

Abstract
Aim
NADPH oxidases are important sources of reactive oxygen species (ROS). Several Nox homologues are present together in the vascular system but whether they exhibit crosstalk at the activity level is unknown. To address this, vessel function of knockout mice for the cytosolic Nox organizer proteins p47phox, NoxO1 and a p47phox-NoxO1-double knockout were studied under normal condition and during streptozotocin-induced diabetes.
Results
In the mouse aorta, mRNA expression for NoxO1 was predominant in smooth muscle and endothelial cells, whereas p47phox was markedly expressed in adventitial cells comprising leukocytes and tissue resident macrophages. Knockout of either NoxO1 or p47phox resulted in lower basal blood pressure. Deletion of any of the two subunits also prevented diabetes-induced vascular dysfunction. mRNA expression analysis by MACE (Massive Analysis of cDNA ends) identified substantial gene expression differences between the mouse lines and in response to diabetes. Deletion of p47phox induced inflammatory activation with increased markers of myeloid cells and cytokine and chemokine induction. In contrast, deletion of NoxO1 resulted in an attenuated interferon gamma signature and reduced expression of genes related to antigen presentation. This aspect was also reflected by a reduced number of circulating lymphocytes in NoxO1-/- mice.
Innovation and conclusion
ROS production stimulated by NoxO1 and p47phox limit endothelium-dependent relaxation and maintain blood pressure in mice. However, NoxO1 and p47phox cannot substitute each other despite their similar effect on vascular function. Deletion of NoxO1 induced an anti-inflammatory phenotype, whereas p47phox deletion rather elicited a hyper-inflammatory response.
Abbreviations: CBA, Cytometric Bead Assay; eNOS, endothelial Nitric Oxide Synthase; Gpx3, Glutathione peroxidase 3; IFNɣ, Interferon gamma; MACE, Massive Analysis of cDNA ends; Nox, NADPH oxidase; PMA, Phorbol Myristate Acetate; ROS, Reactive Oxygen Species; SMCs, Smooth Muscle Cells; STZ, Streptozotocin
Keywords: NADPH oxidase, NoxO1, Nox1, p47phox, Superoxide, Reactive oxygen species
Graphical abstract
1. Introduction
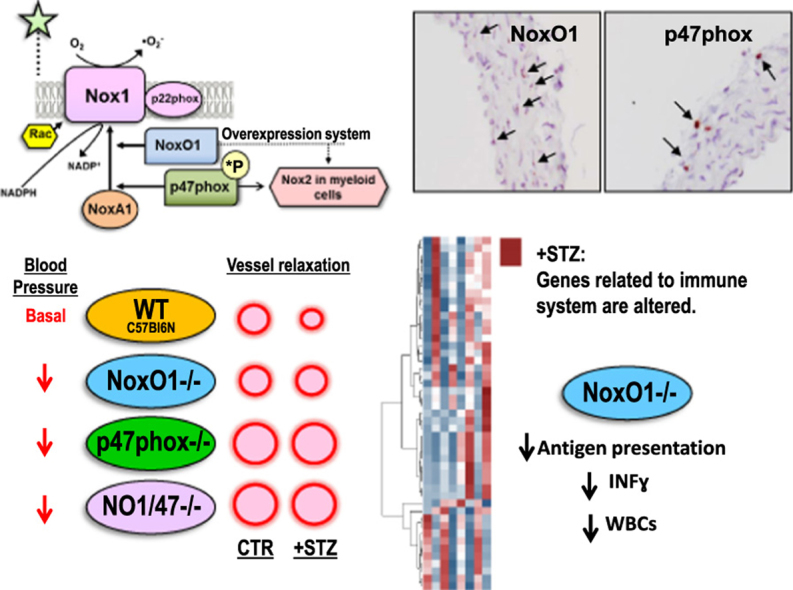
NADPH oxidases of the Nox family are important sources of reactive oxygen species (ROS). In the vascular system, Nox1, Nox2, Nox4 and Nox5 are expressed. Nox1 and Nox2 have been associated with endothelial dysfunction during disease conditions such as diabetes [1], [2], hypertension [3], [4], [5] and atherosclerosis [6], [7]. This is a consequence of the fact that both enzymes produce nitric oxide-inactivating superoxide anions (•O2-). Moreover, Nox1 and Nox2 are induced or activated by forces leading to cardiovascular disease [8]. Nox1 and Nox2 differ from other vascular Nox enzymes in their dependency on cytosolic proteins required for their activation. In the case of Nox2 the organizing protein p47phox tethers the activating protein p67phox to the catalytic subunits which leads to activation. A similar situation exists for Nox1, for which NoxO1 serves as the organizer and NoxA1 as the activator. Protein interactions of p47phox require its serine phosphorylation and displacement of an autoinhibitory region, a mechanism missing in NoxO1. Therefore, in overexpression systems, Nox1-NoxO1-NoxA1 complexes have high basal activity, whereas Nox2-p47phox-p67phox becomes active only after phosphorylation [9]. Overexpression has also demonstrated a degree of exchangeability between the components of the different complexes. The resulting activities, however, were always substantially lower than for the prototypic complexes [10], [11].
To infer the possibility of such mixed complexes occurring within the cell, the relevant proteins have to be present in the same compartment and same cell type. The expression of p47phox predominates in myeloid cells [12], but expression in smooth muscle cells (SMCs) has been reported, albeit at lower level [13], [14], [15]. NoxO1 is highly expressed in colon and testis [16]. Its expression and function in the vascular system has been only recently shown and a quantitative analysis is still missing. NoxO1-/- mice produce lower basal vascular •O2- and show an increased angiogenesis by promoting tip cell formation [17]. Mice transfected with NoxO1 siRNA exhibit attenuated eNOS uncoupling in diabetes [1]. In cultured endothelial cells, NoxO1 expression can be induced by oscillatory flow, leading to eNOS uncoupling [18]. Such findings illustrate a paucity of information concerning cell-specific expression as well as supporting the view that components of the Nox1 and Nox2 systems are expressed and functionally relevant in the vascular system. Considering that Nox1 and Nox2 generate the same product and that the enzymes are similar to some extent, crosstalk between the Nox enzymes could be inferred. This aspect was studied here using p47phox-/-, NoxO1-/- and p47phox-NoxO1-double knockout mice under basal condition and streptozotocin-induced diabetes. Moreover, with the present study we provide a first phenotypic characterization of NoxO1-/- mice in a vascular disease context.
2. Results
2.1. NoxO1 and p47phox can activate an overexpression system of Nox1 and NoxA1
To confirm the function of NoxO1 and p47phox in a complex containing Nox1/NoxA1, different combinations of the proteins were overexpressed in HEK293 cells (Fig. 1). The combination of Nox1/NoxA1/NoxO1 produced 230 fold more •O2-, as measured with L-012 chemiluminescence in intact cells than GFP transfected controls in non-stimulated conditions. PMA (phorbol myristate acetate), which was used to activate the complex, further increased the signal by approx. 30%. The ROS formation of the combination of p47phox with Nox1/NoxA1 had a very different profile: under basal conditions, i.e. in the absence of PMA, only a negligible signal was generated. Upon PMA stimulation, however, this combination yielded a 700% increase in the L-012 signal. These results are in agreement with previous publications [16], [19] and confirm the preference of Nox1 for NoxO1 and the constitutive activity of this complex.
Fig. 1.

Canonical (Nox1/NoxA1/NoxO1) and hybrid (Nox1/NoxA1/p47phox) activation of Nox1 in overexpression system using HEK cells. A: HEK cells were transfected with Nox1 and its activating subunits as indicated. ROS were measured by chemiluminescence with L012 (200 µmol/L). Stimuli-dependent activation of Nox1 was triggered by PMA (phorbol myristate acetate, 100nmol/L). B: quantification of the signal upon normalization to GFP control. n = 5, *p < 0.05 without PMA vs. with PMA.
2.2. NoxO1 is mainly expressed in vascular cells whereas p47phox is mainly present in adventitial myeloid cells
Most of the expression data for p47phox in the vascular system were generated by RT-PCR. This is because this technique is highly sensitive and specific for the target gene. RT-PCR, however, is not the method of choice to identify cell specific expression differences within a tissue. Given the lack of cell-specific expression data, it is plausible that the p47phox mRNA detected in the vascular system is a reflection of a few contaminating myeloid cells [20]. To address this aspect, in situ hybridization by RNAscope® was employed to cell-specifically visualize the mRNA of NoxO1 and p47phox in the aortic wall. By this technique, NoxO1 signal in the adventitia was only 55% of that of the media. In contrast, the average abundance of p47phox in the adventitia was 300% of that in the media, showing a predominant localization of p47phox in adventitial cells whereas NoxO1 was mainly detected in the media (Fig. 2A & C). In the adventitial layer, p47phox but not NoxO1 co-localized with Adgre1 (Adhesion G Protein-Coupled Receptor E1 also called Egf-Like Module Containing, Mucin-Like, Hormone Receptor-Like Sequence (F4/80)), a marker for macrophages. Although RNAscope is limited in sensitivity, the data suggest that high levels of NoxO1 and p47phox are unlikely to reside together in the vascular tissue. Considering that the vascular adventitia is rich in inflammatory cells containing p47phox, the majority of the signal for this activator likely reflects its expression in myeloid cells.
Fig. 2.

In situ hybridization (RNAscope®) showing the expression of NoxO1 and p47phox in aortic tissue. A: (upper panel) detection with fast red and (lower panel) detection with DAB substrate (brown kit). NoxO1 and p47phox are indicated by arrows. (+) CTR: positive control: peptidylprolyl isomerase B. (-) CTR: negative control: B. subtilis dihydrodipicolinate reductase. B: duplex staining for NoxO1 or p47phox with Adgre1 (F4/80, a macrophage marker). The arrows indicate each of the activators labeled in blue. Zoom in of the adventitia layer shows that p47phox but not NoxO1 co-localizes with Adgre1. C: quantification of the expression of NoxO1 and p47phox in aortic tissue as dots by area (mm2) of either media or adventitia. * p < 0.05 media vs. adventitia for each individual activator. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.3. NoxO1 and p47phox contribute to basal blood pressure and vascular tonus
To investigate a contribution of NoxO1 and p47phox to the vascular homeostasis the resting blood pressure was measured in mice and vascular function of the mesenteric artery was determined ex vivo in a wire myograph system. Knockout mice of p47phox or NoxO1 individually or combined exhibited a reduced systolic blood pressure as compared to WT animals. In p47phox-/- and NoxO1/p47phox-double knockout mice, but not in NoxO1-/- mice, was the diastolic pressure also found to be significantly lower. The heart rate and heart to body weight ratio were similar among the groups (Fig. 3A-C, Fig S1).
Fig. 3.

NoxO1 and p47phox contribute to cardiovascular homeostasis. A: systolic (*p < 0.05 NO-/-, p47-/-, O1/47-/- vs. WT), (B) diastolic blood pressure (*p < 0.05 p47-/-, O1/47-/- vs. WT), and (C) heart rate measured by tail cuff. D: organ bath experiment of isolated mesenteric artery pre-constricted with phenylephrine and relaxed with acetylcholine. * p < 0.05 wt vs. p47-/-, # p < 0.05 wt vs. O1/47-/-. n ≥ 8.
The constrictor response to phenylephrine of the isolated mesenteric artery was similar between the lines. In contrast, differences in the endothelium-dependent relaxation in response to acetylcholine were observed; the sensitivity to acetylcholine was significantly higher in vessels of p47phox and p47phox-NoxO1 knockout mice as compared to vessels from NoxO1-/- and WT mice (Fig. 3D).
2.4. NoxO1 and p47phox contribute to diabetes-induced endothelial dysfunction
Diabetes mellitus is considered a condition of oxidative stress and multiple source of ROS, including Nox1, Nox2, mitochondria and eNOS uncoupling, have been implicated [21]. Type I diabetes in response to streptozotocin (STZ) was therefore considered as an ideal model to determine a potential crosstalk among the Nox enzymes. Blood glucose levels and body weight developed similarly in response to STZ in all lines ( Fig. 4A & B and Table 2). 50 days after STZ application, endothelium-dependent relaxation to acetylcholine of mesenteric vessels of WT animals was impaired as compared to non-diabetic WT animals (Fig. 4C-E). Importantly, knockout of either NoxO1 or p47phox prevented the development of endothelial dysfunction and no additive effect was observed in the NoxO1-p47phox double knockout mice (Fig. 4D-E).
Fig. 4.

NoxO1 and p47phox do not contribute to disease onset but to endothelial dysfunction induced by diabetes. A: blood glucose concentration during STZ-induced diabetes (70 mg/kg/day). B: body weight loss over diabetes. C-E: organ bath experiments of mesenteric vessel pre-constricted with phenylephrine and relaxed with acetylcholine. n ≥ 8, black circles: WT CTR and black squares: WT +STZ. * p < 0.05 wt vs. WT +STZ, # p < 0.05 wt +STZ vs. p47-/- +STZ, + p < 0.05 wt +STZ vs. O1/47-/- +STZ.
Table 2.
Average of blood glucose (mmol/dL) (± SEM) at day 50 of diabetes as measured by Accu-Chek.* p < 0.05 comparing CTR and STZ of the respective knockout. # p < 0.05 compared to WT and p47phox-/-.
|
WT |
NOXO1-/- |
p47-/- |
O1/47-/- |
||||
|---|---|---|---|---|---|---|---|
| CTR | STZ | CTR | STZ | CTR | STZ | CTR | STZ |
| 140.5 ± 2.8 | 529.2 ± 21.1* | 119.3 ± 3.4# | 490.9 ± 26.7* | 141.6 ± 11.5 | 536.6 ± 26.6* | 123.8 ± 6.2# | 533.2 ± 33.8* |
2.5. NoxO1 and p47phox do not crosstalk at gene expression level
In order to determine whether the knockout of p47phox or NoxO1 affects expression of Nox homologues or Nox cytosolic proteins, mRNA expression was determined by qPCR and MACE analysis. Importantly, the data, however, were not suggestive towards a crosstalk. Deletion of p47phox or NoxO1 did not result in the induction of the alternative cytosolic organizer under normal conditions and also not in diabetic animals (Table 3). Interestingly, there was also fairly little effect of diabetes on vascular expression of the different Nox subunits. This confirms the notion that diabetes-induced ROS production is largely a consequence of activation of ROS producing systems rather than Nox protein induction [22], [23].
Table 3.
Expression profiling of NADPH oxidases and their activating subunits as determined by qPCR and MACE. Relative expression ± SEM; + p<0.05 compared to WT CTR; * p<0.05 comparing CTR and STZ of the respective knockout.
| Gene: | NoxO1 | p47phox | Nox1 | Nox2 | p67phox | p22phox | Nox4 | Duox1 | Duox2 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MACE | WT | CTR | 1 ± .45 | 1 ± .07 | n.i. | 1 ± .08 | 1 ± .10 | 1 ± .04 | 1 ± .09 | 1 ± .03 | 1 ± .80 | |
| STZ | .62 ± .22 | 1.36 ± .25 | n.i. | .95 ± .03 | .82 ± .01 | .78 ± .05+ | 1.12 ± .14 | .86 ± .32 | 2.14 ± .41 | |||
| NoxO1-/- | CTR | n.d. | .88 ± .07 | n.i. | .96 ± .08 | .71 ± .01+ | .91 ± .06+ | .90 ± .01 | .92 ± .31 | 1.92 ± .14 | ||
| STZ | n.d. | 1.09 ± .14 | n.i. | .66 ± .10 | .75 ± .10 | .73 ± .07+ | 1.06 ± .05* | .65 ± .26 | .59 ± .11* | |||
| p47-/- | CTR | .8 ± .23 | n.d. | n.i. | 1.29 ± .02 | 1.09 ± .02 | .92 ± .003 | .95 ± .01 | .71 ± .14 | 2.16 ± 1.47 | ||
| STZ | .59 ± .19 | n.d. | n.i. | 1.61 ± .06 | 1.06 ± .12 | .82 ± .02 | 1.14 ± .03* | 1.56 ± .37 | 1.62 ± .55 | |||
| p47-/- | CTR | n.d. | n.d. | n.i. | 1.11 ± .05 | .81 ± .03 | .88 ± .01 | .94 ± .06 | .58 ± .22 | 2.71 ± .95 | ||
| STZ | n.d. | n.d. | n.i. | 1.74 ± .24 | 1.29 ± .12* | .97 ± .03 | .98 ± .04 | .39 ± .08 | 1.04 ± .34 | |||
| qPCR | WT | CTR | 1 ± .16 | 1 ± .41 | 1 ± .69 | 1 ± .11 | n.d. | n.d. | n.d. | n.d. | n.d. | |
| STZ | .99 ± .11 | 1.87 ± .16 | 1.07 ± .18 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | |||
| NoxO1-/- | CTR | n.d. | 1.41 ± .11 | 2.02 ± .5 | 1.27 ± .25 | n.d. | n.d. | n.d. | n.d. | n.d. | ||
| STZ | n.d. | 1.35 ± .23 | 1.8 ± .81 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | |||
| p47-/- | CTR | 0.78 ± .18 | n.d. | 2.8 ± .45 | 1.39 ± .13 | n.d. | n.d. | n.d. | n.d. | n.d. | ||
| STZ | 0.54 ± .11+ | n.d. | 2.42 ± 1.08 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | |||
| O1/47-/- | CTR | n.d. | n.d. | n.d. | 1.67 ± .17 | n.d. | n.d. | n.d. | n.d. | n.d. | ||
| STZ | n.d. | n.d. | n.d | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | |||
2.6. Diabetes reduces NoxO1 expression in vascular cells of the media but increases p47phox in vascular and adventitial cells
To investigate changes in the cellular distribution of NoxO1 and p47phox across the vessel under diabetic condition, the expression of these activators was detected by RNAscope technique using the most sensitive DAB detection method. Upon diabetes, the expression of NoxO1 was significantly reduced in the media consistent with the results from the MACE analysis. However, no significant change was observed in the adventitia.
In contrast, p47phox expression significantly increased in both vessel layers but particularly in the adventitia in response to diabetes (Fig. 5). This highly indicates an increased myeloid cell infiltration as a signature of a pro-inflammatory disease.
Fig. 5.

Expression and localization of NoxO1 and p47phox upon diabetes. A: RNAscope (DAB detection, brown kit) for NoxO1 and p47phox (shown by the arrows) in aortic tissue without hematoxylin counter staining. The aortic media comprises the elastic fibers and is delimited by full lines whereas the adventitia by dotted lines. B: quantification of the expression of NoxO1 and p47phox in diabetic aortic tissue represented as relative to CTR * p < 0.05 CTR vs. STZ.
2.7. Knockout of NoxO1 and p47phox leads to specific gene expression patterns
In order to define the impact of p47phox and NoxO1 on vascular gene expression, mRNA expression profiling was performed by the MACE technique (Fig. 6). Functional gene set enrichment confirmed that diabetes induces major changes in metabolic genes, moreover vascular expression of antioxidant genes like catalase and glutathione peroxide 3 (Gpx3) were increased by diabetes (Table S1). Importantly, deletion of either p47phox or NoxO1 attenuated this antioxidant response, suggesting that both proteins contribute to redox-signaling in the vasculature. Similar to the findings for the endothelium-dependent relaxation, the combined knockout of both, p47phox and NoxO1 failed to exert an additive effect. These data indicate that both p47phox and NoxO1 are required to maintain redox-dependent gene expression but speaks against a compensatory function.
Fig. 6.

Gene expression profiling by MACE. Significantly differentially expressed genes of three contrasts (NoxO1-STZ/NoxO1-CTR, WT-STZ/WT-CTR, NoxO1-p47-CTR/NoxO1-p47-STZ) were sorted for descending mean DESeq normalized expression. The top 20 genes of each contrast were merged and transformed to z-scores per gene. Asterisks indicated those genes related to immune response. (For interpretation of the references to color in this figure, the reader is referred to the web version of this article.)
The extensive profiling data obtained by MACE technique also suggest specific differences among the knockout strains. In particular, a large set of genes related to immune response was upregulated in response to the deletion of p47phox in the single, as well as in the double knockout mouse and also in response to diabetes (Fig. 6, red asterisks). This effect is most likely a consequence of the well-known hyperinflammatory phenotype of p47phox-/- mice [24], [25]. Unexpectedly, also deletion of NoxO1 was associated with a specific inflammatory signature; in contrast to the effect of deletion of p47phox, deletion of NoxO1 had an inhibitory impact on diabetes-induced activation of the interferon gamma (IFNγ) system. Of the inflammatory markers upregulated either by diabetes or knockout of NoxO1 or p47phox were Irf7 (Interferon Regulatory Factor 7), Ifitm1 (Interferon Induced Transmembrane Protein 1) and Ifi27l2a (Interferon Alpha Inducible Protein 27), which showed the lowest upregulation in NoxO1-/-. Cytometric bead array subsequently showed a reduction in IFNγ concentration in the plasma of NoxO1-/- diabetic mice (Fig. 7). Similarly, several major histocompatibility complex (class II) molecules were significantly downregulated in diabetic NoxO1-/- as compared to the other groups. In line with this, NoxO1-/- mice represented with attenuated numbers of white blood cells (Fig. 7B-C).
Fig. 7.

Interferon gamma and myeloid cells are changed in NO1-/- and p47-/-. A: Cytometric Bead array from plasma showing changes in interferon gamma concentrations n ≥ 8, *p < 0.05 as indicated. B: blood cell counting (*p < 0.05 NO1-/- vs. WT, #p<0.05 p47-/- vs. WT). C: blood cell profiling.
3. Discussion
In this study we describe a novel vascular function for NoxO1 in maintaining basal blood pressure and mediating endothelial dysfunction in diabetes. Deletion of NoxO1 reduced basal blood pressure and prevented the development of diabetes-induced endothelial dysfunction. Also deletion of p47phox prevented the development of diabetes-induced endothelial dysfunction. We, however, found no evidence for functional redundancy or crosstalk between the two subunits.
The expression of p47phox as observed in the present study was largely restricted to adventitial cells. This suggests a potential role of myeloid NADPH oxidase and myeloid cells in vascular dysfunction. In fact it was previously observed that endothelial dysfunction at least in response to angiotensin II is not observed in mice lacking monocytes [26] or deletion of monocytes [27], [28]. Moreover, a striking similarity to the present data was observed in response to myeloid-specific deletion of Nox2; the animals had have lower blood pressure and increased NO levels [29].
The research on cytosolic Nox interacting proteins has largely focused on p47phox. Not only that p47phox-/- mice are available since 1995 [24], also the initial vascular phenotype of these mice – improved endothelial function and attenuated atherosclerosis - fits well into the established schemes [20]. In contrast to this, little is known about the general function of NoxO1 and its specific role in the vasculature in particular. This is somewhat surprising given that NoxO1 mutant mice have been available for over a decade [30] and that the NoxO1-dependent Nox1 enzyme is well known to mediate vascular dysfunction [5]. It would, however, be an oversimplification to assume that deletion of NoxO1 results just in the same phenotype as deletion of Nox1. Not only because NoxO1 activates additional Nox enzymes, but the subunits might potentially compensate for each other. Finally, pleiotropic effects of the protein appear possible. For p47phox, several publications suggest Nox-independent functions [20] and the similarity between p47phox and NoxO1 implies that almost certainly NoxO1 has targets beyond the catalytically active of the Nox subunit. For example, in fibroblasts NoxO1 mediates TNF-induced necrotic cell death by forming a complex with TRADD, RIP1, and Rac1 [31]. Also the expression of NoxO1 is distinct from that of other subunits. In colon epithelial cell lines, TNFα and IFNγ increase NoxO1 expression and the subsequent production of superoxide formation [32]. The RNA expression data of the present study links NoxO1 to inflammation, and suggest an amplifying function of NoxO1 for interferon γ signaling in diabetes. The altered abundance of HLA-A molecules and leukocyte-specific RNAs as well as reduced leukocytes numbers in the blood indicates that NoxO1 impacts on the leukocyte populations present in the vasculature. Collectively, this might also explain the maintenance of normal endothelium-dependent relaxation in diabetic NoxO1-/- mice as IFNγ is known to induce endothelial dysfunction [33], [34]. Impact of Nox enzymes on the specific immune system is therefore conceivable. The anti-atherosclerotic effect of Nox4 has been linked to lymphocyte signaling [35] and angiotensin II-induced hypertension and vascular dysfunction require specific immune system activation [36]. Importantly, in particular IFNγ appears to be an important mediator in this scenario; smooth muscle-specific overexpression of the Nox subunit p22phox promoted IFNγ production by T-cells [37].
Also p47phox has been linked to the immune system but quiet in an opposite manner. Deletion of this subunit results in a hyper inflammatory complex phenotype with interferon signature, which might be at least in part a consequence of altered T-cell receptor signaling [25], [38], [39]. Why this systemic inflammation has so little impact on endothelium-dependent relaxation is not known. The analogy to Nox2, however, is striking. Deletion of Nox2 also results in a hyper inflammatory phenotype [40] despite the fact that Nox2 knockout mice exhibit enhanced endothelium-dependent relaxation under basal conditions [41] and do not develop endothelial dysfunction in high renin / angiotensin II-induced hypertension [42].
Little evidence, however, for crosstalk or compensation between NoxO1 and p47phox was found. The vascular gene expression profiles of the two knockout strains were different and the association with inflammation was diametric. With respect to vascular function, however, the deletion of any of the two proteins totally normalized endothelium-dependent relaxation in diabetes and there was no additive effect in the double knockout mice. Given that there was no gradual response in the normalization and that the overall degree of dysfunction was weak, the sensitivity of the system might simply not be sufficient to detect additive effects. On the basis of gene expression data however, it is noteworthy that numerous antioxidant enzymes were induced and differentially expressed which impacts on the reaction of the system. Finally, the floodgate hypothesis of signaling should be considered [43]. This concept assumes that any oxidant insult can be compensated as long as antioxidant defense is not overloaded. Once such an overload occurs, a stereotypic response is initiated which is independent of the source of ROS, i.e. mitochondria, uncoupled eNOS or any of the Nox enzymes.
In conclusion, we found little evidence for a direct crosstalk between NoxO1 and p47phox in vivo. By MACE we identified gene expression profiles specific for NoxO1 and p47phox, which are largely linked to modulation of the immune response. Most importantly, we establish NoxO1 as a novel driver of vascular dysfunction. Thus, inhibition of NoxO1 might be an attractive approach to combat vascular disease.
4. Innovation
With the aid of knockout mice for the cytosolic Nox organizers NoxO1 and p47phox and a double knockout (NoxO1/p47phox) we demonstrate that both organizers limit blood pressure and endothelium-dependent relaxation under basal and diabetic condition. However, they show no crosstalk at activity level in vivo but only in overexpression system using HEK293 cells. Moreover, deletion of NoxO1 induced an anti-inflammatory phenotype, whereas p47phox deletion rather elicited a hyper-inflammatory response. Thus, here we describe a novel role for NoxO1 in the vascular system making this molecule a potential pharmacological target.
5. Material and methods
5.1. Overexpression system using HEK293 cells
HEK293 cells were transiently transfected with plasmids (final concentration of 1.5 µg/ 3.5 cm dish) coding for the human sequence of Nox1, NoxO1, NoxA1, p47phox or GFP using lipofectamine according to the manufacturer's instructions.
5.2. ROS measurements with chemiluminescence
ROS production was assessed in intact cells with L-012 (200 µmol/L) in a Berthold TriStar2 microplate reader (LB942, Berthold, Wildbad, Germany) or in a Berthold 6-channel luminometer (LB9505, Berthold, Wildbad, Germany). All measurements were performed in HEPES-Tyrode (HT) buffer containing in mmol/L: 137 NaCl, 2.7 KCl, 0.5 MgCl2, 1.8 CaCl2, 5 glucose, 0.36 NaH2PO4, 10 HEPES.
5.3. Knockout animals and animal procedure
Knockouts for NoxO1 (NoxO1-/-) were generated as previously described [17]. p47phox knockouts (p47phox-/-) were kindly provided by Ajay M. Shah, Kings college London. Double knockouts for NoxO1 and p47phox (O1/47-/-) were generated by crossing NoxO1-/- and p47phox-/- mice. All mice were backcrossed at least for 10 generations into the C57/Bl6N background. All experiments performed with animals were in accordance with German animal protection law and were carried out after approval by the local authorities. Animals were housed in groups with free access to chow and water in a specified pathogen free facility with a 12/12 h day/night cycle. Given the impact of gender on ROS production, only male animals aged 8–16 weeks were used in this study.
5.4. RNAscope® in situ hybridization
In situ hybridizytion by RNAscope® technique was performed according to the manufacturer's instructions (Advanced Cell Diagnostics (ACD), Newark, CA, USA) in paraffin embedded sections of aortic rings previously fixed with 4% PFA (paraformaldehyde). Five micrometers thick sections were deparaffinized and treated with H2O2 followed by antigen retrieval and protease treatment according to kit's instructions (RNAscope H2O2 & Protease Plus reagents ACD #322330). Probes to NoxO1 ACD #466541; p47phox ACD #481991; Adgre1 ACD #460651 and positive/negative controls (peptidylprolyl isomerase B ACD #313911 / B. subtilis dihydrodipicolinate reductase ACD #310043) were hybridized for 2 h followed by 6 amplification steps. The signal was detected with RNAscope 2.5 HD detection kit red (ACD #322360) or kit Brown (ACD #322310) or by duplex chromogenic kit (ACD #322436) and specimens counterstained or not with DAPI or hematoxylin. Slides were analyzed by confocal microscopy in a LSM 800 (Zeiss) in Airyscan mode using the following excitation/emission wavelengths: 505/566 nm (Dy500), 493/517 nm (AF488), 353/465 nm (DAPI).
For the quantification, the areas of the media (where elastic fibers are visualized) and adventitia were delimitated (AxioVision software) and the dots corresponding to RNAscope positive signal were counted. Expression of NoxO1 and p47phox is represented as dots/mm2.
5.5. Streptozotocin model of diabetes
Diabetes was induced in WT and knockout mice (n≥8 per group) by intraperitoneal injection of streptozotocin (STZ) (50 mg/kg body weight) dissolved in 100 mmol/L citrate buffer, pH 4.2 for 4 consecutive days followed by implantation of an insulin pellet (LinBit, LinShin Canada Inc.). Control animals were injected with citrate buffer only. Blood glucose concentration was measured by Accu-Chek strips every 10 days. Animals were diabetic for 50 days and had their body weight controlled every 5 days till day 20 and every 10 days until day 50.
5.6. Blood pressure measurements
Tail cuff measurements were performed with a 6 channel setup (Vistech BP2000). Measurements of 5 days were averaged per mouse.
5.7. Organ chamber experiments
Organ chamber experiments were performed in mesenteric arteries in carbogen-aerated Krebs-Henseleit buffer containing diclofenac (10 µmol/L). The phenylephrine concentration was cumulatively increased (0.03–0.3 µmol/L) to obtain a pre-contraction level of approximately 80% of the initial KCl constriction. Subsequently, acetylcholine (ACh) was cumulatively added and dose-response curves for the relative constrictor response to phenylephrine and the dilator response to acetylcholine were constructed.
5.8. MACE and bioinformatics
For the Massive analysis of cDNA ends (MACE), 150 ng RNA from carotids from 3 different animals were pooled to make n = 1. 3 different pooled aliquots comprising different animals were analyzed for each group (WT and knockouts with and without diabetes). The libraries were prepared using GenXPro “MACE kit v.2.0”. cDNA was produced using oligo dT priming. cDNA was sheared to an average size of 350 bps and fragments were ligated to “TrueQuant” (GenXPro property, contains unique molecule identifiers). MACE- tags were amplified with 10 PCR cycles and the libraries were sequenced on an Illumina NextSeq. 500 machine. This technique was employed over standard RNAseq due to its greater potential to uncover low expressed genes once only the cDNA ends are sequenced.
Reads were aligned versus the Ensembl mouse genome version mm10 (GRCm38) using STAR 2.5.2b with the parameter “--outFilterMismatchNoverLmax 0.1” to increase the maximum ratio of mismatches to mapped length to 10% [44]. The number of reads aligning to genes was counted with feature Counts 1.5.1 tool from the Subread package [45]. Only reads mapping at least partially inside exons were admitted and aggregated per gene. Annotation was performed versus Gencode version vM11. Functional classification and enrichment was obtained by KOBAS 3.0 [46]. Differentially expressed genes were identified using DESeq. 2 version 1.14.1 [47]. The following thresholds were chosen for significantly differentially expressed genes: mean expression ≥ 5, 1 ≤ log2 fold change ≥ −1, Benjamini-Hochberg corrected p-value ≤ 0.05.
5.9. RT-qPCR
Total mRNA from frozen homogenized tissue was isolated with a RNA-Mini-kit (Bio&Sell, Feucht, Germany) according to the manufacturer's protocol. Random hexamer primers (Promega, Madison,WI, USA) and Superscript III Reverse Transcriptase (Invitrogen, Darmstadt, Germany) were used for cDNA synthesis. Semi-quantitative real-time PCR was performed with Mx3000 P qPCR cycler (Agilent Technologie, Santa Clara, CA, USA) using PCR Eva Green qPCR Mix with ROX (Bio&Sell, Feucht, Germany) with appropriate primers. Relative expression of target genes were normalized to eukaryotic translation elongation factor 2 (EF2), analyzed by delta-delta-Ct method and represented as percent of control samples. Primer sequences are listed in Table 1.
Table 1.
Primers.
| mElongation factor 2 | forward | 5′-GACATCACCAAGGGTGTGCAG−3′ |
| reverse | 5′-GCGGTCAGCACACTGGCATA−3′ | |
| mNox1 | forward | 5′-CCTCCTGACTGTGCCAAAGG−3′ |
| reverse | 5′-ATTTGAACAACAGCACTCACCAA−3′ | |
| mNoxO1 | forward | 5′-TGGAGGAGGTAGCAACGTGC−3′ |
| reverse | 5′-AGAGCGACTGCCCTCGTAGG−3′ | |
| p47phox | forward | 5′-TCCCAACTACGCAGGTGAAC−3′ |
| reverse | 5′-CCTGGGTTATCTCCTCCCCA−3′ | |
| Nox2 | forward | 5′-AGCTATGAGGTGGTGATGTTAGTGG−3′ |
| reverse | 5′-CACAATATTTGTACCAGACAGACTTGAG−3′ | |
| Glutathione peroxidase 3 | forward | 5′-TTGGTCATTCTGGGCTTCCC−3′ |
| reverse | 5′-AGGGCAGGAGTTCTTCAGGA−3′ | |
| Adiponectin | forward | 5′-ACGACACCAAAAGGGCTCAG−3′ |
| reverse | 5′-GAGTGCCATCTCTGCCATCA−3′ | |
| mCatalase | forward | 5′-GCATCGAGCCCAGCCCTGAC−3′ |
| forward | 5′-GCACATGGGGCCATCACGCT−3′ |
5.10. Cytometric bead array (CBA)
Cytokines were measured in 25 µL plasma using the CBA mouse Th1/Th2/Th17 kit (BD #560485) according to manufacturer's instructions.
5.11. Blood cell counting
Quantitative hematological analysis was performed with a VetScan HM5. EDTA-treated blood was used for the analysis.
5.12. Statistics
Unless otherwise indicated, data are given as means ± standard error of mean (SEM). Calculations were performed with Prism 5.0 or BiAS.10.12. The latter was also used to test for normal distribution and similarity of variance. In case of multiple testing, Bonferroni correction was applied. For multiple group comparisons ANOVA followed by post hoc testing was performed. Individual statistics of unpaired samples was performed by t-test and if not normal distributed by Mann-Whitney test. A p-value of < 0.05 was considered as significant. Unless otherwise indicated, n indicates the number of individual experiments or animals.
Acknowledgement
We are grateful for excellent technical assistance of Susanne Schütz and Axel Erhardt.
Acknowledgments
Source of funding
The study was supported by the DFG Excellence Cluster “Cardiopulmonary System – ECCPS”, SFB 815 (TP A1 to KS and TP A16 to IF) and SFB 834 (TPA2 to RPB, TP A9N to IF and TP Z1 to MAR), the Faculty of Medicine, Goethe-Universität, Frankfurt am Main, Germany, the Heinrich und Fritz-Riese-Stiftung to FR and DZHK, Partner Site RheinMain, Frankfurt. TF & PPN were supported by SFB118 (A04 & C06).
Disclosures
The authors declare that they have no relevant financial, personal or professional relationships to disclose which could be perceived as a conflict of interest or as potentially influencing or biasing the authors’ work.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.11.014.
Contributor Information
Ralf P. Brandes, Email: brandes@vrc.uni-franfurt.de.
Katrin Schröder, Email: schroeder@vrc.uni-frankfurt.de.
Appendix A. Supplementary material
Supplementary material
References
- 1.Youn J.Y., Gao L., Cai H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia. 2012;55(7):2069–2079. doi: 10.1007/s00125-012-2557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paradis P., Ouerd S., Idris-Khodja N., Trindade M., Coelho S.C., Jandeleit-Dahm K.A., Schiffrin E.L. Vol. 34. 2016. OS 21-01 NOX1 OR NOX4 deletion prevents type 1 diabetes-induced endothelial dysfunction; p. e235. (J. Hypertens.). (ISH 2016 Abstract Book) [Google Scholar]
- 3.Lee M.Y., San Martin A., Mehta P.K., Dikalova A.E., Garrido A.M., Datla S.R., Lyons E., Krause K.-H., Banfi B., Lambeth J.D., Lassegue B., Griendling K.K. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 2009;29(4):480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wind S., Beuerlein K., Armitage M.E., Taye A., Kumar A.H.S., Janowitz D., Neff C., Shah A.M., Wingler K., Schmidt H.H.H.W. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension. 2010;56(3):490–497. doi: 10.1161/HYPERTENSIONAHA.109.149187. [DOI] [PubMed] [Google Scholar]
- 5.Matsuno K., Yamada H., Iwata K., Jin D., Katsuyama M., Matsuki M., Takai S., Yamanishi K., Miyazaki M., Matsubara H., Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112(17):2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 6.Ohara Y., Peterson T.E., Harrison D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993;91(6):2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanic B., Pandey D., Fulton D.J., Miller JR F.J. Increased epidermal growth factor-like ligands are associated with elevated vascular nicotinamide adenine dinucleotide phosphate oxidase in a primate model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012;32(10):2452–2460. doi: 10.1161/ATVBAHA.112.256107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai H., Harrison D.G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 2000;87(10):840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 9.Cheng G., Lambeth J.D. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J. Biol. Chem. 2004;279(6):4737–4742. doi: 10.1074/jbc.M305968200. [DOI] [PubMed] [Google Scholar]
- 10.Kawano M., Miyamoto K., Kaito Y., Sumimoto H., Tamura M. Noxa1 as a moderate activator of Nox2-based NADPH oxidase. Arch. Biochem. Biophys. 2012;519(1):1–7. doi: 10.1016/j.abb.2011.12.025. [DOI] [PubMed] [Google Scholar]
- 11.Geiszt M., Lekstrom K., Brenner S., Hewitt S.M., Dana R., Malech H.L., Leto T.L. NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J. Immunol. 2003;171(1):299–306. doi: 10.4049/jimmunol.171.1.299. [DOI] [PubMed] [Google Scholar]
- 12.Bedard K., Krause K.-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 13.Al Ghouleh I., Meijles D.N., Mutchler S., Zhang Q., Sahoo S., Gorelova A., Henrich Amaral J., Rodriguez A.I., Mamonova T., Song G.J., Bisello A., Friedman P.A., Cifuentes-Pagano M.E., Pagano P.J. Binding of EBP50 to Nox organizing subunit p47phox is pivotal to cellular reactive species generation and altered vascular phenotype. Proc. Natl. Acad. Sci. USA. 2016;113(36):E5308–E5317. doi: 10.1073/pnas.1514161113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Touyz R.M., Chen X., Tabet F., Yao G., He G., Quinn M.T., Pagano P.J., Schiffrin E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ. Res. 2002;90(11):1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 15.Lassegue B., Sorescu D., Szocs K., Yin Q., Akers M., Zhang Y., Grant S.L., Lambeth J.D., Griendling K.K. Novel gp91phox Homologues in Vascular Smooth Muscle Cells: Nox1 Mediates Angiotensin II-Induced Superoxide Formation and Redox-Sensitive Signaling Pathways. Circ. Res. 2001;88(9):888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 16.Banfi B., Clark R.A., Steger K., Krause K.H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 2003;278(6):3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 17.Brandes R.P., Harenkamp S., Schurmann C., Josipovic I., Rashid B., Rezende F., Lowe O., Moll F., Epah J., Eresch J., Nayak A., Kopaliani I., Penski C., Mittelbronn M., Weissmann N., Schroder K. The cytosolic NADPH oxidase subunit NoxO1 promotes an endothelial stalk cell phenotype. Arterioscler., Thromb., Vasc. Biol. 2016;36(8):1558–1565. doi: 10.1161/ATVBAHA.116.307132. [DOI] [PubMed] [Google Scholar]
- 18.Siu K.L., Gao L., Cai H. Differential roles of protein complexes NOX1-NOXO1 and NOX2-p47phox in mediating endothelial redox responses to oscillatory and unidirectional laminar shear stress. J. Biol. Chem. 2016;291(16):8653–8662. doi: 10.1074/jbc.M115.713149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeya R., Ueno N., Kami K., Taura M., Kohjima M., Izaki T., Nunoi H., Sumimoto H. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J. Biol. Chem. 2003;278(27):25234–25246. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- 20.Schroder K., Weissmann N., Brandes R.P. Organizers and activators: cytosolic Nox proteins impacting on vascular function. Free Radic. Biol. Med. 2017 doi: 10.1016/j.freeradbiomed.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 21.Guzik T.J. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105(14):1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 22.Nishikawa T., Edelstein D., Du X.L., Yamagishi S., Matsumura T., Kaneda Y., Yorek M.A., Beebe D., Oates P.J., Hammes H.P., Giardino I., Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 23.Sturza A., Duicu O.M., Vaduva A., Dănilă M.D., Noveanu L., Varró A., Muntean D.M. Monoamine oxidases are novel sources of cardiovascular oxidative stress in experimental diabetes. Can. J. Physiol. Pharmacol. 2015;93(7):555–561. doi: 10.1139/cjpp-2014-0544. [DOI] [PubMed] [Google Scholar]
- 24.Jackson S.H. The p47phox mouse knock-out model of chronic granulomatous disease. J. Exp. Med. 1995;182(3):751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmdahl R., Sareila O., Olsson L.M., Backdahl L., Wing K. Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol. Rev. 2016;269(1):228–247. doi: 10.1111/imr.12378. [DOI] [PubMed] [Google Scholar]
- 26.Ciuceis C.D., Amiri F., Brassard P., Endemann D.H., Touyz R.M., Schiffrin E.L. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II–infused macrophage colony-stimulating factor–deficient mice. Arterioscler., Thromb., Vasc. Biol. 2005;25(10):2106–2113. doi: 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- 27.Wenzel P., Knorr M., Kossmann S., Stratmann J., Hausding M., Schuhmacher S., Karbach S.H., Schwenk M., Yogev N., Schulz E., Oelze M., Grabbe S., Jonuleit H., Becker C., Daiber A., Waisman A., Münzel T. Lysozyme M–positive monocytes mediate angiotensin II–induced arterial hypertension and vascular dysfunction. Circulation. 2011 doi: 10.1161/CIRCULATIONAHA.111.034470. (CIRCULATIONAHA.111.034470) [DOI] [PubMed] [Google Scholar]
- 28.Kossmann S., Hu H., Steven S., Schönfelder T., Fraccarollo D., Mikhed Y., Brähler M., Knorr M., Brandt M., Karbach S.H., Becker C., Oelze M., Bauersachs J., Widder J., Münzel T., Daiber A., Wenzel P. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J. Biol. Chem. 2014;289(40):27540–27550. doi: 10.1074/jbc.M114.604231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sag C.M., Schnelle M., Zhang J., Murdoch C.E., Kossmann S., Protti A., Santos C.X.C., Sawyer G.J., Zhang X., Mongue-Din H., Richards D.A., Brewer A.C., Prysyazhna O., Maier L.S., Wenzel P., Eaton P.J., Shah A.M. Distinct regulatory effects of myeloid cell and endothelial cell Nox2 on blood pressure. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.116.023877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiss P.J., Knisz J., Zhang Y., Baltrusaitis J., Sigmund C.D., Thalmann R., Smith R.J.H., Verpy E., Banfi B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr. Biol. CB. 2006;16(2):208–213. doi: 10.1016/j.cub.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 31.Kim Y.-S., Morgan M.J., Choksi S., Liu Z.-G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell. 2007;26(5):675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 32.Kuwano Y., Tominaga K., Kawahara T., Sasaki H., Takeo K., Nishida K., Masuda K., Kawai T., Teshima-Kondo S., Rokutan K. Tumor necrosis factor alpha activates transcription of the NADPH oxidase organizer 1 (NOXO1) gene and upregulates superoxide production in colon epithelial cells. Free Radic. Biol. Med. 2008;45(12):1642–1652. doi: 10.1016/j.freeradbiomed.2008.08.033. [DOI] [PubMed] [Google Scholar]
- 33.Kossmann S., Schwenk M., Hausding M., Karbach S.H., Schmidgen M.I., Brandt M., Knorr M., Hu H., Kröller-Schön S., Schönfelder T., Grabbe S., Oelze M., Daiber A., Münzel T., Becker C., Wenzel P. Angiotensin II-induced vascular dysfunction depends on interferon-γ-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler. Thromb. Vasc. Biol. 2013;33(6):1313–1319. doi: 10.1161/ATVBAHA.113.301437. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H., Potter B.J., Cao J.-M., Zhang C. Interferon-gamma induced adipose tissue inflammation is linked to endothelial dysfunction in type 2 diabetic mice. Basic Res. Cardiol. 2011;106(6):1135–1145. doi: 10.1007/s00395-011-0212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Craige S.M., Kant S., Reif M., Chen K., Pei Y., Angoff R., Sugamura K., Fitzgibbons T., Keaney J.F. Endothelial NADPH oxidase 4 protects ApoE-/- mice from atherosclerotic lesions. Free Radic. Biol. Med. 2015;89:1–7. doi: 10.1016/j.freeradbiomed.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guzik T.J., Hoch N.E., Brown K.A., McCann L.A., Rahman A., Dikalov S., Goronzy J., Weyand C., Harrison D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007;204(10):2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu J., Saleh M.A., Kirabo A., Itani H.A., Montaniel K.R.C., Xiao L., Chen W., Mernaugh R.L., Cai H., Bernstein K.E., Goronzy J.J., Weyand C.M., Curci J.A., Barbaro N.R., Moreno H., Davies S.S., Roberts L.J., Madhur M.S., Harrison D.G. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J. Clin. Investig. 2016;126(1):50–67. doi: 10.1172/JCI80761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelkka T., Kienhofer D., Hoffmann M., Linja M., Wing K., Sareila O., Hultqvist M., Laajala E., Chen Z., Vasconcelos J., Neves E., Guedes M., Marques L., Kronke G., Helminen M., Kainulainen L., Olofsson P., Jalkanen S., Lahesmaa R., Souto-Carneiro M.M., Holmdahl R. Reactive oxygen species deficiency induces autoimmunity with type 1 interferon signature. Antioxid. Redox Signal. 2014;21(16):2231–2245. doi: 10.1089/ars.2013.5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gelderman K.A., Hultqvist M., Holmberg J., Olofsson P., Holmdahl R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc. Natl. Acad. Sci. USA. 2006;103(34):12831–12836. doi: 10.1073/pnas.0604571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deffert C., Carnesecchi S., Yuan H., Rougemont A.-L., Kelkka T., Holmdahl R., Krause K.-H., Schäppi M.G. Hyperinflammation of chronic granulomatous disease is abolished by NOX2 reconstitution in macrophages and dendritic cells. J. Pathol. 2012;228(3):341–350. doi: 10.1002/path.4061. [DOI] [PubMed] [Google Scholar]
- 41.Gorlach A., Brandes R.P., Nguyen K., Amidi M., Dehghani F., Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ. Res. 2000;87(1):26–32. doi: 10.1161/01.res.87.1.26. [DOI] [PubMed] [Google Scholar]
- 42.Jung O., Schreiber J.G., Geiger H., Pedrazzini T., Busse R., Brandes R.P. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004;109(14):1795–1801. doi: 10.1161/01.CIR.0000124223.00113.A4. [DOI] [PubMed] [Google Scholar]
- 43.Netto L.E.S., Antunes F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol. Cells. 2016;39(1):65–71. doi: 10.14348/molcells.2016.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 46.Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C.-Y., Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–W322. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material