

Abstract
Patient: Male, 11
Final Diagnosis: Haberland syndrome
Symptoms: Seizure
Medication: —
Clinical Procedure: Medical treatment
Specialty: Neurosurgery
Objective:
Rare disease
Background:
Encephalocraniocutaneous lipomatosis (ECCL) was first announced as a new type of ectomesodermal dysgenesis in 1970 by Haberland and Perou. ECCL was first described in 1970, and approximately 60 cases have been reported since then. The classic triad of ECCL are skin, ocular, and central nervous system involvement, including conditions such as unilateral porencephalic cyst, ipsilateral lipomatous hamartoma of the scalp-eyelids-eye globe, cortical atrophy, cranial asymmetry, developmental delay, seizures, mental retardation, and spasticity of the contralateral limbs. The dermatological hallmark is a hairless fatty tissue nevus of the scalp called nevus psiloliparus.
Case Report:
An 11-year-old right-handed boy, born at full term, was referred to our clinic. His family had no consanguinity or history of neurocutaneous disease. The patient’s physical examination revealed a large hairless lesion on the right frontoparietal scalp called nevus psiloliparus. Beginning from the birth, a dermolipoma (an uncommon benign tumor) was reported to have occurred on the conjunctiva, mostly ipsilateral in his right eye and present on the ipsilateral side of the neurological abnormalities shown on magnetic resonance imaging and computed tomography. The patient had muscle weakness in left upper and lower extremities. He had a mild form of mental retardation.
Conclusions:
There is no specific treatment for ECCL. Management of ECCL is usually symptomatic. Surgical correction of a cutaneous lesion can be performed for cosmetic improvement. An early diagnosis of ECCL allows for early symptom treatment and improved patient quality of life.
MeSH Keywords: Epilepsy, Absence; Lipoma; Neurocutaneous Syndromes
Background
Haberland et al. first reported encephalocraniocutaneous lipomatosis (ECCL) in 1970 and defined it as a new type of ectomesodermal dysgenesis [1]. It has also been referred to as Fishman’s syndrome [2]. Although it was first described in 1970, only around 60 cases have been reported since then. The classic triad of ECCL is skin, ocular and central nervous system involvement, which have included specific conditions such as unilateral porencephalic cyst, ipsilateral lipomatous hamartoma of the scalp-eyelids-eye globe, cortical atrophy, cranial asymmetry, developmental delay, seizures, mental retardation and spasticity of the contralateral limbs. Despite most findings being unilateral, a few bilateral cases have been reported [3,4]. The dermatological hallmark is a hairless fatty tissue nevus of the scalp which is also known as nevus psiloliparus [5].
Because differential diagnosis is difficult to define for this specific disease, ECCL can be reported in the literature as multi-organ neuroectodermal syndrome. These patients generally present to dermatology clinics for alopecia and neuropsychiatry departments for problems with school success and education in childhood.
Our case was a child misdiagnosed as hydrocephalus and referred to our clinic for a shunt operation. As there is no specific treatment for ECCL, the management is usually symptomatic, and any surgical correction of the cutaneous lesion is for cosmetic improvement.
Case Report
An 11-year-old right-handed boy, who was born at full term, presented to our clinic. His parents declared no complication at birth and a normal bodyweight. His family reported no consanguinity or neurocutaneous disease history. The patient had first presented to a pediatrician at a neurosurgery department at six months of age due to febrile convulsion. At that time, computed tomography (CT) of the brain showed clear cranial asymmetry and hydrocephalus. A shunt operation for hydrocephalus was recommended if the patient had further seizures and sent the patient was sent home without any treatment. Subsequently, the child had tonic-clonic seizures three times in one day at the age of nine years. At that time, the pediatrician started the patient on carbamazepine 150 mg twice a day. After 15 months, the patient had seizures again and the carbamazepine dose was increased to 300 mg twice a day. However, even under this treatment, there was no control of the patient’s seizures. The patient was referred to our clinic for further evaluations and neurosurgery suggestions.
Our physical examination revealed a large hairless lesion on the right frontoparietal scalp called nevus psiloliparus (Figure 1). Beginning from the birth, a dermolipoma (an uncommon benign tumor) had been developing on the conjunctiva, mostly ipsi-lateral in his right eye as a lipomatous hamartoma (Figure 2); the ipsilateral cranial abnormalities were revealed in magnetic resonance imaging (MRI) and CT scan (Figures 3–5).
Figure 1.
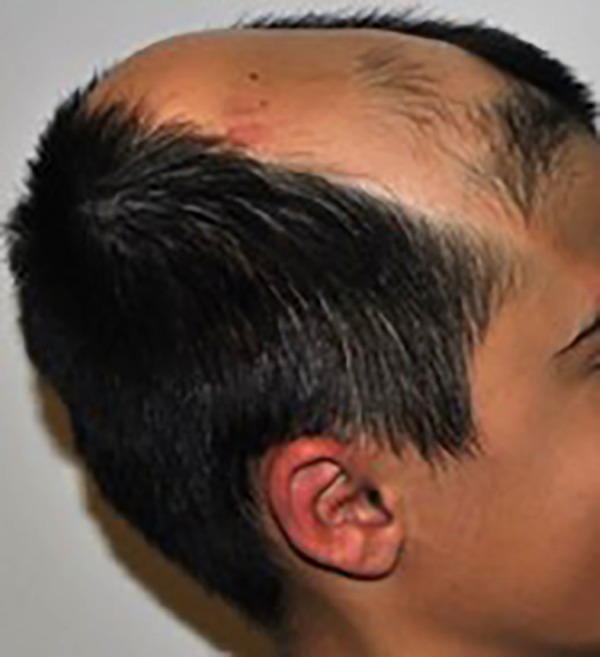
Right frontoparietal nevus psiloliparus.
Figure 2.
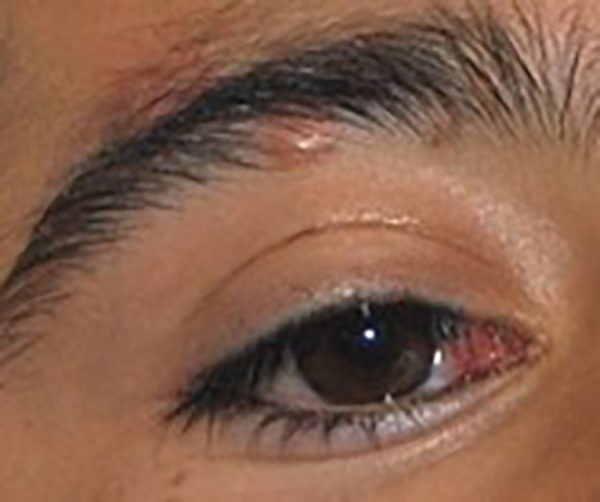
Right eye corneal lipomatous hamartoma.
Figure 3.
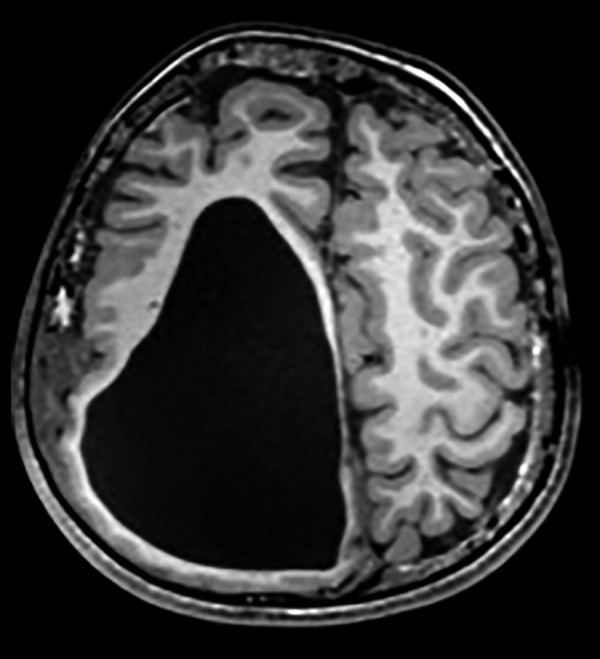
Axial T1A cranial MRI image of asymmetric cystic dilatation of the right lateral ventricle occipital horn, deletion of gyri and sulci irregularity with cortical thinning and diffuse white matter volume decrease, intracranial lipoma under the skin.
Figure 4.
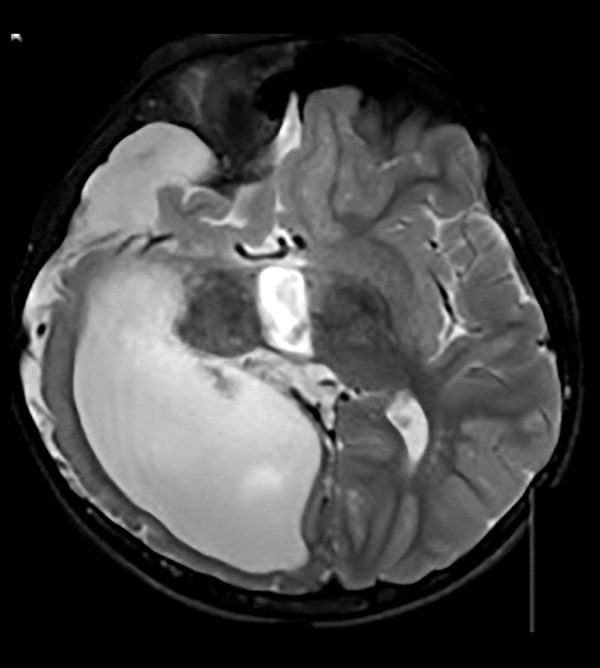
Axial T2A cranial MRI image of asymmetric cystic dilatation of the right lateral ventricle occipital horn, deletion of temporo-occipital gyri and sulci irregularity with cortical thinning and diffuse white matter volume decrease and subdural effusion.
Figure 5.
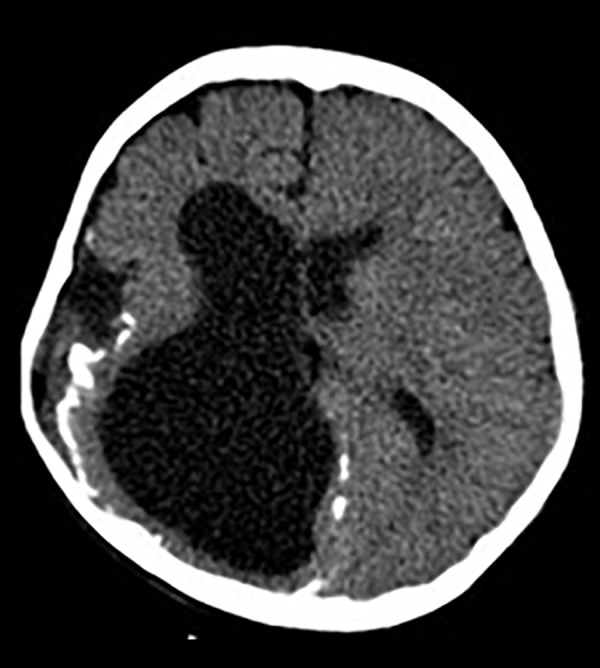
Axial cranial CT images of asymmetric cystic dilatation of the right lateral ventricle occipital horn, deletion of gyri and sulci irregularity with cortical thinning, widening of sylvian fissure, corticopial calcifications.
His eyes were examined and reported as normal. He had a mild form of mental retardation. He had muscle weakness in his left upper and lower extremities. His proximal muscle group strength of the left upper extremity was 4/5 and the distal extremity was 3/5. The muscle strength of left lower extremity was 4/5. There was hypoesthesia on the left extremities. Deep tendon reflexes were brisk on the left extremities and normal on the right extremities. Planter response was positive on the left and negative on the right foot. Cardiovascular and respiratory system examinations were normal. Electroencephalogram (EEG) showed left frontocentral and right frontocentroparietal focal epileptiform activities.
The patient’s antiepileptic medical treatment was organized, and seizures stop. He was referred to the psychiatry department for suggestions of education life.
Discussion
Antenatal diagnosis of ECCL is not usually made because the intracranial malformations noted on an antenatal sonogram are not specific for ECCL [6]. Etiology of ECCL remains unknown and is not associated with gender, race, or geography. A postzygotic gene mutation is suspected in genetic causes with an autosomal mutation in mosaic form [7]. Dysgenesia of the anterior neural tube and cephalic neural crest is thought to occur in embryo-pathogenesis. Neural crest derivatives, dermis and hypodermis of the face and neck, dermal bones of the skull, head mesenchyme, truncoconal septum, odontoblasts and melanocytes are affected by ECCL. Mental retardation and seizures are also seen. The best radio-diagnostic test are CT and MRI to determine the cranial neurological pathologies of ECCL.
The diagnostic triad of ECCL is cutaneous, ocular, and central nervous system (CNS) involvement [8]. The original diagnostic criteria for ECCL proposed by Hunter in 2006, was revised by Moog in 2009 (Table 1) [9,10]. The revised criteria include definite/proven and possible ECCL cases but exclude a third group of probable ECCL as defined by Hunter [9,10]. In the revised Moog criteria for ECCL, there are one major and three minor criteria involving the ocular system, three major and five minor criteria involving the cutaneous system, and three major and six minor criteria for CNS involvement [10]. Three major criteria also involve the other systems. In our patient case, nevus psiloliparus, corneal hamartoma, proencephalic cyst, calcification around the cyst, and asymmetrically dilated ventricles were present and thus it was defined as a definite case based on this classification system (Table 2).
Table 1.
| System | Major criteria | Minor criteria |
|---|---|---|
| Skin |
|
|
| Ocular |
|
|
| Central Nervous System |
|
|
| Other System |
|
Table 2.
ECCL application criteria for diagnosis.
| Definite case | Probable case |
|---|---|
|
|
The pattern of skin and ocular findings is very consistent and recognizable in ECCL. The most important skin anomalies are non-scarring alopecia with or without underlying fatty tissue and subcutaneous fatty masses. Hairless fatty tissue nevus of the scalp is the dermatological hallmark and referred to as nevus psiloliparus. Subcutaneous fatty masses are seen unilateral or bilateral in the frontotemporal or zygomatic region. A rare ECCL with small congenital melanocytic nevus involving 70% of the body surface of the patient was also reported by Ahmed et al. [11]. Ocular anomalies reported in the literature include choristoma, epibulbar or limbal dermoids, ocular and palpebral colobomas, aniridia, micropthalmia, calcification of globe, and irregular or disrupted eyebrows [12].
The most frequently reported intracranial anomaly is intracranial lipoma on radiological examinations. Other abnormalities reported include enlarged lateral ventricle, porencephalic cyst, widened subarachnoid space, arachnoid cyst, corticopial calcifications, cortex with dysplasia, leptomeningeal angiomatosis, tapered corpus callosum, and a lack of normal insular opercularization [13]. Spinal lipomatous lesions as intradural extramedullary lesion have also been reported in ECCL [13]. Skeletal lesions, congenital heart defects, and urogenital anomalies have also been seen, although rare.
Haberland and Perou reported on the histological evaluation of the brain in ECCL and found lamination defects of the cerebrum, calcification on the outer membrane of porencephalic cysts, and polymicrogyria [1]. Histopathological examinations of the skin colored papules and nodular tags represent lipomas, fibromas, fibrolipomas, and cartilage with hamartomatous tissue [14]. Focal dermal fibrosis is associated with subcutaneous fat widening into the reticular dermis in alopecic region of scalp.
The differential diagnosis includes Proteus syndrome, sebaceous nevus syndrome, and oculocerebrocutaneous (OCC) syndrome [7,13]. Sebaceous nevus syndrome is also associated with seizures and mental retardation, but the cutaneous lesions are mainly on the midline of the face. Proteus syndrome presents in the head, limbs, and trunk progressively, bilaterally and asymmetrically, but brain alterations are rarely seen, whereas ECCL is non-progressive, unilateral, and limited to the head. However, some authors consider sebaceous nevus syndrome as a continuum of phenotypic expression of ECCL, and ECCL as a localized form of Proteus syndrome. In differential diagnosis, scalp alopecia and facial lipoma are absent in OCC syndrome [15].
Conclusions
There is no specific treatment for ECCL; management includes symptomatic treatment and surgical correction of cutaneous lesions for cosmetic improvement. If the diagnosis could be made earlier, the care of symptoms and the quality of life of the patient would be improved.
Footnotes
Conflict of interest
None.
References:
- 1.Haberland C, Perou M. Encephalocraniocutaneous lipomatosis: A new example of ectomesodermal dysgenesis. Arch Neurol. 1970;22:144–55. doi: 10.1001/archneur.1970.00480200050005. [DOI] [PubMed] [Google Scholar]
- 2.Fishman MA, Chang CS, Miller JE. Encephalocraniocutaneous lipomatosis. Pediatrics. 1978;61:580–82. [PubMed] [Google Scholar]
- 3.Banta J, Beasley K, Kobayashi T, Rohena L. Encephalocraniocutaneous lipomatosis (Haberland syndrome): A mild case with bilateral cutaneous and ocular involvement. JAAD Case Rep. 2016;2:150–52. doi: 10.1016/j.jdcr.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubegni P, Risulo M, Sbano P, et al. Encephalocraniocutaneous lipomatosis (Haberland syndrome) with bilateral cutaneous and visceral involvement. Clin Exp Dermatol. 2003;28(4):387–90. doi: 10.1046/j.1365-2230.2003.01329.x. [DOI] [PubMed] [Google Scholar]
- 5.Happle R, Kuster W. Nevus psiloliparus: A distinct fatty tissue nevus. Dermatology. 1998;197:6–10. doi: 10.1159/000017968. [DOI] [PubMed] [Google Scholar]
- 6.Nowaczyk MJ, Mernagh JR, Bourgeois JM, et al. Antenatal and postnatal findings in encephalocraniocutaneous lipomatosis. Am J Med Genet. 2000;91(4):261–66. [PubMed] [Google Scholar]
- 7.Thakur S, Thakur V, Sood RG, et al. Encephalocraniocutaneous lipomatosis with calvarial exostosis – Case report and review of literature. Indian J Radiol Imaging. 2013;23:333–36. doi: 10.4103/0971-3026.125607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siddiqui S, Naaz S, Ahmad M, et al. Encephalocraniocutaneous lipomatosis: A case report with review of literature. Neuroradiol J. 2017 doi: 10.1177/1971400917693638. [Epub Ahead of Print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter AG. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: Blind men and an elephant or separate syndromes? Am J Med Genet A. 2006;140:709–26. doi: 10.1002/ajmg.a.31149. [DOI] [PubMed] [Google Scholar]
- 10.Moog U. Encephalocraniocutaneous lipomatosis. J Med Genet. 2009;46:721–29. doi: 10.1136/jmg.2009.066068. [DOI] [PubMed] [Google Scholar]
- 11.Ahmed I, Tope WD, Young TL, et al. Neurocutaneous melanosis in association with encephalocraniocutaneous lipomatosis. J Am Acad Dermatol. 2002;47:S196–200. doi: 10.1067/mjd.2002.110073. [DOI] [PubMed] [Google Scholar]
- 12.MacLaren MJ, Kluijt I, Koole FD. Ophthalmologic abnormalities in encephalocraniocutaneous lipomatosis. Doc Ophthalmol. 1995;90:87–98. doi: 10.1007/BF01203299. [DOI] [PubMed] [Google Scholar]
- 13.Koti K, Bhimireddy V, Dandamudi S, Gunnamreddy R. Encephalocraniocutaneous lipomatosis (Haberland syndrome): A case report and review of literature. Indian J Dermatol. 2013;58:232–34. doi: 10.4103/0019-5154.110835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tekin B, Yücelten AD, Akpınar IN, Ekinci G. Coexistence of aplasia cutis and nevus psiloliparusereport of a novel case. Pediatr Dermatol. 2014;31(6):746–48. doi: 10.1111/pde.12412. [DOI] [PubMed] [Google Scholar]
- 15.Habib F, Elsaid MF, Salem KY, et al. Oculo-ectodermal syndrome: A case report and further delineation of the syndrome. Qatar Med J. 2014;(2):114–22. doi: 10.5339/qmj.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]