

Abstract
Paclitaxel (PTX) is one of the most successful drugs ever used in cancer chemotherapy, acting against a variety of cancer types. Formulating PTX with Cremophor EL and ethanol (Taxol®) realized its clinical potential, but the formulation falls short of expectations due to side effects such as peripheral neuropathy, hypotension, and hypersensitivity. Abraxane®, the albumin bound PTX, represents a superior replacement of Taxol® that mitigates the side effects associated with Cremophor EL. While Abraxane® is now considered a gold standard in chemotherapy, its 21% response rate leaves much room for further improvement. The quest for safer and more effective cancer treatments has led to the development of a plethora of innovative PTX formulations, many of which are currently undergoing clinical trials. In this context, we review recent development of PTX drug delivery systems and analyze the design principles underpinning each delivery strategy. We chose several representative examples to highlight the opportunities and challenges of polymeric systems, lipid-based formulations, as well as prodrug strategies.
Keywords: paclitaxel, drug delivery, nanomedicine, cancer, chemotherapy
Graphic Abstract
Representative platform technologies for paclitaxel delivery currently under preclinical development.
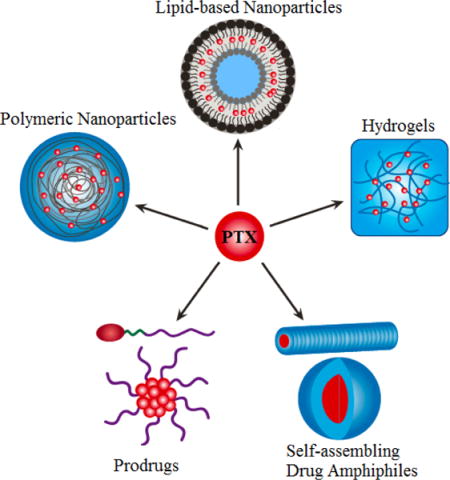
1 Introduction
Paclitaxel (PTX) is a natural compound originally isolated from the Pacific Yew tree by Wall and Wani from Research Triangle Institute in 1967,[1] and is the first taxane used in cancer chemotherapy.[2] Albeit very effective in eradicating cancer cells, the purification of PTX from the tree has been extremely inefficient. It was once estimated that the treatment of one cancer patient would consume eight 100-year-old yew trees. The limitation in PTX resource slowed down the research and clinical translation of PTX. Since the identification of the PTX’s chemical structure in 1971,[1] much effort had been devoted to the total synthesis of PTX. As a result, and the first total synthesis of the drug (overall yield is ca. 4–5%) was achieved by Holton and Nicolaou in 1994.[3–5] However, the relatively low yield prevents the industrial production of PTX through this synthetic approach. Currently, the semi-synthetic method at industrial scale is a combination of the two routes, in which 10-deacetylbaccatin (10-DAB), a relative abundant intermediate extracted from leaves of yew trees, is synthetically converted into PTX. [6]
The key moieties of PTX are composed of four oxetane rings and an ester side chain (Figure 1). For instance, the C3′ amide acyl group in the C13 chain is the active group on the compound, while the hydroxyl group on C2′ enhances its function,[7] both of which are preserved when developing active derivatives of PTX such as docetaxel and cabazitaxel. Further crystallographic study showed that PTX adopts a T-shaped structure when interacting with its target β-tubulin, in a way similar to an extended loop in α-tubulin in α,β-tubulin dimer.[8] The binding of PTX to the β-tubulin located inside of the microtubules stabilizes and promotes polymerization of microtubules,[2] disrupting dynamics of microtubules, which arrests the cells at G2/M phase [9, 10] and triggers apoptosis in proliferating cancer cells.[11] Since cells at G2/M phase are most susceptible to radiation, PTX is also used as a radio sensitizer.[9] With this unique mechanism of action, PTX has shown great potential in inhibiting a broad spectrum of cancer cells of different origins, including ovarian cancer, breast cancer, non-small-cell lung cancer, pancreatic cancer, brain cancer and Kaposi’s sarcoma.[12] Unfortunately, the chemical structure of PTX providing its superior and unique anti-tumor activity also leads to an extremely low water solubility. As such, formulations to improve its water solubility and pharmacokinetics are imperative for PTX’s clinical translation. Over the past two decades, a great diversity of PTX formulations and delivery systems have been developed,[13, 14] among which Cremopor EL solubilized PTX (Taxol®) and albumin stabilized PTX (Abraxane®) are currently in clinical use.[9, 15] Both formulations demonstrated a concentration-dependent pharmacokinetic profile. After intravenous injection, the original formation would dissociate rapidly, resulting in nonspecific free drug distribution throughout the body. This may be accountable for their dose-limiting toxicities. In light of PTX’s superior anti-tumor activity and the unmet needs in cancer chemotherapy, a variety of drug delivery platform technologies have been harnessed to deliver PTX, aiming to achieve more effective cancer chemotherapy. Targeted delivery of PTX is under active development in both academia and industry. One recent review by Allen and coworkers summarized the current industrial interest in the development of PTX formulations that are intended to repeat the clinical successes of Taxol® and Abraxane®. [16] In this review, we focus our discussion on the preclinical development of a number representative PTX delivery systems.
Figure 1.
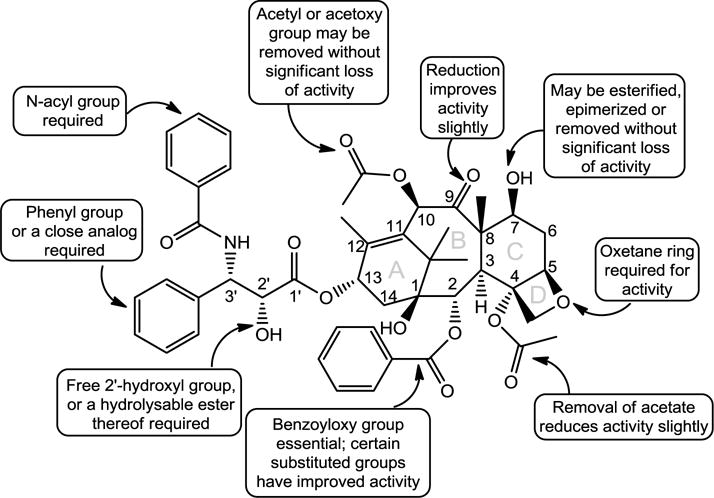
The relationship of PTX’s chemical structure with its bioactivity (Reproduced from ref. [7] with permission).
2 PTX Formulations in Clinic
Taxol® and Abraxane® are the only two PTX-based chemotherapies currently approved by the Food and Drug Administration (FDA) in the United States. Both formulations are intended for intravenous administration but with different formulating agents. PTX is stabilized In Taxol® by the mixture of Cremophor EL and dehydrated ethanol (49.7%, v/v), in contrast to the use of albumin in Abraxane®.[17] In China, liposomal PTX (Lipusu®) has been approved for clinical use.[18, 19]
2.1. Taxol®
Taxol®, marketed by Bristol-Myers-Squibb in 1992, is the first approved formulation of PTX. Each milliliter of the Taxol® formulation contains 6 mg of PTX solubilized by 527 mg Cremophor EL (polyoxyethylated castor oil) and ethanol (49.7%, v/v).[9] Taxol® is typically administrated through 3- or 24-h infusion after dilution with a balanced fluid. The typical pharmacokinetic parameters of Taxol® dosed at 175 mg/m2 after 3 h intravenous fusion are shown in Table 1. As of today, it has been approved to treat primary and metastatic breast cancer, ovarian cancer, non-small cell lung cancer, and AIDS-related Kaposi’s sarcoma. [20]
Table 1.
Comparison of Taxol® and Abraxane®
| Taxol® | Abraxane® | |
|---|---|---|
| Formulation | Cremophor EL and dehydrated ethanol | Albumin NPs |
| Particle size | N/A | ~130 nm |
| MTD | 240 mg/m2 | 300 mg/m2 |
| Clinical Dosage | 175 mg/m2 | 260 mg/m2 |
| Administration | i.v. 3 h | i.v. 30 min |
| Vd (L/m2) | 227 | 632 |
| Clearance (L/h/m2) | 12.2 | 15 |
| Cmax(ng/ml) | 3,650 | 18,741 |
| AUC (h*ng/ml) | 15,007 | 17,434 |
| t1/2 (h) | 20.2 | 27 |
All values acquired from FDA website. MTD: the maximum tolerated dose; Vd: apparent volume of distribution; Cmax: Maximum Concentration; AUC: plasma area under the curve; t1/2: half-life time. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021660s037lbl.pdf https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf
One major shortcoming in the clinical application of Taxol® is its non-linear pharmacokinetics and hypersensitivity, in addition to the nephrotoxicity and neurotoxicity induced by PTX.[9] For instance, when 135 or 175 mg/m2 of Taxol® was administrated through 3 h infusion, 30% increase in the dosage can actually lead to an increase of 68% in Cmax and 89% in AUC0–∞. In way to alleviate severe hypersensitivity reactions, patients treated with Taxol® should be premeditated with corticosteroids, diphenhydramine and H2 antagonists. These issues are believed to be rooted in the use of Cremophor EL: 1) Cremophor EL forms micelles in blood at high concentrations, and traps PTX in the hydrophobic core, resulting in the non-proportional increase of AUC0–∞,[21] and 2) Cremophor EL can elicit complement system activation and release of histamine at high dosages.[22, 23] To minimize the Cremophor EL-rooted side effects, prolonged infusion is often necessary. These limitations had motivated researchers and manufacturers to seek Cremophor EL-free formulations of PTX.
2.2. Abraxane®
Abraxane® was developed by Abraxis BioScience (later obtained by Celgene) and is the second PTX formulation used in clinic. Abraxane® received approval by FDA in 2005 initially for the treatment of metastatic breast cancer, and then for locally advanced or metastatic non-small cell lung cancer, and metastatic adenocarcinoma of the pancreas.[24] In Abraxane®, PTX is formulated with human serum albumin (HAS).[25] HAS is the most abundant plasma protein in the human blood with a half-life of ~19 days. It can reversibly bind hydrophobic chemical substances, transport them in the body and release them at cell surface. [26] In addition, HAS plays several important biological roles in cellular uptake and transcytosis through binding with gp60, and in binding with secreted protein acidic and rich in cysteine (SPARC) that are known to be highly expressed in malignant cells and stromal cells associated with neoplasia. However, it remains elusive how much the biological roles of HAS actually contribute to the improved response rates of Abraxane®. One indisputable fact is that the removal of Cremphor EL permits a higher dose of PTX to be administrated with comparable toxicities. In comparison with Taxol®, the maximum tolerated dose of Abraxane® can be increased to 300 mg/m2, and also allowing for a much shortened administration time (30 min for Abraxane® versus 3 or 24 hr for Taxol®).[11] In addition, the results from Abraxis BioSciences suggested that Abraxane® has a predictable, linear pharmacokinetic profile, and increases intratumor PTX concentration by 33% when compared to equal dose injection of Taxol®. Removing Cremophor EL also eliminates solvent-related hypersensitivity reactions, as well as the medical needs for premedication and special IV tubing for administrating Taxol®. More importantly, Abraxane® showed improved pharmacokinetic profiles (Table 1), improved tolerance, and an increase in overall response rate (ORR). The Phase III clinical trial of Abraxane® directly compared its antitumour activity and tolerability to those of Taxol® in women with measurable metastatic breast cancer. [27] In this designed trial, patients received 3-week cycles of either Abraxane® 260 mg/m2 i.v. over 30 min or Taxol® 175 mg/m2 i.v. over 3 h. The ORR for Abraxane® was almost twice that of Taxol®. Despite the 49% higher PTX dose in the Abraxane® group, the incidence of Grade 4 neutropenia was significantly lower with Abraxane®.
2.3. Other Approved Formulations
Lipusu® developed by Luye Pharmaceutical Co. Ltd. represents the first PTX liposome injection that came onto the China clinical market in 2003. The formulations are composed of PTX, lecithin and cholesterol, and have been approved to treat ovarian cancer, breast cancer and non-small cell lung cancer. Compared to Taxol®, Lipusu® showed comparable activities against breast, gastric cancer and non-small cell lung cancers, but with significantly lower side effects. [28, 29] Hence, Lipusu® could be a useful alternative to Abraxane®. Genexol-PM® is a poly(ethylene glycol)-b-poly(lactic acid) (PEG-b-PLA) block copolymers based formulation of PTX commercialized by Samyang Corporation in 2007. It has been approved in South Korea for the treatment of breast, ovarian and non-small cell lung cancers. Clinical tests revealed that Genexol-PM has dose-dependent pharmacokinetics and is well tolerated by the patients, in particular for those with metastatic breast or advanced pancreatic cancer[30–33].
3 Lipid-Based Nanoparticles
Lipid-based drug-delivery systems have evolved from micro- to nano-scale. Because of their long history being used as drug delivery materials, many lipid-based formulations have been explored for PTX delivery. Liposomal formulations and solid-core nanoparticles are the two most widely utilized lipid-based nanoparticles (Figure 2A), and could be further modified to improve their therapeutic outcomes through the production of long-circulating, tissue-targeted and/or pH-sensitive nanoparticles. More recently, solid lipid nanoparticles have also been engineered to reduce toxicity toward mammalian cells.
Figure 2.
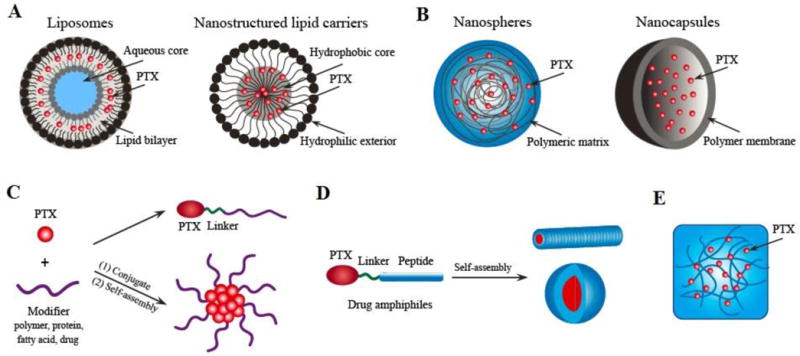
Schematic illustration of several representative platform technologies for target PTX delivery. (A) Lipid-based delivery systems; (B) Polymer-based nanoparticle systems; (C) Polymer-drug conjugates; (D) Self-assembling drug amphiphiles; (E) Hydrogel system for local delivery.
3.1. Liposomes
Liposomes are spherical vesicles with a membrane composed of single or multiple concentric phospholipid bilayers, capable of encapsulating hydrophilic drug in their aqueous core and lipophilic drugs within the bilayer membrane. Liposomal anticancer drugs (such as Doxil® and Dauoxome®) were the first nano-based formulations approved for cancer therapy by the FDA.[34] Liposomes are chosen as carriers for PTX because they can increase solubility and modify pharmacokinetics of PTX to improve its efficacy and also because the excipients are clinically approved. The first PTX liposome injection, Lipusu®, has been put into clinical practice in China for treatment of breast cancer, non-small-cell lung cancer and other malignant tumors.[10] It retained the growth-inhibitory activity of the free drug while significantly reducing the side effects. Another PTX-loaded liposome delivery system, LEP-ETU, was composed of 1,2-Dioleoyl-snglycero-3-phosphocholine (DOPC), cholesterol and cardiolipinin of a 90:5:5 molar ratio.[35] Compared with Taxol®, the plasma pharmacokinetics of LEP-ETU was not much different, but LEP-ETU can be administered rather safely at higher doses, as evidenced by a Phase I study.[36]
The major problems associated with liposomes are their limited long-term stability. Although liposomes can be used to deliver cytotoxic compounds to selected tissues, they can be rapidly cleared by the mononuclear phagocytic system in liver and spleen, unless special modifications, such as PEGylation, are made to the phospholipid surface.[37] PEGylation can increase the average time of liposome circulation within the bloodstream by 10-fold.[38] This could lead to improvement of the half-life and therapeutic outcomes of PTX. Ceruti et al. compared conventional liposome formulations of PTX with the stealth-long circulating liposomes. The PEGylated liposomes demonstrated longer elimination half-life of nearly 50 hr, in contrast to the <10 hr half-life of conventional liposomes.[37] Similar results were observed by Yoshizawa and co-workers when comparing their PEGylated liposomes with conventional ones, resulting in higher intratumoral PTX accumulation and improved antitumor efficacy in colon-26 solid tumor-bearing mice.[39] However, some recent studies indicated that as a result of frequent use of PEG in cosmetics products or multi-dosing of PEGylated therapeutics, some PEGylated liposomes may also been rapidly cleared from blood upon repeated administration.[40] This ‘accelerated blood clearance’ phenomenon was cause by the formation of PEG-specific IgM antibody as a result of repeated dosing, causing the elimination of subsequent doses of PEGylated agents from the circulation.[41] Therefore, the immune response to PEGylated liposomes may also adversely affect their pharmacokinetic and biodistribution profiles.
In theory, the efficacy of PEGylated liposomes could be further enhanced via active targeting strategies.[42, 43] PTX liposomal targeting can be achieved by covalently attaching tissue-specific antibodies,[44] proteins,[45] or peptides[46] to liposome surfaces. One interesting targeting strategy is the incorporation of multifunctional RRRRRRRR-c(RGDfK) (R8-RGD) peptide, which has been applied to the surface of PTX loaded liposomes.[43] Covalently incorporating R8-RGD onto liposomes resulted in increased cellular uptake by 2-fold or nearly 30-fold compared to R8 or RGD decorated ones, respectively. The resultant liposomes displayed effective penetration of three-dimensional glioma spheroids and BBB model in vitro. As a result, the PTX-loaded R8-RGD modified liposomes (PTX-R8-RGD-lipo) induced the strongest inhibition and apoptosis of C6 cells, and achieved the longest survival in intracranial C6 glioma bearing mice. Aside from cancer cell targeting, subcellular organelle targeting can also improve the efficacy of PTX. For example, Torchilin and coworkers incorporated triphenylphosphonium (TPP) into their liposomes and developed a novel mitochondria targeting polyethylene glycol-phosphatidylethanolamine liposome (TPP-PEG-L) for PTX delivery.[42] This TPP-PEG-L demonstrated efficient mitochondrial targeting in cancer cells, as shown by confocal microscopy in co-localization experiments with stained mitochondria (Figure 3). More importantly, PTX-loaded TPP-PEG-L demonstrated enhanced PTX-induced cytotoxicity and anti-tumor efficacy in vitro and in vivo, compared to plain liposomes (PL). These results suggested that targeting moiety decoration can improve the tumor inhibition activity of PEGylated liposome. It should be noted, however, that although the efficiency of targeting could improve cellular uptake, additional modifications may be required to ensure the effective release of the drug within the cytoplasm.
Figure 3.
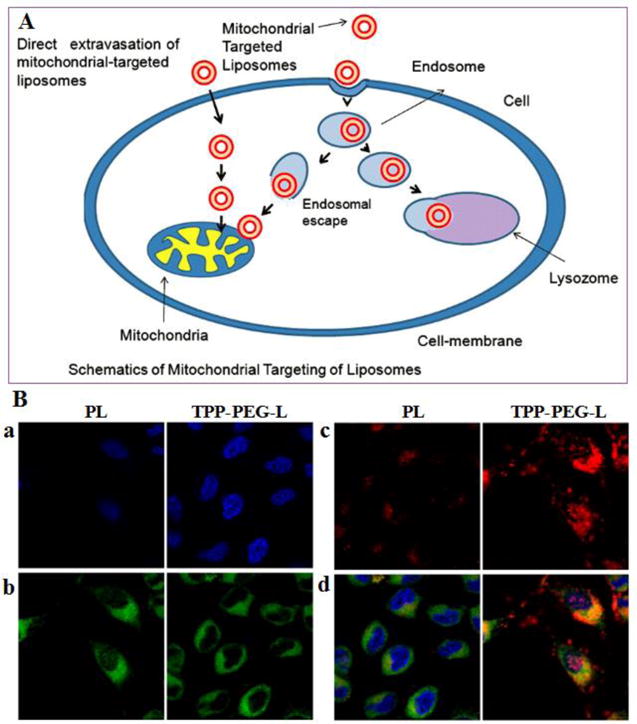
(A) Schematic illustration of mitochondrial targeting of TPP-PEG-L. (B) Mitochondrial colocalization of fluorescently-labeled TPP-PEG-L compared to PL by confocal laser scanning microscopy. HeLa cells were incubated with Rh-PE-labeled TPP-PEG-L and PL for 18 h and then stained with MTG and Hoechst 33342. Yellow spots in the merged pictures denote the co-localization of the liposomes within mitochondrial compartments. a. Images of nuclei stained with Hoechst; b. Mitochondria staining with MTG; c. Uptake of Rh-labeled liposomes; d. Merged picture of all. (Reproduced from ref. [42] with permission)
Liposomal formulations can be degraded by lysozymes within the cellular matrix before the release of the liposome’s therapeutic contents to the cytoplasm [47]. In order to increase the probability that a drug is released into the cytoplasm prior to its degradation, the formulation should be quickly destabilized after endocytosis [48]. This can be achieved by incorporating into liposomes a pH-sensitive lipid, which destabilizes the liposomes at the endosomal pH (pH <6), and induces the release of entrapped drugs in cytosol.[49, 50] For example, Chen et al. prepared a PTX-loaded pH-sensitive liposomes using a hydrazone (Hz)-based pH-sensitive methoxy(polyethylene glycol)-cholesterol (mPEG-Hz-Chol) conjugates.[51] The mPEG2000-Hz-Chol modified pH-sensitive liposomes showed greater accumulative release of PTX at pH 5.5 than that at 7.4. Notably, the delivery of PTX by pH-sensitive liposomes demonstrated more efficient tumor suppression in BALB/c mice when compared to conventional liposomes.[52] Similar to PEGylated liposomes, pH-sensitive liposomes can also be targeted to specific cells and tissues. For instance, the inclusion of a targeting cell-penetrating peptides (CPPs) resulted in high pH-sensitive liposomes binding to B16F10 cells.[53] At a concentration of 20 μg/mL, the killing activity of PTX-loaded TR-Lip (PTX-TR-Lip) against B16F10 cells was 1.80- and 1.15-time higher than that of PTX-loaded PEG-liposomes and free PTX at pH 6.5, respectively. In B16F10 tumor-bearing mice, PTX-TR-Lip showed higher survival rate. This study revealed the importance of combining different strategies to produce multifunctional liposomal formulations.
3.2. Lipid Micelles
The component of lipid micelles are usually low-molecular-weight surfactants. Lipid micelles have a core-shell structure with hydrophilic heads facing the outside aqueous environment and lipophilic tails forming the inner core for drug encapsulation. They are formed spontaneously within aqueous medium above a critical micelle concentration.[54] The lipophilic region of the micelle efficiently encapsulates molecules of poor water solubility, whilst protecting them from degradation by extracellular elements.[55] The properties of lipid micelles are largely determined by the chemical structure of the lipid compounds, which can be tailored through chemical modifications in both the hydrophobic and hydrophilic regions. Similar to liposomes, modifications to the lipid’s hydrophilic region include the addition of polymers and targeting ligands. The addition of PEGylated lipids to the hydrophilic region resulted in long circulating time, due to the steric inhibition of protein binding and decoration.[56]
Recently, a sterically stabilized phospholipid micelles (SSMs) composed of PEG(2000)-grafted distearoyl phosphatidylethanolamine (PEG(2000)-DSPE) was developed as carriers for PTX.[57] This long acyl chains of DSPE-PEG can create a large hydrophobic inner core to entrap PTX. To further increase the hydrophobic space for PTX, another phospholipid, egg phosphatidylcholine (ePC) was incorporated to form sterically stabilized mixed micelles (SSMM).[58] The PTX-loaded SSM and SSMM possess mean particle sizes of 15 ± 1 nm and 13.1 ± 1.1 nm, respectively. In these constructs, SSMM solubilized 1.5-times more PTX than SSM at the same total lipid concentration. This is mainly due to the increased volume of the hydrophobic region of the micelle by the inserted hydrophobic egg phosphatidylcholine. Furthermore, Asn-Gly-Arg (NGR) peptide was covalently bonded to the distal end of PEG chains to produce brain tumor targeted micelle [59]. This NGR peptide-SSM micelle showed superior cellular uptake over passive targeting micelle in murine brain microvascular endothelial cells via receptor-mediated endocytosis, resulting in improved efficacy against C6 glioma both in vitro and in vivo. Collectively, these results suggest that the SSM or SSMM could serve as effective carriers for PTX delivery.
3.3. Solid Lipid Nanoparticles
A majority of the lipids used for the preparation of liposomes and lipid micelles are of low phase transition temperature, which are in their liquid phase and would facilitate leakage of PTX at body temperature. Solid Lipid Nanoparticles (SLNs) are primarily made up of solid lipids, which can be highly purified triglycerides, complex glyceride mixtures or even waxes.[60] A large pool of solid lipids (mono-, di- and tri-glycerides, lipidacids, phospholipids, wax, etc.) and surfactants are available for SLN engineering. Among these excipients, some lipids (e.g., glycerides, phospholipids) and surfactants (e.g., Tween 80, lecithin, Poloxamer188, sodium glycocholate) suitable for i.v. injection, which makes SLN a versatile platform for drug delivery ready for clinical translation.[61] SLNs have several advantages, including ease of preparation and scale-up with low cost, good physical stability, controlled drug release, biocompatibility, low toxicity and versatile chemistry.
Choosing a lipid with high drug solubility and miscibility is a prerequisite for the preparation of SLN with high PTX loading and slow drug release.[34, 62] More importantly, PTX-loaded SLNs composed of different lipid materials may result in different cellular uptake and cytotoxicity effect. Studies demonstrated that the cellular uptake of SLNs was time- and concentration-dependent, and was associated with the lipid material melting point, the length of the hydrocarbon chain, as well as the particle size. Yuan et al. evaluated the cellular uptake of several SLNs composed of different lipid materials including monostearin, stearic acid, glycerol tristearate and ATO888.[63] Cellular uptake studies were performed in A549 cells, leading to internalization in the order of glycerol tristearate>monostearin>stearic acid>ATO 888. This can be explained by differences in affinity between fatty acids and cell membrane. Moreover, the PEGylated stearic acid SLN showed the highest cellular uptake among the materials tested. PTX loaded in these SLNs showed 1.6- to 10-fold higher cytotoxicity relative to free PTX. SLN was also used as drug formulations to overcome P-gp-mediated multidrug resistance (MDR). PTX-loaded SLN exhibited 9-fold lower IC50 values in NCI/ADR-RES cells (P-gp overexpressing human breast cancer cells) than those of free PTX. The SLNs inhibited P-gp activity and transiently depleted ATP as demonstrated from relative calcein acetoxymethyl ester and ATP assays. This might be related to the use of P-gp regulating surfactant during the preparation of SLN. In addition, it is also believed that the efficacy of SLNs against MDR cancer could be further improved by delivering P-gp substrates.[64]
Similar to other nanoparticle design strategies, the long-circulation of SLNs can be achieved by modifying the particle surface with some chemical moieties to evade reticuloendothelial system (RES) clearance[34]. Chen et al. developed two types of long-circulating SLNs as PTX carriers with Brij 78 as surfactant in one formulation (Brij78-SLN) and Poloxamer F68 and DSPE-PEG2000 in the other (F68-SLN).[65] The Brij78-SLN and F68-SLN were found to extend the half-life of circulated PTX up to 10.06 and 4.88 h, respectively, compared to 1.36 h of Taxol®. The longer PEG chain of DSPE-PEG2000 (MW 2000) compared to Brij 78 (MW 1200) may be responsible for the extended circulation and higher AUC of F68-SLN. In addition to long-circulation time, SLNs also have the ability to be targeted to specific organs, tissues or cells through the addition of targeting ligands. For example, in a folate receptor overexpressing A549 cell line, PTX-loaded SLNs modified with folic acid-stearic acid enhanced the cellular uptake and cytotoxicity 2- and 8.8-fold compared to non-targeted SLNs, respectively. Pandey et al. designed SLNs using a lactoferrin (Lf) moiety to target Lf receptors.[66] In ex vivo efficacy studies, the targeted SLNs demonstrated a superior anticancer activity than that of plain SLNs and free PTX. These studies suggested that targeted SLNs could be used as potential carrier for delivering PTX to tumor sites.
4 Polymeric Nanoparticles
Many of the nanoparticle therapeutics in clinical and preclinical evaluations are polymeric nanoparticles. Biodegradable polymeric nanoparticles are highly preferred partially because they are shown to be biocompatible and demonstrate controlled/sustained release of incorporated therapeutic agents,[67] and partially because they can provide large space for the deposition and delivery of various molecules such as drugs, proteins, peptides, or nucleic acids.[68] Compared with lipid materials, polymeric materials are superior for their diverse functionality and robustness in construction. In this section, we discuss several types of PTX-loaded polymeric nanoparticles, such as poly(lactic-co-glycolic acid) (PLGA), poly(lactide) (PLA), poly(ε-caprolactone) (PCL) and chitosan. In these polymeric constructs, the anticancer drug PTX is either bound to surface of nanosphere or encapsulated inside (Figure 2B).
4.1. Synthetic Polymer Nanoparticles
4.1.1 PLGA Nanoparticles
PLGA, a FDA approved polymer,[69] is one of the most successful polymers used for the development of nanomedicines and related biomaterials, because it can hydrolyze into lactic and glycolic acids in the body, the rate of which can be tuned by varying the molecular weights and composition of the polymer. As a result, PLGA showed minimal systemic toxicity, and the release rate of the encapsulated drug can be precisely tailored. PTX-loaded PLGA nanoparticles have been mostly prepared by emulsion solvent evaporation,[70] nanoprecipitation,[71] and interfacial deposition methods.[72] In most cases, PTX was released from PLGA nanoparticles in a biphasic pattern with a fast initial release during the first 1–3 days followed by a slow and continuous release. [70, 72] It has also been demonstrated that incorporation of PTX in the PLGA nanoparticles strongly enhances its anti-tumoral efficacy, as compared to Taxol®. [71, 73]
It is well known that the employed emulsifiers could have strong influence on the properties of produced nanoparticles, such as morphology, size, drug entrapment efficiency, and release behavior, so as to impact their cellular uptake, cytotoxicity, pharmacokinetics, and eventually the therapeutic efficacy.[74] Win et al. conducted a full spectrum of research on tocopheryl PEG succinate (TPGS) emulsified PLGA nanoparticles for PTX formulation to improve its therapeutic index.[75] PTX-loaded, TPGS-emulsified PLGA nanoparticles had a relatively uniform size of approximately 240 nm in diameter. The AUC0-48 for TPGS emulsified PLGA nanoparticles formulation of PTX were found to be three times larger than that of the Taxol®. PTX formulated in the TPGS-emulsified PLGA nanoparticles could also result in a much longer half-life of the drug in the plasma. More importantly, both in vitro and in vivo therapeutic effects of PTX formulated in the nanoparticles were found to be much higher than those of Taxol®. In addition to TPGS, montmorillonite (MMT),[76] 1,2-dilauroylphosphatidylcholine (DLPC),[77] and dipalmitoyl-phosphatidylcholine (DPPC) [78] were also used as emulsifiers in PLGA NPs. These formulations demonstrated greater drug encapsulation efficiency, better controlled drug release kinetics, and enhanced cellular uptake and cytotoxicity.
Rapid opsonization is a major hurdle to achieve effective drug targeting to the site of action by PLGA nanoparticles. To address these limitations, several methods of surface modifications have been developed to produce nanoparticles that can evade opsonization. PEG can form a hydrated outer shell, thereby protecting the nanoparticles from being quickly taken up by the RES, thus extending the half-life of drugs and their tissue distribution.[76, 79] In view of this, diblock or triblock copolymers of PLGA-b-PEG and PLGA-b-PEG-b-PLGA have been synthesized and used for PTX delivery.[80–83] For example, Fabienne et al. prepared PTX-loaded PLGA-b-PEG nanoparticles by simple emulsion and nanoprecipitation.[71] This nanoparticle exhibited high drug loading and a biphasic pattern drug release behavior. When exposed to 25 μg/ml of PTX, the cell viability was lower for PTX-loaded nanoparticles than for Taxol® (IC50 5.5 vs 15.5 μg/ml). PTX-loaded nanoparticles showed greater tumor growth inhibition effect on TLT tumor. Further modification with the RGD peptide enabled targeting of the encapsulated PTX to endothelial cells in the tumor, revealing improved efficacy in inhibiting the growth of tumor and prolonging the survival of animals.[84] In another case, PTX-loaded PLGA-b-PEG-b-PLGA nanoparticles exhibited significantly higher anticancer effects than Taxol®. [85] Targeted delivery of PTX-loaded PLGA-b-PEG NPs was also achieved by surface decoration with epidermal growth factor receptor (EGFR).[86] These targeted nanoparticles enhanced the pharmacokinetics relative to Taxol®, and also improved the plasma and tumor half-life and mean residence time of the drugs. Other targeting ligands, such as folate,[87] RGD,[88] transferrin[89] and aptamer[80, 90] have also been conjugated to PTX-encapsulated PLGA NPs for better antitumor efficacy.
4.1.2 PLA Nanoparticles
PLA has been widely used as matrix material for polymeric NP preparation because of its biodegradability and safe properties.[91] In the context of PTX delivery, the Feng Lab developed the mPEG–b–PLA NPs of 80–90 nm in diameter for controlled delivery of PTX.[92] PTX delivered by the resultant nanoparticles showed 33.3-fold higher activity against MCF-7 cells when compared with Taxol®, with 3.1- and 2.8-fold increase in the AUC and half-life, respectively.[93] These preclinical studies led to human clinical trials and the eventual approval of Genexol-PM, a PTX-loaded PLA-PEG-based micelle, in South Korea.
As an alternative to PEG, the TGPS has been investigated by various groups [94]. TPGS-based nanomedicine of PTX has exhibited increased water solubility and oral bioavailability of PTX. In addition, TPGS was able to overcome P-gp-mediated MDR in cancer cells and effectively inhibit the growth of cancer cells. To utilize the antitumor efficacy and long circulation properties of TPGS, TPGS-b-PLA copolymers were synthesized for PTX delivery. The PLA-TPGS NPs exhibited enhanced cytotoxicity compared to conventional polyvinylalcohol (PVA)-emulsified PLGA NPs,[95] and achieved a 27.4- and 1.6-fold greater half-life and AUC, respectively, relative to Taxol® [96] After surface decoration with folic acid, the efficacy of this NP construct was further improved.[97]
In addition to PEG and TPGS modified PLA polymers, copolymers of PLA-b-PEG-b-PLA,[98] PEG-b-PLA-b-PEG,[99] PLA/Tween 80,[100] Poly(γ-glutamic acid)-poly(lactide) (PGA-b-PLA),[101] and poly[(amine-ester)-co-(d,l-lactide)] (HPAE-co-PLA)[102] were also used for PTX delivery. Moreover, PTX-loaded PLA NPs decorated with targeting or functional molecules, such as HER2,[103] biotin,[104] galactosamine,[101] RGD,[105] and CDX [106] have been reported to greatly improve efficacy both in vitro and in vivo. These reports showed that the PLA based NP formulation can have higher therapeutic effects and lower side effects than Taxol®, revealing great potentials in various biomedical applications.
4.1.3 PCL Nanoparticles
Poly(ε-caprolactone) (PCL) is a homopolymer of 6-hydroxycaproic acid, and can be hydrolyzed under physiological conditions. It was approved by the FDA as materials for drug delivery device. Because of the high crystallinity and strong hydrophobicity, PCL degrades more slowly than PLGA and PLA, and tends to aggregate in aqueous solution. To address these issues, surface modification of the polymer with PEG chains has been developed [108]. Wang and colleagues developed a PTX-loaded mPEG-PCL NPs with a high drug loading (25.6%, w/w) with preferential accumulation in tumor when compared to Taxol®.[109] Discher and colleagues explored the shape effect of filaments formed by self-assembly of PEG-b-PCL (Figure 4).[107] Significantly, they found that filaments showed much prolonged circulation after intravenous injection due to their capability to evade macrophage clearance. Thus, more PTX was delivered into the tumor, the growth of which was greatly retarded when compared with those treated with Taxol®. In another example, the Fang’s group prepared PTX-loaded mPEG-b-PCL NPs for the treatment of brain tumor, and prolonged survival of glioma bearing mice (28 days vs. 20 days) was observed, which was further enhanced after surface modification of the nanoparticles with Angiopep, a peptide targeting the low-density lipoprotein receptor-related proteins over-expressed on blood brain barrier (BBB).[110, 111]
Figure 4.
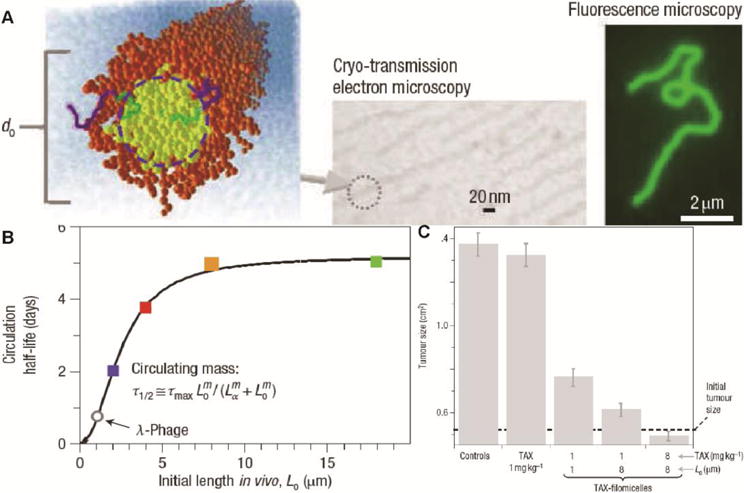
Filamentous micelles of block copolymers for PTX delivery. (A) Characterization of PEG-PCL filamentous micelles. (B) Length-dependent change in the circulation half-life of the filamentous micelles. (C) Tumor growth inhibition effect of Taxol and PTX-loaded filamentous micelles. (Reproduced from ref. [107] with permission)
In addition to PEG-modified PCL NPs, other PCL-based polymers, such as PCL-F68,[112] PCL-PVP,[113] PCL-polyphosphoester,[114] and PCL-PEG-PCL [115], have also been used for PTX delivery. For example, poloxamers was conjugated to PCL because of its capability of sensitizing MDR tumors. Taking advantage of this property, Zhang et al. developed a PTX-loaded PCL/Poloxamer 188.[112] The developed PTX-loaded PCL/Poloxamer188 NPs showed increased cellular uptake and toxicity when compared to PCL NPs and Taxol® in resistant MCF-7/TAX cells. These studies suggested that PTX-loaded, PCL-based delivery systems could afford an effective platform to enhance the cytotoxicity of PTX and to overcome drug resistance in ovarian cancer patients.
Recent effects have also been devoted to biomimetic strategies. One highly innovative design is to coat polymeric nanoparticle with cell membranes by Zhang and colleagues.[116] Li et al. recently developed cell membrane coated PTX-loaded PCL nanoparticles for the treatment of metastatic breast cancer.[117, 118] In these nanoparticles, PCL served as reservoir for PTX, while the cell membranes endowed the nanomedicine with camouflage properties such as long circulation and tumor targeting. Increased accumulation in both primary tumor and their metastasis sites was achieved, leading to greatly improved efficacy, again when compared to Taxol®. The primary challenge for clinical translation of this very promising strategy is that the field is still at its early stages and most studies to date have been performed in small animals. Translation of the results from small animals to larger animals and eventually to humans are not trivial and requires experimental demonstration. The effects of removing cells, modifying them and subsequently reintroducing them can cause unpredictable effects and reproducibility. Scaling up manufacturing can be a significant hurdle as well. [119][120]
4.2. Natural Polymer Nanoparticles
4.2.1 Chitosan Nanoparticles
Chitosan is produced by deacetylation of chitin, the second most abundant natural polysaccharide. Chitosan possesses a number of interesting properties such as biocompatibility, biodegradability, nontoxicity and bioadhesivity, warranting its high potential as an important class of biomaterials.[121, 122] However, the poor solubility of chitosan in biological solution (pH 7.4) is a major drawback for its extended application in drug delivery systems. Over the past decade, a variety of chitosan derivatives have been developed by introducing hydrophilic and/or hydrophobic groups to the chitosan backbone.[122, 123] The resultant chitosan-based amphiphiles can assemble into micellar structures in water as the delivery vehicles for antitumor drugs.
Water-soluble chitosan derivatives could be prepared by various approaches such as quaternization of the amino group, carboxymethylation, and PEGylation. In particular, glycol chitosan is emerging as drug carriers because of their solubility and biocompatibility in vivo.[124, 125] With hydrophobic moiety derived glycol chitosan (HGC), self-assembled nanoparticles were prepared as a carrier for PTX by the Kwon’s lab.[126] The HGC conjugates formed nano-sized particles with a diameter of 200 nm and PTX was efficiently loaded into HGC nanoparticles up to 10 wt.% using a dialysis method. These PTX-HGC nanoparticles showed sustained release with 80% of the loaded PTX released in 8 days at 37 °C in PBS. The anti-tumor effect of PTX-HGC nanoparticles was similar to that of Taxol®, but the former were much less toxic. A hydrotropic oligomer-glycol chitosan (HO-GC) was further synthesized by chemical conjugation of the N,N-diethylnicotinamide-based oligomer to the backbone of glycol chitosan uniquely designed for enhancing the aqueous solubility of PTX (Figure 5). Due to the presence of hydrotropic inner cores, this HO-GC nanoparticle could encapsulate a high quantity (24wt.%) of PTX with a maximum drug-loading efficiency of 78%. This PTX nanoparticle also showed an excellent tumor specificity in MDA-MB-231 tumor-bearing mice due to the enhanced permeability and retention (EPR) effect.[127] In another case, a novel water-soluble chitosan derivative, N-octyl-O-sulfate chitosan (OSC), was synthesized and used for PTX delivery [128]. Although the PTX-loaded micelle based on OSC (PTX-OSC) showed similar antitumor efficacy as Taxol® and significantly reduced the side effect of PTX, the AUC of PTX-OSC was 3.6-fold lower than that of Taxol® and most of the PTX were distributed in the RES. To prolong circulation time and minimize non-specific uptake, mPEG group was introduced into the 2-NH2 of OSC.[129] Compared with OSC, mPEGOSC decreased the adsorption of plasma protein to micelle, resulting in a 2.93-fold increase in the AUC and uterus accumulation compared with PTX-OSC and Taxol®.
Figure 5.
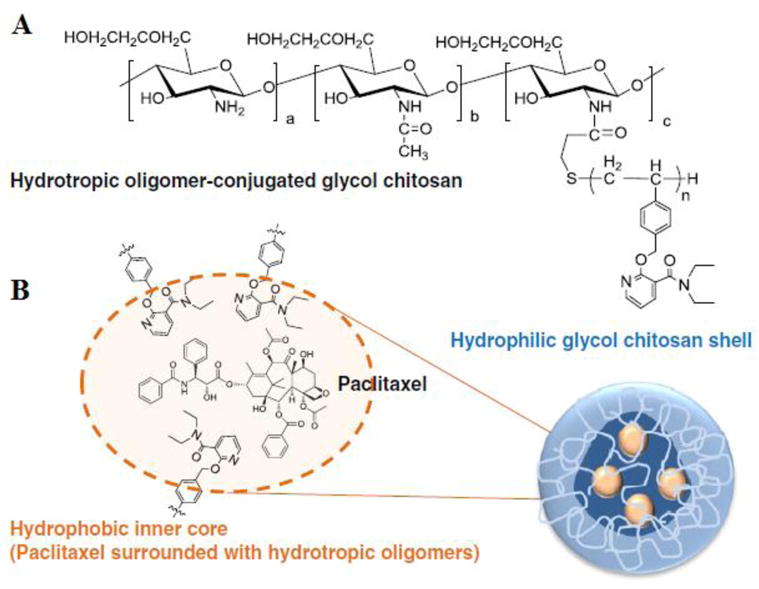
Hydrotropic oligomer-conjugated glycol chitosan (HO-GC) nanoparticles. (A) Chemical structure of HO-CN. (B) Amphiphilic self-assembly of PTX-encapsulated (PTX-HO-GC) nanoparticles. (Reproduced from Ref. [127] with permission)
Hydrophobic derivatives of chitosan such as alkylated chitosan [130] and deoxycholic acid-modified chitosan[121] have been explored as carriers for PTX due to their amphiphilic property and self-assembly nature. For example, TPGS modified chitosan (CS-TOS) was synthesized and used as a micelle system for PTX delivery. This PTX-loaded micelle was about 78 nm with a drug loading of 7.7%.[131] By simply conjugating hydrophobic long alkyl chains to low molecular weight chitosan (LMWC), Zhang et al. developed a novel amphiphilic derivative, N-octyl-N,O-carboxymethyl LMWC (OC-LMWC)[132]. The self-assembled OC-LMWC micelles exhibited very high PTX loading content of 32.17% (w/w), and also the PTX-loaded micelles showed comparable cytotoxic effect to that of Taxol®, with much lower vehicle derived toxicity. Recently, a dual-grafted amphiphilic chitosan derivatives were synthesized by Wang et al to deliver PTX.[111] In their design, O-carboxymethylated chitosan (OCMC) was modified with deoxycholic acid (DOCA), then covalently bound with FA to develop a cancer-targeted PTX delivery system (DOMC-FA). Incorporation of DOCA into the OCMC induced the self-assembly of micelles, and PTX could be physically encapsulated into the core of the micelles. The superior features of this PTX-loaded DOMC-FA (DOMC-FA/PTX) over other polymeric micelles were its high drug-loading efficiency (33.61%) and high encapsulation efficiency (84.43%). More importantly, the cytotoxicity of the DOMC-FA/PTX to cancer cells was much higher than micelles without folate (DOMC/PTX) or Taxol®.[111, 133]
4.2.2 Hyaluronic Acid Nanoparticles
Hyaluronic acid (HA), also known as hyaluronan, is a naturally occurring polysaccharide present in the extracellular matrix and synovial fluids. Owing to its biocompatibility and biodegradability, HA has been extensively investigated for biomedical applications. In particular, since HA can specifically bind to various cancer cells that over-express CD44, many studies have focused on the pharmaceutical applications of HA for anti-cancer therapeutics.[134, 135] Most HA-drug conjugates have been developed for cancer chemotherapy as macromolecular prodrugs.[136] HA has also been conjugated onto various drug-loaded nanoparticles for use as a targeting moiety.[135, 137] These HA NPs containing anti-cancer agents such as PTX [134] exhibited enhanced targeting ability to the tumor and higher therapeutic efficacy compared to the free anti-cancer agents. One kind of modified HA was synthesized by using amine-terminated hydrotropic oligomer, which enhanced PTX solubility and achieved the drug loading up to 20.7%. The NPs were found to selectively target SCC-7 cells which over-expressed CD44, and exhibited enhanced cytotoxicity in this cell line than normal fibroblast CV-1 cells.[138] Recently, a targeted intracellular delivery system of PTX was developed based on redox sensitive HA-SS-DOCA conjugates.[139] The conjugates self-assembled into nano-sized micelles in aqueous media and exhibited excellent drug-loading capacities (34.1%) and entrapment efficiency (93.2%) for PTX. Intracellular release of fluorescent probe Nile red implied that HA-SS-DOCA micelles could provide an effective approach for transport of cargo into the cytoplasm. In vivo studies confirmed that HA-SS-DOCA micelles have much higher tumor targeting capacity than the insensitive control.
5. Prodrug Strategies
Prodrugs are bioreversible derivatives of drug molecules that undergo an enzymatic and/or chemical transformation in vivo to release the active parent drug, which can then exert the desired pharmacological effect.[140] Prodrugs are usually designed to overcome the problems of the parent drug in administration, such as low solubility, low permeability, instability, poor oral absorption, toxicity and non-targeting.[141, 142] Compared to free drug-loaded liposomes, micelles, NPs and other types of formulations, prodrugs are formulated by chemical linkage, with better quality control and small batch-to-batch variation. Furthermore, some polymer- or lipid-based prodrug can self-assemble into well-defined nanoparticles. The PTX prodrugs were generally constructed at C-2′ or 7-OH group of PTX (Figure 1). Compared to C-7-position, 2′ moiety of PTX is more practical for cleaving to keep cytotoxic effects and can be synthesized without protecting the 7-OH position.[142] For conjugation, the 2′-active OH group of PTX was usually first carboxylated by reaction with succinic anhydride, and the formed PTX-2′-hemisuccinate will be conjugated with auxiliary segments (such as polymers, proteins, peptides et al.) (Figure 2C and D) through subsequently reaction between -COOH and -NH2 groups.
5.1. Polymer-based Prodrugs
Pioneering work on the conjugation of hydrophobic drugs to a hydrophilic polymer was initially intended to take advantage of the relatively larger size of the polymer (Rg: radius of fyration) to avoid quick renal clearance and to improve the drug’s water solubility, thus leading to enhanced tumor accumulation.[143] Although such polymer-drug conjugates were widely investigated, only a few polymers, such as PLA,[144, 145] PEG,[146] N-(2-hydroxypropyl)methacrylamide (HPMA) [147] and poly(L-glutamic acid) (PGA) [148] have been systemically examined or have entered clinical trials. In a very innovative approach, Cheng and co-workers used PTX as a drug initiator to formulate PTX-PLA conjugates via living polymerization.[149] Due to the steric hindrance and bulkymetal catalyst, polymerization of LA was preferentially initiated at the 2′-OH group of PTX. These PLA-drug conjugates can be further formulated using the nanoprecipitation method to form uniform particles of less than ~150 nm in size. A water-soluble PEGylated PTX prodrug, conjugated with PEG (MW 5000) through a spacer-succinyl group tethered to the 2′ position of PTX, prepared by Li et al.[146] resulted in an improved solubility and comparable in vitro cytotoxicity to PTX in B16 melanoma cells. Pendriet al. demonstrated that PEG-conjugated PTX-2′-glycinate had increased antitumor activity and less toxicity in a P388 murine leukemia model when compared to Taxol®.[150] A HPMA-conjugated PTX prodrug was synthesized with a self-immolative dendritic linker. The prodrug was found water-soluble and can release a triple payload of PTX as a result of cleavage by the endogenous enzyme cathepsin B. The prodrug also showed higher cytotoxicity on murine prostate adenocarcinoma (TRAMP C2) cells compared to a classic monomeric drug-polymer conjugate. [147] The most promising polymer-PTX conjugate to date is probably the PGA-PTX conjugate (CT-2103), where PGA is conjugated to 2′-OH position of PX via an ester linkage. The conjugate showed significantly better antitumor efficacy compared to Taxol® in several tumor-bearing mouse models, and in some cases it completely eliminated tumors. In addition, the MTD and tumor uptake of CT-2013 were about 2- and 5-fold higher than Taxol®, respectively, and prolonged circulation time was also observed in mouse tumor models.[148, 151]
PTX prodrug can be developed with a pH sensitive, reductive and/or enzyme-sensitive linkage to realize stimuli responsive prodrugs. This strategy would allow for the release of the active ingredient preferentially at the site of action such as cancer cells, thus sparing normal tissue. For example, Shabat’s group designed a PTX prodrug (PGD) by conjugating PTX with a hydrophilic poly(amdioamine) [PAMAM] dendrimer through the cathepsin B-cleavable tetrapeptide.[152] In comparison to PTX, PGD demonstrated a higher cytotoxicity specific to cell lines with moderate-to-high cathepsin B activity. Cells with comparatively lower cathepsin B activity demonstrated an inverse of this relationship. Regression analysis between the magnitude of PGD-induced cytotoxic increase over PTX and cathepsin B expression showed a strong, statistically significant correlation. In another study, PTX was conjugated to triazine dendrimers by a biodegradable disulfide linkage. This linkage was cleaved by the endogenous reductant, such as glutathione, whose intracellular concentration is more than 1,000 higher than that in the vasculature. After intravenous injection of this prodrug in human prostate cancer-bearing SCID mice, significant tumor growth suppression was observed with dosages from 50 to 200 mg PTX/kg over 20 days treatment and the mice treated with 50 mg PTX/kg exhibited high antitumor efficiency and 100% survivor over 10-weektreatment period.[153]
Tumor development is often associated with overexpression of some receptors or ligands which may serve as effective tumor targets, and as such targeted prodrugs were designed to increase the solubility and tumor-targeting properties of the drugs after drug conjugation with specific ligand. An integrin-targeting prodrug, PGA-PTX-E-[c(RGDfK)2], was developed to target the tumor by a combination of the EPR effect and active targeting through integrin binding. In this design, the cyclic RGD peptide mimetic was utilized for the active targeting to the αvβ3 integrin overexpressed on tumor endothelial and epithelial cells. The prodrug demonstrated increased water solubility and selective cytotoxicity against cancer cell lines. It was found that the RGD-conjugated prodrug exhibited preferential tumor accumulation of PTX, enhanced antitumor efficacy and reduced side effects.[154] Along the same lines, a ternary conjugate of heparin-folic acid-PTX was also designed for enhancement of tumor targeting. This conjugate was shown to recognize folate receptor positive KB-3-1 cells and can enhance tumor inhibitory activity in a subcutaneous KB-3-1xenograft model.[155] By combination of EPR and OCT-receptor mediated targeting, sharp redox response, and programmed drug release, a multifunctional prodrug, octreotide(Phe)-polyethene glycol-disulfide bond-PTX [OCT(Phe)-PEG-ss-PTX], was developed by the Zhou lab.[156] This polymer-PTX conjugates exhibited approximately 23,000-fold increase in water solubility. Moreover, the objective OCT(Phe)-PEG-SS-PTX prodrug was confirmed to be selectively internalized into tumor cells by SSTRs-mediated endocytosis. As a result, the in vivo investigations on NCI-H446 tumor xenografts confirmed that the OCT(Phe)-PEG-SS-PTX possessed superior tumor-targeting ability and antitumor activity over both mPEG-PTX and OCT(Phe)-PEG-PTX controls, and reduced systemic cytotoxicity compared with Taxol®. Overall, these prodrug constructs hold great potential for target chemotherapy.
In another study, polymeric prodrugs were shown to self-assemble into nanosized micelles of low polydispersity. Zhang et al. synthesized poly(ethylene oxide)-polyphosphoester-based PTX drug conjugates (PEO-PPE-PTX) in a two-step manner using organo-catalyst promoted ring-opening-polymerization, followed by click reaction based conjugation of a PTX prodrug.[157] The PEO-PPE-PTX formed well-defined core-shell nanoparticles in aqueous solution, containing ultra-high levels of drug loading. The Yu group developed a poly(L-γ-glutamyl glutamine)-PTX (PGG-PTX) conjugate. This conjugate self-assembled into NPs with the particle size of 12–15 nm. The formulation had low toxicity in mice and the in vivo antitumor activity was superior to Abraxane® in multiple tumor models.[39] More recently, the water-insoluble PTX was conjugated to LMWC via a cleavable bond, forming NPs with variable sizes (depending on drug content) that could be delivered to tumors.[158] The resulting LMWC-PTX NPs substantially improved the half-life of PTX in the blood (at least 6-fold) compared with that of the free PTX formulation. Furthermore, LMWC-PTX NPs exhibited excellent antitumor efficacy that was dramatically superior to that of free PTX. In another work, Zhang et al. developed a novel water-soluble polymeric prodrug, PTX-poly(ethyl ethylene phosphate) conjugated with folic acid molecules (PTX-PEEP-FA). This prodrug could self-assemble in aqueous solution to form micelles with PTX in the core and PEEP-FA as the corona, and the average particle size was less than 130 nm. The hydrophobic PTX core could be further used to load more water-insoluble anti-cancer drugs, such as PTX or doxorubicin (DOX), while the hydrophilic PEEP-FA chain endowed micelles with good stability during systemic circulation and significantly improved controlled-release properties compared to free PTX or DOX. In vitro biological evaluations confirmed that PTX-PEEP-FA, simultaneously acted as both a prodrug and drug delivery carrier, could achieve the aims of increased drug loading efficiency and enhanced targeting efficacy.[157]
5.2. Protein-based Prodrugs
Protein-based drugs have been used in the clinic and currently enjoy unprecedented recognition of their therapeutic potential.[159] Albumin is emerging as a versatile natural protein carrier for drug targeting and for improving the pharmacokinetic profile of protein-based drugs. Motivated by the clinical successes of Abraxane®, Ruoslahti and coworkers developed tumor targeted Abraxane® formulations with two respective tumor-homing peptides of CREKA and LyP-1. According to their results, LyP-1-Abraxane demonstrated significantly improved antitumor efficacy compared to untargeted Abraxane® in an MDA-MB-435 xenograft mouse model.[160] In another study, a novel ligand-mediated prodrugs Tf–Glu–PTX, namely, PTX conjugated with Fmoc-L-glutamic acid 5-tert-butyl ester (linker) and transferrin (Tf), to specifically target tumor cells/tissues.[161] Transferrin, a 78 kDa-monomeric serum glycoprotein, was used as both carrier and tumor targeting ligand. The cell studies demonstrated that the prodrug had efficient inhibition of tumor cell proliferation with low toxicity to healthy cells. Tf–Glu–PTX also displayed a potential to overcome PTX resistance in drug-resistant MDA-MB-231 cell lines.
Antibody-drug conjugates (ADCs) represent a hihgly promising class of anticancer therapeutics that combine the specificity of monoclonal antibodies (MAb) and the efficacy of small-molecule therapeutics.[162] The development of MAb specific to tumor antigens enables enhanced selectivity of anti-cancer drugs. A PTX MAb prodrug was developed to improve the drug’s accumulation in tumor. PEG was used to conjugate PTX with the anti-epidermal growth factor receptor MAb, C225 at a molar ratio of up to 12. The prodrug exhibited increased drug solubility, stability and preserved drug cytotoxicity against MDA-MB-468 breast cancer cells.[163] In another case, Ojima and coworker developed a tumor-specific PTX–monoclonal antibody conjugates. In their design, a highly cytotoxic C-10 methyldisulfanylpropanoyl PTX was conjugated to an MAb recognizing the epidermal growth factor receptor (EGFR) expressed in human squamous cancers.[164] These conjugates were shown to possess remarkable target-specific antitumor activity in vivo against EGFR-expressing A431 tumor xenografts in SCID mice, resulting in complete inhibition of tumor growth in the treated mice without showing noticeable toxicities to the animals.
5.3. Peptide-based Prodrugs
Peptide and its derivatives have been exploited for PTX delivery. For instance, Tat-based self-assembling peptide amphiphiles can self-assembled into filaments for intracellular delivery of PTX.[165] In the peptide-based prodrug strategy, a rationally designed peptide sequence is conjugated to PTX via an ester bond or other biodegradable linkers. In some cases, protecting the hydrolytic linker is necessary, which can be achieved by encapsulating the conjugates into polymeric micelles.[166] An alternative strategy is to create peptide-PTX conjugates that could self-assemble into self-delivering prodrug assemblies.[167] Taking advantages of the biological function and assembly potential of peptides, Cui and co-workers developed a novel class of drug amphiphiles (DAs) by covalent linkage of anticancer drugs to a rationally chosen/designed peptide through a biodegradable linker.[168–174] Their core design concept is to consider drugs as molecular building units, not just being functional units. By incorporating a short peptide as both a functionally active and structurally guiding unit, the resulting DAs are actually amphiphilic prodrugs capable of forming a variety of supramolecular morphologies in aqueous solutions (Figure 2D). Expansion of this concept led to the construction of PTX loaded DAs with a drug content up to 41% with activity comparable to free PTX.[175] Conjugation of the bulky PTX to a short peptide does not prevent the peptide from assembling into one-dimensional nanostructures. In vitro study showed that filaments have near identical toxicity to free PTX, indicating that the self-assembling PTX DA does not compromise the drug’s efficacy. Ding and coworkers reported another example in which the conjugation of PTX to a tumor-homing cell-penetrating peptide (CPP) resulted in the formation of spherical nanostructures ~120 nm in diameter.[176] This PTX-CPP nanospheres also do not compromise the PTX’s potency, which can also be utilized as the carriers for co-delivery of another anticancer drug (DOX).
The attachment of drugs to molecular transporters, such as cell-penetrating peptides, can help facilitate cellular uptake through multiple mechanisms other than passive diffusion, which can be helpful in overcoming MDR. In view of this, the Liang Lab proposed a strategy to overcome the MDR of drug-resistant cells by employing a biocompatible condensation reaction.[177] Specifically, they designed and synthesized a PTX derivative Ac-Arg-Val-Arg-Arg-Cys(StBu)-Lys(PTX)-2-cyanobenzothiazole (CBT-PTX), which could be subjected to furin-controlled condensation and self-assembly into PTX nanoparticles (PTX-NPs). In vitro and in vivo studies indicated that CBT-PTX showed a 1.5-fold increase in anti-MDR effects on tumors without significant toxicity to the mice. In another example, Wender and coworkers conjugated PTX to an octaarginine peptide transporter through a bio-reducible linker, producing conjugates with significantly improved activity against malignant cells that were resistant to the therapeutic agent alone.[178] They further demonstrated that this approach was effective in animal models of ovarian cancer and explored the generality and mechanistic basis for this effect with coelenterazine, a P-gp substrate. Although coelenterazine itself does not enter cells because of P-gp efflux, its octaarginine conjugates can do so readily. This general approach helps overcome the MDR elicited by small molecule cancer chemotherapeutics and could improve the prognosis for many cancer patients.
Polypeptide-drug conjugates, albeit less studied relative to polymer-drug conjugates, represent a very promising drug delivery platform. Polypeptidic materials could be designed to possess metabolizable and biodegradable features, bio-inspired functionalities, and stimuli-responsive properties. Polypeptides containing amino acids with functionalizable side chains (e.g. lysine, glutamic acids, cysteine, serine) can be used to conjugate with drug molecules to improve the drug’s bioavailability and efficacy [179]. In some cases, amphiphilic polypeptide-drug conjugates exhibited the ability to aggregate into nanostructures under physiologically relevant conditions.[180] Chilkoti and co-workers reported the conjugation of PTX to recombinant chimeric polypeptides (CPs) that spontaneously self-assemble into ~60 nm near-monodisperse nanoparticles.[181] In their design, the covalent conjugate of PTX is stable at the pH of blood, but the drug can be cleaved off at an appreciable rate at the tumor interstitial pH (~6.5) and at the pH in late endosomes (~5.3). More importantly, the tumor uptake of the CP-PTX nanoparticle was 5-fold higher than the free drug and 2-fold higher than Abraxane®. In a murine cancer model of human triple-negative breast cancer and prostate cancer, CP-PTX induced near-complete tumor regression after a single dose in both tumor models. These results show that a molecularly engineered nanoparticle with precisely engineered design features outperforms Abraxane®, the current gold standard for PTX delivery. To take advantage of the thermal responsive property transition between 37°C and 42°C, Shama et al. developed a thermally responsive elastin-like polypeptide (ELP) for the delivery of PTX.[182] In the presence of hyperthermia, the ELP-PTX prodrug inhibits MCF-7 cell proliferation by stabilizing the microtubule structures, arresting the cells at the G2/M stage and inducing apoptosis in a manner similar to conventional PTX.
5.4. Small Molecule-based Prodrugs
The emergence and development of small molecule prodrugs has been primarily focused on improving the drug’s water solubility, bioavailability, chemical stability, and in some cases brain permeability and targeting, and also on addressing the multiple drug resistance mechanisms.[183] In an effort to develop novel PTX prodrugs, Bradley’s group conjugated a natural fatty acid, docosahexaenoic acid (DHA), through an ester bond to the PTX 2′-oxygen.[184] The resulting PTX fatty acid conjugate (DHA-PTX) does not have microtubule stabilizing activity and is non-toxic. This prodrug can be converted to the active PTX form when metabolized by esterases in the body. Therefore, the conjugate prolonged the exposure of PTX and reduced the peak concentration, which allowed the administration of 4.4-fold higher doses of DHA-PX conjugate over that of PTX alone in mice. Conjugated linoleic acids (CLAs) are structurally unique group of dietary long chain polyunsaturated fatty acids, which have elicited considerable interest because of their significant anti-proliferative effects in cancer cells. This prodrug strategy may provide the synergistic antitumor effect with PTX. A new prodrug, CLA-PTX, was synthesized by covalently linked CLA to the PTX with good stability in PBS and rat plasma. The conjugate exhibited higher cellular uptake efficiency on C6 glioma cells and improved pharmacokinetics compared with PTX. Unlike PTX, CLA-PTX could distribute in brain tissue and retained higher concentrations throughout 360 h, which resulted significant antitumor efficiency in brain tumor-bearing rats.[112]
Polymeric (such as PEG-PLA) micelles has progressed to human trials for PTX as a safer alternative to Cremophor EL and ethanol in Taxol®. For example, Genexol-PM is approved in Asia and in a phase 3 clinical trial. However, the low drug loading and poor stability against precipitation might limit the broader usage of PEG-b-PLA micelles in treating cancers. In addition, PEG-b-PLA micelles release PTX rapidly, resulting in widespread biodistribution and a low tumor exposure.[186] To address these issues, lipophilic acyl ester prodrugs have been synthesized for polymeric micelles. A 2′-acyl prodrug attached on PTX by a diglycolate linkage has also been stably loaded in PEG-poly(styrene) micelles by Mayer et al.[187] While prodrugs were 1–2 orders of magnitude less potent than PTX in vitro, a long-circulating diglycolate PTX prodrug exerted higher antitumor efficacy than PTX at the MTD of 60 and 20 mg/kg, respectively, in a HT29 human colon carcinoma model. More recently, the Kwon lab synthesized a lipophilic PTX derivative, oligo(lactic acid)8-PTX (o(LA)8-PTX), and then incorporated this conjugate into PEG-b-PLA micelles (Figure 6). As expected, o(LA)8-PTX was more compatible with PEG-b-PLA micelles than PTX, increasing drug loading from 11 to 54%. While the in vitro release of PTX from Taxol® was rapid, o(LA)8-PTX release was more gradual with t1/2 = 14 h. Notably, o(LA)8-PTX induced tumor regression in A549 tumor-bearing mice and caused less toxicity than PTX in terms of change in body weight. This PTX delivery system holds great potential for clinical evaluation.[185]
Figure 6.
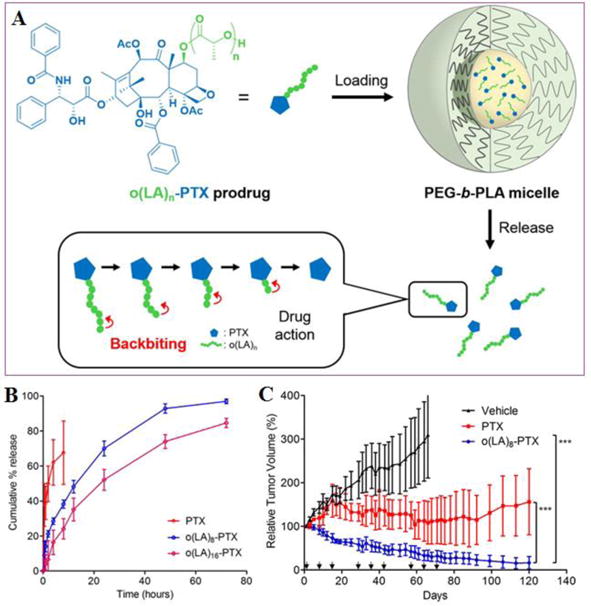
(A) Schematic illustration of Oligo(lactic acid)n-PTX prodrugs for PEG-PLA micelles: Loading, release, and backbiting conversion for anticancer activity. (B) In vitro release of PTX, o(LA)8-PTX, or o(LA)16-PTX prodrug from PEG-b-PLA micelles. (C) In vivo antitumor efficacy PTX or o(LA)8-PTX prodrug (20 mg/kg) as PEG-b-PLA micelles (9% loading) in an A549 xenograft tumor model. Mice received 3 weekly injections followed by 1 week off for 3 cycles. (Reproduced from Ref. [185] with permission)
5.5. Hybrid Prodrugs
Rational combinations of two or more drugs can potentially generate synergistic effect in reducing side effects associated with high dose of single drugs, and also offer a way to overcome multidrug resistance mechanisms that cancer cells may develop during the course of the treatment.[169] Recently, a bifunctional dual-acting prodrug was prepared by conjugating PTX and DOX in the same carrier to realize combinational chemotherapy. The prodrug incorporated a 1,6-self-immolative para-aminobenzyloxycarbonyl (PABC) spacer between the conjugated drug and the lysosomally cleavable dipeptide Phe-Lys. It can bind to cysteine-34 position of albumin and cleavable by cathepsin B, a cysteine protease that is overexpressed in solid tumors, to release the free drugs.[188] Cheetham et al. reported the synthesis and assembly of a mikto-arm star dual drug amphiphile containing both a bulky PTX and a planar CPT.[172] The two different anti-cancer drugs were conjugated to the terminal lysine of a β-sheet forming peptide (Sup35), and the dual DAs could spontaneously associate into a well-defined filamentous morphology containing a fixed 41% total drug loading. The hetero dual DA was found to effectively release the two anticancer agents, exhibiting superior cytotoxicity against PTX-resistant cervical cancer cells (Figure 7). They believed that delivering two drugs of completely different action mechanisms could increase the possibility of overcoming multidrug resistance and reduce the possibility for tumor reoccurrence. In another example, Mao et al. designed a dexamethasone-FFFK(PTX/HCPT)E-SS-EE system based on two complementary anticancer drugs for chemotherapy.[189] In this design, PTX serves as an anticancer agent, while dexamethasone acts as an anti-inflammatory to reduce side effects associated with cancer chemotherapy. This multiple complementary prodrug represents a smart co-delivery system for the disease treatment.
Figure 7.
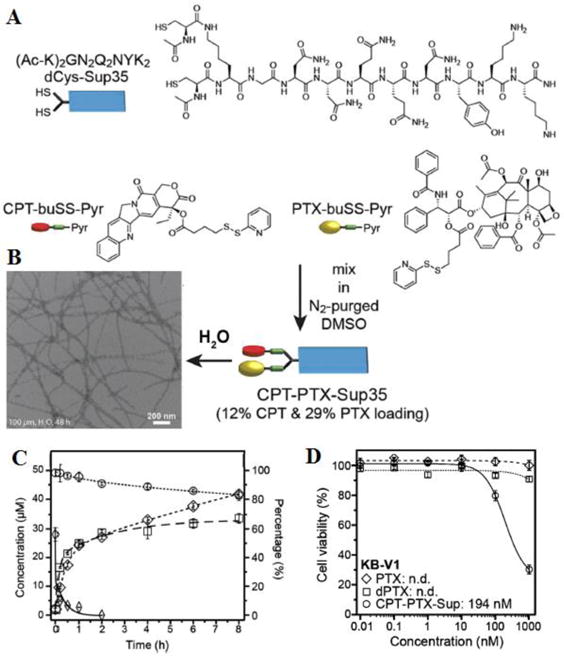
(A) Synthesis of CPT-PTX-Sup35 from the reaction of dCys-Sup35 with a 1:1mixture of activated disulfide drugs, CPT-buSS-Pyr and PTX-buSS-Pyr. (B) Representative TEM images of CPT-PTX-Sup35 in water. (C) Release of CPT and PTX and degradation of CPT-PTX-Sup35 (50 mM in 10mM sodium phosphate solution, pH 7.4) in the presence or absence of 10 mM GSH (○ CPT-PTX-Sup35 without GSH; ◊ CPT-PTX-Sup35 with GSH; ◇ released CPT; □ released PTX). (D). Cytotoxicity of CPT-PTX-Sup35 against PTX-resistant KB-V1 cervical cancer cells (○ CPT-PTX-Sup35; □ PTX; ◇dPTX-Sup35). Cell viability was determined by SRB assay after 48 h incubation with the appropriate drug-containing media. (Adapted from Ref. [172] with permission)
Nucleic acid-based materials have received much attention because of their hydrophilicity, biocompatibility and more importantly their feasibility to be engineered into different sequence, lengths, sizes and shapes.[190] Therefore, nucleic acids have been used both as carriers and therapeutics. One excellent example integrating the two functions together is the spherical nucleic acids self-assembled from conjugates of PTX and oligonucleotide (Figure 8).[191] Zhang and coworkers conjugated ten PTX molecules to one oligonucleotide through disulfide bond, and the resulting amphiphilic conjugates self-assembled into spherical micelles of ~15 nm in diameter in aqueous solution. Upon intracellular uptake, the micelles was disrupted by glutathione, releasing free PTX and functional DNA for microtubule stabilization and gene regulation, respectively.
Figure 8.
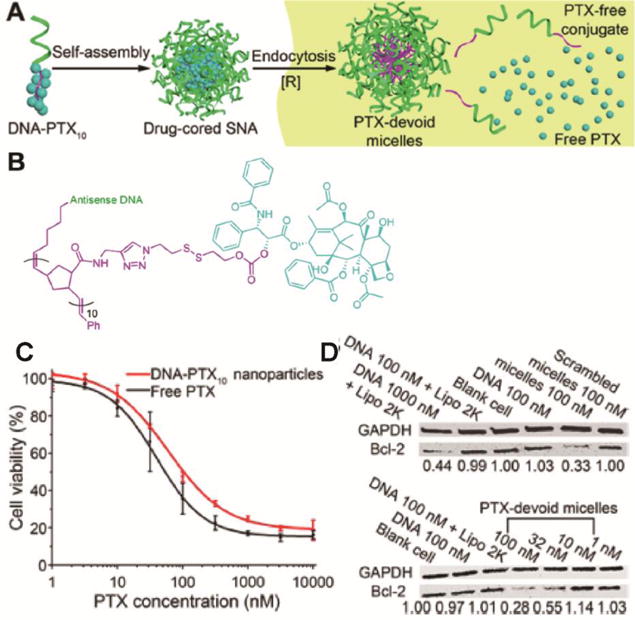
(A) Schematic illustration of the self-assembly of oligonucleotide-PTX conjugates. (B) Molecular structure of the oligonucleotide-PTX conjugates. (C) MTT cytotoxicity assay of nanoparticles self-assembled from oligonucleotide-PTX conjugates. (D) Efficacy for antisense gene knockdown using PTX-devoid nanoparticles. (Reproduced from Ref. [191] with permission)
6. Local Delivery Systems
Surgery represents the primary option to treat solid tumors, followed by radiotherapy and chemotherapy as an adjunct. The postsurgical local placement of a biodegradable system loaded with an anticancer agent can prevent further proliferation of remaining malignant cancer cells.
6.1. Hydrogels
Hydrogels are three-dimensional (3D) polymeric and hydrophilic networks able to imbibe large amounts of water or biological fluid. Hydrogels exhibit a thermodynamic compatibility with water which allows them to swell in aqueous media.[192] Hydrogels emerged as excellent candidates for controlled release, bio-adhesive and/or targeted drug delivery as they are able to encapsulate hydrophilic or hydrophobic drugs.[170, 193–196] A key point in the success of hydrogels development is the ability to gel in situ (Figure 2E). This can be achieved by UV polymerization or via either reversible interactions or chemical reactions. The gelation process can also be time-dependent or be triggered by specific stimulus (e.g. pH, temperature).[197]
Injectable hydrogels have demonstrated numerous advantages compared to conventional delivery systems and have been extensively studied for localized antitumor drug delivery.[198] Local delivery of antitumor drugs provides a high local concentration and decreases the incidence of side effects commonly observed with systemic therapy. A clinical trial recently tested the efficacy of OncoGel® (MacroMed, Inc.) for local tumor treatment.[199] The thermo-responsive gel was composed of PLGA-PEG-PLGA triblock copolymer, which transformed to a water-insoluble hydrogel at body temperature.[200, 201] It had sustained PTX release properties for up to 6 weeks upon injection. OncoGel® was proven to be safe with negligible PTX concentration in plasma after single injection into solid tumor site. In another example, Ruel-Gariepy et al. have recently reported using a thermo-sensitive chitosan gel for the sustained delivery of PTX.[202, 203] This formulation can be injected directly at the site of tumor resection and the liquid uniformly fills the cavity as it sets into the gel. EMT-6 tumor bearing Balb/c mice study demonstrate that a single injection of the PTX loaded chitosan gel results similar to four intravenous injections of Taxol®. In addition, the chitosan based system showed significantly less toxicity when compared with systemic Taxol® administration.[203] Along the same lines, Guo et al. developed a linoleic acid (CLA)-coupled poloxamer thermo-sensitive hydrogel for local delivery of PTX.[198] The PTX-loaded thermo-sensitive hydrogel showed excellent cell cycle arrest and apoptosis in tumor tissue compared with the PTX incorporated poloxamer hydrogel. In a more recent study, Bajaj et al. developed HA-based in situ crosslinkable hydrogel, which served as a carrier of PTX particles to improve their intraperitoneal retention and therapeutic effects.[204]
Intelligent nanogels that exhibit drastic response to various stimuli have been a popular research topic in the rapidly growing fields of smart materials and nanomedicine.[205, 206] Among various intelligent nanogels, the most extensively studied are those responsive to changes in pH, [207] temperature,[198, 203] reduction potential,[208] and enzymatic reactions.[209–211] By taking advantage of the weak acidic environment of tumorous tissue, Zhao et al. developed a pH sensitive injectable hydrogel with hydrophobic microdomains for incorporating both DOX and PTX. Dual drug release from the PTX+DOX loaded gels were observed with pH-sensitive behavior and accelerated drug release rate at the acidic pH environment. After intratumoral administration, the hydrogel formulations of PTX resulted in melanoma B16F10 tumor inhibition on C57/BL6 mouse model.[207] In another example, Yang and coworkers connected the oxidized form of glutathione to PTX via a succinyl linkage which upon reduction could form a hydrogel.[24] The mechanical properties of this hydrogel was suitable for injection, and the injected gels can recover within a few minutes once the stress was removed. Intra-tumoral injection of the gel in a mouse model was found to inhibit tumor growth and metastasis.
By mimicking biomacromolecular self-assembly, the integration of enzymatic reactions with self-assembly of small molecules provides an effective means to form nanofiber networks and results in hydrogels under various conditions.[209–211] Taking advantage of this strategy, Xu and coworkers have developed a PTX-bearing supramolecular hydrogel for chemotherapy using a phosphorylated naphthalene–Phe–Phe–Lys–Tyr precursor with conjugated PTX (Figure 9). Upon treatment with alkaline phosphatase (ALP), cleavage of the phosphate group occurred and a translucent hydrogel spontaneously formed. They found that both the precursor PTX–NapFFKYp and the hydrogelator exhibited comparable anticancer abilities compared with free PTX, indicating that the drug’s activity remains unaffected.[209]
Figure 9.
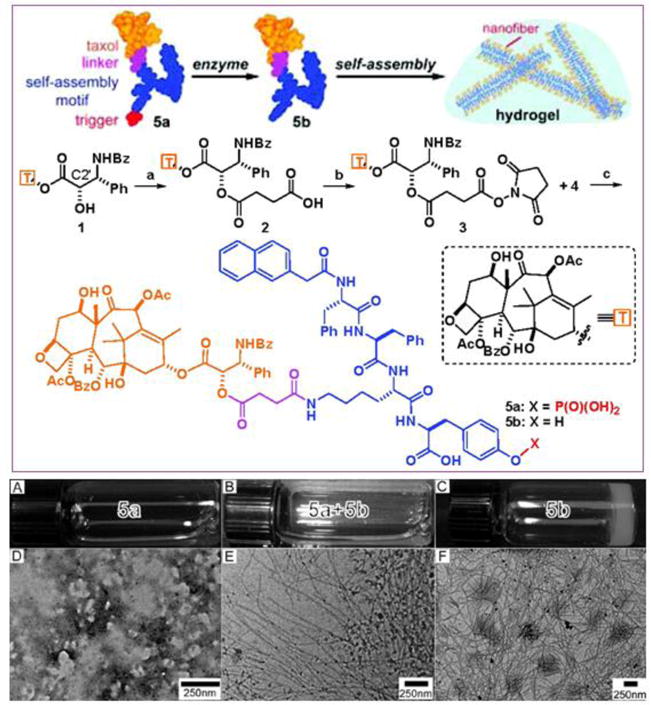
Schematic illustration of Xu’s design of the enzyme-triggered formation of supramolecular hydrogels. This particular precursor consists of a self-assembly promoting motif, an enzyme-cleavable group, and a PTX drug molecule. After the enzymatic reaction, the PTX derivative can spontaneously assemble into nanofibers in water that eventually entangle into a self-supporting hydrogel. In the absence of enzyme (alkaline phosphatase, ALP), the pregelator solution exhibits no defined nanostructure (A and D). Upon addition of ALP, cleavage of the phosphate group triggers self-assembly (B and E), resulting in hydrogel formation (C and F). (Adapted from Ref. [209] with permission)
Although there are many reports on various intelligent micro/nanogels as aforementioned, multi-stimuli responsive micro/nanogels are still limited. Injectable pH- and temperature-sensitive hydrogels that respond to change in pH and temperature, have unique advantages over temperature-sensitive hydrogels. The drug release behavior can be tuned by changing the chemical structure of the pH- and temperature-sensitive polymers, leading to enhanced specificity of drug delivery and less side effects.[212] In view of this, a novel pH- and temperature-sensitive biodegradable block copolymer OSM–PCLA–PEG–PCLA–OSM was synthesized and used as carriers for PTX. A solution of the polymer can rapidly form a strong gel once at physiological body pH/and temperature, showing gradual in vivo degradation over the course of six weeks.[213] PTX-loaded hydrogel showed good anti-tumor effect up to 2 weeks after one injection into tumor-bearing mice.[214] More recently, Qiao et al. developed a type of multi-responsive nanogels by the miniemulsion copolymerization of monomethyl oligo(ethylene glycol) acrylate (OEGA) and another ester-containing acrylic monomer (DMDEA) in the presence of a disulfide-containing cross-linker. The nanogel exhibited thermo-responsive behavior, acid-triggered hydrolysis and reduction-induced degradation and closely relevant to their compositions and crosslinking degrees. These nanogels are capable of encapsulating hydrophobic PTX. The drug release can be accelerated by a cooperative effect of both acid-triggered hydrolysis and DTT-induced degradation. The PTX-loaded nanogels exhibit a concentration-dependent toxicity to MCF-7 cells while the intact unloaded nanogels are non-toxic [208].
6.2. Implants
The production of drug loaded polymeric implants and wafer has introduced an innovative concept in drug administration. Drugs can be delivered to tumor in a sustained, continuous and predictable release fashion using biodegradable polymers as delivery vehicles. Due to their transient nature, biodegradable polymers do not require surgical removal after their intended application and thus can circumvent some of the problems related to the long-term safety of non-degradable implanted devices. Moreover, the postsurgical local placement of a biodegradable device loaded with an anticancer agent can prevent further proliferation of malignant cells while sparing the patient the harmful side effects of chemotherapy. Recently, a novel chitosan-based implantable formulation (chitosan-ePC) was developed to provide controlled, local release of PTX for the treatment of ovarian tumors. A sustained, zero-order release of PTX was observed in vivo over two weeks in mice implanted with 14C-PTX–chitosan-ePC films. More importantly, after 2–4 weeks, mice treated with chitosan-ePC implants did not demonstrate any signs of inflammation or infection during a 4 weeks test. Park et al. developed disc-shaped implants of polyanhydride, P (FAD-SA 50 : 50, w/w), with 10% w/w PTX, from which the drug was slowly released with only 15% of the drug released in 77 days, and requiring 44 months to release the total amount of PTX.[215] Similar formulations were used for localized implantation of drug in the cavity of resected brain tumors.[216]
The most successful drug delivery implant is arguably Gliadel® wafer, as it still remains the only one approved by the FDA for local treatment of brain tumors. The polymer matrix is a biodegradable copolymer formed by reaction of 1,3-bis-(p-carboxyphenoxy)propane (pCPP) and sebacic acid (SA) in a 20:80 ratio (polifeprosan 20). The impregnated active compound is carmustine (BCNU). Gliadel® wafers release the drug in approximately three weeks, but in vivo studies in mammalian models showed that the majority of the drug release takes place in the first 5–7 days.[217] In different animal models, high concentrations of drug were observed adjacent to the polifeprosan20 implants while low drug concentrations were observed in distant regions of the brain.[218] Given the limitations that Gliadel® has revealed, such as poor tissue penetration, fast drug release, implant dislodgement, and large resection cavity size needed for implantation, several research groups attempted to optimize the efficacy of polymer-mediated implants for the controlled release of other chemotherapeutics including PTX. For example, Von Eckardstein et al. developed a PTX/carboplatin liquid crystalline cubic phases.[219] In a pilot study, a total of 12 patients with a recurrence of glioblastoma multiforme underwent re-resection and received an intracavitary application of PTX and carboplatin cubic phases in different dosages. Six of the patients received more than 15 mg PTX and suffered from moderate to severe brain edema, while the remaining patients received only a total of 15 mg PTX. In the latter group, brain edema was markedly reduced and dealt medically. This design shows its feasibility and safety in intracavitary chemotherapy of recurrent glioblastoma. PTX has also been loaded in PLGA microfiber, foams or wafers with different form and geometry, obtaining good pharmacokinetic and diffusion profiles and promising preliminary efficacy in vivo results.[220, 221]
7. Future Perspectives
PTX is one of the most successful small molecule anticancer drugs ever developed to treat a wide spectrum of cancer, including lung, ovarian, breast, head and neck cancer as well as Kaposi’s sarcoma. The clinical and marketing successes of Taxol® (annual peak sale was once $1.6 Billion USD) and its superior replacement Abraxane® (2016 annual sale was close to $1 Billion USD) have motivated heavy academic and industrial interests in the development of a variety of drug delivery systems and platform technologies, including liposomes, micelles, SLNs, biodegradable polymers, prodrug designs, and hydrogels. Currently, Genexol-PM® (PLA-mPEG micelles) and Lipusu® (PTX liposomes) are frontrunners, as they have been approved in South Korea and China, respectively, as an alternative for Taxol®. Table 2 lists PTX formulations currently in clinical trials.
Table 2.
Summary of PTX-based formulations under clinical development
| Product Name | Formulation | Company | Clinical Stage | Therapeutic Indication |
|---|---|---|---|---|
| NK105 | PEG-polyaspartate micelle | NanoCarrier | Phase III complete | Metastatic breast cancer |
| CT-2103 | Poly-(L)-Glutamic Acid-Paclitaxel conjugate | Cell Therapeutics | Phase II complete | Primary peritoneal carcinoma |
| ANG 1005 | Angiopep-2 Peptide conjugate | Angiochem | Phase II | Glioma |
| LEP-ETU | DOPC/cholesterol/cardiolipin liposome | NeoPharm Inc.- Insys Therapeutics | Phase II | Metastatic breast cancer |
| Paxceed | mPEG-PDLLA micelle | Angiotech Pharmaceuticals Inc | Phase II | Rheumatoid Arthritis |
| OncoGel | PLGA-PEG-PLGA Gel | Protherics | Phase II/terminated | Glioblastoma Multiforme |
| LOC-paclitaxel | Linoleyl carbonate-Paclitaxel conjugate | Luitpold Pharmaceuticas | Phase I/terminated | Melanoma |
While most of PTX formulations have demonstrated much reduced toxicity relative to Taxol®, they reveal only marginal improvement in response rate and treatment efficacy. Now, with the bar being raised from Taxol® to Abraxane®, PTX formulations currently under clinical and preclinical development including Genexol-PM® and Lipusu® can at most become competitors (not replacement) of Abraxane®. The disruptive innovation in PTX delivery has yet to come. From the perspectives of drug delivery sciences, the specific accumulation of PTX in the primary and metastasis site of cancer has not been achieved, necessitating much more detailed and systemic investigation on the real and unique advantages of using nanoscale carriers. Likely, the quest for more effective PTX-based formulations will push the limit of the ultimate benefits and effectiveness of cancer chemotherapy based on cytotoxic small molecule drugs. It is very clear that more knowledge on cancer biology and pathology is needed to assist in the PTX formulation design. Despite many challenges, the future of nano-delivery systems is promising and poised to have a role in improving PTX-based cancer chemotherapy.
Acknowledgments
We acknowledge financial support from the National Science Foundation (DMR 1255281) and the National Institutes of Health (NIH/1R21CA191740).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wani MC, Taylor HL, Wall ME, Coggon P, Mcphail AT. Plant Antitumor Agents .6. Isolation and Structure of Taxol, a Novel Antileukemic and Antitumor Agent from Taxus-Brevifolia. J Am Chem Soc. 1971;93:2325–&. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 2.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 3.Holton RA, Somoza C, Kim HB, Liang F, Biediger RJ, Boatman PD, Shindo M, Smith CC, Kim SC, Nadizadeh H, Suzuki Y, Tao CL, Vu P, Tang SH, Zhang PS, Murthi KK, Gentile LN, Liu JH. First Total Synthesis of Taxol .1. Functionalization of the B-Ring. J Am Chem Soc. 1994;116:1597–1598. [Google Scholar]
- 4.Holton RA, Kim HB, Somoza C, Liang F, Biediger RJ, Boatman PD, Shindo M, Smith CC, Kim SC, Nadizadeh H, Suzuki Y, Tao CL, Vu P, Tang SH, Zhang PS, Murthi KK, Gentile LN, Liu JH. First Total Synthesis of Taxol .2. Completion of the C-Ring and D-Ring. J Am Chem Soc. 1994;116:1599–1600. [Google Scholar]
- 5.Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claiborne CF, Renaud J, Couladouros EA, Paulvannan K, Sorensen EJ. Total Synthesis of Taxol. Nature. 1994;367:630–634. doi: 10.1038/367630a0. [DOI] [PubMed] [Google Scholar]
- 6.Liu WC, Gong T, Zhu P. Advances in exploring alternative Taxol sources. Rsc Adv. 2016;6:48800–48809. [Google Scholar]
- 7.Kingston DGI. Taxol - the Chemistry and Structure-Activity-Relationships of a Novel Anticancer Agent. Trends Biotechnol. 1994;12:222–227. doi: 10.1016/0167-7799(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 8.Snyder JP, Nettles JH, Cornett B, Downing KH, Nogales E. The binding conformation of Taxol in beta-tubulin: a model based on electron crystallographic density. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5312–5316. doi: 10.1073/pnas.051309398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marupudi NI, Han JE, Li KW, Renard VM, Tyler BM, Brem H. Paclitaxel: a review of adverse toxicities and novel delivery strategies. Expert Opin Drug Saf. 2007;6:609–621. doi: 10.1517/14740338.6.5.609. [DOI] [PubMed] [Google Scholar]
- 10.Zhang ZP, Mei L, Feng SS. Paclitaxel drug delivery systems. Expert Opin Drug Del. 2013;10:325–340. doi: 10.1517/17425247.2013.752354. [DOI] [PubMed] [Google Scholar]
- 11.Singh S, Dash AK. Paclitaxel in Cancer Treatment: Perspectives and Prospects of its Delivery Challenges. Crit Rev Ther Drug. 2009;26:333–372. doi: 10.1615/critrevtherdrugcarriersyst.v26.i4.10. [DOI] [PubMed] [Google Scholar]
- 12.Rowinsky EK, Donehower RC. The Clinical-Pharmacology of Paclitaxel (Taxol(R)) Semin Oncol. 1993;20:16–25. [PubMed] [Google Scholar]
- 13.Paik PK, James LP, Riely GJ, Azzoli CG, Miller VA, Ng KK, Sima CS, Heelan RT, Kris MG, Moore E, Rizvi NA. A phase 2 study of weekly albumin-bound paclitaxel (Abraxane(A (R))) given as a two-hour infusion. Cancer Chemoth Pharm. 2011;68:1331–1337. doi: 10.1007/s00280-011-1621-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Testa B. Prodrug research: futile or fertile? Biochem Pharmacol. 2004;68:2097–2106. doi: 10.1016/j.bcp.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 15.Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235:179–192. doi: 10.1016/s0378-5173(01)00986-3. [DOI] [PubMed] [Google Scholar]
- 16.Sofias AM, Dunne M, Storm G, Allen C. The battle of “nano” paclitaxel. Advanced Drug Delivery Reviews. 2017 doi: 10.1016/j.addr.2017.1002.1003. [DOI] [PubMed] [Google Scholar]
- 17.Yardley DA. nab-Paclitaxel mechanisms of action and delivery. J Control Release. 2013;170:365–372. doi: 10.1016/j.jconrel.2013.05.041. [DOI] [PubMed] [Google Scholar]
- 18.Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioengineering & Translational Medicine. 2016;1:10–29. doi: 10.1002/btm2.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nature reviews Drug discovery. 2014;13:813–827. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim SC, Kim DW, Shim YH, Bang JS, Oh HS, Kim SW, Seo MH. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. Journal of Controlled Release. 2001;72:191–202. doi: 10.1016/s0168-3659(01)00275-9. [DOI] [PubMed] [Google Scholar]
- 21.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. European journal of cancer. 2001;37:1590–1598. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 22.Sparreboom A, Van Zuylen L, Verweij J, Mross K, Brouwer E, Gianni L. Interrelationships of paclitaxel disposition, infusion duration and Cremophor EL kinetics in cancer patients. Clin Cancer Res. 1999;5:3840s–3840s. doi: 10.1097/00001813-200006000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Lorenz W, Reimann HJ, Schmal A, Dormann P, Schwarz B, Neugebauer E, Doenicke A. Histamine-Release in Dogs by Cremophor-El and Its Derivatives - Oxethylated Oleic-Acid Is Most Effective Constituent. Agents Actions. 1977;7:63–67. doi: 10.1007/BF01964882. [DOI] [PubMed] [Google Scholar]
- 24.Green MR, Manikhas GM, Orlov S, Afanasyev B, Makhson AM, Bhar P, Hawkins MJ. Abraxane((R)), a novel Cremophor((R))-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann Oncol. 2006;17:1263–1268. doi: 10.1093/annonc/mdl104. [DOI] [PubMed] [Google Scholar]
- 25.Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmaco. 2006;7:1041–1053. doi: 10.1517/14656566.7.8.1041. [DOI] [PubMed] [Google Scholar]
- 26.Paal K, Muller J, Hegedus L. High affinity binding of paclitaxel to human serum albumin. Eur J Biochem. 2001;268:2187–2191. doi: 10.1046/j.1432-1327.2001.02107.x. [DOI] [PubMed] [Google Scholar]
- 27.Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 28.Chen Q, Zhang QZ, Liu J, Li LQ, Zhao WH, Wang YJ, Zhou QH, Li L. Multi-center prospective randomized trial on paclitaxel liposome and traditional taxol in the treatment of breast cancer and non-small-cell lung cancer. Zhonghua zhong liu za zhi [Chinese journal of oncology] 2003;25:190–192. [PubMed] [Google Scholar]
- 29.Xu X, Wang L, Xu HQ, Huang XE, Qian YD, Xiang J. Clinical Comparison between Paclitaxel Liposome (Lipusu (R)) and Paclitaxel for Treatment of Patients with Metastatic Gastric Cancer. Asian Pac J Cancer P. 2013;14:2591–2594. doi: 10.7314/apjcp.2013.14.4.2591. [DOI] [PubMed] [Google Scholar]
- 30.Kim TY, Kim DW, Chung JY, Shin SG, Kim SC, Heo DS, Kim NK, Bang YJ. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10:3708–3716. doi: 10.1158/1078-0432.CCR-03-0655. [DOI] [PubMed] [Google Scholar]
- 31.Lim WT, Tan EH, Toh CK, Hee SW, Leong SS, Ang PC, Wong NS, Chowbay B. Phase I pharmacokinetic study of a weekly liposomal paclitaxel formulation (Genexol-PM) in patients with solid tumors. Ann Oncol. 2010;21:382–388. doi: 10.1093/annonc/mdp315. [DOI] [PubMed] [Google Scholar]
- 32.Lee KS, Chung HC, Im SA, Park YH, Kim CS, Kim SB, Rha SY, Lee MY, Ro J. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast cancer research and treatment. 2008;108:241–250. doi: 10.1007/s10549-007-9591-y. [DOI] [PubMed] [Google Scholar]
- 33.Saif MW, Podoltsev NA, Rubin MS, Figueroa JA, Lee MY, Kwon J, Rowen E, Yu J, Kerr RO. Phase II clinical trial of paclitaxel loaded polymeric micelle in patients with advanced pancreatic cancer. Cancer investigation. 2010;28:186–194. doi: 10.3109/07357900903179591. [DOI] [PubMed] [Google Scholar]
- 34.Feng L, Mumper RJ. A critical review of lipid-based nanoparticles for taxane delivery. Cancer Lett. 2013;334:157–175. doi: 10.1016/j.canlet.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehta R, Burke TG. Membrane Biophysical Parameters Influencing Anthracycline Action. Acs Sym Ser. 1995;574:222–240. [Google Scholar]
- 36.Fetterly GJ, Grasela TH, Sherman JW, Dul JL, Grahn A, Lecomte D, Fiedler-Kelly J, Damjanov N, Fishman M, Kane MP, Rubin EH, Tan AR. Pharmacokinetic/pharmacodynamic modeling and simulation of neutropenia during phase I development of liposome-entrapped paclitaxel. Clin Cancer Res. 2008;14:5856–5863. doi: 10.1158/1078-0432.CCR-08-1046. [DOI] [PubMed] [Google Scholar]
- 37.Crosasso P, Ceruti M, Brusa P, Arpicco S, Dosio F, Cattel L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. Journal of Controlled Release. 2000;63:19–30. doi: 10.1016/s0168-3659(99)00166-2. [DOI] [PubMed] [Google Scholar]
- 38.Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic Polyethyleneglycols Effectively Prolong the Circulation Time of Liposomes. Febs Lett. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 39.Yoshizawa Y, Kono Y, Ogawara K, Kimura T, Higaki K. PEG liposomalization of paclitaxel improved its in vivo disposition and anti-tumor efficacy. Int J Pharm. 2011;412:132–141. doi: 10.1016/j.ijpharm.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 40.Abu Lila AS, Kiwada H, Ishida T. The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage. J Control Release. 2013;172:38–47. doi: 10.1016/j.jconrel.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Ishida T, Kiwada H. Anti-PEG IgM elicited by injection of liposomes is involved in the enhanced blood clearance of a subsequent dose of PEGylated liposomes. J Control Release. 2007;119:236–244. doi: 10.1016/j.jconrel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 42.Biswas S, Dodwadkar NS, Deshpande PP, Torchilin VP. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. Journal of Controlled Release. 2012;159:393–402. doi: 10.1016/j.jconrel.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu YY, Ran R, Chen JT, Kuang QF, Tang J, Mei L, Zhang QY, Gao HL, Zhang ZR, He Q. Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials. 2014;35:4835–4847. doi: 10.1016/j.biomaterials.2014.02.031. [DOI] [PubMed] [Google Scholar]
- 44.Buyukkoroglu G, Senel B, Basaran E, Gezgin S. Development of paclitaxel-loaded liposomal systems with anti-her2 antibody for targeted therapy. Trop J Pharm Res. 2016;15:895–903. [Google Scholar]
- 45.Qin L, Wang CZ, Fan HJ, Zhang CJ, Zhang HW, Lv MH, Cui SD. A dual-targeting liposome conjugated with transferrin and arginine-glycine-aspartic acid peptide for glioma-targeting therapy. Oncol Lett. 2014;8:2000–2006. doi: 10.3892/ol.2014.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo LM, Huang Y, Zhao BX, Zhao X, Duan Y, Du R, Yu KF, Song P, Zhao Y, Zhang X, Zhang Q. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34:1102–1114. doi: 10.1016/j.biomaterials.2012.10.029. [DOI] [PubMed] [Google Scholar]
- 47.Dijkstra J, van Galen WJ, Hulstaert CE, Kalicharan D, Roerdink FH, Scherphof GL. Interaction of liposomes with Kupffer cells in vitro. Exp Cell Res. 1984;150:161–176. doi: 10.1016/0014-4827(84)90711-0. [DOI] [PubMed] [Google Scholar]
- 48.Tycko B, Maxfield FR. Rapid acidification of endocytic vesicles containing alpha 2-macroglobulin. Cell. 1982;28:643–651. doi: 10.1016/0092-8674(82)90219-7. [DOI] [PubMed] [Google Scholar]
- 49.Collins D, Litzinger DC, Huang L. Structural and functional comparisons of pH-sensitive liposomes composed of phosphatidylethanolamine and three different diacylsuccinylglycerols. Biochim Biophys Acta. 1990;1025:234–242. doi: 10.1016/0005-2736(90)90102-t. [DOI] [PubMed] [Google Scholar]
- 50.Rybak SL, Murphy RF. Primary cell cultures from murine kidney and heart differ in endosomal pH. J Cell Physiol. 1998;176:216–222. doi: 10.1002/(SICI)1097-4652(199807)176:1<216::AID-JCP23>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 51.Chen DQ, Jiang XQ, Huang YY, Zhang C, Ping QN. pH-Sensitive mPEG-Hz-Cholesterol Conjugates as a Liposome Delivery System. J Bioact Compat Pol. 2010;25:527–542. [Google Scholar]
- 52.Chen DQ, Jiang XQ, Liu J, Jin X, Zhang C, Ping QN. In Vivo Evaluation of Novel pH-sensitive mPEG-Hz-Chol Conjugate in Liposomes: Pharmacokinetics, Tissue Distribution, Efficacy Assessment. Artif Cell Blood Sub. 2010;38:136–142. doi: 10.3109/10731191003685481. [DOI] [PubMed] [Google Scholar]
- 53.Shi KR, Li JP, Cao ZL, Yang P, Qiu Y, Yang B, Wang Y, Long Y, Liu YY, Zhang QY, Qian J, Zhang ZR, Gao HL, He Q. A pH-responsive cell-penetrating peptide-modified liposomes with active recognizing of integrin alpha(v)beta(3) for the treatment of melanoma. Journal of Controlled Release. 2015;217:138–150. doi: 10.1016/j.jconrel.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 54.Torchilin VP. Micellar nanocarriers: Pharmaceutical perspectives. Pharm Res. 2007;24:1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 55.May S, Ben-Shaul A. Molecular theory of lipid-protein interaction and the Lalpha-HII transition. Biophys J. 1999;76:751–767. doi: 10.1016/S0006-3495(99)77241-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Advanced Drug Delivery Reviews. 2004;56:1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 57.Ashok B, Arleth L, Hjelm RP, Rubinstein I, Onyuksel H. In vitro characterization of PEGylated phospholipid micelles for improved drug solubilization: Effects of PEG chain length and PC incorporation. J Pharm Sci-Us. 2004;93:2476–2487. doi: 10.1002/jps.20150. [DOI] [PubMed] [Google Scholar]
- 58.Krishnadas A, Rubinstein I, Onyuksel H. Sterically stabilized phospholipid mixed micelles: In vitro evaluation as a novel carrier for water-insoluble drugs. Pharm Res. 2003;20:297–302. doi: 10.1023/a:1022243709003. [DOI] [PubMed] [Google Scholar]
- 59.Zhao BJ, Ke XY, Huang Y, Chen XM, Zhao X, Zhao BX, Lu WL, Lou JN, Zhang X, Zhang Q. The antiangiogenic efficacy of NGR-modified PEG-DSPE micelles containing paclitaxel (NGR-M-PTX) for the treatment of glioma in rats. J Drug Target. 2011;19:382–390. doi: 10.3109/1061186X.2010.504267. [DOI] [PubMed] [Google Scholar]
- 60.Qi JP, Lu Y, Wu W. Absorption, Disposition and Pharmacokinetics of Solid Lipid Nanoparticles. Curr Drug Metab. 2012;13:418–428. doi: 10.2174/138920012800166526. [DOI] [PubMed] [Google Scholar]
- 61.Shahgaldian P, Da Silva E, Coleman AW, Rather B, Zaworotko MJ. Para-acyl-calix-arene based solid lipid nanoparticles (SLNs): a detailed study of preparation and stability parameters. Int J Pharm. 2003;253:23–38. doi: 10.1016/s0378-5173(02)00639-7. [DOI] [PubMed] [Google Scholar]
- 62.Yegin BA, Benoit JP, Lamprecht A. Paclitaxel-loaded lipid nanoparticles prepared by solvent injection or ultrasound emulsification. Drug Dev Ind Pharm. 2006;32:1089–1094. doi: 10.1080/03639040600683501. [DOI] [PubMed] [Google Scholar]
- 63.Yuan H, Miao J, Du YZ, You J, Hu FQ, Zeng S. Cellular uptake of solid lipid nanoparticles and cytotoxicity of encapsulated paclitaxel in A549 cancer cells. Int J Pharm. 2008;348:137–145. doi: 10.1016/j.ijpharm.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 64.Dong XW, Mattingly CA, Tseng MT, Cho MJ, Liu Y, Adams VR, Mumper RJ. Doxorubicin and Paclitaxel-Loaded Lipid-Based Nanoparticles Overcome Multidrug Resistance by Inhibiting P-Glycoprotein and Depleting ATP. Cancer Res. 2009;69:3918–3926. doi: 10.1158/0008-5472.CAN-08-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen DB, Yang TZ, Lu WL, Zhang Q. In vitro and in vivo study of two types of long-circulating solid lipid nanoparticles containing paclitaxel. Chem Pharm Bull. 2001;49:1444–1447. doi: 10.1248/cpb.49.1444. [DOI] [PubMed] [Google Scholar]
- 66.Pandey V, Gajbhiye KR, Soni V. Lactoferrin-appended solid lipid nanoparticles of paclitaxel for effective management of bronchogenic carcinoma. Drug Deliv. 2015;22:199–205. doi: 10.3109/10717544.2013.877100. [DOI] [PubMed] [Google Scholar]
- 67.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–347. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 68.des Rieux A, Fievez V, Garinot M, Schneider YJ, Preat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116:1–27. doi: 10.1016/j.jconrel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 69.Di Toro R, Betti V, Spampinato S. Biocompatibility and integrin-mediated adhesion of human osteoblasts to poly(DL-lactide-co-glycolide) copolymers. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences. 2004;21:161–169. doi: 10.1016/j.ejps.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 70.Jin C, Wu H, Liu J, Bai L, Guo G. The effect of paclitaxel-loaded nanoparticles with radiation on hypoxic MCF-7 cells. Journal of clinical pharmacy and therapeutics. 2007;32:41–47. doi: 10.1111/j.1365-2710.2007.00796.x. [DOI] [PubMed] [Google Scholar]
- 71.Danhier F, Lecouturier N, Vroman B, Jerome C, Marchand-Brynaert J, Feron O, Preat V. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133:11–17. doi: 10.1016/j.jconrel.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 72.Fonseca C, Simoes S, Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release. 2002;83:273–286. doi: 10.1016/s0168-3659(02)00212-2. [DOI] [PubMed] [Google Scholar]
- 73.Mo Y, Lim LY. Paclitaxel-loaded PLGA nanoparticles: potentiation of anticancer activity by surface conjugation with wheat germ agglutinin. J Control Release. 2005;108:244–262. doi: 10.1016/j.jconrel.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 74.Vandervoort J, Ludwig A. Biocompatible stabilizers in the preparation of PLGA nanoparticles: a factorial design study. Int J Pharm. 2002;238:77–92. doi: 10.1016/s0378-5173(02)00058-3. [DOI] [PubMed] [Google Scholar]
- 75.Win KY, Feng SS. In vitro and in vivo studies on vitamin E TPGS-emulsified poly(D,L-lactic-co-glycolic acid) nanoparticles for paclitaxel formulation. Biomaterials. 2006;27:2285–2291. doi: 10.1016/j.biomaterials.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 76.Dong Y, Feng SS. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26:6068–6076. doi: 10.1016/j.biomaterials.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 77.Liu Y, Pan J, Feng SS. Nanoparticles of lipid monolayer shell and biodegradable polymer core for controlled release of paclitaxel: effects of surfactants on particles size, characteristics and in vitro performance. Int J Pharm. 2010;395:243–250. doi: 10.1016/j.ijpharm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 78.Feng S, Huang G. Effects of emulsifiers on the controlled release of paclitaxel (Taxol) from nanospheres of biodegradable polymers. J Control Release. 2001;71:53–69. doi: 10.1016/s0168-3659(00)00364-3. [DOI] [PubMed] [Google Scholar]
- 79.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 82.Kwon GS, Kataoka K. Block copolymer micelles as long-circulating drug vehicles. Adv Drug Deliv Rev. 1995;16:295–309. [Google Scholar]
- 83.Yoo HS, Park TG. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA-PEG block copolymer. J Control Release. 2001;70:63–70. doi: 10.1016/s0168-3659(00)00340-0. [DOI] [PubMed] [Google Scholar]
- 84.Danhier F, Vroman B, Lecouturier N, Crokart N, Pourcelle V, Freichels H, Jerome C, Marchand-Brynaert J, Feron O, Preat V. Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles loaded with Paclitaxel. Journal of Controlled Release. 2009;140:166–173. doi: 10.1016/j.jconrel.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 85.Gao NS, Chen ZH, Xiao XJ, Ruan CS, Mei L, Liu ZG, Zeng XW. Surface modification of paclitaxel-loaded tri-block copolymer PLGA-b-PEG-b-PLGA nanoparticles with protamine for liver cancer therapy. J Nanopart Res. 2015;17 [Google Scholar]
- 86.Milane L, Duan ZF, Amiji M. Pharmacokinetics and biodistribution of lonidamine/paclitaxel loaded, EGFR-targeted nanoparticles in an orthotopic animal model of multi-drug resistant breast cancer. Nanomedicine: nanotechnology, biology, and medicine. 2011;7:435–444. doi: 10.1016/j.nano.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liang C, Yang Y, Ling Y, Huang Y, Li T, Li X. Improved therapeutic effect of folate-decorated PLGA-PEG nanoparticles for endometrial carcinoma. Bioorganic & medicinal chemistry. 2011;19:4057–4066. doi: 10.1016/j.bmc.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 88.Wang Z, Chui WK, Ho PC. Nanoparticulate delivery system targeted to tumor neovasculature for combined anticancer and antiangiogenesis therapy. Pharm Res. 2011;28:585–596. doi: 10.1007/s11095-010-0308-2. [DOI] [PubMed] [Google Scholar]
- 89.Sahoo SK, Ma W, Labhasetwar V. Efficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancer. International journal of cancer. 2004;112:335–340. doi: 10.1002/ijc.20405. [DOI] [PubMed] [Google Scholar]
- 90.Guo J, Gao X, Su L, Xia H, Gu G, Pang Z, Jiang X, Yao L, Chen J, Chen H. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials. 2011;32:8010–8020. doi: 10.1016/j.biomaterials.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 91.Rancan F, Papakostas D, Hadam S, Hackbarth S, Delair T, Primard C, Verrier B, Sterry W, Blume-Peytavi U, Vogt A. Investigation of polylactic acid (PLA) nanoparticles as drug delivery systems for local dermatotherapy. Pharm Res. 2009;26:2027–2036. doi: 10.1007/s11095-009-9919-x. [DOI] [PubMed] [Google Scholar]
- 92.Dong Y, Feng SS. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25:2843–2849. doi: 10.1016/j.biomaterials.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 93.Dong Y, Feng SS. In vitro and in vivo evaluation of methoxy polyethylene glycol-polylactide (MPEG-PLA) nanoparticles for small-molecule drug chemotherapy. Biomaterials. 2007;28:4154–4160. doi: 10.1016/j.biomaterials.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 94.Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33:4889–4906. doi: 10.1016/j.biomaterials.2012.03.046. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Z, Feng SS. Nanoparticles of poly(lactide)/vitamin E TPGS copolymer for cancer chemotherapy: synthesis, formulation, characterization and in vitro drug release. Biomaterials. 2006;27:262–270. doi: 10.1016/j.biomaterials.2005.05.104. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Z, Lee SH, Gan CW, Feng SS. In vitro and in vivo investigation on PLA-TPGS nanoparticles for controlled and sustained small molecule chemotherapy. Pharm Res. 2008;25:1925–1935. doi: 10.1007/s11095-008-9611-6. [DOI] [PubMed] [Google Scholar]
- 97.Pan J, Feng SS. Targeted delivery of paclitaxel using folate-decorated poly(lactide)-vitamin E TPGS nanoparticles. Biomaterials. 2008;29:2663–2672. doi: 10.1016/j.biomaterials.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 98.He G, Ma LL, Pan J, Venkatraman S. ABA and BAB type triblock copolymers of PEG and PLA: a comparative study of drug release properties and “stealth” particle characteristics. Int J Pharm. 2007;334:48–55. doi: 10.1016/j.ijpharm.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 99.Venkatraman SS, Jie P, Min F, Freddy BY, Leong-Huat G. Micelle-like nanoparticles of PLA-PEG-PLA triblock copolymer as chemotherapeutic carrier. Int J Pharm. 2005;298:219–232. doi: 10.1016/j.ijpharm.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 100.Zhang Z, Feng SS. In vitro investigation on poly(lactide)-Tween 80 copolymer nanoparticles fabricated by dialysis method for chemotherapy. Biomacromolecules. 2006;7:1139–1146. doi: 10.1021/bm050953v. [DOI] [PubMed] [Google Scholar]
- 101.Liang HF, Chen SC, Chen MC, Lee PW, Chen CT, Sung HW. Paclitaxel-loaded poly(gamma-glutamic acid)-poly(lactide) nanoparticles as a targeted drug delivery system against cultured HepG2 cells. Bioconjugate chemistry. 2006;17:291–299. doi: 10.1021/bc0502107. [DOI] [PubMed] [Google Scholar]
- 102.Xu Q, Liu Y, Su S, Li W, Chen C, Wu Y. Anti-tumor activity of paclitaxel through dual-targeting carrier of cyclic RGD and transferrin conjugated hyperbranched copolymer nanoparticles. Biomaterials. 2012;33:1627–1639. doi: 10.1016/j.biomaterials.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 103.Cirstoiu-Hapca A, Buchegger F, Bossy L, Kosinski M, Gurny R, Delie F. Nanomedicines for active targeting: Physico-chemical characterization of paclitaxel-loaded anti-HER2 immunonanoparticles and in vitro functional studies on target cells. European Journal of Pharmaceutical Sciences. 2009;38:230–237. doi: 10.1016/j.ejps.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 104.Patil YB, Toti US, Khdair A, Ma L, Panyam J. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials. 2009;30:859–866. doi: 10.1016/j.biomaterials.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hu Z, Luo F, Pan Y, Hou C, Ren L, Chen J, Wang J, Zhang Y. Arg-Gly-Asp (RGD) peptide conjugated poly(lactic acid)-poly(ethylene oxide) micelle for targeted drug delivery. Journal of biomedical materials research Part A. 2008;85:797–807. doi: 10.1002/jbm.a.31615. [DOI] [PubMed] [Google Scholar]
- 106.Zhan C, Li B, Hu L, Wei X, Feng L, Fu W, Lu W. Micelle-based brain-targeted drug delivery enabled by a nicotine acetylcholine receptor ligand. Angewandte Chemie. 2011;50:5482–5485. doi: 10.1002/anie.201100875. [DOI] [PubMed] [Google Scholar]
- 107.Geng Y, Dalhaimer P, Cai SS, Tsai R, Tewari M, Minko T, Discher DE. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gou M, Wei X, Men K, Wang B, Luo F, Zhao X, Wei Y, Qian Z. PCL/PEG copolymeric nanoparticles: potential nanoplatforms for anticancer agent delivery. Current drug targets. 2011;12:1131–1150. doi: 10.2174/138945011795906642. [DOI] [PubMed] [Google Scholar]
- 109.Wang C, Wang Y, Wang Y, Fan M, Luo F, Qian Z. Characterization, pharmacokinetics and disposition of novel nanoscale preparations of paclitaxel. Int J Pharm. 2011;414:251–259. doi: 10.1016/j.ijpharm.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 110.Xin H, Chen L, Gu J, Ren X, Wei Z, Luo J, Chen Y, Jiang X, Sha X, Fang X. Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(varepsilon-caprolactone) nanoparticles: In vitro and in vivo evaluation. Int J Pharm. 2010;402:238–247. doi: 10.1016/j.ijpharm.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 111.Xin H, Jiang X, Gu J, Sha X, Chen L, Law K, Chen Y, Wang X, Jiang Y, Fang X. Angiopep-conjugated poly(ethylene glycol)-co-poly(epsilon-caprolactone) nanoparticles as dual-targeting drug delivery system for brain glioma. Biomaterials. 2011;32:4293–4305. doi: 10.1016/j.biomaterials.2011.02.044. [DOI] [PubMed] [Google Scholar]
- 112.Ke XY, Zhao BJ, Zhao X, Wang Y, Huang Y, Chen XM, Zhao BX, Zhao SS, Zhang XA, Zhang QA. The therapeutic efficacy of conjugated linoleic acid - Paclitaxel on glioma in the rat. Biomaterials. 2010;31:5855–5864. doi: 10.1016/j.biomaterials.2010.03.079. [DOI] [PubMed] [Google Scholar]
- 113.Zhu Z, Li Y, Li X, Li R, Jia Z, Liu B, Guo W, Wu W, Jiang X. Paclitaxel-loaded poly(N-vinylpyrrolidone)-b-poly(epsilon-caprolactone) nanoparticles: preparation and antitumor activity in vivo. J Control Release. 2010;142:438–446. doi: 10.1016/j.jconrel.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 114.Zhang PC, Hu LJ, Yin Q, Zhang ZW, Feng LY, Li YP. Transferrin-conjugated polyphosphoester hybrid micelle loading paclitaxel for brain-targeting delivery: Synthesis, preparation and in vivo evaluation. Journal of Controlled Release. 2012;159:429–434. doi: 10.1016/j.jconrel.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 115.Hu Y, Xie J, Tong YW, Wang CH. Effect of PEG conformation and particle size on the cellular uptake efficiency of nanoparticles with the HepG2 cells. J Control Release. 2007;118:7–17. doi: 10.1016/j.jconrel.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 116.Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang LF. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Su JH, Sun HP, Meng QS, Yin Q, Tang S, Zhang PC, Chen Y, Zhang ZW, Yu HJ, Li YP. Long Circulation Red-Blood-Cell-Mimetic Nanoparticles with Peptide-Enhanced Tumor Penetration for Simultaneously Inhibiting Growth and Lung Metastasis of Breast Cancer. Adv Funct Mater. 2016;26:1243–1252. [Google Scholar]
- 118.Su JH, Sun HP, Meng QS, Yin Q, Zhang PC, Zhang ZW, Yu HJ, Li YP. Bioinspired Nanoparticles with NIR-Controlled Drug Release for Synergetic Chemophotothermal Therapy of Metastatic Breast Cancer. Adv Funct Mater. 2016;26:7495–7506. [Google Scholar]
- 119.Anselmo AC, Mitragotri S. Cell-mediated delivery of nanoparticles: taking advantage of circulatory cells to target nanoparticles. J Control Release. 2014;190:531–541. doi: 10.1016/j.jconrel.2014.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fang RH, Jiang Y, Fang JC, Zhang L. Cell membrane-derived nanomaterials for biomedical applications. Biomaterials. 2017;128:69–83. doi: 10.1016/j.biomaterials.2017.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim C, Lee SC, Kang SW, Kwon IC, Kim YH, Jeong SY. Synthesis and the micellar characteristics of poly(ethylene oxide)-deoxycholic acid conjugates. Langmuir. 2000;16:4792–4797. [Google Scholar]
- 122.Kim C, Lee SC, Kwon IC, Chung H, Jeong SY. Complexation of poly(2-ethyl-2-oxazoline)-block-poly(epsilon-caprolactone) micelles with multifunctional carboxylic acids. Macromolecules. 2002;35:193–200. [Google Scholar]
- 123.Chen XG, Park HJ. Chemical characteristics of O-carboxymethyl chitosans related to the preparation conditions. Carbohyd Polym. 2003;53:355–359. [Google Scholar]
- 124.Kwon S, Park JH, Chung H, Kwon IC, Jeong SY, Kim IS. Physicochemical characteristics of self-assembled nanoparticles based on glycol chitosan bearing 5 beta-cholanic acid. Langmuir. 2003;19:10188–10193. [Google Scholar]
- 125.Sinha VR, Singla AK, Wadhawan S, Kaushik R, Kumria R, Bansal K, Dhawan S. Chitosan microspheres as a potential carrier for drugs. Int J Pharm. 2004;274:1–33. doi: 10.1016/j.ijpharm.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 126.Kim JH, Kim YS, Kim S, Park JH, Kim K, Choi K, Chung H, Jeong SY, Park RW, Kim IS, Kwon IC. Hydrophobically modified glycol chitosan nanoparticles as carriers for paclitaxel. J Control Release. 2006;111:228–234. doi: 10.1016/j.jconrel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 127.Koo H, Min KH, Lee SC, Park JH, Park K, Jeong SY, Choi K, Kwon IC, Kim K. Enhanced drug-loading and therapeutic efficacy of hydrotropic oligomer-conjugated glycol chitosan nanoparticles for tumor-targeted paclitaxel delivery. J Control Release. 2013;172:823–831. doi: 10.1016/j.jconrel.2013.08.297. [DOI] [PubMed] [Google Scholar]
- 128.Zhang C, Qu G, Sun Y, Wu X, Yao Z, Guo Q, Ding Q, Yuan S, Shen Z, Ping Q, Zhou H. Pharmacokinetics, biodistribution, efficacy and safety of N-octyl-O-sulfate chitosan micelles loaded with paclitaxel. Biomaterials. 2008;29:1233–1241. doi: 10.1016/j.biomaterials.2007.11.029. [DOI] [PubMed] [Google Scholar]
- 129.Qu G, Yao Z, Zhang C, Wu X, Ping Q. PEG conjugated N-octyl-O-sulfate chitosan micelles for delivery of paclitaxel: in vitro characterization and in vivo evaluation. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences. 2009;37:98–105. doi: 10.1016/j.ejps.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 130.Liu WG, Zhang X, Sun SJ, Sun GJ, Yao KD, Liang DC, Guo G, Zhang JY. N-alkylated chitosan as a potential nonviral vector for gene transfection. Bioconjugate chemistry. 2003;14:782–789. doi: 10.1021/bc020051g. [DOI] [PubMed] [Google Scholar]
- 131.Liang N, Sun S, Li X, Piao H, Piao H, Cui F, Fang L. alpha-Tocopherol succinate-modified chitosan as a micellar delivery system for paclitaxel: preparation, characterization and in vitro/in vivo evaluations. Int J Pharm. 2012;423:480–488. doi: 10.1016/j.ijpharm.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 132.Zhang Y, Huo MR, Zhou JP, Yu D, Wu YP. Potential of amphiphilically modified low molecular weight chitosan as a novel carrier for hydrophobic anticancer drug: Synthesis, characterization, micellization and cytotoxicity evaluation. Carbohyd Polym. 2009;77:231–238. [Google Scholar]
- 133.Wang F, Chen Y, Zhang D, Zhang Q, Zheng D, Hao L, Liu Y, Duan C, Jia L, Liu G. Folate-mediated targeted and intracellular delivery of paclitaxel using a novel deoxycholic acid-O-carboxymethylated chitosan-folic acid micelles. International journal of nanomedicine. 2012;7:325–337. doi: 10.2147/IJN.S27823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Auzenne E, Ghosh SC, Khodadadian M, Rivera B, Farquhar D, Price RE, Ravoori M, Kundra V, Freedman RS, Klostergaard J. Hyaluronic acid-paclitaxel: antitumor efficacy against CD44(+) human ovarian carcinoma xenografts. Neoplasia. 2007;9:479–486. doi: 10.1593/neo.07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eliaz RE, Szoka FC., Jr Liposome-encapsulated doxorubicin targeted to CD44: a strategy to kill CD44-overexpressing tumor cells. Cancer Res. 2001;61:2592–2601. [PubMed] [Google Scholar]
- 136.Lee H, Lee K, Park TG. Hyaluronic acid-paclitaxel conjugate micelles: synthesis, characterization, and antitumor activity. Bioconjugate chemistry. 2008;19:1319–1325. doi: 10.1021/bc8000485. [DOI] [PubMed] [Google Scholar]
- 137.Platt VM, Szoka FC., Jr Anticancer therapeutics: targeting macromolecules and nanocarriers to hyaluronan or CD44, a hyaluronan receptor. Molecular pharmaceutics. 2008;5:474–486. doi: 10.1021/mp800024g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Saravanakumar G, Choi KY, Yoon HY, Kim K, Park JH, Kwon IC, Park K. Hydrotropic hyaluronic acid conjugates: Synthesis, characterization, and implications as a carrier of paclitaxel. Int J Pharm. 2010;394:154–161. doi: 10.1016/j.ijpharm.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 139.Li J, Huo M, Wang J, Zhou J, Mohammad JM, Zhang Y, Zhu Q, Waddad AY, Zhang Q. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33:2310–2320. doi: 10.1016/j.biomaterials.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 140.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Prodrugs: design and clinical applications. Nature Reviews Drug Discovery. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 141.Ettmayer P, Amidon GL, Clement B, Testa B. Lessons learned from marketed and investigational prodrugs. J Med Chem. 2004;47:2393–2404. doi: 10.1021/jm0303812. [DOI] [PubMed] [Google Scholar]
- 142.Skwarczynski M, Hayashi Y, Kiso Y. Paclitaxel prodrugs: Toward smarter delivery of anticancer agents. J Med Chem. 2006;49:7253–7269. doi: 10.1021/jm0602155. [DOI] [PubMed] [Google Scholar]
- 143.Khandare J, Minko T. Polymer-drug conjugates: Progress in polymeric prodrugs. Prog Polym Sci. 2006;31:359–397. [Google Scholar]
- 144.Yu Y, Chen CK, Law WC, Mok J, Zou J, Prasad PN, Cheng C. Well-Defined Degradable Brush Polymer-Drug Conjugates for Sustained Delivery of Paclitaxel. Molecular pharmaceutics. 2013;10:867–874. doi: 10.1021/mp3004868. [DOI] [PubMed] [Google Scholar]
- 145.Yu Y, Zou J, Yu L, Jo W, Li YK, Law WC, Cheng C. Functional Polylactide-g-Paclitaxel-Poly(ethylene glycol) by Azide-Alkyne Click Chemistry. Macromolecules. 2011;44:4793–4800. [Google Scholar]
- 146.Li C, Yu DF, Inoue T, Yang DJ, Milas L, Hunter NR, Kim EE, Wallace S. Synthesis and evaluation of water-soluble polyethylene glycol-paclitaxel conjugate as a paclitaxel prodrug. Anti-Cancer Drug. 1996;7:642–648. doi: 10.1097/00001813-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 147.Erez R, Segal E, Miller K, Satchi-Fainaro R, Shabat D. Enhanced cytotoxicity of a polymer-drug conjugate with triple payload of paclitaxel. Bioorganic & medicinal chemistry. 2009;17:4327–4335. doi: 10.1016/j.bmc.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 148.Singer JW. Paclitaxel poliglumex (XYOTAX (TM), CT-2103): A macromolecular taxane. Journal of Controlled Release. 2005;109:120–126. doi: 10.1016/j.jconrel.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 149.Tong R, Cheng JJ. Paclitaxel-initiated, controlled polymerization of lactide for the formulation of polymeric nanoparticulate delivery vehicles. Angew Chem Int Edit. 2008;47:4830–4834. doi: 10.1002/anie.200800491. [DOI] [PubMed] [Google Scholar]
- 150.Pendri A, Conover CD, Greenwald RB. Antitumor activity of paclitaxel-2′-glycinate conjugated to poly(ethylene glycol): a water-soluble prodrug. Anti-Cancer Drug Des. 1998;13:387–395. [PubMed] [Google Scholar]
- 151.Galic VL, Herzog TJ, Wright JD, Lewin SN. Paclitaxel poliglumex for ovarian cancer. Expert Opin Inv Drug. 2011;20:813–821. doi: 10.1517/13543784.2011.576666. [DOI] [PubMed] [Google Scholar]
- 152.Satsangi A, Roy SS, Satsangi RK, Vadlamudi RK, Ong JL. Design of a Paclitaxel Prodrug Conjugate for Active Targeting of an Enzyme Upregulated in Breast Cancer Cells. Molecular pharmaceutics. 2014;11:1906–1918. doi: 10.1021/mp500128k. [DOI] [PubMed] [Google Scholar]
- 153.Lim J, Lo ST, Hill S, Pavan GM, Sun XK, Simanek EE. Antitumor Activity and Molecular Dynamics Simulations of Paclitaxel-Laden Triazine Dendrimers. Molecular pharmaceutics. 2012;9:404–412. doi: 10.1021/mp2005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Eldar-Boock A, Miller K, Sanchis J, Lupu R, Vicent MJ, Satchi-Fainaro R. Integrin-assisted drug delivery of nano-scaled polymer therapeutics bearing paclitaxel. Biomaterials. 2011;32:3862–3874. doi: 10.1016/j.biomaterials.2011.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang X, Li J, Wang YQ, Cho KJ, Kim G, Gjyrezi A, Koenig L, Giannakakou P, Shin HJC, Tighiouart M, Nie SM, Chen Z, Shin DM. HFT-T, a Targeting Nanoparticle, Enhances Specific Delivery of Paclitaxel to Folate Receptor-Positive Tumors. Acs Nano. 2009;3:3165–3174. doi: 10.1021/nn900649v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yin TJ, Wu Q, Wang L, Yin LF, Zhou JP, Huo MR. Well-Defined Redox-Sensitive Polyethene Glycol-Paclitaxel Prodrug Conjugate for Tumor-Specific Delivery of Paclitaxel Using Octreotide for Tumor Targeting. Molecular pharmaceutics. 2015;12:3020–3031. doi: 10.1021/acs.molpharmaceut.5b00280. [DOI] [PubMed] [Google Scholar]
- 157.Zhang SY, Zou J, Elsabahy M, Karwa A, Li A, Moore DA, Dorshow RB, Wooley KL. Poly(ethylene oxide)-block-polyphosphester-based paclitaxel conjugates as a platform for ultra-high paclitaxel-loaded multifunctional nanoparticles. Chem Sci. 2013;4:2122–2126. doi: 10.1039/C3SC50252J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kim H, Lee E, Lee IH, Lee J, Kim J, Kim S, Lee Y, Kim D, Choi M, Kim YC, Jon S. Preparation and therapeutic evaluation of paclitaxel-conjugated low-molecular-weight chitosan nanoparticles. Macromol Res. 2014;22:805–808. [Google Scholar]
- 159.Leader B, Baca QJ, Golan DE. Protein therapeutics: A summary and pharmacological classification. Nature Reviews Drug Discovery. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 160.Karmali PP, Kotamraju VR, Kastantin M, Black M, Missirlis D, Tirrell M, Ruoslahti E. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomed-Nanotechnol. 2009;5:73–82. doi: 10.1016/j.nano.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shan LL, Shan X, Zhang T, Zhai KF, Gao GZ, Chen XY, Gu YQ. Transferrin-conjugated paclitaxel prodrugs for targeted cancer therapy. Rsc Adv. 2016;6:77987–77998. [Google Scholar]
- 162.Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345–352. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- 163.Quiles S, Raisch KP, Sanford LL, Bonner JA, Safavy A. Synthesis and Preliminary Biological Evaluation of High-Drug-Load Paclitaxel-Antibody Conjugates for Tumor-Targeted Chemotherapy. J Med Chem. 2010;53:586–594. doi: 10.1021/jm900899g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wu X, Ojima I. Tumor specific novel taxoid-monoclonal antibody conjugates. Curr Med Chem. 2004;11:429–438. doi: 10.2174/0929867043455963. [DOI] [PubMed] [Google Scholar]
- 165.Zhang PC, Cheetham AG, Lin YA, Cui H. Self-Assembled Tat Nanofibers as Effective Drug Carrier and Transporter. Acs Nano. 2013;7:5965–5977. doi: 10.1021/nn401667z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang PC, Hu LJ, Yin Q, Feng LY, Li YP. Transferrin-Modified c[RGDfK]-Paclitaxel Loaded Hybrid Micelle for Sequential Blood-Brain Barrier Penetration and Glioma Targeting Therapy. Molecular pharmaceutics. 2012;9:1590–1598. doi: 10.1021/mp200600t. [DOI] [PubMed] [Google Scholar]
- 167.Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Deliv Rev. 2016 doi: 10.1016/j.addr.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen ZP, Zhang PC, Cheetham AG, Moon JH, Moxley JW, Lin YA, Cui HG. Controlled release of free doxorubicin from peptide-drug conjugates by drug loading. Journal of Controlled Release. 2014;191:123–130. doi: 10.1016/j.jconrel.2014.05.051. [DOI] [PubMed] [Google Scholar]
- 169.Su H, Koo JM, Cui HG. One-component nanomedicine. Journal of Controlled Release. 2015;219:383–395. doi: 10.1016/j.jconrel.2015.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chakroun RW, Zhang P, Lin R, Schiapparelli P, Quinones-Hinojosa A, Cui H. Nanotherapeutic systems for local treatment of brain tumors. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2017 doi: 10.1002/wnan.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cheetham AG, Lin Y-a, Lin R, Cui H. Molecular design and synthesis of self-assembling camptothecin drug amphiphiles. Acta Pharmacologica Sinica. 2017;38:874. doi: 10.1038/aps.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cheetham AG, Zhang P, Lin YA, Lin R, Cui H. Synthesis and self-assembly of a mikto-arm star dual drug amphiphile containing both paclitaxel and camptothecin. J Mater Chem B. 2014;2:7316–7326. doi: 10.1039/C4TB01084A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lock LL, LaComb M, Schwarz K, Cheetham AG, Lin Y-a, Zhang P, Cui H. Self-assembly of natural and synthetic drug amphiphiles into discrete supramolecular nanostructures. Faraday discussions. 2013;166:285–301. doi: 10.1039/c3fd00099k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ma W, Su H, Cheetham AG, Zhang W, Wang Y, Kan Q, Cui H. Synergistic antitumor activity of a self-assembling camptothecin and capecitabine hybrid prodrug for improved efficacy. Journal of Controlled Release. 2017 doi: 10.1016/j.jconrel.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 175.Lin R, Cheetham AG, Zhang PC, Lin YA, Cui HG. Supramolecular filaments containing a fixed 41% paclitaxel loading. Chem Commun. 2013;49:4968–4970. doi: 10.1039/c3cc41896k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tian R, Wang HM, Niu RF, Ding D. Drug delivery with nanospherical supramolecular cell penetrating peptide-taxol conjugates containing a high drug loading. J Colloid Interf Sci. 2015;453:15–20. doi: 10.1016/j.jcis.2015.04.028. [DOI] [PubMed] [Google Scholar]
- 177.Yuan Y, Wang L, Du W, Ding ZL, Zhang J, Han T, An LN, Zhang HF, Liang GL. Intracellular Self-Assembly of Taxol Nanoparticles for Overcoming Multidrug Resistance. Angew Chem Int Edit. 2015;54:9700–9704. doi: 10.1002/anie.201504329. [DOI] [PubMed] [Google Scholar]
- 178.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12128–12133. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Furgeson DY, Dreher MR, Chilkoti A. Structural optimization of a “smart” doxorubicin-polypeptide conjugate for thermally targeted delivery to solid tumors. Journal of Controlled Release. 2006;110:362–369. doi: 10.1016/j.jconrel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 180.Chilkoti A, Christensen T, MacKay JA. Stimulus responsive elastin biopolymers: applications in medicine and biotechnology. Curr Opin Chem Biol. 2006;10:652–657. doi: 10.1016/j.cbpa.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li XH, Lin CC, Chi JTA, Chilkoti A. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer models. Nat Commun. 2015;6 doi: 10.1038/ncomms8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Moktan S, Ryppa C, Kratz F, Raucher D. A thermally responsive biopolymer conjugated to an acid-sensitive derivative of paclitaxel stabilizes microtubules, arrests cell cycle, and induces apoptosis. Invest New Drug. 2012;30:236–248. doi: 10.1007/s10637-010-9560-x. [DOI] [PubMed] [Google Scholar]
- 183.Mahato R, Tai WY, Cheng K. Prodrugs for improving tumor targetability and efficiency. Advanced Drug Delivery Reviews. 2011;63:659–670. doi: 10.1016/j.addr.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bradley MO, Swindell CS, Anthony FH, Witman PA, Devanesan P, Webb NL, Baker SD, Wolff AC, Donehower RC. Tumor targeting by conjugation of DHA to paclitaxel. Journal of Controlled Release. 2001;74:233–236. doi: 10.1016/s0168-3659(01)00321-2. [DOI] [PubMed] [Google Scholar]
- 185.Tam YT, Gao JM, Kwon GS. Oligo(lactic acid)(n)-Paclitaxel Prodrugs for Poly(ethylene glycol)-block-poly(lactic acid) Micelles: Loading, Release, and Backbiting Conversion for Anticancer Activity. J Am Chem Soc. 2016;138:8674–8677. doi: 10.1021/jacs.6b03995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Owen SC, Chan DPY, Shoichet MS. Polymeric micelle stability. Nano Today. 2012;7:53–65. [Google Scholar]
- 187.Ansell SM, Johnstone SA, Tardi PG, Lo L, Xie SW, Shu Y, Harasym TO, Harasym NL, Williams L, Bermudes D, Liboiron BD, Saad W, Prud’homme RK, Mayer LD. Modulating the therapeutic activity of nanoparticle delivered paclitaxel by manipulating the hydrophobicity of prodrug conjugates. J Med Chem. 2008;51:3288–3296. doi: 10.1021/jm800002y. [DOI] [PubMed] [Google Scholar]
- 188.Abu Ajaj K, Biniossek ML, Kratz F. Development of Protein-Binding Bifunctional Linkers for a New Generation of Dual-Acting Prodrugs. Bioconjugate chemistry. 2009;20:390–396. doi: 10.1021/bc800429q. [DOI] [PubMed] [Google Scholar]
- 189.Mao LN, Wang HM, Tan M, Ou LL, Kong DL, Yang ZM. Conjugation of two complementary anti-cancer drugs confers molecular hydrogels as a co-delivery system. Chem Commun. 2012;48:395–397. doi: 10.1039/c1cc16250k. [DOI] [PubMed] [Google Scholar]
- 190.Bath J, Turberfield AJ. DNA nanomachines. Nat Nanotechnol. 2007;2:275–284. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]
- 191.Tan XY, Lu XG, Jia F, Liu XF, Sun YH, Logan JK, Zhang K. Blurring the Role of Oligonucleotides: Spherical Nucleic Acids as a Drug Delivery Vehicle. J Am Chem Soc. 2016;138:10834–10837. doi: 10.1021/jacs.6b07554. [DOI] [PubMed] [Google Scholar]
- 192.Peppas NA, Bures P, Leobandung W, Ichikawa H. Hydrogels in pharmaceutical formulations. Eur J Pharm Biopharm. 2000;50:27–46. doi: 10.1016/s0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 193.Lin CC, Metters AT. Hydrogels in controlled release formulations: Network design and mathematical modeling. Advanced Drug Delivery Reviews. 2006;58:1379–1408. doi: 10.1016/j.addr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 194.Anderson CF, Cui H. Protease-Sensitive Nanomaterials for Cancer Therapeutics and Imaging. Industrial & Engineering Chemistry Research. 2017;56:5761–5777. doi: 10.1021/acs.iecr.7b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hu Y, Lin R, Patel K, Cheetham AG, Kan C, Cui H. Spatiotemporal control of the creation and immolation of peptide assemblies. Coordination Chemistry Reviews. 2016;320:2–17. [Google Scholar]
- 196.Li Y, Wang F, Cui H. Peptide-Based Supramolecular Hydrogels for Delivery of Biologics. Bioengineering & Translational Medicine. 2016 doi: 10.1002/btm2.10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Van Tomme SR, Storm G, Hennink WE. In situ gelling hydrogels for pharmaceutical and biomedical applications. Int J Pharm. 2008;355:1–18. doi: 10.1016/j.ijpharm.2008.01.057. [DOI] [PubMed] [Google Scholar]
- 198.Guo DD, Xu CX, Quan JS, Song CK, Jin H, Kim DD, Choi YJ, Cho MH, Cho CS. Synergistic anti-tumor activity of paclitaxel-incorporated conjugated linoleic acid-coupled poloxamer thermosensitive hydrogel in vitro and in vivo. Biomaterials. 2009;30:4777–4785. doi: 10.1016/j.biomaterials.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 199.Elstad NL, Fowers KD. OncoGel (ReGel/paclitaxel) - Clinical applications for a novel paclitaxel delivery system. Advanced Drug Delivery Reviews. 2009;61:785–794. doi: 10.1016/j.addr.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 200.Jeong B, Bae YH, Lee DS, Kim SW. Biodegradable block copolymers as injectable drug-delivery systems. Nature. 1997;388:860–862. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- 201.Zentner GM, Rathi R, Shih C, McRea JC, Seo MH, Oh H, Rhee BG, Mestecky J, Moldoveanu Z, Morgan M, Weitman S. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. Journal of Controlled Release. 2001;72:203–215. doi: 10.1016/s0168-3659(01)00276-0. [DOI] [PubMed] [Google Scholar]
- 202.Ruel-Gariepy E, Leclair G, Hildgen P, Gupta A, Leroux JC. Thermosensitive chitosan-based hydrogel containing liposomes for the delivery of hydrophilic molecules. Journal of Controlled Release. 2002;82:373–383. doi: 10.1016/s0168-3659(02)00146-3. [DOI] [PubMed] [Google Scholar]
- 203.Ruel-Gariepy E, Shive M, Bichara A, Berrada M, Le Garrec D, Chenite A, Leroux JC. A thermosensitive chitosan-based hydrogel for the local delivery of paclitaxel. Eur J Pharm Biopharm. 2004;57:53–63. doi: 10.1016/s0939-6411(03)00095-x. [DOI] [PubMed] [Google Scholar]
- 204.Bajaj G, Yeo Y. Hyaluronic acid-based hydrogel for regional delivery of paclitaxel to intraperitoneal tumors (vol 158, pg 386, 2012) Journal of Controlled Release. 2012;162:257–257. doi: 10.1016/j.jconrel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hendrickson GR, Smith MH, South AB, Lyon LA. Design of Multiresponsive Hydrogel Particles and Assemblies. Adv Funct Mater. 2010;20:1697–1712. [Google Scholar]
- 206.Oishi M, Nagasaki Y. Stimuli-responsive smart nanogels for cancer diagnostics and therapy. Nanomedicine: nanotechnology, biology, and medicine. 2010;5:451–468. doi: 10.2217/nnm.10.18. [DOI] [PubMed] [Google Scholar]
- 207.Zhao LL, Zhu LJ, Liu FY, Liu CY, Shan-Dan, Wang Q, Zhang CL, Li JL, Liu JG, Qu XZ, Yang ZZ. pH triggered injectable amphiphilic hydrogel containing doxorubicin and paclitaxel. Int J Pharm. 2011;410:83–91. doi: 10.1016/j.ijpharm.2011.03.034. [DOI] [PubMed] [Google Scholar]
- 208.Qiao ZY, Zhang R, Du FS, Liang DH, Li ZC. Multi-responsive nanogels containing motifs of ortho ester, oligo(ethylene glycol) and disulfide linkage as carriers of hydrophobic anti-cancer drugs. Journal of Controlled Release. 2011;152:57–66. doi: 10.1016/j.jconrel.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 209.Gao Y, Kuang Y, Guo ZF, Guo ZH, Krauss IJ, Xu B. Enzyme-Instructed Molecular Self-assembly Confers Nanofibers and a Supramolecular Hydrogel of Taxol Derivative. J Am Chem Soc. 2009;131:13576–+. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- 210.Toledano S, Williams RJ, Jayawarna V, Ulijn RV. Enzyme-triggered self-assembly of peptide hydrogels via reversed hydrolysis. J Am Chem Soc. 2006;128:1070–1071. doi: 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]
- 211.Williams RJ, Smith AM, Collins R, Hodson N, Das AK, Ulijn RV. Enzyme-assisted self-assembly under thermodynamic control. Nat Nanotechnol. 2009;4:19–24. doi: 10.1038/nnano.2008.378. [DOI] [PubMed] [Google Scholar]
- 212.Singh NK, Lee DS. In situ gelling pH- and temperature-sensitive biodegradable block copolymer hydrogels for drug delivery. Journal of Controlled Release. 2014;193:214–227. doi: 10.1016/j.jconrel.2014.04.056. [DOI] [PubMed] [Google Scholar]
- 213.Shim WS, Kim JH, Park H, Kim K, Kwon IC, Lee DS. Biodegradability and biocompatibility of a pH- and thermo-sensitive hydrogel formed from a sulfonamide-modified poly(epsilon-caprolactone-co-lactide)-poly(ethylene glycol)-poly(epsilon-caprolactone-co-lactide) block copolymer. Biomaterials. 2006;27:5178–5185. doi: 10.1016/j.biomaterials.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 214.Shim WS, Kim JH, Kim K, Kim YS, Park RW, Kim IS, Kwon IC, Lee DS. pH- and temperature-sensitive, injectable, biodegradable block copolymer hydrogels as carriers for paclitaxel. Int J Pharm. 2007;331:11–18. doi: 10.1016/j.ijpharm.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 215.Park ES, Maniar M, Shah JC. Biodegradable polyanhydride devices of cefazolin sodium, bupivacaine, and taxol for local drug delivery: preparation, and kinetics and mechanism of in vitro release. Journal of Controlled Release. 1998;52:179–189. doi: 10.1016/s0168-3659(97)00223-x. [DOI] [PubMed] [Google Scholar]
- 216.Wu MP, Tamada JA, Brem H, Langer R. In-Vivo Versus in-Vitro Degradation of Controlled-Release Polymers for Intracranial Surgical Therapy. J Biomed Mater Res. 1994;28:387–395. doi: 10.1002/jbm.820280314. [DOI] [PubMed] [Google Scholar]
- 217.Bota DA, Desjardins A, Quinn JA, Affronti ML, Friedman HS. Interstitial chemotherapy with biodegradable BCNU (Gliadel) wafers in the treatment of malignant gliomas. Ther Clin Risk Manag. 2007;3:707–715. [PMC free article] [PubMed] [Google Scholar]
- 218.Fleming AB, Saltzman WM. Pharmacokinetics of the carmustine implant. Clin Pharmacokinet. 2002;41:403–419. doi: 10.2165/00003088-200241060-00002. [DOI] [PubMed] [Google Scholar]
- 219.von Eckardstein KL, Reszka R, Kiwit JCW. Intracavitary chemotherapy (paclitaxel/carboplatin liquid crystalline cubic phases) for recurrent glioblastoma - clinical observations. J Neuro-Oncol. 2005;74:305–309. doi: 10.1007/s11060-004-7559-x. [DOI] [PubMed] [Google Scholar]
- 220.Ranganath SH, Wang CH. Biodegradable microfiber implants delivering paclitaxel for post-surgical chemotherapy against malignant glioma. Biomaterials. 2008;29:2996–3003. doi: 10.1016/j.biomaterials.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 221.Ong BYS, Ranganath SH, Lee LY, Lu F, Lee HS, Sahinidis NV, Wang CH. Paclitaxel delivery from PLGA foams for controlled release in post-surgical chemotherapy against glioblastoma multiforme. Biomaterials. 2009;30:3189–3196. doi: 10.1016/j.biomaterials.2009.02.030. [DOI] [PubMed] [Google Scholar]