

Abstract
With over 1,900 variants reported in the cystic fibrosis transmembrane conductance regulator (CFTR), enhanced understanding of cystic fibrosis (CF) genotype-phenotype correlation represents an important and expanding area of research. The potentiator Ivacaftor has proven an effective treatment for a subset of individuals carrying missense variants, particularly those that impact CFTR gating. Therapeutic efforts have recently focused on correcting the basic defect resulting from the common F508del variant, as well as many less frequent missense alleles. Modest enhancement of F508del-CFTR function has been achieved by combining Ivacaftor with Lumacaftor, a compound that aids maturational processing of misfolded CFTR. Continued development of in silico and in vitro models will facilitate CFTR variant characterization and drug testing, thereby elucidating heterogeneity in the molecular pathogenesis, phenotype, and modulator responsiveness of CF.
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive disorder with nearly 90,000 reported cases worldwide, and highest incidence occurs among white individuals of Northern European descent. The disease results from loss-of-function of the CF transmembrane conductance regulator (CFTR), a chloride and bicarbonate channel expressed at the apical surface of exocrine secretory epithelia. In the absence of functional CFTR, hyperviscous luminal secretions accumulate within respiratory, pancreatic, gastrointestinal and reproductive systems, ultimately leading to chronic inflammation, severe tissue damage, and multi-organ destruction [1].
The CFTR gene was mapped in 1989, allowing extensive study and characterization of the biochemistry and functional role of CFTR protein. More than 1,900 variants within CFTR have been observed to date, many of which have been shown to elicit one or more defects of steps comprising biogenesis, ion transport, or plasma membrane (PM) turnover [2,3,4]. CFTR variants have traditionally been grouped into six classes based on features associated with molecular pathogenesis: class I – defective protein synthesis (e.g. premature termination codons); class II – aberrant protein maturation and premature degradation (e.g. ‘processing’ defects); class III – abnormal channel regulation (e.g. ‘gating’ defects); class IV – improper formation of the channel pore (e.g. ‘conductance’ defects); class V – decreased levels of protein synthesis (e.g. splicing defects); and class VI – accelerated internalization or faulty recycling from the PM (e.g. ‘turnover’ defects) [2,3,4; www.cftr2.org] (Fig. 1).
Figure 1. Classification scheme and cellular localization of CFTR variants.
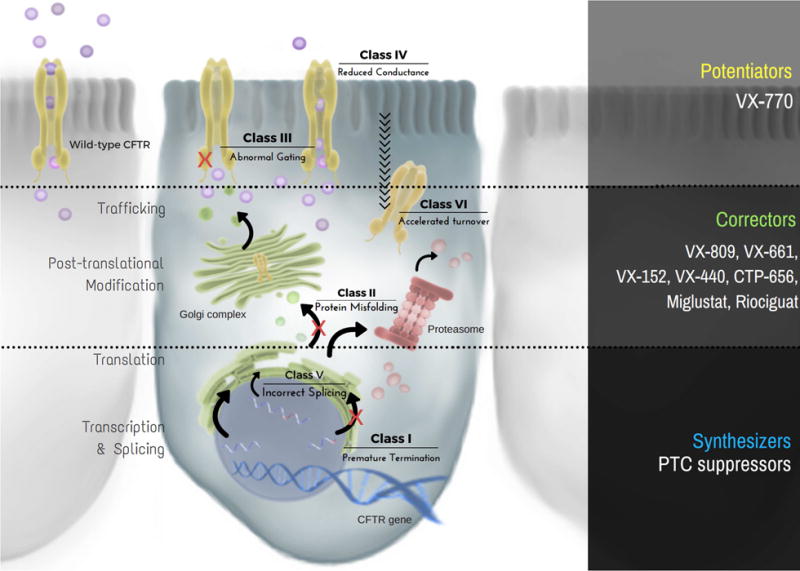
Class I and V defects result in diminished protein production, whereas class II and VI yield reduced stability of CFTR. In addition, class III and IV variants inhibit channel function or activity of cell surface associated CFTR. Molecular-based therapeutic strategies target each of these categories and include the following: (1) ‘synthesizers’, which rescue CFTR protein production (e.g. suppression of premature truncation codons, or PTCs), (2) ‘correctors’, which augment maturation and decelerate turnover of CFTR (e.g. VX-809, VX-661, VX-152, VX-440, CTP-656, Miglustat, Riociguat), and (3) ‘potentiators’, which increase open channel probability and/or gating potential of apically localized CFTR (e.g. VX-770).
Although these subcategories provide a valuable means of profiling the panoply of CFTR abnormalities, molecular complexity of individual CFTR variants has become increasingly evident. CFTR defects exist across a spectrum of severity at the molecular level, which correlate with a range of clinical presentations. For example, mild variants result in male infertility and deleterious variants result in chronic lung infection with mucus obstruction and progressive deterioration, whereas the most severe variants often lead to pancreatic and/or hepatic insufficiency. The challenge, therefore, is to understand mechanisms underlying various CFTR variants and develop precision medicine approaches tailored to all forms of the disease. This review will focus on missense variants (i.e. amino acid substitutions), which represent the largest category of CFTR defects (~40%) [1]. We will discuss strategies that have proven successful for rescuing CFTR function, methods for predicting therapeutic responsiveness, challenges faced with the current design of clinical trials, and cutting-edge tools utilized to develop more efficient interventions.
SUCCESS STORY FOR p.G551D
The first CFTR-targeted compound resulted from a revolutionary partnership between the U.S. CF Foundation and the pharmaceutical industry. Using high-throughput, cell-based fluorescence membrane potential assays in recombinant Fischer rat thyroid (FRT) cells, the small molecule VX-770 (Ivacaftor, Kalydeco™) was discovered as a robust potentiator of gating activity in the CFTR class III variant, p.G551D. Ivacaftor was shown to improve multiple measures of clinical utility in patients carrying at least one G551D-CFTR allele, including: (1) enhanced forced expiratory volume in one second (FEV1), (2) increased body mass index (BMI), (3) fewer respiratory infections, (4) decreased hospitalizations, and (5) reduced sweat chloride levels [5,6]. The effectiveness of this drug was made possible by the strong understanding of the molecular mechanisms underlying the G551D variant. Advances in our understanding of the genetics underlying CF, at the level of individual sequence variants as well as modifier genes, will lead to development of effective therapies for more individuals of different genotypes [1].
The Food and Drug Administration (FDA) approved Ivacaftor in 2012 as a pharmacologic treatment for CF patients 12 years and older who carry at least one copy of the p.G551D variant. In the years following, trials conducted in younger patients with p.G551D (i.e. 6–12 years old), suggested that starting Ivacaftor at an earlier age may slow or even prevent lung disease progression [7]. Today, children two years and older carrying certain CFTR gating variants [2,3,4] are approved to receive the drug, and longevity studies among all ages indicate clinical benefits can be maintained for years [6,8,9].
IVACAFTOR SPECTRUM OF ACTIVITY
Based on the compelling response of G551D-CFTR achieved with Ivacaftor, additional studies were undertaken to determine whether this compound could rescue other CFTR molecular phenotypes similar to p.G551D (i.e. class III or IV defects, including p.R117H, p.G178R, p.S549N, p.S549R, p.G551S, p.G1244E, p.G1349D, p.S1251N, and p.S1255P). In vitro analysis conducted on this cohort of gating/conductance variants revealed the compound greatly augmented resident channel activity [10], and subsequent clinical trial results showed significant improvement in FEV1, BMI, and sweat chloride levels [11,12]. As a consequence, Ivacaftor was FDA-approved in 2014 for individuals two years and older carrying R117H-CFTR, and the following year, gained approval for the eight other variants listed above.
While this strategy has proven highly successful for a number of CFTR variants, similar approaches may not be sufficient to identify therapeutic interventions for all missense mutations. Many CF variants exist at extremely low frequencies in the patient population, presenting a significant challenge to clinical evaluation. Informative human trials may require large numbers of subjects and controls, and are impractical for rare variants occurring in only 2 or 3 individuals worldwide. In general, ultra-orphan diseases such as CF may require alternative strategies for establishing clinical efficacy [13].
IMPROVED UNDERSTANDING OF CFTR VARIANT COMPLEXITY
To bring molecular-based therapy and precision medicine to CF patients of all genotypes, two significant hurdles must be overcome: (1) the large number of variants known to exist in CFTR, and (2) the low frequency at which many of these occur. Of the 796 missense variants reported in the CF mutation database [www.genet.sickkids.on.ca/app], 81 have been functionally described, and 57 of those have been classified as disease-causing [www.cftr2.org]. The primary goal of the CFTR2 project is to characterize and determine disease liability for all mutant CFTR alleles [14].
Distinguishing CF disease phenotypes through use of traditional categories has proven beneficial for studying individual CFTR variants (Fig. 1). However, variants that affect several processes require a more complex classification protocol involving multiple categories, each of which might require a separate class of compound [15]. Missense variants frequently fall into the aforementioned group. In silico tools such as molecular modeling and dynamics simulations [16] can help predict functional consequences of missense variants, but are far from comprehensive.
Sequencing of the entire CFTR coding region has become nearly commonplace due to substantial reduction in cost [17]. The increasing number of individuals sequenced by whole-exome, whole-genome, and carrier screening, has dramatically enhanced the volume of sequence data derived from CF patients, asymptomatic carriers, and the general population. As with other genetic diseases [18], this has led to significant increases in the number of CFTR variants identified, necessitating more sophisticated methods for variant interpretation. Sequence analysis of all CFTR exons has augmented discovery of complex alleles, which contain multiple variants in cis. Complex alleles may behave differently than those with single variants in terms of function and/or drug response. This can lead to incorrect labeling of a variant as disease-causing until segregation analysis and/or functional studies exclude deleterious effects, as occurred with p.I148T [19]. Consequently, it may become imperative to identify all variants within CFTR when considering appropriate therapy for a particular CF patient. The establishment of large general databanks for genetic variants (ClinVar, ExAC, etc.), as well as expertly curated databases (e.g. CFTR2), provides important tools for differentiating detrimental versus benign variants based on population frequency [20].
CHALLENGES EVALUATING THERAPEUTIC RESPONSIVENESS
As the number of reported rare CFTR variants increases, model systems will become significantly more valuable for experimental evaluation of underlying molecular defects. The ability of cell-based platforms to generate robust, reliable data in an efficient manner will determine the rate at which new therapies can be delivered to patients. Investigations using an immortalized cell line (FRT) contributed to the FDA-approval process for Ivacaftor [10,21] and Orkambi™ (combination of Ivacaftor with the corrector Lumacaftor) [22], and have been useful for interpreting variant phenotypes studied thus far [23].
In vitro systems can be utilized to identify additional variants that respond well to Ivacaftor alone or with Lumacaftor. For example, p.P67L is a rare variant for which clinical trials are less likely to occur, since there are ~240 patients worldwide [www.cftr2.org] who carry this defect. Notably, studies conducted in FRT cells have shown p.P67L responds robustly to both compounds, reaching nearly wild-type levels of CFTR activity [24], suggesting that patients would benefit from these drugs. Conversely, the p.N1303K variant is 8 times more common (reported in over 2,000 individuals), but it is unresponsive to VX-770 or VX-809 in FRT cells [23] and primary airway epithelia [25]. Although sufficient numbers of patients may be available for a robust clinical study, preclinical evidence strongly suggests against clinical improvement in this setting.
Of the missense variants reported to CFTR2 still requiring clinical classification, more than 500 have been noted in 10 individuals or fewer worldwide (personal communication, Karen S. Raraigh, Johns Hopkins). The rarity of these patients precludes routine collection of primary cells for functional and drug studies. Thus, in vitro studies are expected to be primary generators of preliminary data for classification and analysis of the variants. Development of cell-based systems that more closely approximate primary human airway cells provides an opportunity to study rare CFTR defects and their response to FDA-approved compounds in a near-native context [26]. Novel strategies must also be employed to perform clinical efficacy trials in individuals with ultra-rare variants, and “N-of-1” or “N-of-a-few” are among methods currently under consideration [27].
In vitro experiments, in silico predictions, and pre-clinical testing are distinct branches of research employed for characterizing individual variants and predicting response to pharmacological agents. As CFTR missense variants under study are increasingly rare and functionally complex, strengthening the quality and quantity of evidence generated by these strategies will be paramount.
RECENT ADVANCES IN MODEL SYSTEMS
Characterization of disease-associated CFTR variants, including assessment of therapeutic responsiveness, has been performed in cell-based models for decades. Many immortalized mammalian cell lines have proven essential for distinguishing specific features of CFTR biogenesis, as well as mechanisms invoked by investigational compounds [10,28]. Recently, primary human nasal or bronchial epithelia [25,29] and induced pluripotent stem cells [30,31,32] have emerged as strong predictive tools. Organoids generated from patients represent another topical area of progress with the potential to predict individual response to a therapeutic strategy (i.e. precision medicine), but the validity of this postulate remains to be determined [33,34]. Additionally, six animal models expressing a variety of CFTR variants are now available (e.g. zebrafish, mouse, rat, rabbit, ferret, pig), although each has limitations regarding ease of use or degree to which human disease is recapitulated [35,36]. Finally, yeast phenomic screening has emerged as a means for discovery of gene-gene interaction networks and other features of CFTR class II and III variants [37,38,39], including identification and targeting of novel CFTR modulators in patient-derived epithelia [40].
FUTURE DIRECTIONS RELEVANT TO CF THERAPEUTICS
Based on the success of Ivacaftor, pharmaceutical companies have begun developing other small molecules that partially restore CFTR function, the most advanced of which are second generation correctors that improve intracellular processing and cell surface activity of class II variants (P Grootenhuis et al, abstract 188, 30th North American Cystic Fibrosis Conference, Orlanda FL, October 2016), i.e. to levels above those achieved by the combination of Ivacaftor and Lumacaftor. There are at least 20 clinical trials underway that utilize such pharmacological interventions, with examples of phase II and III studies currently under enrollment shown in Fig. 2 [www.clinicaltrials.gov].
Figure 2. Current and upcoming CFTR modulator clinical trials.
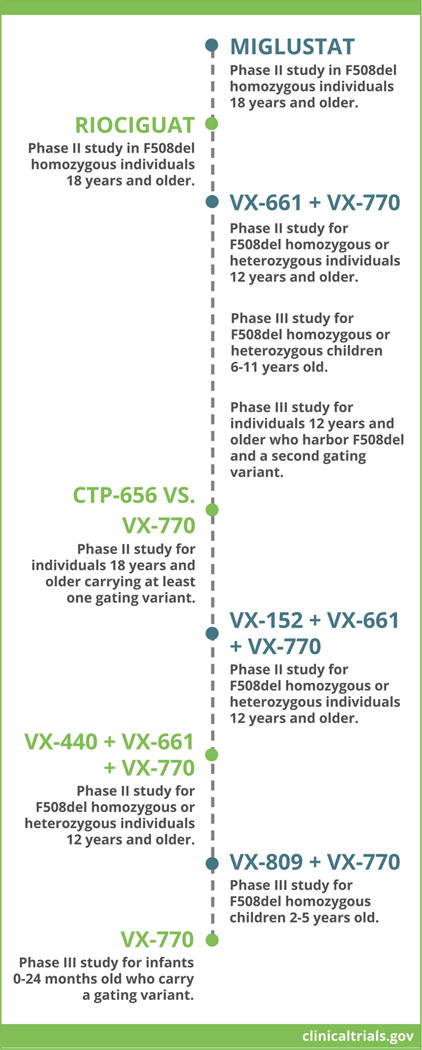
Examples of phase II or III clinical trials under enrollment in the United States and Europe are listed (see also www.clinicaltrials.gov). Various strategies outlined above intend to test safety, tolerability, and efficacy of CFTR modulators administered as monotherapy (e.g. Miglustat), combination therapy with two agents (e.g. VX-661 + VX-770), or combination therapy with three agents (e.g. VX-152 + VX-661 + VX-770). In the majority of cases, eligible patients must be homozygous or heterozygous for the most prevalent CFTR variant, F508del, or carry a gating or partial function defect.
The overarching goal of translational CF science is to develop therapeutic strategies that will benefit all individuals with CF, irrespective of genotype. Basic and clinical investigations are in progress to explore feasibility of innovative genetic and genomic medicine technologies, including transfer of nucleic acids by airway stem/progenitor cells [41,42], zinc finger nuclease-or CRISPR/Cas9-edited human pluripotent stem cells [30,31,32], and nanoparticles [43,44,45], as well as protein replacement via mRNA transfer [46]. Recently, enhanced adenoviral and lentiviral vectors were used to show functional CFTR gene delivery to airways of the CF porcine model [47,48], and the first lentivirus-based clinical trial is scheduled for 2017 [49].
CONCLUSIONS
Therapeutic benefit for individuals with CF harboring missense variants can be achieved by improving function of existing, partially-processed CFTR protein. As such, missense alleles represent “low hanging fruit” for small molecule intervention. Variants that result in complete loss of CFTR protein share a potential therapeutic mechanism, in that they require insertion and expression of an entirely new or repaired CFTR allele. This is no small task, as gene transfer therapy has been in development for CF and other monogenic diseases since the 1990s. While the medical genetics community awaits technological progress to allow for sufficient CFTR gene delivery, effective therapy for most individuals with CF carrying missense variants is expected much sooner.
HIGHLIGHTS.
CFTR missense alleles exhibit diverse mechanisms of dysfunction.
Compounds targeting specific classes of CFTR defects have been variably successful.
Methods and models utilized for studying CFTR variants are continually improving.
Characterizing each CFTR variant will facilitate approaches to precision medicine.
Genetic tools under development may lead to future treatments for all forms of CF.
Acknowledgments
We sincerely thank Ms. Carleen M. Sabusap for assistance with computer generation of Figure 1 (i.e. formatting of resources provided by the “Library of Science and Medical Illustrations”; https://gumroad.com/l/library).
Support for this research was provided by R01DK44009 (NIH/NIDDK), CUTTIN15XX0 (CF Foundation) and CUTTIN16IO (CF Foundation) to GRC; P30DK072482 (NIH/NIDDK), SORSCH13XX0 (CF Foundation) and SORSCH14XX0 (CF Foundation) to EJS; and F31HL131231 (NIH/NHLBI) and OLIVER17F0 (CF Foundation) to KEO.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT(S) OF INTEREST DISCLOSURE
The authors whose names are listed immediately below report the following details of affiliation or involvement in an organization or entity with a financial or non-financial interest in the subject matter or materials discussed in this manuscript. Please specify the nature of the conflict on a separate sheet of paper if the space below is inadequate
| Author names: | Details of the conflict(s) of interest: |
| Garry R. Cutting | Consultant for Vertex Pharmaceuticals Company Research funded by Cystic Fibrosis Foundation |
| Sangwoo T. Han | Research funded by Cystic Fibrosis Foundation |
| Eric J. Sorscher | Board Member and Medical Advisory Council Chair, Cystic Fibrosis Foundation. Research contracts for drug development from Pfizer and Proteostasis. Research funded by Cystic Fibrosis Foundation and National Institutes of Health. |
| Kathryn E. Oliver | Research funded by Cystic Fibrosis Foundation and National Institutes of Health. |
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the period of review, have been highlighted as:
*of special interest
**of outstanding interest
- **1.Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16(1):45–56. doi: 10.1038/nrg3849. A topical manuscript dealing with the relationships between molecular and clinical findings in cystic fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Welsh MJ. Smith AE: Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73(7):1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 3.Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. doi: 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
- 4.Haardt M, Benharouga M, Lechardeur D, Kartner N, Lukacs GL. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis. A novel class of mutation. J Biol Chem. 1999;274(31):21873–21877. doi: 10.1074/jbc.274.31.21873. [DOI] [PubMed] [Google Scholar]
- 5.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, Ratjen F, Sermet-Gaudelus I, Plant B, Munck A, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST) Lancet Respir Med. 2014;2(11):902–910. doi: 10.1016/S2213-2600(14)70218-8. [DOI] [PubMed] [Google Scholar]
- 7.Dryden C, Wilkinson J, Young D, Brooker RJ. Scottish Paediatric Cystic Fibrosis Managed Clinical Network (SPCFMCN): The impact of 12 months treatment with ivacaftor on Scottish paediatric patients with cystic fibrosis with the G551D mutation: a review. Arch Dis Child. 2016;0:1–3. doi: 10.1136/archdischild-2015-310420. [DOI] [PubMed] [Google Scholar]
- *8.Sawicki GS, McKone EF, Pasta DJ, Millar SJ, Wagener JS, Johnson CA, Konstan MW. Sustained Benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am J Respir Crit Care Med. 2015;192(7):836–842. doi: 10.1164/rccm.201503-0578OC. Provides evidence for a durable responsiveness to Ivacaftor among patients with G551D-related CF. [DOI] [PubMed] [Google Scholar]
- 9.Fila L, Valentova Bartakova L, Grandcourtova A, Marel M, Drnek R, Bilkova A, Macek M, Drevinek P. Ivacaftor in cystic fibrosis adults: Czech experience with six years of follow-up. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2016;160(2):276–279. doi: 10.5507/bp.2016.029. [DOI] [PubMed] [Google Scholar]
- 10.Yu H, Burton B, Huang CJ, Worley J, Cao D, Johnson JP, Jr, Urrutia A, Joubran J, Seepersaud S, Sussky K, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros. 2012;11(3):237–245. doi: 10.1016/j.jcf.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 11.De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, Higgins M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–680. doi: 10.1016/j.jcf.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, Rubenstein RC, Higgins M, VX11-770-110 (KONDUCT) Study Group Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524–533. doi: 10.1016/S2213-2600(15)00201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gagne JJ, Thompson L, O’Keefe K, Kesselheim AS. Innovative research methods for studying treatments for rare diseases: methodological review. BMJ. 2014;349:g6802. doi: 10.1136/bmj.g6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, Ramalho AS, Amaral MD, Dorfman R, Zielenski J, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013;45(10):1160–1167. doi: 10.1038/ng.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **15.Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, Hong JS, Pollard HB, Guggino WB, Balch WE, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27(3):424–433. doi: 10.1091/mbc.E14-04-0935. Highlights challenges associated with molecular sub-grouping of rare CFTR variants, including theratype designation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Callebaut I, Hoffmann B, Lehn P, Mornon JP. Molecular modelling and molecular dynamics of CFTR. Cell Mol Life Sci. 2017;74(1):3–22. doi: 10.1007/s00018-016-2385-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19(2):192–203. doi: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claustres M, Altiéri JP, Guittard C, Templin C, Chevalier-Porst F, Des Georges M. Are pI148T, p.R74W and p.D1270N cystic fibrosis causing mutations? BMC Med Genet. 2004;5:19. doi: 10.1186/1471-2350-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brookes AJ, Robinson PN. Human genotype-phenotype databases: aims, challenges and opportunities. Nat Rev Genet. 2015;16(12):702–715. doi: 10.1038/nrg3932. [DOI] [PubMed] [Google Scholar]
- 21.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106(44):18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108(46):18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros. 2014;13(1):29–36. doi: 10.1016/j.jcf.2013.06.008. [DOI] [PubMed] [Google Scholar]
- **24.Sabusap C, Wang W, McNicholas-Bevensee C, Chung J, Fu L, Wen H, Mazur M, Kirk K, Collawn J, Hong J, Sorscher E. Analysis of cystic fibrosis-associated P67L CFTR illustrates barriers to personalized therapeutics for orphan diseases. JCI Insight. 2016;1(14):e86581. doi: 10.1172/jci.insight.86581. Case study describing a traditionally classified conductance (class IV) CFTR defect that instead exhibits processing (class II) and gating (class III) abnormalities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Awatade NT, Uliyakina I, Farinha CM, Clarke LA, Mendes K, Solé A, Pastor J, Ramos MM, Amaral MD. Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis. EBioMedicine. 2015;2(2):147–153. doi: 10.1016/j.ebiom.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottschalk LB, Vecchio-Pagan B, Sharma N, Han ST, Franca A, Wohler ES, Batista DA, Goff LA, Cutting GR. Creation and characterization of an airway epithelial cell line for stable expression of CFTR variants. J Cyst Fibros. 2016;15(3):285–294. doi: 10.1016/j.jcf.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGarry ME, Illek B, Ly NP, Zlock L, Olshansky S, Moreno C, Finkbeiner WE, Nielson DW. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr Pulmonol. 2017;52(4):472–479. doi: 10.1002/ppul.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruscia E, Sangiuolo F, Sinibaldi P, Goncz KK, Novelli G, Gruenert DC. Isolation of CF cell lines corrected at DeltaF508-CFTR locus by SFHR-mediated targeting. Gene Ther. 2002;9(11):683–685. doi: 10.1038/sj.gt.3301741. [DOI] [PubMed] [Google Scholar]
- *29.Terlizzi V, Castaldo G, Salvatore D, Lucarelli M, Raia V, Angioni A, Carnovale V, Cirilli N, Casciaro R, Colombo C, et al. Genotype-phenotype correlation and functional studies in patients with cystic fibrosis bearing CFTR complex alleles. J Med Genet. 2017;54(4):224–235. doi: 10.1136/jmedgenet-2016-103985. Underscores pathogenic importance of complex CFTR alleles in terms of disease phenotype and clinical course. [DOI] [PubMed] [Google Scholar]
- *30.Crane AM, Kramer P, Bui JH, Chung WJ, Li XS, Gonzalez-Garay ML, Hawkins F, Liao W, Mora D, Choi S, et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports. 2015;4(4):569–77. doi: 10.1016/j.stemcr.2015.02.005. Introduces potential use of iPS cells for theratyping and/or cell-based treatment following CFTR gene editing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu Z, Verma N, González F, Shi ZD, Huangfu D. A CRISPR/Cas-Mediated Selection-free Knockin Strategy in Human Embryonic Stem Cells. Stem Cell Reports. 2015;4(6):1103–1111. doi: 10.1016/j.stemcr.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *32.Firth AL, Menon T, Parker GS, Qualls SJ, Lewis BM, Ke E, Dargitz CT, Wright R, Khanna A, Gage FH, Verma IM. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015;12(9):1385–1390. doi: 10.1016/j.celrep.2015.07.062. Describes protocols for generating respiratory epithelia from individual patient-derived iPSCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssen s HM, de Winter-de Groot KM, Brandsma AM, de Jong NW, Bijvelds MJ, Scholte BJ, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–945. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- **34.Dekkers JF, Berkers G, Kruisselbrink E, Vonk A, de Jonge HR, Janssens HM, Bronsveld I, van de Graaf EA, Nieuwenhuis EE, Houwen RH, et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med. 2016;8(344):344ra84. doi: 10.1126/scitranslmed.aad8278. Establishes use of patient-derived organoids as a robust theratyping tool. [DOI] [PubMed] [Google Scholar]
- 35.Keiser NW, Engelhardt JF. New animal models of cystic fibrosis: what are they teaching us? Curr Opin Pulm Med. 2011;17(6):478–483. doi: 10.1097/MCP.0b013e32834b14c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *36.Lavelle GM, White MM, Browne N, McElvaney NG, Reeves EP. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. Biomed Res Int. 2016;2016:5258727. doi: 10.1155/2016/5258727. Recent review of CF animal models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pagant S, Halliday JJ, Kougentakis C, Miller EA. Intragenic suppressing mutations correct the folding and intracellular traffic of misfolded mutants of Yor1p, a eukaryotic drug transporter. J Biol Chem. 2010;285(47):36304–36314. doi: 10.1074/jbc.M110.142760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Louie RJ, Guo J, Rodgers JW, White R, Shah N, Pagant S, Kim P, Livstone M, Dolinski K, McKinney BA, et al. A yeast phenomic model for the gene interaction network modulating CFTR-∆F508 protein biogenesis. Genome Med. 2012;4(12):103. doi: 10.1186/gm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei S, Roessler BC, Chauvet S, Guo J, Hartman JL, 4th, Kirk KL. Conserved allosteric hot spots in the transmembrane domains of cystic fibrosis transmembrane conductance regulator (CFTR) channels and multidrug resistance protein (MRP) pumps. J Biol Chem. 2014;289(29):19942–19957. doi: 10.1074/jbc.M114.562116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *40.Veit G, Oliver K, Apaja PM, Perdomo D, Bidaud-Meynard A, Lin ST, Guo J, Icyuz M, Sorscher EJ, Hartman Iv JL, Lukacs GL. Ribosomal Stalk Protein Silencing Partially Corrects the ∆F508-CFTR Functional Expression Defect. PLoS Biol. 2016;14(5):e1002462. doi: 10.1371/journal.pbio.1002462. Elucidates novel participants during CFTR maturational processing, particularly the importance of ribosomal components. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghosh M, Ahmad S, White CW, Reynolds SD. Transplantation of Airway Epithelial Stem/Progenitor Cells: A Future for Cell-Based Therapy. Am J Respir Cell Mol Biol. 2017;56(1):1–10. doi: 10.1165/rcmb.2016-0181MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer DJ, Grove NC, Ing J, Crane AM, Venken K, Davis BR, Ng P. Homology Requirements for Efficient, Footprintless Gene Editing at the CFTR Locus in Human iPSCs with Helper-dependent Adenoviral Vectors. Mol Ther Nucleic Acids. 2016;5(10):e372. doi: 10.1038/mtna.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McNeer NA, Anandalingam K, Fields RJ, Caputo C, Kopic S, Gupta A, Quijano E, Polikoff L, Kong Y, Bahal R, et al. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat Commun. 2015;6:6952. doi: 10.1038/ncomms7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS, Zhu H, Siegwart DJ. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew Chem Int Ed Engl. 2017;56(4):1059–1063. doi: 10.1002/anie.201610209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dahlman JE, Kauffman KJ, Xing Y, Shaw TE, Mir FF, Dlott CC, Langer R, Anderson DG, Wang ET. Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proc Natl Acad Sci USA. 2017;114(8):2060–2065. doi: 10.1073/pnas.1620874114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bangel-Ruland N, Tomczak K, Fernández E, Leier G, Leciejewski B, Rudolph C, Rosenecker J, Weber WM. Cystic fibrosis transmembrane conductance regulator-mRNA delivery: a novel alternative for cystic fibrosis gene therapy. J Gene Med. 2013;15(11–12):414–426. doi: 10.1002/jgm.2748. [DOI] [PubMed] [Google Scholar]
- **47.Steines B, Dickey DD, Bergen J, Excoffon KJ, Weinstein JR, Li X, Yan Z, Abou Alaiwa MH, Shah VS, Bouzek DC, et al. CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight. 2016;1(14):e88728. doi: 10.1172/jci.insight.88728. Describes efficient CFTR delivery to airways of the porcine CF model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **48.Cooney AL, Abou Alaiwa MH, Shah VS, Bouzek DC, Stroik MR, Powers LS, Gansemer ND, Meyerholz DK, Welsh MJ, et al. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight. 2016;1(14):e88730. doi: 10.1172/jci.insight.88730. Demonstrates effective CFTR gene transfer to CF pig lung using leading-edge lentiviral constructs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **49.Alton EW, Beekman JM, Boyd AC, Brand J, Carlon MS, Connolly MM, Chan M, Conlon S, Davidson HE, Davies JC, et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax. 2017;72(2):137–147. doi: 10.1136/thoraxjnl-2016-208406. Presents scientific basis for a highly efficient lentivirus and CFTR delivery to human airways. [DOI] [PMC free article] [PubMed] [Google Scholar]