

Abstract
Following myocardial infarction (MI), degradation of extracellular matrix (ECM) by upregulated matrix metalloproteinases (MMPs) especially MMP-2 decreases tissue mechanical properties, leading to cardiac function deterioration. Attenuation of cardiac ECM degradation at the early stage of MI has the potential to preserve tissue mechanical properties, resulting in cardiac function increase. Yet the strategy for efficiently preventing cardiac ECM degradation remains to be established. Current preclinical approaches have shown limited efficacy due to low drug dosage allocated to the heart tissue, dose-limiting side effects, and cardiac fibrosis. To address these limitations, we have developed a MMP-2 inhibitor delivery system that can be specifically delivered into infarcted hearts at early stage of MI to efficiently prevent MMP-2-mediated ECM degradation. The system was based on an injectable, degradable, fast gelation and thermosensitive hydrogel, and a MMP-2 specific inhibitor, peptide CTTHWGFTLC (CTT). The use of fast gelation hydrogel allowed to completely retain CTT in the heart tissue. The system was able to release low molecular weight CTT over 4 weeks possibly due to the strong hydrogen bonding between the hydrogel and CTT. The release kinetics was modulated by amount of CTT loaded into the hydrogel, and using chondroitin sulfate and heparin that can interact with CTT and the hydrogel. Both glycosaminoglycans augmented CTT release while heparin more greatly accelerated the release. After injecting into the infarcted hearts for 4 weeks, the released CTT efficiently prevented cardiac ECM degradation as it not only increased tissue thickness, but also preserved collagen composition similar to that in the normal heart tissue. In addition, the delivery system significantly improved cardiac function. Importantly, the delivery system did not induce cardiac fibrosis. These results demonstrate that the developed MMP-2 inhibitor delivery system has potential to efficiently reduce adverse myocardial remodeling and improve cardiac function.
Keywords: Myocardial remodeling, MMP-2, MMP-2 inhibitor, thermosensitive hydrogel, drug delivery
1. Introduction
Myocardial infarction (MI) affects millions of people in the world. Following MI, the matrix metalloproteinases (MMPs) especially MMP-2 is upregulated,1, 2 causing the imbalance of MMPs and tissue inhibitors of MMPs (TIMPs) in the cardiac tissue. This leads to the degradation of cardiac extracellular matrix (ECM). After degradation, cardiac tissue thickness decreases, resulting in cardiac function deterioration.3–5 MMP-2 degrades not only ECM, but also intracellular proteins such as tropoin-I6, titin7, α-actinin8, and myosin light chain9. MMP-2 upregulation occurs as early as one hour after MI10 and can last for over three weeks11.
One of the therapeutic goals of post-MI treatment is to minimize the extent of cardiac ECM degradation to attenuate cardiac function decrease. As MMP-2 plays a major role in cardiac ECM degradation, decreasing its activity in the infarcted region represents a therapeutic target. Studies have shown that MMP-2 knockout mice experienced less cardiac ECM degradation after MI12, 13. The most common approaches to decreasing MMP-2 activity include using pharmacologic MMP inhibitors and transgenic constructs.1, 14–22 Majority of current MMP-2 inhibitors are small organic molecules with reactive zinc-chelating groups such as thiol or hydroxamate.23, 24 These inhibitors have poor selectivity. They inhibit most of MMP family members instead of mainly MMP-2.23–25 In addition, they may have toxic effects.26, 27 Use of recombinant TIMPs may address these disadvantages.16, 20 A potential concern for MMP inhibitors is that they may cause cardiac fibrosis. Studies have demonstrated that various MMP inhibitors increase the density of myofibroblasts that are responsible for cardiac fibrosis.16, 19, 20, 22, 28 Deletion of specific MMP/TIMP genes by transgenic constructs has been shown to change the course of cardiac remodeling. For example, deletion of MMP-2 gene reduced the degree of adverse remodeling when tested in a mouse MI model.13 The major limitation of transgenic constructs is the persistent rather than temporal expression of MMPs and/or TIMPs at different stages of post MI.29 This may lead to beneficial effects in some stages while adverse effects in other stages.29
Systemic delivery represents the most common approach to administrate MMP-2 inhibitors. While this approach has been shown to reduce cardiac ECM degradation to an extent, its low efficacy makes it impractical for wide clinical application.1, 14–22 The inferior efficacy is a result of the systemic delivery approach not being able to distribute sufficient amount of inhibitors to the infarcted region. Those inhibitors distributed to healthy tissue may unbalance the balanced MMPs and TIMPs, causing excessive ECM production and tissue fibrosis.1, 14–22
In this work, we aimed to address these limitations by specifically delivering a MMP-2 inhibitor release system into the infarcted region. Upon delivery, the inhibitor can be released locally, thus reducing MMP activity specifically in the infarcted tissue instead of healthy tissues. In addition, continuous release will make repeated administration of the inhibitor unnecessary. We used a peptide-based, nontoxic MMP-2 inhibitor, CTTHWGFTLC (CTT). The advantages of using CTT include: (1) CTT is an inhibitor specific to MMP-2 and MMP-930. Both MMPs are upregulated after MI while MMP-2 expression is more than 30 times greater than that of MMP-9 in the damaged region.31 Therefore, CTT will mainly inhibit the activity of MMP-2; (2) CTT is a peptide that has a low toxic effect on cells compared to small organic MMP inhibitors;30 and (3) CTT does not affect cell attachment and spreading on the matrix.30 To deliver CTT, a thermosensitive and degradable hydrogel based on poly(N-isopropylacrilamide) (PNIPAAm) copolymer was used. PNIPAAm copolymers have been widely utilized for drug delivery application due to their good biocompatibility and capability to solidify at 37°C without using crosslinkers.32, 33 In this work, the hydrogel was designed to be degradable. Before degradation, it had gelation temperature below 37°C so that the hydrogel can solidify at body temperature. After degradation, the gelation temperature was evaluated to above 37°C. The degradation product can thus dissolve in the body fluid and be removed from body. The results of this work demonstrated that the developed CTT delivery system effectively preserved cardiac ECM from degradation, leading to a significant increase of cardiac function. Importantly, the CTT delivery system decreased cardiac fibrosis.
2. Materials and methods
2.1 Materials
All reagents were purchased from Sigma-Aldrich unless otherwise stated. N-isopropylacrylamide (NIPAAm) was recrystallized in hexane for 3 times before polymerization. 2-hydroxyethyl methacrylate (HEMA) was used after passing through inhibitor remover column to eliminate the inhibitors. Sodium methoxide, D,L-lactide, heparin sodium (Alfa Aesar), chondroitin sulfate, peptide NH2-C-T-T-H-W-G-F-T-L-C-K (FITC)-COOH (CTT, Celtek Bioscience), MMP Substrate III (Calbiochem), and recombinant human MMP-2 (Peprotech) were used as received.
2.2 Synthesis of hydrogel
The hydrogel was synthesized by free radical polymerization of NIPAAm, HEMA and a macromer acrylate-oligolactide (AOLA, Figure 1a). To synthesize AOLA, D,L-lactide (0.347 mol) and NaOCH3 (0.019 mol) were dissolved in 100 mL of CH2Cl2 and 15 mL of methanol, respectively. The two solutions were then mixed and cooled to 0°C. After 2 h of reaction under stirring, the mixture was rinsed with 0.1M HCl and deionized water, respectively. The collected organic layer was dried over anhydrous MgSO4, followed by evaporation of the solvent under reduced pressure. The obtained crude product (0.141 mol) was dissolved in 100 mL CH2Cl2 solution. 0.169 mol of triethylamine was then added. After the mixture was cooled to 0°C, acryloyl chloride (0.169 mol) was added dropwise for 1 h. The mixture was further stirred at room temperature overnight. It was then filtered, and the filtrate was rinsed by 0.2 M Na2CO3, 0.1 M HCl and deionized water sequentially. After the collected organic layer was dried over anhydrous MgSO4, the solvent was evaporated under reduced pressure to obtain crude AOLA. It was then purified by chromatography. Ethyl acetate and hexane at the ratio of 2:1 was used eluent. The structure of the final product was confirmed by 1H-NMR (300 MHz, CDCl3): 6.48–6.53 (d, J=6.5 Hz, CH2=), 6.18–6.25 (dd, J=6.2Hz, −CH =CH2), 5.91–5.93 (d, J=5.9Hz, CH2 =), 5.15–5.28 (m, −CH−), 3.76(s, −OCH3), 1.51–1.64 (m, −CH3).
Figure 1.
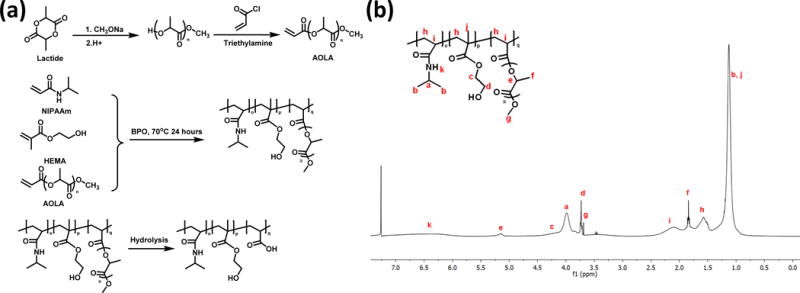
(a) Synthesis route for macromer AOLA and hydrogel, and structure of the hydrogel after degradation. (b) 1H-NMR spectrum of synthesized poly (NIPAAm-co-HEMA-co-AOLA) hydrogel.
To synthesize hydrogel, NIPAAm, HEMA, and AOLA with molar ratio of 86/10/4 were dissolved in 100 mL of dioxane. The initiator benzoyl peroxide was then added. The polymerization was conducted at 70°C for 24 h under the protection of nitrogen. The solution was then precipitated in hexane. The polymer was purified twice by dissolving in THF and precipitating with ethyl ether.
2.3 Characterization of hydrogel properties
Chemical composition of the hydrogel was determined by 1H-NMR using CDCl3 as the solvent. The hydrogel solution (20%) was prepared by dissolving the polymer in Dulbecco’s modified phosphate buffer saline (DPBS) at 4°C. Gelation temperature of the hydrogel solution was measured by DSC (TA instrument) over a temperature range of 0–60°C with a heating rate of 10°C/min. The endothermal peak was recorded as the gelation temperature.34 The hydrogel solution injectability was determined by injecting the 4°C solution through a 26-gauge needle.35 The solid hydrogel was obtained by incubating the hydrogel solution at 37°C. Hydrogel mechanical properties were tested by an Instron tensile tester equipped with a 37°C water bath. An elongation speed of 50 mm/min was used.36 Hydrogel degradation was conducted in DPBS at 37°C for 8 weeks following our previously described method.35 Weight loss was then determined.
2.4 Development and characterization of hydrogel based CTT delivery system
The CTT release system was developed by encapsulation of CTT into hydrogel. The hydrogel solution with concentration of 20% was used. CTT solution in PBS was thoroughly mixed with 1 mL of hydrogel solution at 4°C overnight. To control CTT release kinetics, chondroitin sulfate (CS) and heparin (HP) were added respectively with a final concentration of 1 mg/mL. Total of 5 groups were prepared: hydrogel without CTT, hydrogel encapsulated with 20 μg/mg (CTT mass/hydrogel mass) CTT (denoted as CTT20), hydrogel encapsulated with 20 μg/mg CTT with addition of CS (denoted as CTT20CS), hydrogel encapsulated with 20 μg/mg CTT with addition of HP (denoted as CTT20HP), and hydrogel encapsulated with 40 μg/mg CTT with addition of CS (denoted as CTT40CS).
To characterize CTT release kinetics, FITC labeled CTT was used in order to spectrometrically quantify amount of released CTT. 200 μL of the mixture was transferred into a 1.7 mL Eppendorf tube. The tube was placed in a 37°C water bath for gelation for 30 min. The supernatant was then removed and 200 μL of release medium (DPBS supplemented with 1% penicillin-streptomycin) was added. The release was conducted in a 37°C water bath for 28 days (n=5 for each group). At each time point, 200 μL of supernatant was collected and the same amount of fresh release medium was added. The fluorescent intensity of released CTT was measured by Flexstation 3 benchtop multimode microplate reader (Molecular Device). An excitation wavelength of 495 nm and an emission wavelength of 518 nm were used. The CTT concentration was determined according to calibration curve.
2.5 Characterization of bioactivity of released CTT
Bioactivity of the released CTT was measured in terms of its efficacy in inhibiting MMP-2 to cleavage MMP-2 substrate.37 To determine the effect of CTT concentration on inhibiting MMP-2 to cleavage MMP-2 substrate, different concentrations of freshly prepared CTT were used during the measurement. In brief, recombinant human MMP-2 (rhMMP-2) was diluted to 80 nM with buffer A (50 mM Tris, 5 mM CaCl2, 150 mM NaCl, 1 μM ZnCl2, and 0.01% Brij35). MMP-2 substrate III was dissolved in DMSO to obtain a stock solution with concentration of 0.244 mM. The rhMMP-2 solution was added to each well of a 96-well plate with a final concentration of 12 nM. Different concentrations of CTT were then added into the wells. After incubation at 37°C for 1 h, MMP-2 substrate III solution was added with a final concentration of 12.5 μM. The mixture was then incubated at 37°C for 3 h. The fluorescent intensity was measured using a microplate reader with an excitation wavelength of 340 nm and an emission wavelength of 485 nm. To measure bioactivity of the released CTT, the same protocol was used except that the CTT release medium, 60 μL of rhMMP-2 solution, and 30 μL of MMP-2 substrate III were added into each well. In the control group, DPBS supplemented with 1% penicillin-streptomycin, 60 μL of rhMMP-2 solution, and 30 μL of MMP-2 substrate III were added into each well. The data were then normalized to the control group.
2.6 Myocardial infarction and implantation of CTT release system
All in vivo experiments were conducted in accordance with the National Institutes of Health Guide for the care and use of laboratory animals. Animal protocol was administrated by the Institutional Animal Care and Use Committee (IACUC) of The Ohio State University. Male Sprague-Dawley rats aged from 10 to 12 weeks were used for the study. All rats were divided into four groups, i.e., sham, non-treatment (MI), hydrogel injection (Gel), and CTT40CS injection (n ≥ 6 for each group). The rats were anesthetized with 2.5% isoflurane inhalation followed by endotracheal intubation and connection to a mechanical ventilator. The rat hearts were exposed through a left thoracotomy. After identifying left anterior branch of the coronary artery, it was carefully ligated with 6–0 polypropylene suture. Myocardial infarction was verified by regional cyanosis and ST-segment elevation. Thirty minutes after ligation, 200 μL of Gel or CTT40CS were injected into the apical, proximal, lateral, and septal wall regions bordering the infarct, and the center of the infarct (40 μL/injection). Controls were sham and MI groups.
2.7 Histological analyses
Four weeks after surgery, the rat hearts were harvested. The hearts were then perfused with PBS to rinse out blood followed by fixing in 4% paraformaldehyde overnight. The heart tissues were embedded in paraffin, and sequentially sectioned along the transverse direction with a thickness of 4 μm. The sections at the level of center of injection sites were collected for histological analysis. The sections were stained with Hematoxylin and Eosin (H&E) and Picrosirius Red (PSR) as previously described.38, 39 Wall thickness was calculated from H&E staining using Image J.40 Thickness of five different fields was measured for each sample. The relative thickness of the infarcted area was calculated as the ratio of thickness at infarcted area to that of normal area. Type I and III collagen were identified from PSR staining as yellow-red and green respectively using polarized light microscopy with appropriate band-pass filters.41, 42 The percent area of collagen within the remote and MI regions was determined from images (n≥5) using threshold image analysis with Image J software.41, 42 The ratio of type III to type I collagen was also quantified.41, 42
Immunohistochemical stainings were performed for markers alpha smooth muscle actin (αSMA) and vWF. Before staining, the sections were deparaffinized with xylene followed by rehydrating with serial concentrations of ethanol. The sections were blocked with goat serum and incubated with antibodies against αSMA and vWF. Alexa Fluor 488 and Alexa Fluor 647 were used as secondary antibodies. Nuclei were stained with DAPI. The stained sections were imaged using a confocal laser scanning microscope. Blood vessels were recognized as vWF positive lumen. Myofibroblasts were identified as αSMA positive cells not co-localized with vWF positive endothelial cells.43 Images (n≥5) of the infarcted area were analyzed.
2.8 Analysis of cardiac function by echocardiography
Echocardiography was performed 4 weeks after the surgery following an established protocol.44 The rats were anesthetized with 2.5% isoflurane inhalation. Standard transthoracic echocardiography was performed using a Vevo-2100 high-resolution ultrasound imaging system (Visualsonics; Toronto, Canada). The M-mode ultrasound images were acquired. Ejection fraction (EF) and fractional shortening (FS) were calculated using VevoStrain™ Advanced Cardiac Analysis Software.
2.9 Statistical analysis
Statistical analysis was performed using JMP software. One way ANOVA with post-hoc Tukey HSD test was utilized for data analysis. Data were presented as mean ± standard deviation. Statistical significance was defined as p<0.05.
3. Results
3.1 Characterization of hydrogel properties
The hydrogel was synthesized by free radical polymerization of NIPAAm, HEMA and macromer AOLA. Among the three components, NIPAAm imparts the hydrogel with themosensitivity. Hydrophilic HEMA increases hydrogel solubility in aqueous condition. The degradable and hydrophobic AOLA allows the sol-gel temperature to be lower than 37°C before degradation (Figure 1a). 1H-NMR spectrum confirmed the chemical structure of the hydrogel as the characteristic peaks of each component were seen in the spectrum (Figure 1b). The ratio of NIPAAm/HEMA/AOLA was 86.0/10.4/3.6, consistent with the feed ratio. The hydrogel solution exhibited a gelation temperature of 26.5°C before degradation. The 4°C hydrogel solution was flowable and can be readily injected through a 26G needle (Figure 2 A and B). At 37°C, the hydrogel solution solidified within 7 s to form a flexible hydrogel (Figure 2C) with Young’s modulus of 35 ± 5 kPa. The hydrogel degraded slowly in PBS at 37°C with 7% of weight loss after 8 weeks. The degradation product had a gelation temperature of 41.2°C and was water soluble at 37°C.
Figure 2.

Hydrogel solution was flowable (a) and injectable by 26G needle (b) at 4°C. It solidified at 37°C (c).
3.2 Release kinetics of CTT
CTT delivery systems were developed by encapsulating CTT into the synthesized hydrogel. Glycosaminoglycans CS and HP were used to modulate release kinetics. Figure 3 demonstrates that the small molecular weight CTT was able to gradually release from the hydrogel for 28 days. For all 4 groups, burst release was seen in the first 8 hours followed by a slower release pattern. Interestingly, all groups showed nearly linear release after 4 days. The release kinetics was dependent on the type of glycosaminoglycan added to the hydrogel, and CTT loading amount. Compared to the CTT20 group where no glycosaminoglycan was added, the groups containing CS and HP (CTT20CS and CTT20HP) released significantly higher amount of CTT at each time point (Figure 3, p<0.05 for CTT20CS vs. CTT20, and CTT20HP vs. CTT20). By comparing release kinetics of the groups added with CS and HP, it was found that significantly higher amount of CTT was released from the group with HP (CTT20HP) than the group with CS (CTT20CS) at each time point (p<0.05). When the CTT loading amount was increased from 20 μg/mg to 40 μg/mg, the amount of released CTT was significantly increased (p<0.05 for all time points).
Figure 3.
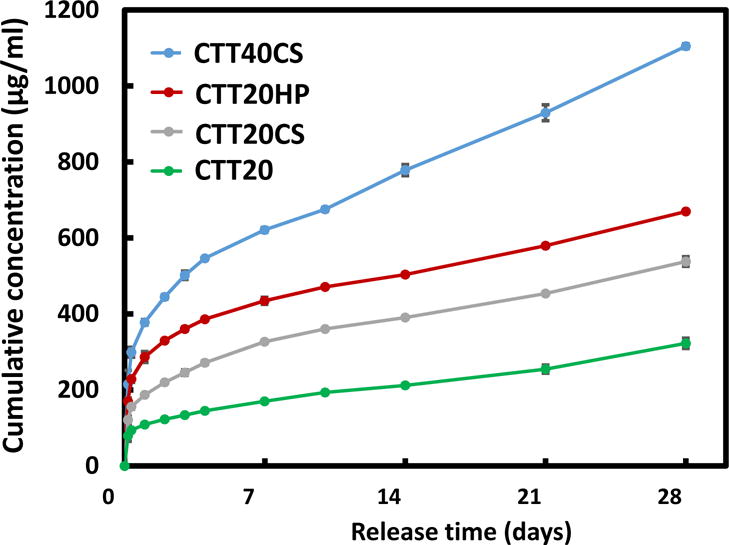
Release kinetics of CTT encapsulated in the hydrogel with or without chondroitin sulfate and heparin. Two different CTT concentrations were loaded into the hydrogel (20 and 40 μgCTT/mgHydrogel). The error bars are small.
3.3 Bioactivity of released CTT
Bioactivity of the released CTT was examined in terms of its ability to inhibit MMP-2 to degrade MMP-2 substrate. Prior to testing released CTT, the efficacy of fresh CTT in inhibiting MMP-2 mediated MMP-2 substrate degradation was examined. Figure 4A shows that the relative MMP-2 bioactivity was decreased when the CTT concentration was increased from 1 μM to 100 μM. The calculated half maximal effective concentration (EC50) was 16 μM or 18.7 μg/mL. These results suggest that CTT is an efficient MMP-2 inhibitor.
Figure 4.
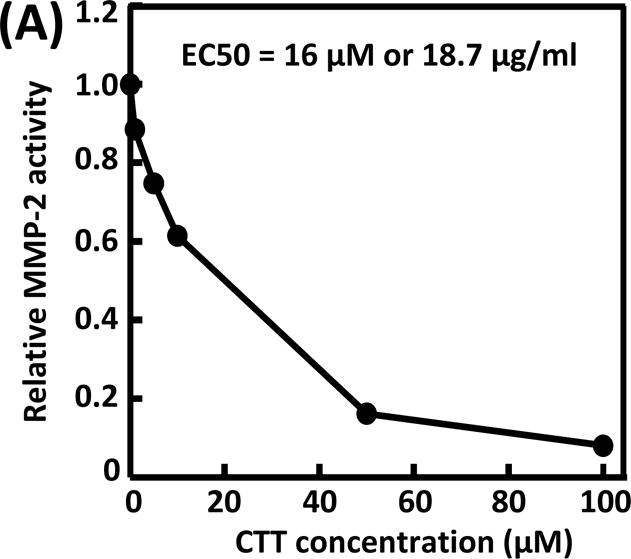
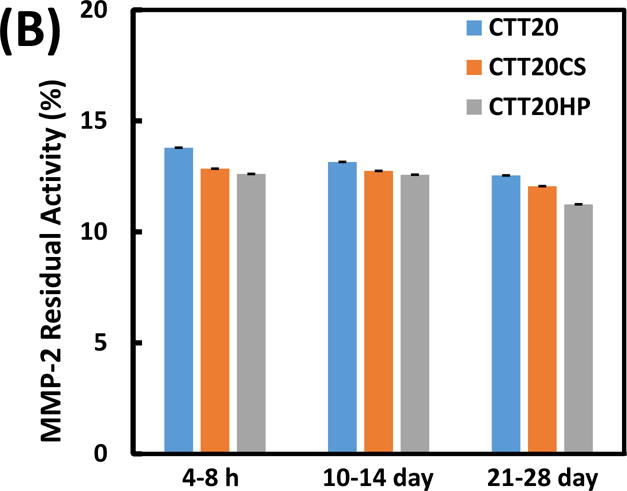
Bioactivity of CTT in inhibiting MMP-2 from degrading MMP-2 substrate. (A) concentration-dependence of CTT; (B) bioactivity of CTT released at 8 hours, days 14 and 28. The error bars are small.
CTT released at 3 time points (4–8 h, 10–14 day, and 21–28 day) from groups CTT20, CTT20CS and CTT20HP were selected for bioactivity tests. The 3 time points represent early, middle and late stages of the release kinetics. The release medium without CTT was used as control. The relative MMP-2 bioactivity was 100% for the control group, indicating that the substrate can be completely degraded by MMP-2 when there was no CTT. For the CTT20, CTT20CS and CTT20HP groups, the relative MMP-2 bioactivity was decreased to less than 15% (Figure 4B). This demonstrates that all of the released CTT was able to efficiently prevent MMP-2 from degrading MMP-2 substrate. Therefore, the released CTT remained bioactive.
3.4 Efficacy of CTT release in preserving cardiac ECM after MI
To examine the efficacy of CTT release in attenuating cardiac ECM degradation by upregulated MMPs especially MMP-2, the hydrogel encapsulated with CTT and CS (CTT40CS group) was injected into infarcted rat hearts 30 min after MI. Sham, infarcted hearts without injection (MI group) and infarcted hearts injected with hydrogel without CTT and CS (Gel group) were used as controls. H&E images demonstrated that substantial left ventricle dilation was occurred for the MI and Gel groups after 4 weeks of injection (Figure 5). In contrast, the CTT40CS group appeared to have less dilation. The wall thickness of infarcted area was calculated from H&E images (Figure 6). The wall thickness of MI group was decreased to 29% of the Sham group after 4 weeks. Injection of hydrogel significantly increased the wall thickness to 46% of the Sham group (p<0.01, Gel group vs. MI group). A remarkable wall thickness increase was observed for the CTT40CS group where relative wall thickness was 76% of the Sham group (p<0.01, CTT40CS group vs. Gel group). These results demonstrate that the released CTT decreased MMP-2 bioactivity, leading to the attenuation of cardiac ECM degradation. The higher magnification H&E images of the infarcted area indicate that cell infiltration into the hydrogel occurred in both Gel and CTT40CS groups (Figure 5).
Figure 5.
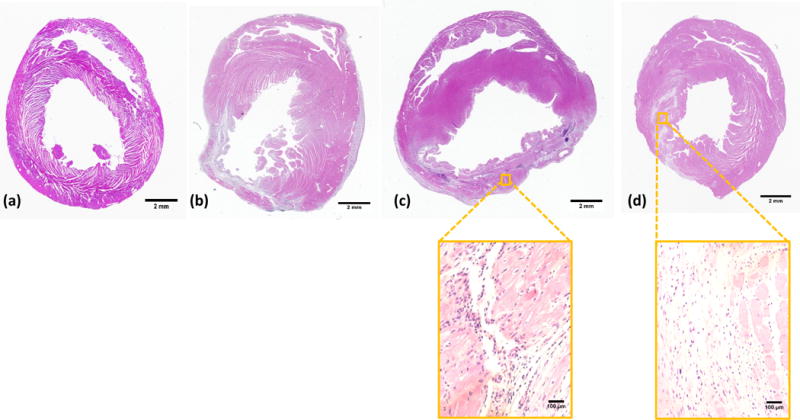
H&E staining of the rat hearts after 4 weeks of injection. (a) Sham; (b) MI; (c) Gel; and (d) CTT40CS. Scale bar = 2 mm for whole heart view, and scale bar = 100 μm for higher magnification view. The wall thickness of MI group was decreased compared to the Sham group. Injection of hydrogel and CTT40CS group increased wall thickness. The higher magnification H&E images indicated that cell infiltration into the hydrogel occurred.
Figure 6.
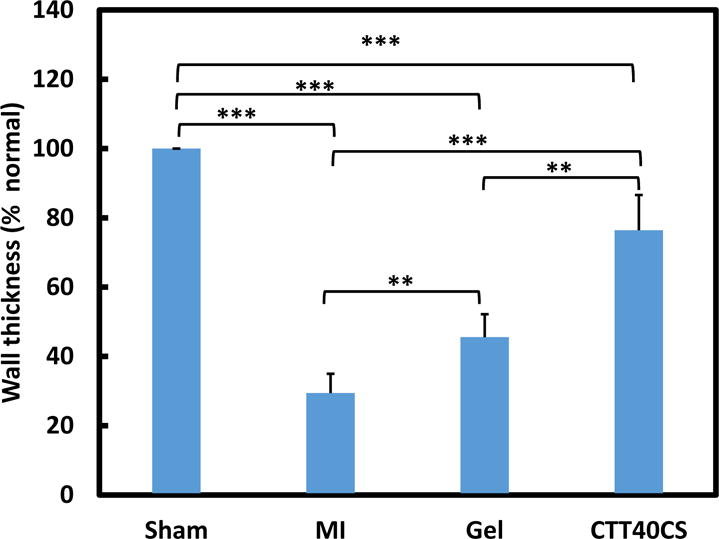
Relative left ventricle wall thickness at infarcted region of the Sham, MI, Gel, and CTT40CS groups. The hearts were harvested 4 weeks after injection. ** p<0.01. *** p<0.001.
Picrosirius red staining was used to characterize collagen types I and III in the infarcted area (Figure 7). Both are major components in the cardiac ECM. Collagen type I is more rigid than type III. In the MI group, the ratio was less than 1/1 (Figure 7d). Injection of hydrogel (Gel group) did not change the ratio (p>0.05, Gel group vs. MI group). Interestingly, the injection of CTT40CS group significantly increased the ratio (p<0.05, CTT40CS group vs. Gel and MI groups). The greater collagen type III ratio may make the infarcted tissue more compliant.
Figure 7.
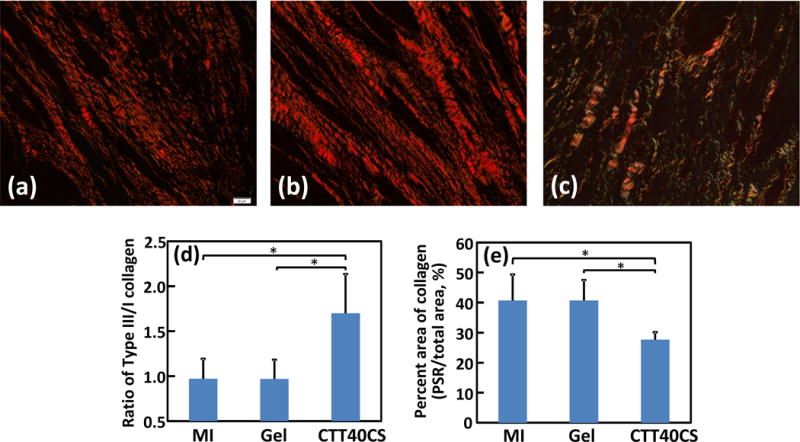
Picrosirius red staining of the hearts harvested 4 weeks after surgery. Views were taken at the infarcted region of the MI (a), Gel (b), and CTT40CS (c) groups. Collagen type III/I ratio (d) and total area of collagen (e) were analyzed from the images. * p<0.05.
3.5 Efficacy of CTT release in reducing cardiac fibrosis
Cardiac fibrosis naturally forms after MI. It is rich in type I collagen. To determine whether CTT release can reduce cardiac fibrosis, total collagen content and myofibroblast density in the infarcted area were characterized. Figure 7e shows that total collagen content in the MI and Gel groups were significantly higher than the CTT40CS group (p<0.05, CTT40CS group vs. MI and Gel groups). There was no significant difference between MI and Gel groups (p>0.05). These results in combination with collagen type III/I ratios in Figure 7d demonstrate that MI and Gel group had higher type collagen I content than the CTT40CS group. As myofibroblasts are mainly responsible for cardiac fibrosis, their densities in different groups were determined from immunohistochemical staining images (Figure 8). Myofibroblasts are identified as αSMA+ cells in the infarct area that are not co-localized with vWF+ endothelial cells in the vessels. Consistent with type I collagen content, the myofibroblast density was significantly higher in the MI and Gel groups than in the CTT40CS group (Figure 9A. p<0.01, CTT40CS group vs. MI and Gel groups). The MI and Gel groups had similar myofibroblast density. The above results demonstrate that the released CTT significantly decreased cardiac fibrosis.
Figure 8.
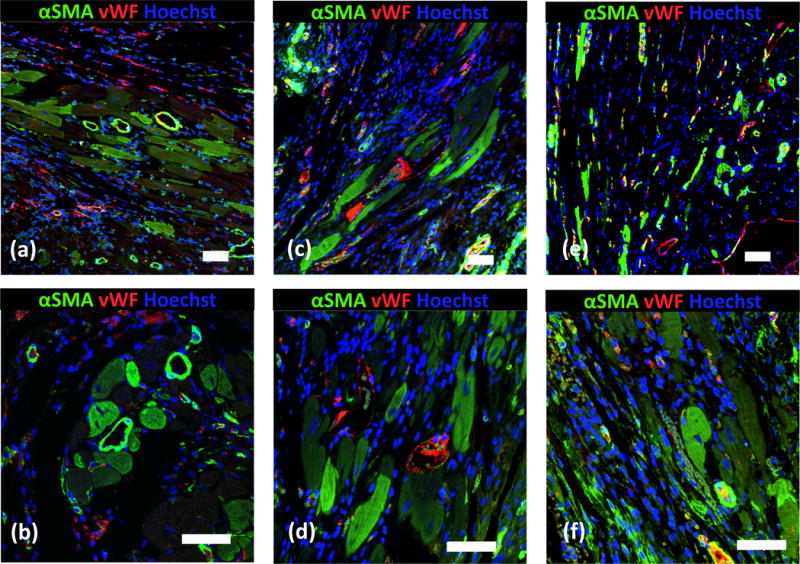
Immunohistological analysis of the infarcted region after 4 weeks of injection. (a, b) MI; (c, d) Gel; and (e, f) CTT40CS. Scale bar = 60 μm.
Figure 9.
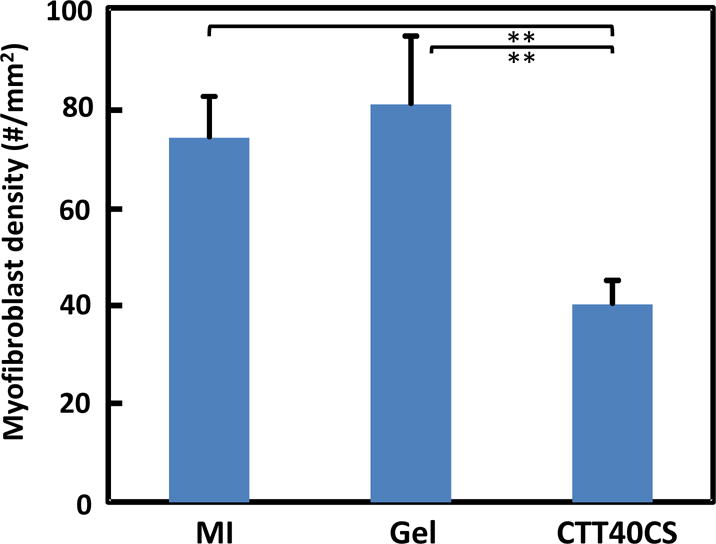
Myofibroblast density determined from immunohistological images for MI, Gel, and CTT40CS groups. ** p<0.01.
3.6 Efficacy of CTT release in improving cardiac function
To determine whether cardiac function can be increased when using CTT to inhibit cardiac ECM degradation, left ventricular ejection fraction (EF), fractional shortening (FS), end-systolic volume (ESV), and end-diastolic volume (EDV) were measured by echocardiography (Figure 10 A–D). After 4 weeks of injection, the Gel group exhibited similar EF, FS, EDV and ESV as the MI group (p>0.05). Compared to the Gel group, the CTT40CS group had significantly higher EF and FS (p<0.01 and p<0.05 for EF and FS respectively), and lower ESV and EDV (p<0.05 for ESV and EDV). These results suggest that using CTT to inhibit cardiac ECM degradation significantly increased cardiac function.
Figure 10.
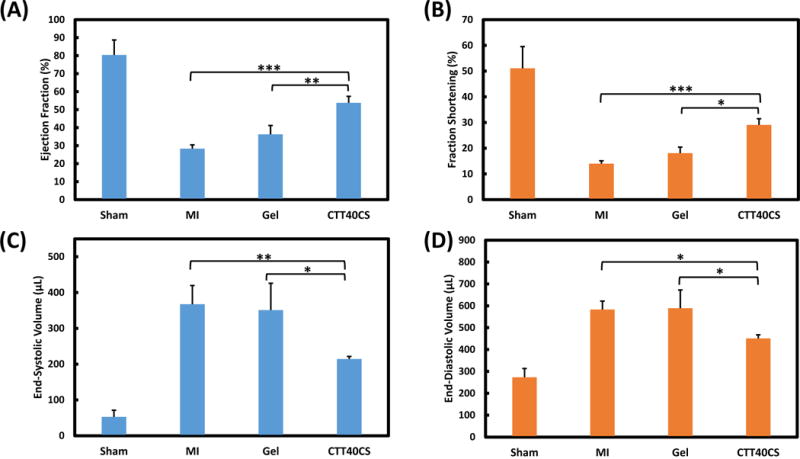
Echocardiographic analysis of Sham, MI, Gel, and CTT40CS groups. (A) Ejection Fraction; (B) Fractional Shortening; (C) End-Systolic Volume, and (D) End-Diastolic Volume. * p<0.05, ** p<0.01, and *** p<0.001.
4. Discussion
The objective of this work was to develop a CTT-based MMP inhibitor release system that can be locally delivered into infarcted heart to decrease activity of upregulated MMP-2, thus inhibiting cardiac ECM from degrading after MI. The preserved cardiac ECM provides mechanical support to the infarcted tissue so as to augment cardiac function. It also provides a native-like platform for further vascularization and cell therapies in order to regenerate new myocardium. As the CTT was delivered specifically into the infracted tissue, it can efficiently reduce MMP activity in the damaged area with limited effect on healthy tissues. Previous studies have demonstrated that local injection of hydrogels capable of releasing tissue inhibitor of MMPs constrained the released inhibitor in the infarcted tissue.16, 20 The continuous release of CTT makes repeated administration of the inhibitor unnecessary. Importantly, CTT used in this work decreased myofibroblast density and cardiac fibrosis (Figures 8–10).
To develop CTT release systems, a thermosensitive, fast gelation, and degradable hydrogel was used as CTT carrier. The thermal sensitivity and degradability are contributed by NIPAAm and AOLA units, respectively. The role of HEMA unit is to increase the hydrophilicity of the polymer. The hydrogel solution had a gelation temperature around room temperature (26.5°C), and can solidify within 7 s at 37°C (Figure 2C). The use of thermosensitive hydrogel for CTT delivery allows solidification process to be simply initiated by temperature without using chemical crosslinkers that may raise cytotoxic concerns. The advantage of fast gelation hydrogel is that it quickly immobilizes in the heart upon injection to largely retain CTT in the tissue. This is evidenced by our finding that no hydrogel was leaked out during and after injection. The injected hydrogel not only serves as CTT carrier but also provides mechanical support to the infarcted heart tissue. After MI, the left ventricular wall stress is gradually increased leading to the decrease of cardiac function.45 Therefore, the injection of hydrogel with similar modulus as that of the heart tissue may efficiently decrease elevated wall stress to increase cardiac function. In this work, the developed hydrogel exhibited Young’s modulus of 35 ± 5 kPa, within in the modulus range of heart tissue.46, 47 The hydrogel had a slow degradation rate with 7% of weight loss after 8 weeks. The degradation product possessed a gelation temperature above body temperature. It can thus dissolve in the body fluid and be removed from the body.
The CTT encapsulated in the hydrogel was able to gradually release for 4 weeks at 37°C in PBS (Figure 3). Sustained CTT release was achieved when simply encapsulating it in the hydrogel (Figure 3). This is interesting because CTT has a low molecular weight (1297 Da), and small molecules usually release completely in a short period. The sustained CTT release over 4 weeks is possibly resulted from hydrogen bonding between the hydrogel and CTT, which delayed the CTT to release out. Specifically, amide and hydroxyl groups in the hydrogel may form hydrogen bonding with amide groups in the CTT.
The incorporation of glycosaminoglycans CS and HP in the hydrogel facilitated CTT release (Figure 3). This is likely because CS and HP increased the hydrogel hydrophilicity as both are highly hydrophilic and can interact with the hydrogel via hydrogen bonding.35 In addition, HP may more greatly increase hydrogel hydrophilicity than CS since it has more sulfate groups. As a result, the CTT released faster in the hydrogel with HP than that with CS. The release profiles can also be tuned by CTT loading dosage. Figure 3 showed that a higher loading dosage released more CTT (CTT20CS vs. CTT40CS). The released CTT remained bioactive as it attenuated MMP-2 to degrade MMP-2 substrate (Figure 4B). The above results demonstrate that the developed CTT release system can effectively release CTT to decrease MMP-2 bioactivity. Previous studies showed that MMP-2 activity was increased as soon as 1 h after MI10, and peaked around 2–3 weeks post-MI11. Therefore, the developed CTT release system has the potential to effectively attenuate MMP-2 activity at different stages of MMP-2 upregulation. CTT release in vivo may be faster than in PBS as the hydrogel may degrade quicker due to presence of enzymes in the tissue. Yet it is possible that CTT release can last for 4 weeks in the infarcted region since the hydrogel was observed after 4 weeks of implantation.
To examine the efficacy of CTT release in attenuating cardiac ECM degradation, three groups were used for comparison, i.e., MI, Gel, and CTT40CS. After 4 weeks of post-MI, the Gel group had greater wall thickness than the MI group (Figures 5 and 6). This is resulted from bulking effect of the injected hydrogel.48 The injection of CTT40CS group significantly increased wall thickness compared to the Gel group (Figures 5 and 6). This indicates that the released CTT inhibited MMP-2 mediated cardiac ECM degradation. Further analysis demonstrated that the preserved cardiac ECM had similar type III/I collagen ratio as the healthy heart tissue (Figure 7d). These results clearly show that the released CTT prevented cardiac ECM from degradation.
A potential concern when using MMP inhibitor for cardiac therapy is that cardiac fibrosis may concurrently occur.16, 20, 49 The formed scar tissue is rich in type I collagen, which stiffens the infarcted heart tissue leading to cardiac function deterioration. MMP inhibitors may initiate cardiac fibrosis in two aspects. First, they may stimulate cardiac fibroblasts to differentiate into myofibroblasts to facilitate the secretion of type I collagen. Second, over dose of MMP inhibitor may break the balance of collagen secretion and degradation, leading to the increase in collagen content. In this work, the developed CTT release system actually reduced cardiac fibrosis (Figures 7–10). Figures 9 and 10 demonstrate that the released CTT decreased myofibroblast density compared to MI and Gel groups. In addition, the type I collagen ratio in the CTT40CS group was decreased (Figure 7d). These results suggest that CTT itself did not induce cardiac fibrosis, and the dosage of released CTT was appropriate. The decrease in myofibroblast density is likely due to the decrease of MMP-9 activity by CTT. MMP-9 has been shown to promote myofibroblast formation though epithelial mesenchymal transition mechanism.50 CTT is an inhibitor specific to MMP-2 and MMP-9.30 Both are upregulated after MI although MMP-2 expression is more than 30 times greater than that of MMP-9 in the damaged region.31
The injection of the CTT40CS group into infarcted hearts augmented cardiac function (EF, FS, ESV and EDV) compared to the MI and Gel groups (Figure 10). In addition, the Gel group had slightly higher EF and FS than MI group. This is likely due to mechanical support of the hydrogel.51 In present work, the hydrogel had similar modulus as the healthy heart tissue. It can thus efficiently decrease wall stress. The increase in cardiac function in the CTT40CS group is possibly resulted from the mechanical support of hydrogel, the preservation of cardiac ECM, and the decrease in cardiac fibrosis. The results in this study are consistent with previous studies where delivery of recombinant tissue inhibitor of MMPs into the infarcted hearts significantly increased cardiac function.16, 20 Various studies demonstrated that increase of tissue vascularization may lead to cardiac function increase. In this work, we found that injection of hydrogel or hydrogel encapsulated with CTT did not increase vessel density in the infarcted area (MI: 37.1±15.3 lumens/mm2; Gel: 30.1±7.2 lumens/mm2; CTT40CS: 39.6±8.4 lumens/mm2).
Overall, the above results demonstrate the benefits of using CTT based MMP inhibitor release system to preserve cardiac ECM while decreasing cardiac fibrosis. CTT has a low molecular weight and EC50 (16 μM or 18.4 μg/ml, Figure 4A). Therefore, a relatively low concentration of CTT can efficiently inhibit MMP activity. This is advantageous over those high molecular weight MMP inhibitor where a relatively higher concentration may be needed.16, 20 The developed CTT release system is translational. It may be delivered into infarcted hearts in the context of surgery, such as during coronary revascularization after MI.16 The release of CTT also improved cellularization in the infarcted region compared to the groups without CTT release (Figure 5). This is consistent with previous studies where recombinant tissue inhibitor of MMPs was delivered into the infarcted hearts.16, 20 It is possible that the growth factors in the preserved ECM played important roles in this process. Therefore, the preserved cardiac ECM may provide a platform for cell therapy as these growth factors may promote cell survival, proliferation and differentiation. Endogenous cells like cardiac progenitor/stem cells, bone marrow stem cells and other stem cell types may migrate into the cardiac ECM. These cells may differentiate into endothelial cells, cardiomyocytes and other cell types for the regeneration of new cardiac tissue.
In this work, acute MI model was used to investigate the effect of CTT release on cardiac ECM degradation and cardiac function. Many previous studies have used this model to study the efficacy of hydrogel therapy.16, 20, 51–53 The major limitation of the model is that it is not highly clinically relevant as there are concerns that patients may not be able to receive treatment after such a short time following MI.
5. Conclusions
An injectable, degradable, thermosensitive and fast gelation hydrogel was synthesized to deliver MMP-2 specific inhibitor CTT into infarcted hearts. The developed CTT delivery system was able to release bioactive CTT over 4 weeks. The use of glycosaminoglycans such CS and HP accelerated the release. Delivery of the CTT release system into infarcted hearts efficiently prevented cardiac ECM from degradation, attenuated cardiac fibrosis, and improved cardiac function.
Acknowledgments
This work was supported by US National Institutes of Health (R01HL138353, R01EB022018, R01HL124122, and R21EB021896), US National Science Foundation (1708956), and National Science Foundation of China (81471788).
References
- 1.Spinale FG. Physiol Rev. 2007;87(4):1285–342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 2.DeCoux A, Lindsey ML, Villarreal F, Garcia RA, Schulz R. J Mol Cell Cardiol. 2014;77c:64–72. doi: 10.1016/j.yjmcc.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cleutjens JPM, Creemers EEJM. J Card Fail. 2002;8(6, Part B):S344–S348. doi: 10.1054/jcaf.2002.129261. [DOI] [PubMed] [Google Scholar]
- 4.Creemers EEJM, Cleutjens JPM, Smits JFM, Daemen MJAP. Matrix Metalloproteinase Inhibition after Myocardial Infarction: A New Approach to Prevent Heart Failure? Circ Res. 2001;89(3):201–210. doi: 10.1161/hh1501.094396. [DOI] [PubMed] [Google Scholar]
- 5.Jugdutt BI, Joljart MJ, Khan MI. Circulation. 1996;94(1):94–101. doi: 10.1161/01.cir.94.1.94. [DOI] [PubMed] [Google Scholar]
- 6.Wang W, Schulze CJ, Suarez-Pinzon WL, Dyck JRB, Sawicki G, Schulz R. Circulation. 2002;106(12):1543–1549. doi: 10.1161/01.cir.0000028818.33488.7b. [DOI] [PubMed] [Google Scholar]
- 7.Ali MAM, Cho WJ, Hudson B, Kassiri Z, Granzier H, Schulz R. Circulation. 2010;122(20):2039–2047. doi: 10.1161/CIRCULATIONAHA.109.930222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sung MM, Schulz CG, Wang W, Sawicki G, Bautista-López NL, Schulz R. J Mol Cell Cardiol. 2007;43(4):429–436. doi: 10.1016/j.yjmcc.2007.07.055. [DOI] [PubMed] [Google Scholar]
- 9.Sawicki G, Leon H, Sawicka J, Sariahmetoglu M, Schulze CJ, Scott PG, Szczesna-Cordary D, Schulz R. Circulation. 2005;112(4):544–552. doi: 10.1161/CIRCULATIONAHA.104.531616. [DOI] [PubMed] [Google Scholar]
- 10.Herzog E, Gu A, Kohmoto T, Burkhoff D, Hochman JS. Cardiovasc Pathol. 1998;7(6):307–312. doi: 10.1016/s1054-8807(98)00008-8. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Tung C-H, Allport JR, Chen S, Weissleder R, Huang PL. Circulation. 2005;111(14):1800–1805. doi: 10.1161/01.CIR.0000160936.91849.9F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashidani S, Tsutsui H, Ikeuchi M, Shiomi T, Matsusaka H, Kubota T, Imanaka-Yoshida K, Itoh T, Takeshita A. Am J Physiol Heart Circ Physiol. 2003;285:H1229–H1235. doi: 10.1152/ajpheart.00207.2003. [DOI] [PubMed] [Google Scholar]
- 13.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. J Clin Invest. 2005;115(3):599–609. doi: 10.1172/JCI22304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abbenante G, Fairlie DP. Med Chem. 2005;1(1):71–104. doi: 10.2174/1573406053402569. [DOI] [PubMed] [Google Scholar]
- 15.Dorman G, Cseh S, Hajdu I, Barna L, Konya D, Kupai K, Kovacs L, Ferdinandy P. Drugs. 2010;70(8):949–64. doi: 10.2165/11318390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 16.Eckhouse SR, Purcell BP, McGarvey JR, Lobb D, Logdon CB, Doviak H, O’Neill JW, Shuman JA, Novack CP, Zellars KN, Pettaway S, Black RA, Khakoo A, Lee T, Mukherjee R, Gorman JH, Gorman RC, Burdick JA, Spinale FG. Sci Transl Med. 2014;6(223):223ra21. doi: 10.1126/scitranslmed.3007244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fingleton B. Curr Pharm Des. 2007;13(3):333–46. doi: 10.2174/138161207779313551. [DOI] [PubMed] [Google Scholar]
- 18.Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Baskin JM, Deschamps AM, Lowry AS, Escobar GP, Lucas DG, Yarbrough WM, Zile MR, Spinale FG. Circulation. 2003;107(4):618–25. doi: 10.1161/01.cir.0000046449.36178.00. [DOI] [PubMed] [Google Scholar]
- 19.Ngu JM, Teng G, Meijndert HC, Mewhort HE, Turnbull JD, Stetler-Stevenson WG, Fedak PW. Cardiovasc Pathol. 2014;23(6):335–43. doi: 10.1016/j.carpath.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell BP, Lobb D, Charati MB, Dorsey SM, Wade RJ, Zellars KN, Doviak H, Pettaway S, Logdon CB, Shuman JA, Freels PD, Gorman JH, 3rd, Gorman RC, Spinale FG, Burdick JA. Nat Mater. 2014;13(6):653–61. doi: 10.1038/nmat3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turk B. Nat Rev Drug Discov. 2006;5(9):785–99. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 22.Uchinaka A, Kawaguchi N, Mori S, Hamada Y, Miyagawa S, Saito A, Sawa Y, Matsuura N. Tissue Eng Part A. 2014;20(21–22):3073–84. doi: 10.1089/ten.tea.2013.0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beckett RP, Davidson AH, Drummond AH, Huxley P, Whittaker M. Drug Discovery Today. 1996;1(1):16–26. [Google Scholar]
- 24.Talbot BC, Brown PD. Eur J Cancer. 1996;32(14):2528–2533. doi: 10.1016/s0959-8049(96)00398-x. [DOI] [PubMed] [Google Scholar]
- 25.Santos O, McDermott C, Daniels R, Appelt K. Clin Exp Metastasis. 1997;15(5):499–508. doi: 10.1023/a:1018418725453. [DOI] [PubMed] [Google Scholar]
- 26.Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P, Lyon R, Quinones M, Theroux P, Sydlowski D, Kim HE, Garcia MJ, Jaber WA, Weaver WD. J Am Coll Cardiol. 2006;48(1):15–20. doi: 10.1016/j.jacc.2006.02.055. [DOI] [PubMed] [Google Scholar]
- 27.Guo Y-H, Gao W, Li Q, Li P-F, Yao P-Y, Chen K. Life Sci. 2004;75(20):2483–2493. doi: 10.1016/j.lfs.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Fan D, Takawale A, Basu R, Patel V, Lee J, Kandalam V, Wang X, Oudit GY, Kassiri Z. Cardiovasc Res. 2014;103(2):268–80. doi: 10.1093/cvr/cvu072. [DOI] [PubMed] [Google Scholar]
- 29.Zavadzkas JA, Stroud RE, Bouges S, Mukherjee R, Jones JR, Patel RK, McDermott PJ, Spinale FG. Circ Res. 2014;114(9):1435–45. doi: 10.1161/CIRCRESAHA.114.303634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koivunen E, Arap W, Valtanen H, Rainisalo A, Medina OP, Heikkila P, Kantor C, Gahmberg CG, Salo T, Konttinen YT, Sorsa T, Ruoslahti E, Pasqualini R. Nat Biotechnol. 1999;17(8):768–74. doi: 10.1038/11703. [DOI] [PubMed] [Google Scholar]
- 31.Wilson EM, Moainie SL, Baskin JM, Lowry AS, Deschamps AM, Mukherjee R, Guy TS, St John-Sutton MG, Gorman JH, 3rd, Edmunds LH, Jr, Gorman RC, Spinale FG. Circulation. 2003;107(22):2857–63. doi: 10.1161/01.CIR.0000068375.40887.FA. [DOI] [PubMed] [Google Scholar]
- 32.GhavamiNejad A, SamariKhalaj M, Aguilar LE, Park CH, Kim CS. Sci Rep. 2016;6:33594. doi: 10.1038/srep33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aguilar LE, GhavamiNejad A, Park CH, Kim CS. Nanomedicine. 2017;13(2):527–538. doi: 10.1016/j.nano.2016.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Li Z, Fan Z, Xu Y, Lo W, Wang X, Niu H, Li X, Xie X, Khan M, Guan J. ACS Appl Mater Interfaces. 2016;8(17):10752–60. doi: 10.1021/acsami.6b01374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang F, Li Z, Khan M, Tamama K, Kuppusamy P, Wagner WR, Sen CK, Guan J. Acta Biomater. 2010;6(6):1978–91. doi: 10.1016/j.actbio.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Li Z, Fan Z, Xu Y, Niu H, Xie X, Liu Z, Guan J. ACS Appl Mater Interfaces. 2016;8(25):15948–15957. doi: 10.1021/acsami.6b04932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morarescu D, West-Mays JA, Sheardown HD. Biomaterials. 2010;31(8):2399–2407. doi: 10.1016/j.biomaterials.2009.11.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rich L, Whittaker P. Braz J Morphol Sci. 2005;22(2):97–104. [Google Scholar]
- 39.Kmiec Z, Kiernan JA. Cytobiol. 2016;54(1):58–9. [Google Scholar]
- 40.Deng B, Shen L, Wu Y, Shen Y, Ding X, Lu S, Jia J, Qian J, Ge J. J Biomed Mater Res A. 2015;103(3):907–18. doi: 10.1002/jbm.a.35232. [DOI] [PubMed] [Google Scholar]
- 41.Namba T, Tsutsui H, Tagawa H, Takahashi M, Saito K, Kozai T, Usui M, Imanaka-Yoshida K, Imaizumi T, Takeshita A. Circulation. 1997;95(10):2448–2454. doi: 10.1161/01.cir.95.10.2448. [DOI] [PubMed] [Google Scholar]
- 42.Junqueira LC, Bignolas G, Brentani R. Histochem J. 1979;11(4):447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- 43.Thomson JA, West M. In: Essentials of Stem Cell Biology. Lanza R, Gearhart J, Hogan B, Melton D, Pedersen R, Thomas, editors. Elsevier Academic Press; Burlington, MA: 2005. pp. 537–539. [Google Scholar]
- 44.Fujimoto KL, Tobita K, Merryman WD, Guan J, Momoi N, Stolz DB, Sacks MS, Keller BB, Wagner WR. J Am Coll Cardiol. 2007;49(23):2292–300. doi: 10.1016/j.jacc.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wall ST, Walker JC, Healy KE, Ratcliffe MB, Guccione JM. Circulation. 2006;114(24):2627–35. doi: 10.1161/CIRCULATIONAHA.106.657270. [DOI] [PubMed] [Google Scholar]
- 46.Al-Haque S, Miklas JW, Feric N, Chiu LL, Chen WL, Simmons CA, Radisic M. Macromol Biosci. 2012;12(10):1342–53. doi: 10.1002/mabi.201200042. [DOI] [PubMed] [Google Scholar]
- 47.Bhana B, Iyer RK, Chen WL, Zhao R, Sider KL, Likhitpanichkul M, Simmons CA, Radisic M. Biotechnol Bioeng. 2010;105(6):1148–60. doi: 10.1002/bit.22647. [DOI] [PubMed] [Google Scholar]
- 48.Fujimoto KL, Ma Z, Nelson DM, Hashizume R, Guan J, Tobita K, Wagner WR. Biomaterials. 2009;30(26):4357–68. doi: 10.1016/j.biomaterials.2009.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGarvey JR, Pettaway S, Shuman JA, Novack CP, Zellars KN, Freels PD, Echols RL, Jr, Burdick JA, Gorman JH, 3rd, Gorman RC, Spinale FG. J Pharmacol Exp Ther. 2014;350(3):701–9. doi: 10.1124/jpet.114.215798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J, Shultz RW, Mars WM, Wegner RE, Li Y, Dai C, Nejak K, Liu Y. J Clin Invest. 2002;110(10):1525–38. doi: 10.1172/JCI16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshizumi T, Zhu Y, Jiang H, D’Amore A, Sakaguchi H, Tchao J, Tobita K, Wagner WR. Biomaterials. 2016;83:182–193. doi: 10.1016/j.biomaterials.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tous E, Ifkovits JL, Koomalsingh KJ, Shuto T, Soeda T, Kondo N, Gorman JH, 3rd, Gorman RC, Burdick JA. Biomacromolecules. 2011;12(11):4127–35. doi: 10.1021/bm201198x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacArthur JW, Jr, Purcell BP, Shudo Y, Cohen JE, Fairman A, Trubelja A, Patel J, Hsiao P, Yang E, Lloyd K, Hiesinger W, Atluri P, Burdick JA, Woo YJ. Circulation. 2013;128(11 Suppl 1):S79–86. doi: 10.1161/CIRCULATIONAHA.112.000343. [DOI] [PMC free article] [PubMed] [Google Scholar]