

Abstract
The most common and deadly form of primary brain cancer, glioblastoma (GBM), is characterized by significant intratumoral heterogeneity, microvascular proliferation, immune system suppression, and brain tissue invasion. Delivering effective and sustained treatments to the invasive GBM cells intermixed with functioning neural elements is a major goal of advanced therapeutic systems for brain cancer. Previously, we investigated the nanoparticle characteristics that enable targeting of invasive GBM cells. This revealed the importance of minimizing non-specific binding within the relatively adhesive, ‘sticky’ microenvironment of the brain and brain tumors in particular. We refer to such nanoformulations with decreased non-specific adhesivity and receptor targeting as ‘DART’ therapeutics. In this work, we applied this information towards the design and characterization of biodegradable nanocarriers, and in vivo testing in orthotopic experimental gliomas. We formulated particulate nanocarriers using poly(lactic-co-glycolic acid) (PLGA) and PLGA-polyethylene glycol (PLGA-PEG) polymers to generate sub-100 nm nanoparticles with minimal binding to extracellular brain components and strong binding to the Fn14 receptor – an upregulated, conserved component in invasive GBM. Multiple particle tracking in brain tissue slices and in vivo testing in orthotopic murine malignant glioma revealed preserved nanoparticle diffusivity and increased uptake in brain tumor cells. These combined characteristics also resulted in longer retention of the DART nanoparticles within the orthotopic tumors compared to non-targeted versions. Taken together, these results and nanoparticle design considerations offer promising new methods to optimize therapeutic nanocarriers for improving drug delivery and treatment for invasive brain tumors.
Keywords: glioblastoma, invasive malignant glioma, biodegradable nanoparticles, targeted therapeutics, fibroblast growth factor-inducible 14, multiple particle tracking, surface plasmon resonance
Graphical Abstract
Fn14-targeted nanoparticles bind specifically to Fn14 receptor but not to brain ECM and are retained in invasive intracranial tumors over significantly longer periods than non-targeted nanoparticles.
Introduction
The most common and deadly form of adult brain cancer, glioblastoma (GBM), features brain invasion, immune evasion, molecular and cellular heterogeneity, rapid proliferation, and angiogenesis. This high degree of structural and biological complexity within the context of the central nervous system sets up a challenging, risky scenario for therapeutic delivery and GBM treatments [1]. The current standard of care for GBM includes maximal surgical removal when safe and feasible followed by radiation and chemotherapy to address the residual, invading tumor cells [2]. Most patients succumb to this disease in less than 18 months even with the most aggressive treatments [3]. Delivering effective and sustained treatments to the invading tumor cells residing within the functioning neural networks without worsening brain injury is a central goal of brain cancer nanomedicine.
Over the years, several strategies have emerged to deliver therapeutics more directly to the unresectable GBM components [4]. One of these strategies, carmustine interstitial wafer (CIW), was shown to improve survival in both primary and recurrent GBM patients, leading to FDA approval of CIW in 1997 and 2003 respectively [5, 6]. Despite this significant advance, the limited distribution and rapid clearance of the free drug once it is released from the biodegradable polymeric wafer [7, 8] led many groups to explore alternative formulations to further enhance therapeutic effects. In particular, these newer formulations were engineered to improve brain tissue penetration [9–11] and enable structure-specific targeting, such as tumor cells or extracellular components [11–15]. Pre-clinical testing of these newer drug formulations have shown early promise toward improving efficacy and safety of otherwise neurotoxic drugs like paclitaxel and cisplatin [10, 16, 17]. Lessons learned from these design modifications can also be applied to other therapeutic classes, namely antibodies and fusion proteins, antibody-drug conjugates, and viral and non-viral vectors.
Our prior work focused on the development of new nanoparticle characterization methods [18, 19] and studied the behavior of tumor-targeted model polystyrene (PS) nanoparticles designed to minimize off-target binding and adhesive brain interactions while maintaining specific GBM binding via the cell surface receptor fibroblast growth factor-inducible 14 (Fn14) [11, 18]. Notably, Fn14, which is a member of the tumor necrosis factor receptor (TNFR) superfamily [20], is frequently upregulated on primary and recurrent tumor cells in the invaded brain regions [21, 22], making this cell surface receptor a promising GBM element for targeted therapeutics [21].
In this study, we applied nanoparticle design considerations to formulate biodegradable decreased non-specific adhesivity, receptor targeted (DART) nanocarriers directed to invasive glioma cells via Fn14. We characterized and tested these new formulations in vitro, ex vivo, and in vivo using surface plasmon resonance (SPR), multiple particle tracking (MPT), flow cytometry, confocal microscopy, and near-infrared fluorescence imaging.
Materials and Methods
Materials
Methoxy terminated poly(lactic-co-glycolic acid)-polyethylene glycol (PLGA-PEG, 10:5 kDa), PLGA-PEG with maleimide end group (PLGA-PEG-Mal, 10:5 kDa), and PLGA-Rhodamine B (PLGA-Rhod, 10:30 kDa) were purchased from Polyscitech (West Lafayette, IN). Poly vinyl alcohol (PVA, 25 kDa) was purchased from Polysciences (Warrington, PA). Lab-Tek glass-bottom tissue culture plates and Zeba Spin Columns (7 kDa cut-off) were purchased from ThermoFisher Scientific (Rochester, NY). The ITEM4 monoclonal antibody was provided by Dr. Hideo Yagita (Juntendo University School of Medicine, Tokyo, Japan). Hoechst 33342 trihydrochloride and 100 nm PS FluoroSpheres were purchased from Invitrogen (Carlsbad, CA). D-Luciferin was purchased from Promega (Madison, WI). Cell culture materials, including Dulbecco’s modified eagles’s medium (DMEM), 0.25% trypsin, fetal bovine serum and penicillin-streptomycin, were purchased from Corning (Manassas, VA). PLGA (7–17 kDa, 50:50), cardiogreen (indocyanine green, ICG), chloroform-d (CDCl3), phosphate buffer solution (PBS), 2-iminothiolane hydrochloride, and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification.
Preparation of ITEM4-SH
ITEM4 was thiol-modified via reaction of free amines with 2-iminothiolane as described previously [11]. Briefly, ITEM4 (0.5 mg/mL) was mixed with 2-iminothiolane (140x molar excess to ITEM4) in 100 mM phosphate buffer with EDTA (pH 7.2, 150 mM NaCl, 5 mM EDTA). The reaction was allowed to proceed for 2 h at room temperature to yield thiolated ITEM4 (ITEM4-SH). After the reaction, resulting solution was purified with Zeba Spin Columns (7 kDa MW cut-off) and frozen immediately to avoid potential disulfide bond formation between newly generated thiol groups.
Nanoparticle preparation
To formulate biodegradable PLGA and PLGA-PEG nanoparticles, either single emulsion (for empty nanoparticles and Rhodamine-labeled nanoparticles) or double emulsion (for ICG-loaded nanoparticles) solvent evaporation technique was used. For single emulsion (Table S1), all the polymers were dissolved in dichloromethane (DCM) to form organic/oil phase. PVA (5% w/v) was dissolved in water and passed through 0.2 μm filter to form water phase. The oil phase was added to the water phase to form oil-in-water emulsion. For double emulsion (Table S1), aqueous ICG solution (0.5 mg/ml) was added dropwise to the polymer solution under vigorous stirring to form primary water-in-oil emulsion. After 30 min stirring, the primary emulsion was added to the aqueous PVA solution to form water-in-oil-in-water emulsion. All the emulsions were sonicated in an ice bath using ultrasonication probe (Sonics Vibra-Cell, Newton, CT) at 30% amplitude for 3 min with 20 sec on-off pulser. The sonicated emulsions were immediately transferred to magnetic stirring for 4 h at room temperature to allow organic solvent evaporation. The formed nanoparticles were washed by microcentrifugation at 21,100 × g for 10 min with ultrapure water (4 washes total). The nanoparticles were resuspended in ultrapure water and used fresh for experiments.
To formulate Fn14-targeted biodegradable PLGA-PEG nanoparticles, ITEM4-SH was conjugated onto the surface of PLGA-PEG nanoparticles containing maleimide functional groups by maleimide-thiol chemistry as described previously [11]. Briefly, PLGA-PEG-Mal (~1 wt% maleimide groups) nanoparticles were mixed with ITEM4-SH (1.2× excess ITEM4-SH to maleimide) in 100 mM phosphate buffer (pH 7.2, 150 mM NaCl) and allowed to react overnight at 4 °C. The maleimide-thiol linkage covalently conjugates the ITEM4 molecules to PEG molecules. This reaction was performed immediately following PLGA-PEG-Mal nanoparticle formulation to avoid hydrolysis of the maleimide groups due to longer incubation times. After the reaction, ITEM4-conjugated nanoparticles were purified from unconjugated free ITEM4-SH via microcentrifugation at 21,100 × g for 10 min with ultrapure water (3 washes total). The nanoparticles were resuspended and used fresh for experiments.
Physicochemical characterization of nanoparticles
The physicochemical characteristics of nanoparticles were measured in 15× diluted PBS (~10 mM NaCl, pH 7.4). Hydrodynamic diameter, polydispersity index (PDI) and ζ-potential (surface charge) were determined by dynamic light scattering and laser Doppler anemometry using Zetasizer NanoZS (Malvern Instruments, South Borough, MA). Particle size measurements were performed at 25 °C at a scattering angel of 173° and are reported as the number-average mean. The surface charge on the particles was calculated using the Smoluchowski equation and is reported as the mean ζ-potential. Nanoparticle structure and morphology was imaged using FEI Tecnai T12 transmission electron microscope (TEM, FEI, Hillsboro, OR) operated at 80 kV. For TEM analysis, nanoparticles were first mixed with methylcellulose and then stained with 0.3% uranyl acetate. Nanoparticle stability was analyzed by incubating in artificial cerebrospinal fluid (ACSF, Tocris Bioscience, Minneapolis, MN) at 37 °C and hydrodynamic size was measured at different time points.
The surface density of PEG (# of PEG chains/100 nm2) and Γ/Γ*, where Γ is the PEG surface coverage over the total surface area (Γ*), was calculated from the 1H integrals of the ethylene oxide peak of PEG using a previously described method [9, 19, 23]. Briefly, nanoparticles were lyophilized, weighed and dissolved in CDCl3 containing 0.1% (v/v) trimethylsilane as an internal standard. Nuclear magnetic resonance (NMR) spectra were obtained at 500 MHz using Agilent DD2 500 MHz Spectrometer. A calibration curve was obtained by plotting the 1H NMR integrals of various concentrations of 5 kDa PEG (~ 3.6 ppm) in CDCl3 solvent containing 0.1% (v/v) trimethylsilane. The average PEG surface density (# of PEG chains/100 nm2) on the surface of the nanoparticles was calculated by taking the total quantity of PEG detected by NMR and the total nanoparticle surface area. The surface area of nanoparticles was calculated assuming that the particles are made of individual particles of diameter equal to that measured by the Zetasizer and using a density of 1.34 g/cm3 for PLGA.
The surface concentration of ITEM4 (# of ITEM4 molecules/nanoparticle) was quantified via the LavaPep protein assay (Gel Company, San Francisco, CA) using free ITEM4 as a standard [11]. A calibration curve was generated by plotting the fluorescence from different concentrations of ITEM4 molecules. The average ITEM4 surface density (# of ITEM4 molecules/nanoparticle) on the surface of the nanoparticles was calculated by taking the total quantity of ITEM4 measured by the LavaPep protein assay and the total number of nanoparticles in 1 ml solution. The number of nanoparticles in the sample was calculated assuming that the nanoparticles are made of individual nanoparticles of diameter equal to that measured by the Zetasizer and using a density of 1.34 g/cm3 for PLGA.
Nanoparticle binding to brain extracellular matrix proteins
Non-specific binding of the nanoparticles was analyzed on brain extracellular matrix (ECM) proteins using a high throughput SPR-based Biacore 3000 instrument (GE Healthcare, Marlborough, MA) at 25 °C as previously described [11, 18, 19]. Brain ECM proteins were isolated from freshly collected mouse brain as previously described [24]. Briefly, resected whole mouse brain was frozen for at least 24 h at −80 °C and subsequently thawed and decellularized in a series of steps: ultrapure water (16 h at 4 °C), 0.02% trypsin/0.05% EDTA (1 h at 37 °C), 3% Triton-X 100 (1 h), 1 M sucrose (15 min), ultrapure water (15 min), 4% deoxycholate (1 h), 0.1% periacetic acid in 4% ethanol (2 h), 1× PBS (15 min), ultrapure water (15 min), and 1× PBS (15 min). The decellularized proteins were filtered (0.2 μm filter) to remove insoluble proteins and then frozen and stored at −80 °C until use.
The isolated ECM proteins were conjugated to one of the flow paths of a CM5 Biacore chip with ligand response units (RU) ranging from 140 to 250. The other flow path was activated and blocked with ethanolamine to serve as a reference for each binding run, as suggested per manufacturer’s protocol. The running buffer, 10 mM HEPES buffer (pH 7.4) containing 150 mM NaCl, 0.05% surfactant P-20 with 50 μM EDTA (HBS-P), was degassed prior to use. For binding experiments, samples (PLGA, PLGA-PEG, PLGA-PEG-ITEM4 nanoparticles and PS nanoparticles as a positive control) were assayed at a flow rate of 20 μl/min with an injection time of 3 min followed by a 2.5 min wait for dissociation, before chip regeneration with 10 mM glycine, pH 1.75. Nanoparticle binding was assayed with nanoparticle concentrations of 1 mg/ml diluted in running buffer.
Nanoparticle binding to Fn14 extracellular domain
Fn14-specific binding of the nanoparticles was analyzed on recombinant Fn14 extracellular domain (Cell Sciences, Canton, MA) conjugated to a CM5 Biacore chip, with RU values ranging from 300 to 1700. The first flow path was activated and blocked with ethanolamine to serve as a reference for each binding run, as suggested per manufacturer’s protocol. The running buffer, 10 mM HEPES buffer (pH 7.4) containing 150 mM NaCl, 0.05% surfactant P-20 with 50 mM EDTA (HBS-P), was degassed prior to use. Similar to the non-specific binding experimental procedure, the samples (PLGA-PEG, PLGA-PEG-ITEM4 nanoparticles and ITEM4 as a positive control) were assayed at a flow rate of 20 μl/min with an injection time of 3 min followed by a 2.5 min wait for dissociation, before chip regeneration with 10 mM glycine, pH 1.75. Nanoparticle binding was assayed with nanoparticle concentrations of 1 mg/mL diluted in running buffer. Data were analyzed using Biacore 3000 Evaluation Software, where data from reference flow path was subtracted from the experimental flow path data to give the final sensorgrams. In addition, binding isotherm of the nanoparticle was generated by analyzing binding at various nanoparticle concentrations. The data was analyzed by fitting to a pseudo-first order process to determine the maximum change in response units (RUeq). RUeq values were then plotted versus nanoparticle concentration and the equilibrium binding affinities (KD) were calculated by fitting the binding isotherm data into a single class of binding sites using non-linear regression analysis employing GraphPad Prism 7.03 software (GraphPad Software, Inc., La Jolla, CA) [11, 18, 25].
Cell culture and evaluation of Fn14 expression
The invasive mouse malignant glioma cell line KR158 was obtained from Dr. Tyler Jacks (Massachusetts Institute of Technology, Cambridge, MA). These cells were initially generated from NF1 and p53 mutant mice and were engineered to express firefly luciferase (KR158-Luc) [26]. Cells were cultured at 37 °C in a humidified incubator (95% air, 5% CO 2) in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (1000 units/l).
To examine Fn14 surface expression by the KR158 cell line, flow cytometry analysis was performed. Briefly, cells were seeded in 24-well plates at a density of 105 cells per well and allowed to attach overnight. The media was then replaced with serum-free DMEM along with no antibody, IgG isotype-PE, or ITEM4-PE. After 1 h incubation, cells were washed 3 times with PBS, detached with trypsin, and diluted in cold PBS for flow cytometry analysis. Mean fluorescence intensity was analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lake, NJ). Data from 104 events were gated using forward and side scatter parameters to exclude dying cells and debris.
Nanoparticle cellular association and internalization in KR158 cells
Cellular association and uptake of rhodamine-labeled fluorescent nanoparticles by Fn14-positive KR158 cells was determined by flow cytometry and confocal microscopy. For nanoparticle cellular association analysis by flow cytometry, cells were seeded in 24-well plates at a density of 105 cells per well and allowed to attach overnight. The media was then replaced with serum-free DMEM containing non-targeted or Fn14-targeted fluorescent nanoparticles (100 μg per well). To confirm a specific interaction between ITEM4 and Fn14, the cells were incubated with excess free ITEM4 (500 μg/ml) for 30 min to block the Fn14 binding sites, prior to adding Fn14-targeted fluorescent nanoparticles. To confirm whether the enhanced PLGA-PEG-ITEM4 uptake was the result of a specific interaction between ITEM4 and Fn14, we performed a competitive inhibition assay with free ITEM4 antibody. Addition of excess free ITEM4 to Fn14-positive KR158 cells, prior to particle addition, significantly inhibited the uptake of PLGA-PEG-ITEM4 nanoparticles to similar levels as that observed for non-targeted PLGA-PEG nanoparticles (Figure 4C). The experimental details and results are added in the revised manuscript.
Figure 4. Analysis of Fn14 expression and nanoparticle uptake in KR158 cells.

(A) Flow cytometry analysis of mouse KR158 cells for Fn14 surface expression with mouse IgG isotype control (blue) and ITEM4 antibody (red). (B) Flow cytometry analysis of PLGA-PEG and PLGA-PEG-ITEM4 nanoparticle uptake in KR158 cells. (C) Inhibition of nanoparticle uptake with pre-incubation of excess free ITEM4. In B and C, values shown are mean +/− SD (n=3). Data analyzed for significance using Student’s t-test. *P < 0.05, **P < 0.01. (D) Confocal microscopy images of KR158 cells showing association of PLGA-PEG or PLGA-PEG-ITEM4 nanoparticles. Scale bars = 25 μm. Cell marker with asterisk was analyzed in E. (E) Representative 3-D projection of series of images from z-stack scan of cell in PLGA-PEG-ITEM4 image, confirming that the nanoparticles (red) are inside the cell. The nucleus is stained with Hoechst 33342 (blue).
After 1 h incubation, cells were washed 3 times with PBS, detached with trypsin, and diluted in cold PBS. Mean fluorescence intensity was analyzed using a FACSCalibur flow cytometer.
The internalization of the nanoparticles in KR158 cells was confirmed by live-cell confocal microscopy. Briefly, cells were seeded onto Lab-Tek glass-bottom culture plates at a density of 105 cells per plate and allowed to attach overnight. The media was then replaced with fresh media containing non-targeted or Fn14-targeted nanoparticles (100 μg per well). After 1 h incubation, cells were treated for 15 min with Hoechst 33342 (5 μg/ml) to stain the nuclei and then washed 3 times with PBS. Clear Opti-MEM (Invitrogen Corp., Carlsbad, CA) media was added to the plates and the cells and nanoparticles were imaged under a LSM5 Duo slit scanning confocal microscope (Carl Zeiss Inc., Thornwood, NY) with a 63x Plan-Apo/1.4 NA oil-immersion objective.
Nanoparticle penetration in brain slices
Brain tissue penetration of nanoparticles was analyzed using MPT assays in ex vivo rat brain slices as described previously [11, 23, 27]. MPT allows simultaneous measurement of particle trajectories for hundreds of individual nanoparticles, facilitating relatively high throughput measurements. Briefly, Sprague-Dawley rats were euthanized and brains were harvested. Brains were sliced into 1.5 mm coronal sections using a Zivic brain matrix slicer (Zivic Instruments, Pittsburgh, PA) and placed on custom-made microscope slide chambers. Rhodamine-labeled fluorescent nanoparticles were then injected (0.5 μl, 100 μg/ml) into the middle of cortical tissue of the brain slices using a Hamilton syringe aided by a stereotactic frame. Cover slips were placed on the slide chambers and sealed with super glue. Slices were incubated at 37 °C for 10 min prior to undergoing imaging at a frame rate of 20 frames/s for a total of 400 frames (20 s) using LSM5 Duo slit scanning confocal microscope with a 63x Plan-Apo/1.4 NA oil-immersion objective. Particle movement movies were analyzed using a custom written MATLAB automated tracking code to extract x, y-coordinates of the nanoparticles over time [27]. The geometric mean of the mean squared displacement (MSD) was calculated per sample and the average MSD was plotted as a function of time scale. The theoretical MSD values of nanoparticles in water were calculated from the Stokes-Einstein equation using the mean particle diameters, measured by DLS.
Intracranial implantation of KR158-Luc tumors and bioluminescence imaging
All animal procedures were approved by the University of Maryland Institutional Animal Care and Use Committee and the Office of Animal Welfare Assurance. C57BL/6 mice (age, 6–8 weeks) were purchased from Envigo RMS, Inc. (Indianapolis, IN). Animals were anesthetized via continuous flow of 2.5% isoflurane through a nose cone and were secured to a stereotactic frame. Using a handheld drill, a burr hole was drilled into the left frontal lobe of the brain 2 mm lateral to the sagittal suture and 1 mm anterior to the coronal suture at a depth of 3 mm below the dura of all animals. Using a Hamilton syringe attached to the stereotactic frame, 3×105 KR158-Luc cells were injected at a rate of 1 μl/min over 5 min through the burr hole. Mice were given the analgesic Rimadyl (Carpofen, 3 mg/kg) subcutaneously after the surgery. Animals were observed daily for any signs of deterioration or neurological dysfunction. If the symptoms persisted and resulted in debilitation, animals were euthanized according to protocol.
For the bioluminescence imaging of the tumors, animals were anesthetized in an induction chamber with 2.5% isoflurane and injected with D-luciferin (150 mg/kg, dissolved in PBS) intraperitoneally. After 10 min, animals were moved to a Xenogen IVIS system (Caliper Life Sciences, Hopkinton, MA) maintained at 2.5% isoflurane and imaged for tumor bioluminescence. Photons emitted from live mice were acquired as photons/s/cm2/steradian (p/s/cm2/cm2/sr) and analyzed using LivingImage software (PerkinElmer, MA).
Brain tissue collection and histopathological analyses
Animals were euthanized with induction of general anesthesia followed by exsanguination using transcardiac perfusion of cold PBS. The brain tissues were rapidly extracted and fixed in 4% formalin for 24 h and transferred to 70% ethanol for immunohistochemistry. Fixed tissues were mounted in paraffin blocks using the Leica EG 1160 embedding center (Leica Microsystems, Buffalo Grove, IL) and then sectioned in 5 μm slices oriented in the coronal plane. Sections were stained with hematoxylin & eosin (H&E) and photographed. Immunohistochemical staining for Fn14 was performed using an anti-Fn14 rabbit monoclonal antibody (Epitomics clone EPR3179) on a Leica BOND-III™ autostainer (Leica Microsystems) and peroxidase/DAB Bond™ Polymer Refine Detection System (Leica Microsystems) was used for visualization.
Intracranial injection of nanoparticles
At day 7 after the implantation of KR158 cells, bioluminescent imaging was performed on each animal. Once tumor signal was confirmed, the animals were anesthetized as described above and rhodamine-labeled or ICG-loaded fluorescent nanoparticles suspended in normal saline were administered into mouse brain through the same burr hole using a stereotactic system. Rhodamine-labeled (10 μl, 1 mg/ml) or ICG-loaded (5 μl, 0.3 mg/ml of ICG equivalent) fluorescent nanoparticles in PBS were loaded into a sterile 30-gauge Hamilton syringe needle, lowered to a depth of 3.5 mm inside brain and injected slowly at a rate of 1 μl/min.
Nanoparticle distribution in KR158 intracranial tumors
The distribution of fluorescent nanoparticles injected into KR158 tumors was evaluated by imaging brain cryosections [11]. The animals were euthanized with an overdose of isoflurane 4 h after the injection of nanoparticles. The euthanized animals were perfused with 30 ml of PBS after which the brains were carefully removed, embedded in Optimal Cutting Temperature (OCT), and stored at −80 °C. A cryostat (Leica CM3050 S) was used to cut serial 10 mm sagittal brain sections and mounted on positively charged microscope slides. The brain sections were stained with Prolong Gold antifade with DAPI (Invitrogen, Carlsbad, CA), sealed with coverslips, and imaged for cell nuclei (dark blue) and fluorescent nanoparticles (red) using a Nikon epifluorescence microscope under 10x and 20x magnification. High resolution stitched images (6 × 6) were obtained by using the montage imaging feature in the Nikon NX 2 software.
Nanoparticle co-localization with tumor cells
The co-localization of fluorescent nanoparticles with KR158 tumors was evaluated by flow cytometry. Briefly, the animals were euthanized with an overdose of isoflurane 4 h after the injection of nanoparticles. The brains were carefully removed and tumor tissues from the brains were harvested. The tumor tissues were dissociated to get single-cell suspension using a GentleMACS Tissue Dissociator and Brain Tumor Dissociation kit (Miltenyl Biotech, Bergisch Gladbach, Germany) following manufacturer’s protocol. Tissue debris was then removed using a Debris Removal Kit (Miltenyl Biotech). Tumor single-cell suspensions were fixed and cells were permeabilized using a fixation/permeabilization kit (Ebiosciences, San Diego, CA). Cell suspensions were stained for luciferase using a fluorescein isoisothiocyanate (FITC)-conjugated anti-luciferase antibody (Abcam, Cambridge, MA) and analyzed by flow cytometry. Tumor cells were detected by gating on anti-luciferase FITC staining. FITC stained cell populations in each sample were then analyzed for Rhodamine fluorescence. Mean fluorescence intensity was analyzed using a FACSCalibur flow cytometer.
Nanoparticle retention in KR158 intracranial tumors
Nanoparticle retention and clearance in tumor tissue was analyzed by imaging nanoparticles in brains of live animals using Xenogen IVIS system. Similar to described above, free ICG, ICG-loaded PLGA-PEG or ICG-loaded PLGA-PEG-ITEM nanoparticles in PBS (5 μl, 0.3 mg/ml ICG equivalent) were slowly administrated, at a rate of 1 μl/min over 5 min, through the same burr hole as the tumor cell implantation using stereotactic frame. Animals were then imaged at various time points with 2 sec exposure at excitation wavelength of 745 nm and emission wavelength of 820 nm to record fluorescence intensity of ICG from the brains. The images were then processed using LivingImage software to calculate fluorescence intensities from region of interest drawn on all the animals with the same dimensions. The percentage relative fluorescence intensities were calculated and plotted against time to estimate retention time and half-life of nanoparticles.
Statistical analysis
Statistical analysis of data was performed by a two-tailed Student’s t test assuming unequal variances or one-way analysis of variance (ANOVA) followed by Tukey HSD using GraphPad software. Differences were considered to be statistically significant at a level of P < 0.05.
Results
Synthesis and characterization of biodegradable nanoparticles
A variety of biodegradable PLGA, PLGA-PEG, and PLGA-PEG-ITEM4 nanoparticles, with or without fluorescent dyes, were synthesized using emulsion solvent evaporation techniques. Empty nanoparticles and Rhodamine-labeled nanoparticles were formulated by single emulsion method, while ICG-loaded nanoparticles were formulated by double emulsion method (Table S1). PLGA-PEG-Mal nanoparticles (~1 wt% maleimide groups) were then conjugated with ITEM4 via maleimide-thiol chemistry to formulate PLGA-PEG-ITEM4 nanoparticles. Approximately ~1% of PLGA-PEG-ITEM4 nanoparticle surface was covered by ITEM4, with the rest of the particle surface area exposed to hydration and eventual degradation. We were able to formulate sub-100 nm sized biodegradable nanoparticles. With the addition of PEG, Rhodamine or ICG, and ITEM4, hydrodynamic diameter of nanoparticles increased from ~53 nm (Z-average of ~72 nm) for empty PLGA nanoparticles to ~95 nm (Z-average of ~108 nm) for the bulkiest ICG-loaded PLGA-PEG-ITEM4 nanoparticles (Table 1). Nanoparticle analysis by TEM showed round morphology and sub-100 nm size of PLGA, PLGA-PEG, and PLGA-PEG-ITEM4 nanoparticles (Fig. 1). All nanoparticle formulations had ζ-potential values close to the neutral surface charge, due to the PEG coatings and/or use of PVA in the synthesis procedure. The nanoparticle suspensions were stable in ACSF at 37 °C for up to 24 h (Fig. S1). We quantified the surface density of PEG and number of ITEM4 molecules on the surface of nanoparticles. There were ~14 PEG molecules per 100 nm2 area of nanoparticles as estimated from the NMR data (Fig. S2) and there were ~6 ITEM4 molecules per particle as estimated from the LavaPep protein assays (Table 2).
Table 1.
Physicochemical characterization of nanoparticles
| Formulation | Hydrodynamic diameter (nm)a | PDI b | ζ-potential (mV) c | |
|---|---|---|---|---|
| Number mean | Z-average | |||
| PLGA | 52.8 ± 2.0 | 72.3 + 1.2 | 0.14 ± 0.01 | −6.0 ± 0.6 |
| PLGA-Rhod | 68.2 ± 2.3 | 78.8 + 5.3 | 0.15 ± 0.02 | −5.5 ± 1.7 |
| PLGA-PEG | 68.8 ± 3.3 | 75.4 + 3.3 | 0.02 ± 0.01 | −1.3 ± 0.2 |
| PLGA-PEG-Rhod | 78.2 ± 4.5 | 89.8 + 5.2 | 0.04 ± 0.01 | −2.1 ± 0.3 |
| PLGA-PEG-ICG | 81.5 ± 8.3 | 94.8 + 2.7 | 0.12 ± 0.03 | −2.4 ± 0.4 |
| PLGA-PEG-ITEM4 | 79.0 ± 5.4 | 90.2 + 1.6 | 0.16 ± 0.01 | −2.6 ± 0.3 |
| PLGA-PEG-ITEM4-Rhod | 89.8 ± 5.2 | 102.4 + 3.7 | 0.18 ± 0.01 | −3.6 ± 0.6 |
| PLGA-PEG-ITEM4-ICG | 94.2 ± 8.5 | 108.2 + 3.4 | 0.17 ± 0.02 | −4.8 ± 0.4 |
Physicochemical characterization data represents the average of 3 independent experiments +/− SD.
Hydrodynamic diameter (number mean and Z-average) measured by dynamic light scattering.
Polydispersity index indicates the distribution of individual molecular masses in a batch of nanoparticles, measured by dynamic light scattering.
Surface charge measured at 25 °C in 15x diluted PBS with ~10 mM NaCl, pH 7.4.
Figure 1. Nanoparticle structure and size.

(A) Schematic representation of nanoparticles depicting PEG and ITEM4 functionalized PLGA nanoparticle surfaces. (B) TEM images show well dispersed round shaped nanoparticles. Scale bars = 300 nm. Insets: magnified images of nanoparticles. Scale bars = 100 nm.
Table 2.
Surface characterization and diffusion behaviors of nanoparticles
| Formulation | Surface density of PEG (#/100nm2) a | PEG conformation [Γ/Γ*] b | Surface density of ITEM4 (#/particle) c | KD, (nM) d | MSDwater/MSDbrain |
|---|---|---|---|---|---|
| PLGA | NA | NA | NA | NA | 374 |
| PLGA-PEG | 13.9 | 3.16 | NA | NA | 89 |
| PLGA-PEG-ITEM4 | 13.7 | 3.10 | 6.13 | 0.75 | 99 |
| ITEM4 | NA | NA | NA | 1.62 | NA |
PEG surface density determined by NMR.
PEG surface coverage/total surface area (value <1 indicates mushroom coverage [low density], whereas >1 indicates brush regime [high density]).
Surface density reported from LavaPep fluorescent protein assay.
KD values determined on a per nanoparticle basis from fit of Biacore data.
The ratio indicates by what multiple the average particle transport rate is slowed in brain tissue compared to in pure water. The larger the ratio, the higher the degree of hindrance to particle motion.
Non-specific binding of nanoparticles to brain ECM proteins
Non-specific binding of nanoparticles was assessed using SPR Biacore assays, as we previously described [11, 19]. For these assays, mouse brain ECM proteins were functionalized on surface of a Biacore chip and the non-specific binding of nanoparticles to this brain ECM was analyzed. As a positive control, non-specific binding of hydrophobic and adhesive PS nanoparticles was measured. PS particles have been shown previously to be nearly completely immobilized when delivered into the rodent brain [9, 11] and as expected they bound strongly to the surface of the brain ECM Biacore chip (Fig. 2A). PLGA-PEG and PLGA-PEG-ITEM4 nanoparticles did not bind appreciably to the brain ECM chip, suggesting minimal non-specific interactions between the nanoparticles and the brain ECM proteins. Similarly, PLGA-PEG-IgG nanoparticles did not bind to the brain ECM proteins (data not shown). In contrast, PLGA nanoparticles without PEG coating showed some binding to the brain ECM chip.
Figure 2. Non-specific binding of nanoparticles to off-target structures.

(A) Non-specific binding of nanoparticles to brain ECM proteins-coated Biacore chip using surface plasmon resonance (SPR) assays (RU: Response Units). (B) Multiple particle tracking (MPT) analysis of nanoparticles in brain slices ex vivo showing ensemble-averaged mean square displacements (MSD) as a function of time scale over 1 sec period and (C) at 1 sec time point (ns: not significant). Values shown are mean +/− SD (n=5). Data analyzed for significance using Student’s t-test. *P < 0.05.
Nanoparticle diffusion in brain slices
Diffusion rates of individual nanoparticles were analyzed ex vivo using rat brain slices by MPT assays as described previously [11, 19]. PLGA nanoparticles without PEG coatings did not diffuse appreciably in brain tissue, whereas PLGA-PEG and PLGA-PEG-ITEM4 nanoparticles exhibited more diffusive Brownian-like trajectories. This can be seen as an upward shift in the MSD vs time scale (τ) curve as compared to PLGA nanoparticles (Fig. 2B). The calculated MSD at a time scale (τ) = 1 s for PLGA-PEG and PLGA-PEG-ITEM4 nanoparticles was significantly higher than PLGA nanoparticles (Fig. 2C). The diffusion rates of PLGA, PLGA-PEG, and PLGA-PEG-ITEM4 nanoparticles were 374-fold, 89-fold, and 99-fold lower, respectively, compared to their theoretical diffusion rates in water at τ = 1 s (Table 2). Although the calculated MSD of PLGA-PEG-ITEM4 nanoparticles was reduced slightly compared to PLGA-PEG nanoparticles, there was no statistical difference between these two formulations. The MPT results suggest that dense PEG-coating on nanoparticles reduces the non-specific interactions between PLGA-PEG and PLGA-PEG-ITEM4 nanoparticles and the brain ECM proteins. Moreover, conjugation of ITEM4 to the PLGA-PEG nanoparticles does not promote binding to brain ECM.
Specific binding of nanoparticles to the Fn14 extracellular domain
Fn14-specific binding of nanoparticles was assessed using SPR Biacore assays. The Fn14 extracellular domain was functionalized on surface of a Biacore chip and binding of nanoparticles to Fn14 was analyzed. As a positive control, Fn14-specific binding of ITEM4 was measured, which bound strongly to the surface of the Fn14 Biacore chip (data not shown). PLGA-PEG-ITEM4 nanoparticles bound strongly to the Fn14 chip; however, PLGA-PEG nanoparticles did not show any appreciable Fn14 binding (Fig. 3A). Similarly, PLGA-PEG-IgG nanoparticles did not bind to the Fn14 chip (data not shown). To determine the equilibrium binding affinity of PLGA-PEG-ITEM4 nanoparticles to the Fn14 extracellular domain, we measured the binding of various nanoparticle concentrations (Fig. 3B). We then obtained equilibrium binding (RUeq) values and plotted against nanoparticle concentration as binding isotherm (Fig. 3C). The equilibrium binding affinity (KD) was then calculated by fitting the binding isotherm data in to a ‘one site specific binding’ model equation. The measured KD for PLGA-PEG-ITEM4 nanoparticles was 0.75 nM, which was ~2-fold lower than that of ITEM4 alone, 1.62 nM (Table 2).
Figure 3. Structure-specific targeting of nanoparticles.

(A) Specific binding of nanoparticles to Fn14 extracellular domain-coated Biacore chip using SPR assays. (B) Kinetic binding analysis of ITEM4-conjugated nanoparticles showing binding curves at various concentrations. These curves were fit to a first order process to determine RUeq values at each concentration. (C) Binding isotherm of ITEM4-conjugated nanoparticles showing RUeq values determined from kinetic binding analysis in B. The data was fit to a single class of binding sites by non-linear regression analysis using GraphPad software (AU: Arbitrary Units).
Nanoparticle cellular association and uptake by Fn14-positive KR158 cells
First, we confirmed that KR158 cells expressed Fn14 by flow cytometry using PE-labeled ITEM4 (Fig. 4A). Cellular association of nanoparticles was then measured by flow cytometry. A significantly higher cellular association of PLGA-PEG-ITEM4 nanoparticles by KR158 cells was observed compared to PLGA-PEG nanoparticles without Fn14 targeting ligand (Fig. 4B). The cellular association efficiency of PLGA-PEG-ITEM4 nanoparticles was ~2-fold higher compared to PLGA-PEG nanoparticles. To confirm whether the enhanced PLGA-PEG-ITEM4 uptake was the result of a specific interaction between ITEM4 and Fn14, we performed a competitive inhibition assay with free ITEM4. Addition of excess free ITEM4 to Fn14-positive KR158 cells, prior to particle addition, significantly inhibited the cellular association of PLGA-PEG-ITEM4 nanoparticles (Fig. 4C). To confirm nanoparticle internalization within cells, live cell confocal microscopy imaging was performed. We found higher uptake of PLGA-PEG-ITEM4 nanoparticles by KR158 cells compared to PLGA-PEG nanoparticles (Fig. 4C). In addition, we captured 3-D projection of series of images from z-stack scan of cells, to confirm that the nanoparticles were not just at the surface but were inside of the cells (Fig. 4D).
Nanoparticle distribution and co-localization with tumor cells in vivo
First we confirmed that our KR158-Luc glioma model was invasive and expressed Fn14 in vivo. Luciferase-positive KR158 tumors were evident in the brain 7 days after tumor implantation (Fig. 5A). The mice were then euthanized and brain tumor tissues were harvested. Tumor tissues were sectioned and stained with H&E and anti-Fn14 mAb. The histology shows an invasive tumor with high Fn14 expression (Fig. 5B).
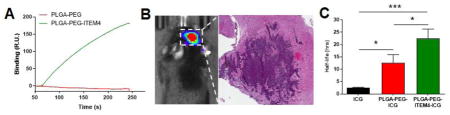
Figure 5. Analysis of nanoparticle co-localization with KR158 cells in vivo.

(A) Bioluminescence image of mouse bearing KR158 intracranial tumor indicated by dotted white circle. (B) H&E (left) and Fn14 (right) staining of KR158 tumor. Fn14 staining is shown in the expanded view of boxed region from H&E stain. (C) Flow cytometry analysis of PLGA-PEG and PLGA-PEG-ITEM4 nanoparticle co-localization in vivo with luciferase-positive KR158 cells in intracranial tumors. Values shown are mean +/− SD (n=3). Data analyzed for significance using Student’s t-test. *P < 0.05.
To determine if Fn14-targeted nanoparticles distribute uniformly in the brain and co-localize with tumor cells in vivo, we injected rhodamine-labeled nanoparticles into intracranial KR158-Luc tumors. The mice were euthanized at 4 h after nanoparticle injection; brains were harvested, processed, and analyzed by epifluorescence microscopy and flow cytometry. Fluorescence microscopy images showed that PLGA-PEG-ITEM4 nanoparticles (red) were distributed in and around the tumor region (outlined by white dotted line) as well as deeper inside the brain tissue (Fig. S3). These results demonstrate that our PLGA-PEG-ITEM4 nanoparticles can penetrate within brain tissue and selectively target invading glioma tumors. To determine if Fn14-targeted nanoparticles co-localize with tumor cells in vivo, we performed flow cytometry. A significantly higher co-localization of PLGA-PEG-ITEM4 nanoparticles with luciferase-positive KR158 cells was observed compared to PLGA-PEG nanoparticles without Fn14 targeting ligand (Fig. 5C). The tumor cell co-localization efficiency of PLGA-PEG-ITEM4 nanoparticles was ~3.5-fold higher compared to PLGA-PEG nanoparticles.
Nanoparticle retention in intracranial KR158 tumors
To measure nanoparticle retention and clearance in intracranial KR158 tumors ICG, ICG-loaded PLGA-PEG nanoparticles, or ICG-loaded PLGA-PEG-ITEM4 nanoparticles were injected into KR158 tumors and ICG fluorescence was measured over time using a Xenogen imaging system. We found decreasing fluorescence intensities from tumors of the animals in all the groups (Fig. 6A). The percentage fluorescence intensities relative to 100% at time 0 were quantified and plotted against time. We observed that free ICG was cleared the fastest (100% clearance in 4 days) followed by non-targeted PLGA-PEG nanoparticles (100% clearance in 6 days) and PLGA-PEG-ITEM4 nanoparticles (100% clearance in 8 days) (Fig. 6B). In addition, half-life of the free ICG, ICG-loaded PLGA-PEG, and ICG-loaded PLGA-PEG-ITEM4 nanoparticles was calculated to determine the time required for 50% clearance. The Fn14-targeted PLGA-PEG-ITEM4 nanoparticles had the longest half-life of ~23 h and were retained significantly longer in the tumors of mice as compared to the non-targeted PLGA-PEG nanoparticles (half-life of ~10.5 h) and free ICG (half-life of ~2.5 h) (Fig. 6C). Therefore, the retention of nanoparticles in the brain depends on the particle surface properties and not affected by the ICG release profiles.
Figure 6. Analysis of nanoparticle retention in mice bearing KR158 tumors.

(A) Images of ICG-loaded nanoparticle fluorescence in tumor-bearing brains over time. (B) Line graph of averaged fluorescence for each group at all time points. (C) Averaged nanoparticle fluorescence half-life in tumor-bearing brains. Values shown are mean +/− SD (n=3). Data analyzed for significance using one-way ANOVA. *P < 0.05, *** P < 0.001.
Discussion
In this study, we formulated particulate nanocarriers using PLGA and PLGA-PEG polymers to generate sub-100 nm nanoparticles with DART characteristics. SPR analysis revealed minimal binding to extracellular brain components and strong binding to the Fn14 receptor – an upregulated, conserved component in invasive GBM. The association of Fn14-directed DART nanoparticles with Fn14-positive tumor cells was significantly greater than non-adhesive but non-targeted particles. Ex vivo MPT in fresh rodent brain tissues and in vivo testing in orthotopic murine glioma revealed preserved nanoparticle diffusivity and increased uptake in brain tumor cells. The DART characteristics also resulted in longer retention of the nanoparticles within the intracranial tumors compared to non-targeted versions and the free fluorescent dye. Collectively, these results and nanoparticle design considerations offer promising new methods to optimize therapeutic nanocarriers for improving drug delivery and treatment for invasive brain tumors.
Biodegradable polymeric nanoscale drug formulations offer the potential to augment and control the biological behavior of multiple therapeutic agent types and classes including chemotherapies, oligonucleotides, proteins, and others [28–31]. Such formulations enable minimizing off-target effects or toxicities, rapid clearance or degradation, and poor distribution within the pathological site or tissues [12]. Augmenting brain penetration is one strategy that has shown some promise in pre-clinical testing by improving the efficacy and safety of drugs with otherwise low therapeutic ratios, such as paclitaxel, cisplatin, and others [9, 10, 16, 17, 32]. Another strategy has been to target nanotherapeutics in order to focus the desired treatment effects directly to disease sites. This too has shown early evidence toward improved delivery and therapeutic efficacy including targets to epidermal growth factor receptor mutant EGFRvIII [13], transferrin receptor [14], interleukin-13 (IL-13) receptor [15, 33], and tumor-associated ECM components such as Tenascin C [34]. Despite these beneficial properties, separated penetration and targeting strategies may still be limited by rapid degradation, clearance or partitioning, and other phenomena that can lead to off-target effects. In order to fully harness the potential benefits of nanotherapeutic formulations, effectively balancing non-specific adhesivity within the body with specific binding to disease targets is a crucial consideration.
Previous studies exploring delivery improvements and limitations to the brain, and brain tumors in particular, have revealed that therapeutic agents not designed to avoid body clearance and degradation mechanisms, resistance to dispersion within tissue(s), or partitioning in non-disease sites, lead to undesirable biological behavior and potential toxicities [3, 12]. While targeting therapeutics to specific disease components may decrease some of these limitations, in order to fully capitalize on the potential benefits of targeting, low levels of non-specific adhesivity and off-target binding must be maintained in setting of an effective level of target specific binding. This balance is often quite challenging to attain as many targeting moieties (antibodies and related fragments, peptides, carbohydrates, and others) may also result in non-specific binding to cellular, extracellular and intravascular components. This study represents an important step towards identifying the nanotherapeutic design and characterization considerations to effectively achieve this balance. Our version of these considerations features decreased non-specific adhesivity to brain interstitial components and Fn14 receptor targeting, which we abbreviate to ‘DART’ characteristics.
Of note, Fn14 is a promising molecular target for GBM [20, 21, 35] and perhaps a more appealing target than other cell surface receptors mentioned above. Fn14 is minimally expressed in the uninjured human brain but highly expressed in high grade gliomas including GBM. Fn14 gene expression levels correlate directly with increasing glioma grade and worse patient survival [21, 22]. Importantly, elevated Fn14 transcript and protein levels have been identified in the margin of GBM tissues where invading tumor cells and reactive processes are present. Lower Fn14 levels are present in tumor core regions, where surgery is often successful [21]. Importantly, Fn14 undergoes constitutive receptor internalization, which could facilitate therapeutic agent entry into target cells [36]. These findings warrant further exploration of Fn14 for invasive GBM-targeted therapeutics as well as inhibition of brain cancer invasion mechanisms.
This study identified three possible mechanisms by which beneficial augmentation of Fn14 DART therapeutics may occur, specifically enhanced brain tissue dispersion, increased tumor cell uptake, and improved tumor retention. The latter less expected finding related to the in vivo duration of nanoparticles at the target site suggests that a given therapeutic dose may have an even greater therapeutic effect than we had estimated previously. We anticipate that future applications of DART characteristics to other therapeutic formulations will lead to significant improvements in therapeutic ratio and the application of therapeutic agents or drug classes that were not considered safe or feasible for brain cancer and other diseases with challenging delivery considerations.
An important limitation of this study is the lack of a larger animal model where invasion distance and brain and tumor volumes more closely mimic humans. While the KR158 malignant glioma model used here is highly invasive [37], numerous clinical trials testing delivery strategies to improve distribution of therapeutics in the brain [33, 38] have highlighted the difficulty with translating results from smaller pre-clinical models to humans. In the future, we plan to test this therapeutic formulation strategy in larger mammals (pigs and tumor-bearing dogs) in order to better understand critical limitations to human translation. Another potential limitation of this study is the lack of a tested therapeutic or drug to ascertain actual improvements in efficacy and safety. We opted not to include a specific therapeutic component in this study as we felt it was important to first investigate the design of biodegradable DART nanocarriers and potential value of this therapeutic platform. As such, we sought to understand the polymeric formulation and characterization considerations prior to inserting the complexities of therapeutic testing. Our findings motivate the study and testing of drug-loaded, Fn14 DART nanotherapeutics and suggest that the effective balance of specific and non-specific binding of nanotherapeutics within the brain tumor milieu may significantly improve treatment efficacy while minimizing off-target toxicities.
Conclusion
In summary, we have developed biodegradable nanoformulations with balanced decreased non-specific adhesivity and receptor targeting. These DART nanocarriers showed improved brain tissue dispersion, tumor cell uptake, and tumor retention in in vitro, ex vivo, and in vivo testing. These results and nanoparticle design considerations offer promising new methods to optimize therapeutic nanocarriers for improving drug delivery and treatment for invasive brain tumors, and warrant further investigation with drug-loaded versions.
Supplementary Material
Acknowledgments
This research was supported in part by the National Institutes of Health [K25 EB018370 (A.J.K.), K08 NS09043 (G.F.W.), and R35 HL135743 (D.K.S.)]; a Research Scholar Grant RSG-16-012-01 (G.F.W.) and an Institutional Research Grant IRG-97-153-10 (A.S.W.) from the American Cancer Society; an Elsa U. Pardee Foundation Research Grant (A.J.K. and J.A.W.); and a AAPS Foundation New Investigator Grant Award (A.J.K.). A.S.W., J.G.D., and N.P.C. are trainees on the NIH Grant T32 CA154274. J.G.D. was also supported by an NIGMS Initiative for Maximizing Student Development Grant (R25GM55036). We thank Dr. Yinghua Zhang at the Biosensor Core at the University of Maryland School of Medicine for her expertise. We thank Dr. Ru-Ching Tsai at the Electron Microscopy Core at the University of Maryland School of Dentistry for her expertise. We also thank Dr. Kellie Hom at the Nuclear Magnetic Resonance (NMR) Facility at the University of Maryland School of Pharmacy for her help with NMR measurements.
Footnotes
Author Contibutions
ASW, JGD, JAW, GFW, and AJK conceived and designed the experiments. ASW, JGD, NBR, NPC, and AJK performed the experiments. ASW, JGD, NBR, and DKS analyzed the data. ASW, JGD, DKS, JAW, GFW, and AJK wrote the paper. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tate MC, Aghi MK. Biology of angiogenesis and invasion in glioma. Neurotherapeutics. 2009;6(3):447–57. doi: 10.1016/j.nurt.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Woodworth GF, et al. Emerging insights into barriers to effective brain tumor therapeutics. Front Oncol. 2014;4:126. doi: 10.3389/fonc.2014.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakroun RW, et al. Nanotherapeutic systems for local treatment of brain tumors. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2017:e1479. doi: 10.1002/wnan.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valtonen S, et al. Interstitial chemotherapy with carmustine-loaded polymers for high-grade gliomas: a randomized double-blind study. Neurosurgery. 1997;41(1):44–8. doi: 10.1097/00006123-199707000-00011. discussion 48–9. [DOI] [PubMed] [Google Scholar]
- 6.Westphal M, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5(2):79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grossman SA, et al. The intracerebral distribution of BCNU delivered by surgically implanted biodegradable polymers. J Neurosurg. 1992;76(4):640–7. doi: 10.3171/jns.1992.76.4.0640. [DOI] [PubMed] [Google Scholar]
- 8.Westphal M, et al. Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien) 2006;148(3):269–75. doi: 10.1007/s00701-005-0707-z. discussion 275. [DOI] [PubMed] [Google Scholar]
- 9.Nance EA, et al. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med. 2012;4(149):149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou J, et al. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc Natl Acad Sci U S A. 2013;110(29):11751–6. doi: 10.1073/pnas.1304504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider CS, et al. Minimizing the non-specific binding of nanoparticles to the brain enables active targeting of Fn14-positive glioblastoma cells. Biomaterials. 2015;42:42–51. doi: 10.1016/j.biomaterials.2014.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wadajkar AS, et al. Tumor-targeted nanotherapeutics: overcoming treatment barriers for glioblastoma. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016 doi: 10.1002/wnan.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hadjipanayis CG, et al. EGFRvIII antibody-conjugated iron oxide nanoparticles for magnetic resonance imaging-guided convection-enhanced delivery and targeted therapy of glioblastoma. Cancer Res. 2010;70(15):6303–12. doi: 10.1158/0008-5472.CAN-10-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pang Z, et al. Enhanced intracellular delivery and chemotherapy for glioma rats by transferrin-conjugated biodegradable polymersomes loaded with doxorubicin. Bioconjug Chem. 2011;22(6):1171–80. doi: 10.1021/bc200062q. [DOI] [PubMed] [Google Scholar]
- 15.Madhankumar AB, et al. Interleukin-13 receptor-targeted nanovesicles are a potential therapy for glioblastoma multiforme. Mol Cancer Ther. 2006;5(12):3162–9. doi: 10.1158/1535-7163.MCT-06-0480. [DOI] [PubMed] [Google Scholar]
- 16.Nance E, et al. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano. 2014;8(10):10655–64. doi: 10.1021/nn504210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C, et al. Convection enhanced delivery of cisplatin-loaded brain penetrating nanoparticles cures malignant glioma in rats. J Control Release. 2017 doi: 10.1016/j.jconrel.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider CS, et al. Surface plasmon resonance as a high throughput method to evaluate specific and non-specific binding of nanotherapeutics. J Control Release. 2015;219:331–44. doi: 10.1016/j.jconrel.2015.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dancy JG, et al. Non-specific binding and steric hindrance thresholds for penetration of particulate drug carriers within tumor tissue. J Control Release. 2016;238:139–48. doi: 10.1016/j.jconrel.2016.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov. 2008;7(5):411–25. doi: 10.1038/nrd2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez JG, et al. The TWEAK receptor Fn14 is a potential cell surface portal for targeted delivery of glioblastoma therapeutics. Oncogene. 2016;35(17):2145–55. doi: 10.1038/onc.2015.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran NL, et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-kappaB and correlate with poor patient outcome. Cancer Res. 2006;66(19):9535–42. doi: 10.1158/0008-5472.CAN-06-0418. [DOI] [PubMed] [Google Scholar]
- 23.Kim AJ, et al. Use of single-site-functionalized PEG dendrons to prepare gene vectors that penetrate human mucus barriers. Angew Chem Int Ed Engl. 2013;52(14):3985–8. doi: 10.1002/anie.201208556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medberry CJ, et al. Hydrogels derived from central nervous system extracellular matrix. Biomaterials. 2013;34(4):1033–40. doi: 10.1016/j.biomaterials.2012.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown SA, et al. TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem J. 2006;397(2):297–304. doi: 10.1042/BJ20051362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reilly KM, et al. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26(1):109–13. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 27.Schuster BS, et al. Overcoming the cystic fibrosis sputum barrier to leading adeno-associated virus gene therapy vectors. Mol Ther. 2014;22(8):1484–93. doi: 10.1038/mt.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langer R. Drug delivery and targeting. Nature. 1998;392(6679 Suppl):5–10. [PubMed] [Google Scholar]
- 29.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 30.Cheng CJ, et al. A holistic approach to targeting disease with polymeric nanoparticles. Nat Rev Drug Discov. 2015;14(4):239–47. doi: 10.1038/nrd4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lesniak MS, Brem H. Targeted therapy for brain tumours. Nat Rev Drug Discov. 2004;3(6):499–508. doi: 10.1038/nrd1414. [DOI] [PubMed] [Google Scholar]
- 32.Strohbehn G, et al. Imaging the delivery of brain-penetrating PLGA nanoparticles in the brain using magnetic resonance. J Neurooncol. 2015;121(3):441–9. doi: 10.1007/s11060-014-1658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunwar S, et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010;12(8):871–81. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reardon DA, et al. Phase II trial of murine (131)I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2002;20(5):1389–97. doi: 10.1200/JCO.2002.20.5.1389. [DOI] [PubMed] [Google Scholar]
- 35.Cheng E, et al. TWEAK/Fn14 Axis-Targeted Therapeutics: Moving Basic Science Discoveries to the Clinic. Front Immunol. 2013;4:473. doi: 10.3389/fimmu.2013.00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurunathan S, et al. Regulation of fibroblast growth factor-inducible 14 (Fn14) expression levels via ligand-independent lysosomal degradation. J Biol Chem. 2014;289(19):12976–88. doi: 10.1074/jbc.M114.563478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ouyang M, et al. Metronomic Doses of Temozolomide Enhance the Efficacy of Carbon Nanotube CpG Immunotherapy in an Invasive Glioma Model. PLoS One. 2016;11(2):e0148139. doi: 10.1371/journal.pone.0148139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sampson JH, et al. Poor drug distribution as a possible explanation for the results of the PRECISE trial. J Neurosurg. 2010;113(2):301–9. doi: 10.3171/2009.11.JNS091052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.