of CAC could have diminished the ability to detect genetic association [49, 50].
Among the significant associations identified, SNP rs16879003 resides in the ATXN1 gene, a member of the spinocerebellar ataxia (SCA) family. Although this protein is ubiquitously expressed, its function remains unknown. However, recent studies have linked variants in this gene with adiposity, MI and CHD [35, 53]. Based on these observations, there is support for a potential role in which variation at this locus serves as a modifier, either indirectly, i.e. metabolic syndrome, or directly as a transcriptional repressor in the development of CAC more prominently in individuals who are genetically susceptible to atherosclerosis [54].
rs113533135 is located in the LRP1B gene, a member of the LDL receptor family. LRP1B is a multi-ligand receptor that binds urokinase plasminogen activator and plasminogen activator inhibitor-1, suggesting a role in fibrinolysis and extracellular matrix remodeling [55]. The LRP1B gene has also been associated with insulin resistance and tumor suppression in humans and LDL cholesterol levels in rats [56–58].
rs10978777 is located in the 9q31 region upstream of the ABCA1 gene. This gene encodes a transmembrane protein that is expressed in most cells in the body and is critical for cellular cholesterol efflux and HDL particle formation [59]. Mutations in ABCA1 cause Tangier disease, a rare genetic disorder that is characterized by plasma HDL cholesterol concentrations <5% of normal, ~50% reduction in plasma LDL cholesterol concentrations, cholesterol accumulation in macrophages, peripheral neuropathy, and hepatosplenomegaly. Some Tangier disease kindreds have premature CHD, likely due to very low plasma HDL cholesterol levels, but some are spared, presumably due to significant reductions in plasma LDL cholesterol.
rs77934287 is located on 12p12.1, a genomic region that contains the LOX-1 gene and ABCC9, a gene that has been associated with the Cantu syndrome, cardiac conduction disturbances and possibly cardiomyopathy [60, 61]. LOX-1 encodes the lectin-like oxidized LDL receptor, which is expressed on the surface of macrophages, smooth muscle cells, and endothelial cells. The LOX-1 receptor binds and internalizes proinflammatory oxidized LDL, an initiator of macrophage lipid accumulation and endothelial cell dysfunction. LOX-1 expression has been documented in human atherosclerotic plaques and its expression is increased by proinflammatory cytokines. ABCC9 is a member of the superfamily of ATP-binding cassette (ABC) transporters. The ABCC9 protein is thought to form ATP-sensitive potassium channels in cardiac, skeletal, and vascular and non-vascular smooth muscle.
Multiple SNPs in the 18q12.1 region were associated with CAC in this study (Table 3). These SNPs are in an intergenic region between DSC1 and DSG1, both of which belong to the cadherin superfamily of cell-cell adhesion genes that form a cluster on chromosome 18. DSC1 and DSG1 are calcium-dependent transmembrane glycoproteins involved in desmosome formation between cells. Atherogenic lipoproteins, such as oxidized LDL, that initiate atherogenesis and endothelial cell inflammation reportedly downregulate desmocollin and desmoglein expression. This may lead to endothelial barrier dysfunction and increased influx of atherogenic lipoproteins into the arterial intima of atherosclerotic lesions [62].
This study has limitations, including the small sample size and limited generalizability of results observed to AAs with longstanding T2D. Regarding the concern over sample size, it is important to note that the replication provides the ultimate protection against type 1 errors. Unfortunately, CAC in AA populations has not been studied as intensively as in EAs. When presence of T2D is considered as an additional inclusion criterion, the present study is the largest GWAS of which we are aware in AAs with T2D, and it is unlikely that many genome-wide association datasets will soon become available in this minority population.
The 90 HU CAC variable was used instead of the 130 HU CAC in the AA-DHS because of its improved sensitivity for detection calcified plaques. A sample size weighted meta-analysis was then performed to combine the observed results with JHS. An inverse variance weighted meta-analysis combining results observed with 130 HU CAC measure that is available in both studies as a sensitivity analysis. Results from these analyses are presented in Additional file 4. Overall, they show similar patterns as the main analyses, although the meta-analysis effect sizes are usually stronger when the 90 HU CAC variable is used in AA-DHS.
Conclusion
This report contains the first GWAS results for CAC in the understudied African American population with T2D. Potential roles for LRB1, ATXN1, MAGI2, DSC1 and DSG1 were reported and additional support for genomic regions identified by admixture mapping was identified. Hyperglycemia increases CAC, both in prevalence and severity, and may provide a necessary environmental trigger to detect the genetic basis of coronary atherosclerosis in populations with recent African ancestry, who seem to be biologically protected from developing CP. Future functional studies of the roles of variation in these genes in human tissues/cells and transgenic animals will contribute to our understanding of genetic susceptibility to coronary atherosclerosis.
Additional files
Meta-analysis of 130 HU CAC scores in AA-DHS and JHS. (XLSX 230 kb)
Replication of AA-DHS admixture mapping results in JHS. (XLSX 11 kb)
List of SNPs with meta-analysis p-value less than 10–4. (XLSX 190 kb)
Joint test of association with local ancestry and genotype. (XLSX 389 kb)
Acknowledgements
The authors thank the participants and data collection staff of the AA-DHS and Jackson Heart Studies.
Funding
This project was supported by NIH R01 DK071891 (BIF), NIH R01 AR48797 (JJC), NIH R01 HL67348 (DWB) and NIH P20MD006899 (SGB).
The Jackson Heart Study is supported by contracts HHSN268201300046C, HHSN268201300047C, HHSN268201300048C, HHSN268201300049C, HHSN268201300050C from the National Heart, Lung, and Blood Institute and the National Institute on Minority Health and Health Disparities.
Availability of data and materials
The dataset generated in AA-DHS are available from the senior author based on reasonable request. JHS data are available on dbGap and/or direct request addressed to the JHS leadership.
Disclaimer
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Institutes of Health; or the U.S. Department of Health and Human Services.
Abbreviations
- AA
African Americans
- AA-DHS
African American-Diabetes Heart Study
- CAC
coronary artery calcified atherosclerotic plaque
- CP
Calcified Plaque
- CVD
cardiovascular disease
- GEMMA
Genome-wide Efficient Mixed Model Analysis
- GWAS
genome-wide association study
- HDL
high density lipoprotein
- HU
Hounsfield Unit
- JHS
Jackson Heart Study
- LD
linkage disequilibrium
- LDL
low density lipoprotein
- MOI
mode of inheritance
- REAP
Relatedness Estimation in Admixed Populations
- SNP
Single nucleotide polymorphism
- T2D
type 2 diabetes
Authors’ contributions
JD established the statistical quality control (QC) plan, performed some analyses, summarized and helped interpret the results, and drafted the manuscript. NDP performed the genotyping, lead the laboratory QC, helped interpret the results, and contributed in the manuscript preparation. CDL participated in the design of the study, contributed his expertise in the QC process, helped interpret results, and reviewed the manuscript. WMB and LL performed most of the statistical analyses and generated the graphs. PJH prepared the DNA for genotyping. SCS and JX managed the databases and created the analysis dataset. JGT and JJC measured CAC levels and revised the manuscript. TCR, LEW, JSP, LM, and GCC are AA-DHS investigators, providing scientific input for the project, genotyping, writing and revising the final manuscript. SGB, AC, SM, JGW, and HAT provided Jackson Heart Study data and assisted in data analysis and manuscript preparation. DWB and BIF developed this project, BIF recruited and phenotyped all AA-DHS participants, and both authors supervised genotyping, statistical analysis and manuscript preparation. All authors contributed to revising the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
AA-DHS was approved by the WFSM Institutional Review Board, and all participants provided written informed consent.
JHS is a population-based cohort study that recruited participants from various Institutions in the Jackson, MI metropolitan area. Study protocols and consent forms for JHS were approved by the responsible research ethics committees and institutional research boards. All JHS participants provided written informed consent.
Consent for publication
Not applicable – all reported data have been aggregated. This manuscript does not provide participant level data.
Competing interests
The authors have no competing interests to disclose.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12863-017-0572-9) contains supplementary material, which is available to authorized users.
Contributor Information
Jasmin Divers, Phone: 336-716-5778, Email: jdivers@wakehealth.edu.
Nicholette D. Palmer, Email: nallred@wakehealth.edu
Carl D. Langefeld, Email: clangefe@wakehealth.edu
W. Mark Brown, Email: wmbrown@wakehealth.edu.
Lingyi Lu, Email: llu@wakehealth.edu.
Pamela J. Hicks, Email: panderso@wakehealth.edu
S. Carrie Smith, Email: suscsmit@wakehealth.edu.
Jianzhao Xu, Email: jxu@wakehealth.edu.
James G. Terry, Email: james.g.terry@vanderbilt.edu
Thomas C. Register, Email: register@wakehealth.edu
Lynne E. Wagenknecht, Email: lwgnkcht@wakehealth.edu
John S. Parks, Email: jparks@wakehealth.edu
Gary C. Chan, Email: drgchan@gmail.com
Sarah G. Buxbaum, Email: sarah.g.buxbaum@jsums.edu
Adolfo Correa, Email: acorrea@umc.edu.
Solomon Musani, Email: smusani@umc.edu.
James G. Wilson, Email: jgwilson2@umc.edu
Herman A. Taylor, Email: htaylor@msm.edu
Donald W. Bowden, Email: dbowden@wakehealth.edu
John Jeffrey Carr, Email: j.jeffrey.carr@vanderbilt.edu.
Barry I. Freedman, Email: bfreedma@wakehealth.edu
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després J-P, Fullerton HJ, Howard VJ, et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–e322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Yeboah J, McClelland RL, Polonsky TS, Burke GL, Sibley CT, O'Leary D, Carr JJ, Goff DC, Greenland P, Herrington DM. Comparison of novel risk markers for improvement in cardiovascular risk assessment in intermediate-risk individuals. JAMA. 2012;308(8):788–795. doi: 10.1001/jama.2012.9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, Liu K, Shea S, Szklo M, Bluemke DA, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358(13):1336–1345. doi: 10.1056/NEJMoa072100. [DOI] [PubMed] [Google Scholar]
- 4.Anand DV, Lim E, Darko D, Bassett P, Hopkins D, Lipkin D, Corder R, Lahiri A. Determinants of progression of coronary artery calcification in type 2 diabetes: role of glycemic control and inflammatory/vascular calcification markers. J Am Coll Cardiol. 2007;50(23):2218–2225. doi: 10.1016/j.jacc.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 5.Budoff MJ, McClelland RL, Chung H, Wong ND, Carr JJ, McNitt-Gray M, Blumenthal RS, Detrano RC. Reproducibility of coronary artery calcified plaque with cardiac 64-MDCT: the multi-ethnic study of atherosclerosis. AJR Am J Roentgenol. 2009;192(3):613–617. doi: 10.2214/AJR.08.1242. [DOI] [PubMed] [Google Scholar]
- 6.Freedman BI, Hsu FC, Langefeld CD, Rich SS, Herrington DM, Carr JJ, Xu J, Bowden DW, Wagenknecht LE. The impact of ethnicity and sex on subclinical cardiovascular disease: the diabetes heart study. Diabetologia. 2005;48(12):2511–2518. doi: 10.1007/s00125-005-0017-2. [DOI] [PubMed] [Google Scholar]
- 7.Wagenknecht LE, Divers J, Bertoni AG, Langefeld CD, Carr JJ, Bowden DW, Elbein SC, Shea S, Lewis CE, Freedman BI. Correlates of coronary artery calcified plaque in blacks and whites with type 2 diabetes. Ann Epidemiol. 2011;21(1):34–41. doi: 10.1016/j.annepidem.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee TC, O’Malley PG, Feuerstein I, Taylor AJ. The prevalence and severity of coronaryartery calcification on coronary arterycomputed tomography in black and white subjects. J Am Coll Cardiol. 2003;41(1):39–44. doi: 10.1016/S0735-1097(02)02618-9. [DOI] [PubMed] [Google Scholar]
- 9.Jain T, Peshock R, McGuire DK, Willett D, Yu Z, Vega GL, Guerra R, Hobbs HH, Grundy SM. African Americans and Caucasians have a similar prevalence of coronary calcium in the Dallas heart study. J Am Coll Cardiol. 2004;44(5):1011–1017. doi: 10.1016/j.jacc.2004.05.069. [DOI] [PubMed] [Google Scholar]
- 10.Carson AP, Howard G, Burke GL, Shea S, Levitan EB, Muntner P. Ethnic differences in hypertension incidence among middle-aged and older adults: the multi-ethnic study of atherosclerosis. Hypertension. 2011;57(6):1101–1107. doi: 10.1161/HYPERTENSIONAHA.110.168005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Divers J, Wagenknecht LE, Bowden DW, Carr JJ, Hightower RC, Xu J, Langefeld CD, Freedman BI. Ethnic differences in the relationship between albuminuria and calcified atherosclerotic plaque: the African American-diabetes heart study. Diabetes Care. 2010;33(1):131–138. doi: 10.2337/dc09-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frank ATH, Zhao B, Jose PO, Azar KMJ, Fortmann SP, Palaniappan LP. Racial/ethnic differences in dyslipidemia patterns. Circulation. 2014;129(5):570–579. doi: 10.1161/CIRCULATIONAHA.113.005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wassel CL, Pankow JS, Peralta CA, Choudhry S, Seldin MF, Arnett DK, Genetic Ancestry I. Associated with subclinical cardiovascular disease in African-Americans and Hispanics from the multi-ethnic study of atherosclerosis. Circulation: Cardiovascular Genetics. 2009;2(6):629–636. doi: 10.1161/CIRCGENETICS.109.876243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Divers J, Palmer ND, Lu L, Register TC, Carr JJ, Hicks PJ, Hightower RC, Smith SC, Xu J, Cox AJ, et al. Admixture mapping of coronary artery calcified plaque in African Americans with type 2 diabetes mellitus. Circulation: Cardiovascular Genetics. 2013;6(1):97–105. doi: 10.1161/CIRCGENETICS.112.964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wade AN, Fedyna S, Mehta NN, Clair C, Ginwala N, Krishna RK, Qasim AN, Braunstein S, Iqbal N, Schutta MH, et al. Type 2 diabetes does not attenuate racial differences in coronary calcification. Diabetes Res Clin Pract. 2011;91(1):101–107. doi: 10.1016/j.diabres.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parra EJ, Pfaff CL, Hiester K, Sosnoski D, Jien Y, Gulden F, McKeigue P, Shriver MD. Pattern and dynamics of admixture in US African-American populations: a global perspective. Am J Hum Genet. 2000;67(4):221–1. [Google Scholar]
- 17.Taylor HA, Jr, Wilson JG, Jones DW, Sarpong DF, Srinivasan A, Garrison RJ, Nelson C, Wyatt SB. Toward resolution of cardiovascular health disparities in African Americans: design and methods of the Jackson heart study. Ethn Dis. 2005;15(4 Suppl 6):S6. [PubMed] [Google Scholar]
- 18.Carr JJ, Nelson JC, Wong ND, McNitt-Gray M, Arad Y, Jacobs DR, Jr, Sidney S, Bild DE, Williams OD, Detrano RC. Calcified coronary artery plaque measurement with cardiac CT in population-based studies: standardized protocol of multi-ethnic study of atherosclerosis (MESA) and coronary artery risk development in young adults (CARDIA) study. Radiology. 2005;234(1):35–43. doi: 10.1148/radiol.2341040439. [DOI] [PubMed] [Google Scholar]
- 19.Carr JJ, Crouse JR, 3rd, Goff DC, Jr, D'Agostino RB, Jr, Peterson NP, Burke GL. Evaluation of subsecond gated helical CT for quantification of coronary artery calcium and comparison with electron beam CT. AJR Am J Roentgenol. 2000;174(4):915–921. doi: 10.2214/ajr.174.4.1740915. [DOI] [PubMed] [Google Scholar]
- 20.Wagenknecht LE, Bowden DW, Carr JJ, Langefeld CD, Freedman BI, Rich SS. Familial aggregation of coronary artery calcium in families with type 2 diabetes. Diabetes. 2001;50(4):861–866. doi: 10.2337/diabetes.50.4.861. [DOI] [PubMed] [Google Scholar]
- 21.The Genomes Project C A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sankararaman S, Sridhar S, Kimmel G, Halperin E. Estimating local ancestry in admixed populations. Am J Hum Genet. 2008;82(2):290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price AL, Tandon A, Patterson N, Barnes KC, Rafaels N, Ruczinski I, Beaty TH, Mathias R, Reich D, Myers S. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5(6):e1000519. doi: 10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reilly MP, Wolfe ML, Localio AR, Rader DJ. Coronary artery calcification and cardiovascular risk factors: impact of the analytic approach. Atherosclerosis. 2004;173(1):69–78. doi: 10.1016/j.atherosclerosis.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 26.Zhou X, Stephens M. Genome-wide efficient mixed model analysis for association studies. Nat Genet. 2012;44(7):821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thornton T, Tang H, Hoffmann Thomas J, Ochs-Balcom Heather M, Caan Bette J, Risch N. Estimating kinship in admixed populations. Am J Hum Genet. 2012;91(1):122–138. doi: 10.1016/j.ajhg.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q, Zheng G, Li Z, Yu K. Efficient approximation of P-value of the maximum of correlated tests, with applications to genome-wide association studies. Ann Hum Genet. 2008;72(Pt 3):397–406. doi: 10.1111/j.1469-1809.2008.00437.x. [DOI] [PubMed] [Google Scholar]
- 29.Lin DY, Zeng D, Millikan R. Maximum likelihood estimation of haplotype effects and haplotype-environment interactions in association studies. Genet Epidemiol. 2005;29(4):299–312. doi: 10.1002/gepi.20098. [DOI] [PubMed] [Google Scholar]
- 30.So HC, Sham PC. Robust association tests under different genetic models, allowing for binary or quantitative traits and covariates. Behav Genet. 2011;41(5):768–775. doi: 10.1007/s10519-011-9450-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pasaniuc B, Zaitlen N, Lettre G, Chen GK, Tandon A, Kao WHL, Ruczinski I, Fornage M, Siscovick DS, Zhu X, et al. Enhanced statistical tests for GWAS in admixed populations: assessment using African Americans from CARe and a breast cancer consortium. PLoS Genet. 2011;7(4):e1001371. doi: 10.1371/journal.pgen.1001371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70(2):425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minelli C, De Grandi A, Weichenberger CX, Gogele M, Modenese M, Attia J, Barrett JH, Boehnke M, Borsani G, Casari G, et al. Importance of different types of prior knowledge in selecting genome-wide findings for follow-up. Genet Epidemiol. 2013;37(2):205–213. doi: 10.1002/gepi.21705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou L, Zhao H, Review A. Of post-GWAS prioritization approaches. Front Genet. 2013;4 [DOI] [PMC free article] [PubMed]
- 35.Dehghan A, Bis JC, White CC, Smith AV, Morrison AC, Cupples LA, Trompet S, Chasman DI, Lumley T, Volker U, et al. Genome-wide association study for incident myocardial infarction and coronary heart disease in prospective cohort studies: the CHARGE consortium. PLoS One. 2016;11(3):e0144997. doi: 10.1371/journal.pone.0144997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pajukanta P, Cargill M, Viitanen L, Nuotio I, Kareinen A, Perola M, Terwilliger JD, Kempas E, Daly M, Lilja H, et al. Two loci on chromosomes 2 and X for premature coronary heart disease identified in early- and late-settlement populations of Finland. Am J Hum Genet. 2000;67(6):1481–1493. doi: 10.1086/316902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulminski AM, He L, Culminskaya I, Loika Y, Kernogitski Y, Arbeev KG, Loiko E, Arbeeva L, Bagley O, Duan M, et al. Pleiotropic associations of allelic variants in a 2q22 region with risks of major human diseases and mortality. PLoS Genet. 2016;12(11):e1006314. doi: 10.1371/journal.pgen.1006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasstedt SJ, Highland HM, Elbein SC, Hanis CL, Das SK. The American Diabetes Association GSG: five linkage regions each harbor multiple type 2 diabetes genes in the African American subset of the GENNID study. J Hum Genet. 2013;58(6):378–383. doi: 10.1038/jhg.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer ND, Langefeld CD, Ziegler JT, Hsu F, Haffner SM, Fingerlin T, Norris JM, Chen YI, Rich SS, Haritunians T, et al. Candidate loci for insulin sensitivity and disposition index from a genome wide association analysis of Hispanics in the IRAS family study. Diabetologia. 2010;53(2):281–289. doi: 10.1007/s00125-009-1586-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Donnell CJ, Cupples LA, D'Agostino RB, Fox CS, Hoffmann U, Hwang SJ, Ingellson E, Liu CY, Murabito JM, Polak JF, et al. Genome-wide association study for subclinical atherosclerosis in major arterial territories in the NHLBI's Framingham heart study. BMC Med Genet. 2007;8(Suppl 1):S4. doi: 10.1186/1471-2350-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koller DL, Zheng HF, Karasik D, Yerges-Armstrong L, Liu CT, McGuigan F, Kemp JP, Giroux S, Lai D, Edenberg HJ, et al. Meta-analysis of genome-wide studies identifies WNT16 and ESR1 SNPs associated with bone mineral density in premenopausal women. J Bone Miner Res. 2013;28(3):547–558. doi: 10.1002/jbmr.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beazley KE, Nurminsky D, Lima F, Gandhi C, Nurminskaya MV. Wnt16 attenuates TGFbeta-induced chondrogenic transformation in vascular smooth muscle. Arterioscler Thromb Vasc Biol. 2015;35(3):573–579. doi: 10.1161/ATVBAHA.114.304393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.St Hilaire C, Liberman M, Miller JD. Bidirectional translation in cardiovascular calcification. Arterioscler Thromb Vasc Biol. 2016;36(3):e19–e24. doi: 10.1161/ATVBAHA.115.307056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li YB, Zhang QH, Chen Z, He ZJ, Yi GH. Oxidized low-density lipoprotein attenuated desmoglein 1 and desmocollin 2 expression via LOX-1/ca(2+)/PKC-beta signal in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2015;468(1–2):380–386. doi: 10.1016/j.bbrc.2015.10.079. [DOI] [PubMed] [Google Scholar]
- 45.Harmon RM, Desai BV, Green KJ. Regulatory roles of the cadherin superfamily. F1000 Biology Reports. 2009;1:13. doi: 10.3410/B1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karter AJ, Ferrara A, Liu JY, Moffet HH, Ackerson LM, Selby JV. EThnic disparities in diabetic complications in an insured population. JAMA. 2002;287(19):2519–2527. doi: 10.1001/jama.287.19.2519. [DOI] [PubMed] [Google Scholar]
- 47.Young BA, Rudser K, Kestenbaum B, Seliger SL, Andress D, Boyko EJ. Racial and ethnic differences in incident myocardial infarction in end-stage renal disease patients: the USRDS. Kidney Int. 2006;69(9):1691–1698. doi: 10.1038/sj.ki.5000346. [DOI] [PubMed] [Google Scholar]
- 48.Young BA, Maynard C, Boyko EJ. Racial differences in diabetic nephropathy, cardiovascular disease, and mortality in a national population of veterans. Diabetes Care. 2003;26(8):2392–2399. doi: 10.2337/diacare.26.8.2392. [DOI] [PubMed] [Google Scholar]
- 49.Wojczynski MK, Li M, Bielak LF, Kerr KF, Reiner AP, Wong ND, Yanek LR, Qu L, White CC, Lange LA, et al. Genetics of coronary artery calcification among African Americans, a meta-analysis. BMC Med Genet. 2013;14:75. doi: 10.1186/1471-2350-14-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gomez F, Wang L, Abel H, Zhang Q, Province MA, Borecki IB. Admixture mapping of coronary artery calcification in African Americans from the NHLBI family heart study. BMC Genet. 2015;16:42. doi: 10.1186/s12863-015-0196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong ND, Sciammarella MG, Polk D, Gallagher A, Miranda-Peats L, Whitcomb B, Hachamovitch R, Friedman JD, Hayes S, Berman DS. The metabolic syndrome, diabetes, and subclinical atherosclerosis assessed by coronary calcium. J Am Coll Cardiol. 2003;41(9):1547–1553. doi: 10.1016/S0735-1097(03)00193-1. [DOI] [PubMed] [Google Scholar]
- 52.Hoff JA, Quinn L, Sevrukov A, Lipton RB, Daviglus M, Garside DB, Ajmere NK, Gandhi S, Kondos GT. The prevalence of coronary arterycalcium among diabetic individuals without known coronary artery disease. J Am Coll Cardiol. 2003;41(6):1008–1012. doi: 10.1016/S0735-1097(02)02975-3. [DOI] [PubMed] [Google Scholar]
- 53.Chu AY, Deng X, Fisher VA, Drong A, Zhang Y, Feitosa MF, Liu C-T, Weeks O, Choh AC, Duan Q, et al. Multiethnic genome-wide meta-analysis of ectopic fat depots identifies loci associated with adipocyte development and differentiation. Nat Genet. 2017;49(1):125–130. doi: 10.1038/ng.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cvetanovic M, Patel JM, Marti HH, Kini AR, Opal P. Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nat Med. 2011;17(11):1445–1447. doi: 10.1038/nm.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C-X, Li Y, Obermoeller-McCormick LM, Schwartz AL, Bu G. The putative tumor suppressor LRP1B, a novel member of the low density lipoprotein (LDL) receptor family, exhibits both overlapping and distinct properties with the LDL receptor-related protein. J Biol Chem. 2001;276(31):28889–28896. doi: 10.1074/jbc.M102727200. [DOI] [PubMed] [Google Scholar]
- 56.Burgdorf KS, Gjesing AP, Grarup N, Justesen JM, Sandholt CH, Witte DR, Jorgensen T, Madsbad S, Hansen T, Pedersen O. Association studies of novel obesity-related gene variants with quantitative metabolic phenotypes in a population-based sample of 6,039 Danish individuals. Diabetologia. 2012;55(1):105–113. doi: 10.1007/s00125-011-2320-4. [DOI] [PubMed] [Google Scholar]
- 57.Beer AG, Zenzmaier C, Schreinlechner M, Haas J, Dietrich MF, Herz J, Marschang P. Expression of a recombinant full-length LRP1B receptor in human non-small cell lung cancer cells confirms the postulated growth-suppressing function of this large LDL receptor family member. Oncotarget. 2016; [DOI] [PMC free article] [PubMed]
- 58.Hodulova M, Sedova L, Krenova D, Liska F, Krupkova M, Kazdova L, Tremblay J, Hamet P, Kren V, Seda O. Genomic determinants of triglyceride and cholesterol distribution into lipoprotein fractions in the rat. PLoS One. 2014;9(10):e109983. doi: 10.1371/journal.pone.0109983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Assman G; von Eckardstein A; Brewer HB J: Familial Analphalipoproteinemia: Tangier Disease. In: The Metabolic and Molecular Bases of Inherited Disease. Edited by BF SCBASWVDCBKKV. New York: McGraw-Hill; 2001: 2937–2960.
- 60.Chistiakov DA, Orekhov AN, Bobryshev YV. LOX-1-mediated effects on vascular cells in atherosclerosis. Cell Physiol Biochem. 2016;38(5):1851–1859. doi: 10.1159/000443123. [DOI] [PubMed] [Google Scholar]
- 61.Xu S, Ogura S, Chen J, Little PJ, Moss J, Liu P. LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cell Mol Life Sci. 2013;70(16):2859–2872. doi: 10.1007/s00018-012-1194-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8(11):1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Meta-analysis of 130 HU CAC scores in AA-DHS and JHS. (XLSX 230 kb)
Replication of AA-DHS admixture mapping results in JHS. (XLSX 11 kb)
List of SNPs with meta-analysis p-value less than 10–4. (XLSX 190 kb)
Joint test of association with local ancestry and genotype. (XLSX 389 kb)
Data Availability Statement
The dataset generated in AA-DHS are available from the senior author based on reasonable request. JHS data are available on dbGap and/or direct request addressed to the JHS leadership.