

Abstract
Carbon-11 labelled carbon dioxide is the most common feedstock for the synthesis of positron emission tomography radiotracers, and can be directly used for 11C-carbonylation. Herein, we report the development of an apparatus that takes advantage of “in-loop” technologies to facilitate robust and reproducible syntheses of 11C-carbonyl-based radiotracers by [11C]CO2-fixation. Our “in-loop” [11C]CO2-fixation method is simple, efficient, and proceeds smoothly at ambient pressure and temperature. We selected model 11C-carbonyl labelled carbamates as well as symmetrical and unsymmetrical ureas based on their widespread use in radiotracer design and our clinical research interests for proof-of-concept. Utility of this method is demonstrated by the synthesis of a reversible radiopharmaceutical for monoamine oxidase B, [11C]SL25.1188, as well as two novel fatty acid amide hydrolase inhibitors. These radiotracers were isolated and formulated (>3.5 GBq; 100 mCi) with radiochemical purities (>99%) and molar radioactivity (≥80 GBq/μmol; ≥2162 mCi/μmol).
Keywords: CO2-fixation, FAAH, MAO-B, Carbonylation, Carbon-11, Loop
Introduction
Carbon-11 (11C; t1/2 = 20.4 min) is a valuable radionuclide in positron emission tomography (PET).1–4 The high positron emission decay mode (β+; 99.8%) and low energy protons (0.96 keV) allow for high resolution in-vivo imaging. Another advantage is that 11C can be incorporated into bioactive or endogenous molecules without any structural modification (isotopologue of carbon-12) and with minimal effect on the (bio)chemical properties of the compound. Moreover, the short half-life of 11C allows for multiple in vivo PET imaging studies to be undertaken in the same subject or animal on a single day. The most widely used reaction in 11C-radiopharmaceutical production is 11C-methylation, via [11C]CH3I or [11C]CH3OTf. However 11C-carbonylation is also a viable strategy using [11C]CO2, [11C]CO, [11C]COCl2 or [11C]HCN.5
[11C]CO2 is a highly attractive building block for radiolabelling, since it is produced directly in target. However, due to low chemical reactivity and trapping efficiency, the direct incorporation of [11C]CO2 into organic molecules poses a significant challenge. The traditional methods for “[11C]CO2-fixation” rely on Grignard reagents, organolithiums or organosilanamines.5 All such reagents require the rigorous exclusion of atmospheric moisture and CO2 during storage and manipulation. The direct formation of 11C-labelled carbonyl groups by [11C]CO2-fixation, has been recently established in radiopharmaceutical production,5–6 where strong organic bases, e.g. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU)7 or 2-tert-Butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorline (BEMP),8–9 are utilized to reversibly capture [11C]CO2 in solution. While initial work focused on the synthesis of 11C-labelled carbamates, the scope of the method rapidly extended to syntheses of unsymmetrical [11C]ureas and [11C]oxazolidinones via POCl3 dehydration to generate 11C-labelled isocyanates as a key intermediates,9 as well as 11C-carboxylic acids via metal-catalyzed aryl boronation reactions.10–11
Despite evident advantages of [11C]CO2-fixation for radiotracer production, the radiochemistry has been generally restricted to semi-manual vial-based methods. Automation of this methodology would be highly desirable, not only from a user’s perspective, as it would simplify the radiosynthesis process and reduce exposure to radioactivity, but also to improve its reproducibility and compliance of routine radiopharmaceutical production under current good manufacturing practices (cGMP). The aim of this study was two-fold: 1) to develop a simple and easily automated [11C]CO2-fixation platform; and 2) to carry out proof-of-concept radiosyntheses of 11C-carbonyl labelled compounds with diverse scaffolds.
The traditional vial-based procedure was abandoned and we sought to take advantage of the captive solvent or “in-loop” technologies, that have been established for Grignard reactions12–15, or 11C-methylation,16–17 specifically, ease of automation, reliability, efficiency (minimal radioactive loss to the surrounding atmosphere, needles, septa, or to transfer from vessels), and in particular, its high surface-to-volume ratio, and should be ideally suited for [11C]CO2-fixation radiochemistry. We selected model 11C-carbonyl labelled carbamates as well as symmetrical and unsymmetrical ureas based on their widespread use in radiotracer design and our clinical research interests for the proof-of-concept radiolabelling by “in loop [11C]CO2-fixation” (Table 1 and Scheme 1). The radiotracers that were isolated in this study include, two new and structurally similar 11C-isotopologues of a fatty acid amide hydrolase (FAAH) inhibitor, [11C]JNJ1661010 ([11C]5) and [11C]7, as well as the monoamine oxidase B (MAO-B) radiopharmaceutical, [11C]SL25.1188 ([11C]2). 18–20
Table 1.
“In-loop” formation of [11C]11 and [11C]12

| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Benzylamine (8) (μmol) | Trapped [11C]CO2 (%)c | RCY of [11C]11 (%)d | RCY of [11C]12 (%)d |
| 1b | 4.6 | >99 | 98±1h | - |
| 2a | 4.6 | >99 | 16 | 68 |
| 3a | 1.3 | >99 | 37 | 46 |
| 4a | 0.6 | >99 | 52 | 29 |
| 5a | 0.3 | >99 | 83±2h | 4 |
| 6a | 0.15 | >99 | 70 | 1 |
| 7a, e | 0.3 | >99 | 91 | 4 |
| 8a, f | 0.3 | >99 | 2 | 10 |
| 9a, g | 0.3 | >99 | 13 | 52 |
Reaction conditions: Using Route 1, benzylamine (0.15–4.6 μmol), BEMP (2.5 μL, 8.6 μmol), DMF (40 μL), 0.2% POCl3 (v/v) in MeCN (100 μL, 1.1 μmol), 50% MeOH (v/v) in DMF (50 μL, 1.2 mmol).
Using Route 2, with 10% iodomethane (v/v) in MeCN (100 μL) as alkylating agent.
Percentage [11C]CO2 left in loop after entrapment.
Non-isolated radiochemical yield (RCY), based on radio-HPLC analysis of the crude product.
MeCN was used as solvent.
DMSO was used as solvent.
DBU was used as fixation base.
(n = 3); all other entries unless noted represent single experiments.
Scheme 1.
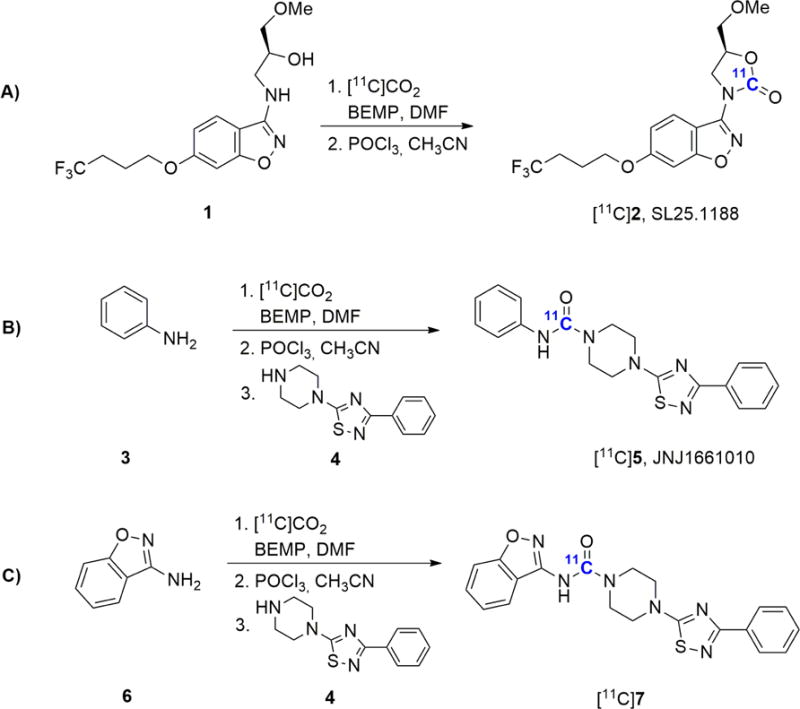
Compounds radiolabelled at room temperature using the “in-loop” [11C]CO2-fixation module.
Results and Discussion
The “in-loop” [11C]CO2-fixation apparatus is designed as a standalone system, where crude 11C-carbonylated products are generated for subsequent purification by chromatography. The method described herein consists of trapping and reacting [11C]CO2 directly in a stainless steel loop (as commonly used for sample injection in HPLC). The system is divided into two main parts: 1) the gas-phase entrapment and purification of in-target produced [11C]CO2; and 2) the “in-loop” reactor system, where [11C]CO2 is fixated and reacted to furnish the final 11C-carbonyl labelled product (Figure 1). For easy adoption of this method into a standard hot cell, we integrated the “in loop” system to a commercial radiosynthesis module (GE TracerLab FXFN) to carry out automated HPLC purification and formulation of the labelled compounds.
Figure 1.

Schematic drawing of the “in-loop” [11C]CO2-fixation module.
Our initial focus was to optimize conditions for the “in-loop” [11C]CO2-fixation reaction. The synthesis of [11C]methyl-N-benzylcarbamate ([11C]11) was selected as a model reaction (Table 1). The experiments were carried out by bubbling [11C]CO2 in a stream of N2 (20 mL/min) into the reactor loop with benzylamine (4.6 μmol) and the CO2 fixating base, BEMP (2.5 μL, 8.6 μmol), dissolved in DMF, which was pre-loaded in section A of the reactor loop (Figure 1). The radiochemical yields (RCY) shown in Table 1 is non-isolated and determined by radio-HPLC. Initially, the intermediate [11C]carbamate ion ([11C]9) was treated with methyl iodide (Table 1, Route 2) and to our delight, the desired 11C-labelled carbamate product was obtained in nearly quantitative yield (Table 1, entry 1). Next, we set to explore the [11C]isocyanide formation ([11C]10, Table 1, Route 1). The [11C]11 formation was studied using different amounts of benzylamine (0.15 – 4.6 μmol), with POCl3 (0.2% v/v in MeCN) as the dehydrating reagent, and the reaction mixture was quenched with an excess of methanol (50% in DMF) pre-loaded in section B of the reactor loop (Figure 1). As anticipated, the reaction was strongly dependent on the amount of benzylamine (8), as larger amounts of 8 led to formation of the undesirable symmetrical product, [11C]dibenzylurea ([11C]12). For example, using 4.6 μmol of 8, only 16% of [11C]11 was obtained with [11C]12 as the main 11C-labelled product (Table 1, entry 2). However, by reducing the amount of 8 to 0.3 μmol, a high and reproducible non-isolated RCY of 83 ± 2% (n = 3) was obtained. Further reduction of 8 by 1-fold resulted in a reduced yield (Table 1, entry 6).
One potential concern with 11C-reactions via captive solvent methods is the general dependency on solvents with high viscosity or boiling points, for example, DMF and DMSO. MeCN is the most common solvent in [11C]CO2-fixation radiochemistry.21–25 Our “in-loop” reactions demonstrated excellent compatibility with MeCN as a solvent in the reaction to prepare [11C]11, which resulted in excellent non-isolated RCY (91%, Table 1, entry 7). Quantitative trapping efficiency was achieved for all examined solvents (MeCN, DMSO and DMF). However, a large variation in selectivity to the desired product was observed under these conditions (Table 1, entries 6–8). Despite superior yield obtained with MeCN, DMF was selected as the optimal solvent for the subsequent reactions, due to evaporation of MeCN that occurred while degassing with helium during our solution preparation. It is noteworthy that this degassing step was crucial in our hands for isolating 11C-labelled compounds with high molar radioactivity. Finally, in a last attempt to further increase the yield, BEMP was replaced with another commonly used fixating base, DBU. High trapping efficiency of [11C]CO2 was obtained with DBU (>99%); however, the overall yield to the desired 11C-labelled product was reduced. In summary, our model reaction showed that ideal “in-loop” 11C-labelling conditions were achieved with BEMP (8.6 μmol), benzylamine (0.3 μmol) dissolved in DMF (40 μL), followed by elution with 0.2% v/v POCl3 in MeCN (100 μL) and finally quenching with an excess of 50% v/v methanol in DMF (50 μL).
To further exemplify the utility of this newly developed method, using the established conditions for the “in-loop” [11C]CO2-fixation reaction (Table 1, entry 5), we set out to prepare three radiotracers (vide supra), namely [11C]SL25.1188 ([11C]2), [11C]JNJ1661010 ([11C]5), and [11C]7 (Scheme 1). The radiotracers were synthesized using both the standard vial-based method and our novel “in-loop” method (Table 2). In all cases, the “in-loop” methodology attained higher non-isolated RCYs compared with conventional reactions in a vial. For example, [11C]SL2511.88 ([11C]2), was obtained in a two-fold higher non-isolated RCY using the “in-loop” methodology (Table 2, entry 1). It is important to note that our previous synthesis of [11C]2 resulted in comparable RCYs (60–70%) when using a vial and MeCN as the solvent.15 However, the experimental conditions (including the solvent) for both methods were kept constant, to facilitate a direct comparison. Moreover, a staggering 15-fold improvement in non-isolated RCY was obtained for both FAAH inhibitor radioligands, [11C]JNJ1661010 ([11C]5) and [11C]7, using the “in-loop” method (Table 2, entries 2 and 3). It is noteworthy that higher amounts of the sparingly reactive primary amines (3 and 6, 55 μmol and 75 μmol, respectively) were necessary to reach the high RCYs (Table 2, entries 2 and 3), indicating that the [11C]carbamate ion formation is promoted by higher amine concentration. The difference in RCYs between the two methods could be explained by the high surface-to-volume ratio inside the loop. Consequently, the “in-loop” method generates an ideal environment for efficient [11C]CO2 trapping and promotes the formation of the [11C]carbamate ion.
Table 2.
Radiotracers synthesized using vial and “in-loop” [11C]CO2-fixation
| Entry | Compound | Vial methoda | “In-loop” methodb | RCP (%)e | Synthesis time (min) | Radioactivity at EOS (GBq (mCi))f | Molar radioactivity (GBq/μmol)g |
|---|---|---|---|---|---|---|---|
|
| |||||||
| RCY(%)c, d | RCY(%)c, d | ||||||
| 1 | [11C]2 | 33 | 70 (43)h | >99 | 35 | 10.2±2.8 (276±74) | 134±21 |
| 2 | [11C]5 | 5 | 72 (54)h | >99 | 33 | 14.3 (387) | 84 |
| 3 | [11C]7 | 2 | 30 (18)h | >99 | 38 | 3.8 (102) | 80 |
Conditions for vial reaction. (1) Precursor 1 (0.6 μmol); primary amines 3 and 6 (110 and 150 μmol, respectively); BEMP (5 μL, 34.6 μmol), DMF (80 μL); (2) 0.2% POCl3 (v/v) in MeCN (100 μL, 1.1 μmol); (3) secondary amine (4) in DMF (100 μL).
Conditions for “in-loop” reaction. (1) Precursor 1 (0.3 μmol); primary amines 3 and 6 (55 and 75 μmol, respectively); BEMP (2.5 μL, 17.3 μmol), DMF (40 μL); (2) 0.2% POCl3 (v/v) in MeCN (100 μL, 1.1 μmol); (3) secondary amine (4) in DMF (50 μL).
Non-isolated radiochemical yield (RCY), based on radio-HPLC analysis of the crude product.
Experiments were performed once (n=1).
Radiochemical purity (RCP) of the isolated product.
Total amount of the isolated product at the end-of-synthesis.
Molar radioactivity of the isolated product established by HPLC.
The decay-corrected radiochemical yield of the isolated product obtained after semi-preparative HPLC and formulation.
Carbon-11 labelled PET radiopharmaceuticals for in-vivo human studies are typically produced in GBq quantities. Therefore, to establish the utility of the current method, all three compounds were prepared on a large scale, starting from 110 GBq (3 Ci) of [11C]CO2. Compounds were isolated using semi-preparative HPLC followed by solid-phase extraction (SPE) to generate the final formulated product (for HPLC traces of [11C]2, [11C]5 and [11C]7 see Figures 2 – 4, respectively).
Figure 2.

A) Semi-preparative HPLC trace (55:45 MeCN:10 mM NH4HCO2, at a flow rate 7 mL/min, ACE 5 C18, 5 μm, 10 × 250 mm) of a typical radiosynthesis of [11C]2, purified using a GE TracerLab FXFN radiosynthesis module. Gamma (tR = 14.7 minutes; top), and UV (λ = 254 nm; bottom). Units on the y-axis are arbitrary. B) Analytical HPLC trace (50:50 MeCN:H2O, at a flow rate of 1 mL/min, XBridge C-18, 3.5 μm, 150 × 4.6 mm) of a typical analysis of [11C]2. Gamma (tR ~ 5 minutes; top), and UV (λ = 254 nm; bottom). Units on the y-axis are arbitrary.
Figure 4.
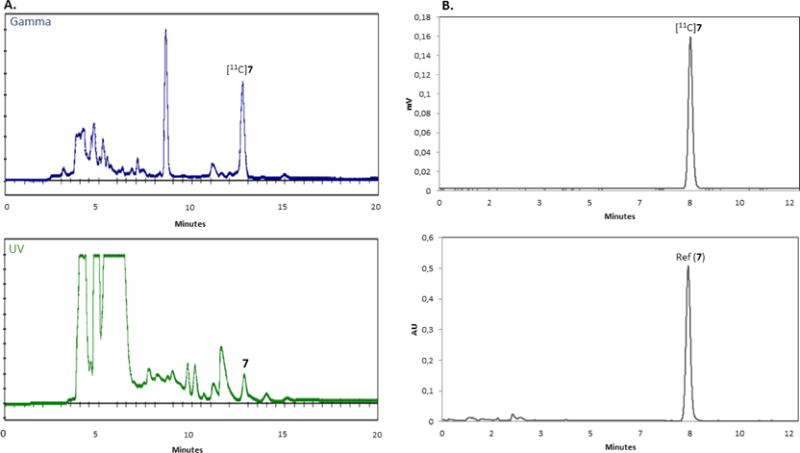
A) Semi-preparative HPLC trace (55:45 MeCN:10 mM NH4CH3CO2, at a flow rate 5 mL/min, ACE 5-C18, 5 μm, 10 × 250 mm) of a typical radiosynthesis of [11C]7, purified using a GE TracerLab FXFN radiosynthesis module. Gamma (tR = 12.7 minutes; top), and UV (λ = 254 nm; bottom). Units on the y-axis are arbitrary. B) Analytical HPLC trace (50:50 MeCN:H2O, at a flow rate of 1.5 mL/min, XBridge C-18, 3.5 μm, 150 × 4.6 mm) of a typical analysis of [11C]7. Gamma (tR ~ 8 minutes; top), and UV (λ = 254 nm; bottom). Units on the y-axis are arbitrary.
Table 2 shows that all compounds were produced in high RCP (>99%) and molar radioactivities (80–134 GBq/μmol (2162–3621 mCi/μmol)). Carbon-11 labelled 7 was isolated and formulated in sufficient quantities for in-vivo PET imaging studies (3.8 GBq; 103 mCi; 18% decay-corrected RCY, relative to starting [11C]CO2). Compounds [11C]2 and [11C]5 were obtained in very high (>50%) isolated RCYs, with 10.2 and 14.3 GBq (276 and 387 mCi), respectively, ready for injection. PET imaging studies with all of these radiotracers are underway in our laboratories, and we are presently designing a fully-automated version of the “in-loop” apparatus for cGMP radiopharmaceutical production. We anticipate that the simplicity of this “in-loop” methodology will facilitate widespread use and will provide comparable radiochemical yields and specific radioactivities to our previously developed 11C-urea- and carbamate-based radiotracers.21–23 Our future work will focus on 11C-carboxylation and amidation as innovative methodologies to achieve these reactions in radiopharmaceutical production are unveiled.
Conclusion
We describe herein the development and design of a novel “in-loop” [11C]CO2-fixation apparatus, which offers an efficient method for the production of a wide range of 11C-carbonylated radiotracers including 11C-oxazolidinones, 11C-ureas, and 11C-carbamates. The “in-loop” methodology was applied in the preparation of three 11C-carbonyl labelled PET radiotracers ([11C]2, [11C]5, [11C]7) in high isolated yields (>3.5 GBq) and radiochemical purity (>99%). The molar radioactivity of all 3 compounds were above 80 GBq/μmol (≥2162 Ci/mmol) at the end-of-synthesis. Given its simplicity, we anticipate the current “in-loop” [11C]CO2 fixation methodology to be widely implemented in the development and routine production of PET radiopharmaceuticals.
Experimental section
Materials and general methods
All reagents and solvents were obtained from commercially available sources and used without further purification, unless specified otherwise. Anhydrous grade MeCN, MeOH and DMF was purchased from Acros organics (USA); BEMP, DBU, aniline (3), benzylamine, methyl iodide, 1,2-benzisoxazol-3-amine (6), 3-phenyl-5-piperazino-1,2,4-thiadiazole (4), JNJ-1661010 (5) as well as phosphorus(V) oxychloride (POCl3) were all obtained from Aldrich (USA). A Tracerlab FXFN radiofluorination module (GE, Sweden) installed with a C18 semi-preparative HPLC column (ACE 5, C18-HL, 250 × 10 mm, Waters) was used for automated purification of the radiolabelled compounds. A HLB C18 light (30 mg, Waters) cartridge was used for SPE. An analytical HPLC system included a high-pressure gradient pump (Shimadzu LC-10AD pump), variable wavelength UV-detector (Shimadzu SPD-10AV), and a radioactivity detector. The system was controlled by Clarity™ 6.1 chromatography software. The radiochemical purity (RCP) of the isolated products was determined by reverse phase HPLC. Analysis was monitored using 254 nm as the detection wavelength. Identification of all radioactive products was confirmed by co-elution with the corresponding non-radioactive compound. The specific radioactivity (SA) was determined by HPLC.
Prototype design
A schematic of the [11C]CO2 “in-loop” synthesizer is shown in Figure 1. All components were purchased from commercial sources. The operation of the module was controlled remotely via electrical switches from outside the hot cell. All reagents and solvents were conveniently stored in loops and transferred using controlled N2 gas flow (2 Bar) generated by a standard mass-flow controller (MFC, 0–2000 mL/min, OMEGA). The radioactive entrapment and release were monitored using scintillation detectors (Caroll & Ramsey). [11C]CO2 was trapped from the target using a stainless steel coil (0.5-m, 1/16″ o.d., 0.04″ i.d.) immersed into a vertically driven (air-pressure) Dewar vessel filled with nitrogen. Two 3-port, two-way valves (V1 and V2, P/N 009-0933-900, Parker) were used to direct N2 flow or target gas flow onto the liquid nitrogen trap. An 8-port, two-way HPLC injection valve (V3, P/N 170-0160H, Vici) was equipped with a reagent loop (1 mL (2-m), Tefzel™, 1/16″ o.d., 0.03″ i.d.) as well as a reactor loop (3.5 mL (7-m), stainless-steel, 1/16″ o.d., 0.03″ i.d.), which was applied as reaction vessel. Importantly, the reactor loop is dividable into two different sections: section A (1 mL (2-m)) and section B (2.5 mL (5-m)). Two union-connectors (U1 and U2, P/N ZU1M, Valco) were used to combine the sections. This dual-loop system makes it possible to pre-load two solutions at the same time. A sodium hydroxide-coated silica trap (Ascarite II, 20–30 mesh) was connected to the waste port of the HPLC valve and placed in a well counter to measure unreacted [11C]CO2.
Model reaction for the “in-loop” [11C]CO2-fixation
(a) Set-up
A schematic of the reactor loop is displayed in Figure 5. Prior to the start-of-synthesis with the injection valve in position A and by disconnecting the finger tight luer adaptors (Figure 5, U1, P-655 and P-660, Idex Health & Science), a solution of alkylating (10 μL, methyl iodide) or dehydrating (0.2 μL, POCl3) agent in MeCN (100 μL) were loaded into the reagent loop using a 1 mL syringe. Furthermore, to the same loop, additional amount of MeCN (800 μL) was added. A solution of appropriate amine, BEMP (2.5 μL) in dimethylformamide (40 μL, DMF), was loaded onto the reactor loop (Figure 5, section A). If “Route 1” (Table 1, scheme) are to be used, a solution of methanol (25 μL) dissolved in DMF (25 μL) was also loaded onto the reactor loop (Figure 5, section B). To enable reagent injection to the different sections of the reactor loop, the inlet nut on union-connectors (Figure 5, U2 and U3) were replaced by a needle injection port (#VISF-2, Valco). It is important to note that prior to use, all solutions were de-gassed with helium flow for 15 min and kept over 4Å molecular sieves to minimize atmospheric CO2.
Figure 5.

General “in-loop” [11C]CO2-fixation radiosynthesis procedure. Step 1: The “in-loop” [11C]CO2 fixation in a pre-loaded trapping-solution (BEMP, amine dissolved in DMF) to form the intermediate product, [11C]carbamate ion (reactor loop, section A). Step 2: Elution and “in-loop” reaction using a pre-loaded solution (POCl3 or methyl iodide in reagent loop) in section A of the reactor loop, followed by a second in-loop reaction with a pre-loaded nucleophile (alcohol or amine) in section B of the reactor loop.
(b) Trapping and reaction
No-carrier-added [11C]CO2 was prepared using a PET trace cyclotron (GE medical system, Sweden) via irradiation of a nitrogen target using 16.4 MeV protons. After 1-min irradiation at 50 μA, typically 5 GBq (140 mCi) of [11C]CO2 was obtained at end-of-bombardment (EOB). The [11C]CO2 was transferred from the target chamber by the target gas (1% O2 in N2) and concentrated in the liquid nitrogen trap of the [11C]CO2-fixation apparatus (Figure 1). The stainless-steel tube was removed from the liquid nitrogen by an air-pressure driven lift, whereby the [11C]CO2 was passed through the coated reaction loop (Figure 5, Step 1) in a controlled stream of N2 (20 mL/min). When radioactivity peaked in the reaction loop, as measured by the proximal radiation detector (typically within 2 min), the flow of N2 was stopped and the reaction was allowed to proceed for an additional 2 min. Following this reaction, the position of the injection valve was switched to position B (Figure 5, Step 2) and the content of the reactor loop was eluted out with a second reagent (alkylating or dehydration reagent in MeCN) by a stream of nitrogen (80 mL/min) into an empty receiving vial (5 mL, glass v-vial, Alltech). Moreover, the pre-loaded MeCN was immediately flushed through the reactor loop to maximize recovery. The crude radioactive product was dissolved in 3.5 mL of water and injected onto a GE TracerLab FXFN radiofluorination module for HPLC purification and formulation.
(c) Clean-up
The [11C]CO2-fixation module was easily cleaned by passing 5 mL volumes of water, MeCN and acetone through both loops (reactor and reagent loop) with the injection valve in position B (Figure 5), and flushed using N2 gas (500 mL/min) for 15 min.
Precursor and non-radioactive standard synthesis
Precursors and the non-radioactive standards for the [11C]CO2-fixation reactions were either synthesized in-house (1, 2 and 7) or were obtained from commercially available sources (3, 4, 5 and 6). Compounds included in this study are depicted in Scheme 1. SL25.1188 reference standard (2) and the corresponding precursor compound (1) were synthesized in 5- and 6-steps, respectively, starting from 4-(benzyloxy)-2-fluorobenzonitrile via reduction, alkylation, and hydrolysis (Scheme 2).18, 26 Moreover, the non-radioactive standard 7 was synthesized in two-steps starting from 6 (Scheme 3).
Scheme 2.
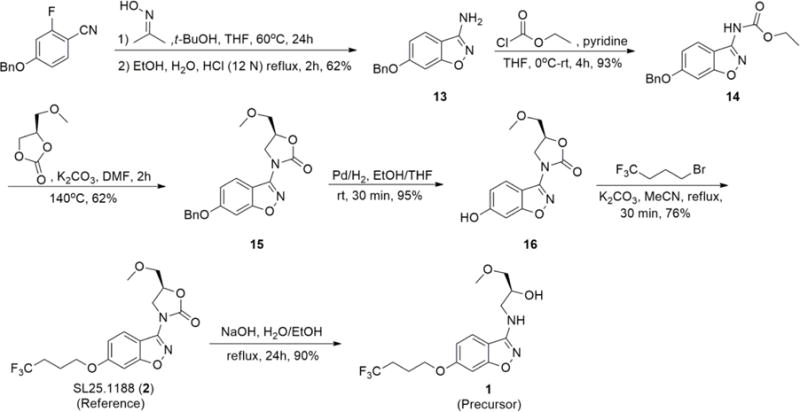
Synthesis procedure for compounds 1 and 2.
Scheme 3.
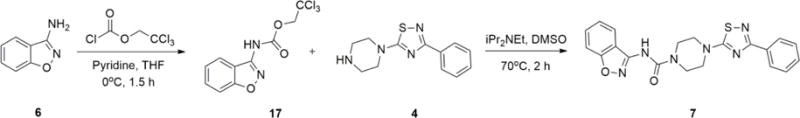
Synthesis procedure for compound 7. (32% yield over 2 steps).
Synthesis of SL25.1188 reference (2) and precursor (1) compounds
6-(Benzyloxy)benzo[d]isoxazol-3-amine (13)
Acetone oxime (20 mmol) was added to a solution of 1 M potassium tert-butoxide in THF (20 mL, 20 mmol). The solution was kept at room temperature (r.t.) for 20 min, after which a solution of 4-(benzyloxy)-2-fluorobenzonitrile (18.5 mmol) in THF (15 mL) was slowly added using an addition funnel. After stirring for 3 h at r.t., the mixture was heated at 60°C overnight. A dark brown solution was obtained. The reaction mixture was quenched with water (10 mL) and partitioned between saturated NaHCO3 solution (5 mL) and ethyl acetate (75 mL). The organic layers were separated and washed with water (3 × 30 mL). A brown solid was obtained upon solvent removal using a rotary evaporator. The solid was then treated with a mixture of EtOH (40 mL), H2O (26 mL) and HCl (12 N, 14 mL) at reflux for 2 h. After cooling, the reaction mixture was basified with solid sodium carbonate and NaOH (1 N, 15 mL), followed by extraction with ethyl acetate (2 × 100 mL). The combined organic layers were washed with brine (50 mL), dried over MgSO4, and the solvent was removed under reduced pressure. The residue was purified using chromatography on silica gel by eluting with a 1:3 mixture of ethyl acetate and hexane. The product was obtained as a white solid. Yield: 62%; 1H NMR (300 MHz, CDCl3) δ 7.31 – 7.48 (m, 6H), 6.90 – 6.97 (m, 2H), 5.12 (s, 2H), 4.30 (s, 2H).
Ethyl 6-(benzyloxy)benzo[d]isoxazol-3-ylcarbamate (14)
Ethyl chloroformate (1.2 mmol) was added dropwise to a solution of 13 (1 mmol) and pyridine (2 mmol) maintained at 0 °C. The mixture was stirred at 0 °C for 4 h, poured into water and extracted with ethyl acetate (2 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, and the solvent was evaporated in vacuo. The residue was purified by chromatography on silica gel by eluting with a 1:3 mixture of ethyl acetate and hexane. The product was obtained as a white solid. Yield: 93%; 1H NMR (300 MHz, CDCl3) δ 8.08 (d, J = 9.6 Hz, 1H), 7.86 (s, 1H), 7.30 – 7.49 (m, 5H), 6.95 – 7.03 (m, 2H), 5.13 (s, 2H), 4.32 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 166.1, 161.5, 153.1, 153.0, 135.9, 128.7, 128.3, 127.5, 125.2, 114.6, 108.9, 93.5, 70.5, 62.4, 14.4.
(S)-3-(6-(Benzyloxy)benzo[d]isoxazol-3-yl)-5-(methoxymethyl)oxazolidin-2-one (15)
A solution of 14 (0.9 mmol) in 1 mL of DMF was added to a solution of (R)-4-(methoxymethyl)-1,3-dioxolan-2-one (1.7 mmol) and potassium carbonate (0.085 mmol) in 2 mL DMF at 140 °C. The mixture was stirred at 140 °C for 2 h. After cooling the solution was evaporated in vacuo. The residue was purified by chromatography on silica gel by eluting with a 1:3 mixture of ethyl acetate and hexane. The product was obtained as a white solid. Yield: 62%; 1H NMR (300 MHz, CDCl3) δ 8.41 (d, J = 9.7 Hz, 1H), 7.30 – 7.49 (m, 5H), 6.95 – 7.02 (m, 2H), 5.14 (s, 2H), 4.85 – 4.96 (m, 1H), 4.23 (dd, J = 9.9, 8.9 Hz, 1H), 4.11 (dd, J = 10.0, 6.5 Hz, 1H), 3.69 (qd, J = 10.8, 4.1 Hz, 2H), 3.45 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 166.4, 161.5, 153.8, 152.9, 135.9, 128.7, 128.3, 127.5, 126.5, 114.5, 108.2, 93.4, 73.8, 72.4, 70.5, 59.6, 46.6.
(S)-3-(6-Hydroxybenzo[d]isoxazol-3-yl)-5-(methoxymethyl)oxazolidin-2-one (16)
A solution of 15 (8.47 mmol) and Pd/C (500 mg) in 100 mL EtOH/THF (1:1) under hydrogen atmosphere was stirred at r.t. for 60 min. Upon completion of the reaction, the mixture was filtered through Celite® and the filtrate was evaporated in vacuo. The residue was purified by chromatography on silica gel by eluting with a 1:2 mixture of ethyl acetate and hexane. The product was obtained as a white solid. Yield: 95%; 1H NMR (300 MHz, DMSO) δ 10.46 (s, 1H), 8.19 (d, J = 8.9 Hz, 1H), 6.89 (d, J = 2.0 Hz, 1H), 6.83 (dd, J = 8.9, 2.1 Hz, 1H), 4.88 – 5.06 (m, 1H), 4.16 (t, J = 9.3 Hz, 1H), 3.85 (dd, J = 9.6, 6.3 Hz, 1H), 3.54 – 3.72 (m, 2H). 13C NMR (75 MHz, DMSO) δ 166.3, 161.0, 154.2, 153.1, 126.5, 114.4, 106.8, 95.0, 74.5, 72.6, 59.1, 46.5.
(S)-5-(Methoxymethyl)-3-(6-(4,4,4-trifluorobutoxy)benzo[d]isoxazol-3-yl)oxazolidin-2-one (SL25.1188, 2)
16 (7.6 mmol) was added to a solution of 1,1,1-trifluorobutane (10.5 mmol) and potassium carbonate (15.2 mmol) in 28 mL MeCN. The mixture was heated to reflux for 2 h. The mixture was than cooled, filtered and evaporated under reduced pressure. The residue was purified by chromatography on silica gel by eluting with a 1:3 mixture of ethyl acetate and hexane. The product was obtained as a white solid. Yield: 76%. 1H NMR (300 MHz, CDCl3) δ 8.41 (d, J = 9.7 Hz, 1H), 6.82 – 6.95 (m, 2H), 4.84 – 5.00 (m, 1H), 4.20 – 4.27 (m, 1H), 4.03 – 4.17 (m, 3H), 3.69 (qd, J = 10.8, 4.1 Hz, 2H), 3.45 (s, 3H), 2.25 – 2.45 (m, 2H), 2.04–2.20 (m, 2H). 19F NMR (282 MHz, CDCl3) δ -66.3 (t, J = 10.8 Hz).
(S)-1-Methoxy-3-(6-(4,4,4-trifluorobutoxy)benzo[d]isoxazol-3-ylamino)propan-2-ol (1)
To a solution of 2 (1.4 mmol) in EtOH (7 mL), aq. NaOH (1.3 N, 5.4 mL, 7 mmol) was added. The mixture was stirred at r.t. overnight. The resulting solution was diluted with water (50 mL) and EtOAc (50 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, and the solvent was evaporated in vacuo. The residue was purified by chromatography on silica gel by eluting with a 1:1 mixture of ethyl acetate and hexane. The product was obtained as a white solid. Yield: 90%; 1H NMR (300 MHz, DMSO) δ 7.73 (d, J = 8.7 Hz, 1H), 6.99 (d, J = 1.9 Hz, 1H), 6.78 – 6.88 (m, 2H), 4.96 (d, J = 5.2 Hz, 1H), 4.08 (t, J = 6.2 Hz, 2H), 3.89 (dq, J = 10.5, 5.3 Hz, 1H), 3.21 – 3.34 (m, 5H), 3.04 – 3.16 (m, 1H), 2.30 – 2.46 (m, 2H), 1.86 – 2.01 (m, 2H). 13C NMR (75 MHz, DMSO) δ 164.1, 161.1, 158.7, 128.1 (q, J = 277.0 Hz), 122.5, 112.4, 110.1, 94.0, 75.5, 67.8, 66.8, 58.8, 46.8, 29.9 (q, J = 30.0 Hz), 21.9 (d, J = 3.0 Hz). 19F NMR (282 MHz, DMSO) δ -64.8 (t, J = 11.6 Hz).
Synthesis of reference compound (7)
N-(benzo[d]isoxazol-3-yl)-4-(3-phenyl-1,2,4-thiadiazol-5-yl)piperazine-1-carboxamide (7)
2,2,2-Trichloroethyl chloroformate (15mg, 72 μmol) was added to a solution of benzo[d]isoxazol-3-amine (6, 8.4 mg, 60 μmol) and pyridine (5.7 mg, 72 μmol) in THF (0.6 mL) maintained at 0 °C. The mixture was kept at 0 °C for 1.5 h under stirring. The resulting solution was quenched with water and subsequently extracted with EtOAc (3 × 5 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, and the solvent was removed in vacuo to yield the crude intermediate product (17). Next, N-ethyldiisopropylamine (4.2 mg, 32 μmol) was added to a solution of 17 (8.0 mg, 26 μmol) and 3-phenyl-5-(piperazin-1-yl)-1,2,4-thiadiazole (4, 8.5 mg, 34 μmol) in DMSO (0.6 mL). The mixture was stirred at 70 °C for 2 h, quenched with water and subsequently extracted with EtOAc (2 × 5 mL). The combined organic layer was washed with brine and dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by preparative TLC by eluting with a 10:1 mixture of dichloromethane and methanol. The product was obtained as a yellow solid. Yield: 32% over two steps; 1H NMR (300 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.14 – 8.09 (m, 2H), 7.85 (d, J = 7.8 Hz, 1H), 7.65 – 7.55 (m, 2H), 7.48 – 7.45 (m, 3H), 7.33 – 7.27 (m, 1H), 3.72 (s, 4H), 3.68 (s, 4H). 13C NMR (75 MHz, DMSO-d6) δ 185.2, 169.5, 163.4, 155.8, 154.3, 133.3, 130.7, 130.5, 129.1, 128.0, 125.1, 123.3, 117.3, 110.2, 48.8, 43.7.
Figure 3.
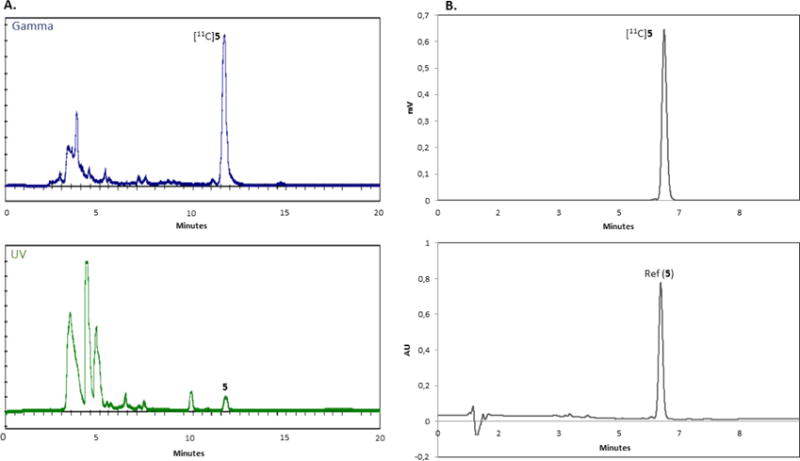
A) Semi-preparative HPLC trace (55:45 MeCN:10 mM NH4CH3CO2, at a flow rate of 5 mL/min, ACE 5-C18, 5 μm, 10 × 250 mm) of a typical radiosynthesis of [11C]5, purified using a GE TracerLab FXFN radiosynthesis module. Gamma (tR = 11.8 minutes; top), and UV (λ = 254 nm; bottom). Units on the y-axis are arbitrary. B) Analytical HPLC trace (50:50 MeCN:H2O, at a flow rate of 1.5 mL/min, XBridge C-18, 3.5 μm, 150 × 4.6 mm) of a typical analysis of [11C]5. Gamma (tR ~ 6.5 minutes; top), and UV (λ = 254 nm; bottom). Units on the y-axis are arbitrary.
Acknowledgments
We would like to thank the staff at the Radiochemistry Program, Massachusetts General Hospital and Harvard Medical School for their support. R.C. is supported by China Scholarship Council (201506250036). S.H.L. is a recipient of an NIH career development award (DA038000). N.V. thanks National Institute on Ageing of the NIH for funding this work (R01AG054473).
References
- 1.Miller PW, Long NJ, Vilar R, Gee AD. Angew Chem Int Ed. 2008;47:8998. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 2.Ametamey SM, Honer H, Schubiger PA. Chem Rev. 2008;108:1501. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 3.Pike VW. Curr Med Chem. 2016;23:1818. doi: 10.2174/0929867323666160418114826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahl K, Halldin C, Schou M. Clin Transl Imaging. 2017;5:257. doi: 10.1007/s40336-017-0223-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rotstein BH, Liang SH, Placzek MS, Hooker JM, Gee AD, Dollé F, Wilson AA, Vasdev N. Chem Soc Rev. 2016;45:4708. doi: 10.1039/c6cs00310a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rotstein BH, Liang SH, Holland JP, Collier TL, Hooker JM, Wilson AA, Vasdev N. Chem Comm. 2013;49:5621. doi: 10.1039/c3cc42236d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hooker JM, Reibel A, Hill S, Schueller M, Fowler JS. Angew Chem Int Ed. 2009;48:3482. doi: 10.1002/anie.200900112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson AA, Garcia A, Houle S, Vasdev N. Org Biomol Chem. 2010;8:428. doi: 10.1039/b916419g. [DOI] [PubMed] [Google Scholar]
- 9.Wilson AA, Garcia A, Houle S, Sadovski O, Vasdev N. Chem Eur J. 2011;17:259. doi: 10.1002/chem.201002345. [DOI] [PubMed] [Google Scholar]
- 10.Riss PJ, Lu S, Telu S, Aigbirhio FI, Pike VW. Angew Chem Int Ed. 2012;51:2698. doi: 10.1002/anie.201107263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rotstein BH, Hooker JM, Woo J, Collier TL, Brady TJ, Liang SH, Vasdev N. ACS Med Chem Lett. 2014;5:668. doi: 10.1021/ml500065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davenport RJ, Pike VW, Dowsett K, Turton DR, Poole K. Appl Radiat Isot. 1997;48:917. doi: 10.1016/s0969-8043(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 13.Davenport RJ, Dowsett K, Pike VW. Appl Radiat Isot. 1997;48:1117. doi: 10.1016/s0969-8043(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 14.Matarrese M, Sudati F, Soloviev D, Todde S, Turolla EA, Kienle MG, Fazio F. Appl Radiat Isot. 2002;57:675. doi: 10.1016/s0969-8043(02)00182-3. [DOI] [PubMed] [Google Scholar]
- 15.Zhang MR, Ogawa M, Yoshida Y, Suzuki K. Appl Radiat Isot. 2006;64:216. doi: 10.1016/j.apradiso.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 16.Wilson AA, Garcia A, Jin L, Houle S. Nucl Med Biol. 2000;27:529. doi: 10.1016/s0969-8051(00)00132-3. [DOI] [PubMed] [Google Scholar]
- 17.Wilson AA, Garcia A, Houle S, Vasdev N. J Label Compd Radiopharm. 2009;52:490. [Google Scholar]
- 18.Saba W, Valette H, Peyronnea M, Bramoulle Y, Coulon C, Curet O, George P, Dolle F, Bottlaender M. Synapse. 2010;64:61. doi: 10.1002/syn.20703. [DOI] [PubMed] [Google Scholar]
- 19.Vasdev N, Sadovski O, Garcia A, Dollé F, Meyer JH, Houle S, Wilson AA. J Label Compd Radiopharm. 2011;54:678. [Google Scholar]
- 20.Rusjan P, Wilson AA, Miler L, Fan I, Mizrahi R, Houle S, Vasdev N, Meyer JH. J Cereb Blood Flow Metab. 2014;34:883. doi: 10.1038/jcbfm.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hicks JW, Parkes J, Sadovski O, Tong J, Houle S, Vasdev N, Wilson AA. Nucl Med Biol. 2013;40:740. doi: 10.1016/j.nucmedbio.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hicks JW, Wilson AA, Rubie EA, Woodgett JR, Houle S, Vasdev N. Bioorg Med Chem Lett. 2012;22:2099. doi: 10.1016/j.bmcl.2011.12.139. [DOI] [PubMed] [Google Scholar]
- 23.Wilson AA, Hicks JW, Sadovski O, Parkes J, Tong J, Houle S, Fowler C, Vasdev N. J Med Chem. 2013;56:201. doi: 10.1021/jm301492y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dheere A, Yusuf N, Gee AD. Chem Comm. 2013;49:8193. doi: 10.1039/c3cc44046j. [DOI] [PubMed] [Google Scholar]
- 25.Mossine AV, Brooks AF, Jackson IM, Quesada CA, Sherman P, Cole EL, Donnelly DJ, Scott PJH, Shao X. Bioconjugate Chem. 2016;27:1382. doi: 10.1021/acs.bioconjchem.6b00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jegham S, Puech F, Burnier P. PCT Int Appl. 1996 WO9638444 A1. [Google Scholar]