

Abstract
Alterations in the balance between neuronal excitation and inhibition (E:I balance) have been implicated in the neural circuit activity–based processes that contribute to autism phenotypes. We investigated whether acutely reducing E:I balance in mouse brain could correct deficits in social behavior. We used mice lacking the CNTNAP2 gene, which has been implicated in autism, and achieved a temporally precise reduction in E:I balance in the medial prefrontal cortex (mPFC) either by optogenetically increasing the excitability of inhibitory parvalbumin (PV) neurons or decreasing the excitability of excitatory pyramidal neurons. Surprisingly, both of these distinct, real-time, and reversible optogenetic modulations acutely rescued deficits in social behavior and hyperactivity in adult mice lacking CNTNAP2. Using fiber photometry, we discovered that native mPFC PV neuronal activity differed between CNTNAP2 knockout and wild-type mice. During social interactions with other mice, PV neuron activity increased in wild-type mice compared to interactions with a novel object, whereas this difference was not observed in CNTNAP2 knockout mice. Together, these results suggest that real-time modulation of E:I balance in the mouse prefrontal cortex can rescue social behavior deficits reminiscent of autism phenotypes.
INTRODUCTION
The autism spectrum disorders are characterized by highly heterogeneous clinical presentations and complex genetics. For this reason, a fundamental goal of autism research is to identify a biological final common pathway wherein the disparate genetic and pathophysiological pathways may converge. In part, because of our ongoing failure to identify such a final common pathway, currently available pharmacological interventions used in autism patients have not been able to address the underlying disorder itself—instead addressing only comorbid symptoms with varying degrees of success (1). There remain no approved pharmacological interventions for treating the core symptoms of autism, namely, communication difficulties, social challenges, and restricted, repetitive behaviors (2).
Recently developed tools for circuit-level interventions (3) may open up new possibilities for identifying and targeting shared mechanisms in autism. An altered balance in neuronal excitation and inhibition (E:I balance) is commonly invoked as a possible final common pathway in autism (4), with clues coming from anatomy (5–7), as well as from genetics, physiology, and behavior in humans and rodents. Supporting evidence for this concept includes observations in postmortem human brain tissue of a decreased number of parvalbumin (PV) neurons in the medial prefrontal cortex (mPFC) (8), a decreased expression of mRNAs encoding receptors for the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (9), a decreased ratio of GABA to glutamate (an excitatory neurotransmitter) (10), increased resting-state activity in the frontal cortex (11, 12), and behavioral or epidemiological changes that suggest increased excitation including hypersensitivity to sensory stimuli and comorbidity with epilepsy (1). Mouse models carrying mutations known to cause autism in humans also point to a decreased number of cortical inhibitory interneurons (especially PV neurons) and decreased GABAergic transmission (1), as well as increased gamma frequency rhythmicity under baseline (nonstimulus-evoked) conditions. The latter phenomenon may be relevant to PV neurons (13, 14) and has also been observed in patients with autism (15–20). However, this E:I balance must be interpreted in the context of specific cells and circuits (21). Causal demonstration of improvements in core behavioral deficits after restoration of cellular E:I balance with real-time modulation of specific circuitry in a genetic mouse model of autism needs to be achieved.
In previous work that may help to guide attention to particular brain circuits relevant to social behavior, it has been found that an increase in E:I balance in the rodent mPFC leads to social behavior deficits (22), which can be partially rescued by stimulation of PV neurons (22). This previous work was entirely conducted in wild-type (WT) animals and thus may have limited translational significance. Here, we begin with a genetic mouse model lacking CNTNAP2 that exhibits autism-like phenotypes, including social behavior deficits, together with the common comorbidities of hyperactivity and seizures and reduced interneuron density in cortical and striatal brain regions (23). For real-time interrogation of neuronal circuits during social behavior, we used engineered microbial opsin tools, specifically the stabilized step-function opsin (SSFO) (22) or the step-function inhibitory opsin SwiChR++ (24). We used these tools to genetically target inhibitory PV or excitatory pyramidal (PYR) neurons, respectively, together with fiber photometry, to record real-time neural activity signals in the prefrontal cortex of CNTNAP2 knockout (KO) mice.
RESULTS
Characterization of the engineered microbial opsin SSFO in mPFC PV neurons
To specifically and stably decrease cellular E:I balance in mPFC, we first sought to target PV neurons in mice lacking CNTNAP2, which required generation of new mouse lines. We bred PV::Cre homozygous female mice with CNTNAP2 KO male mice; after several generations, PV::Cre homozygous/CNTNAP2 homozygous KO and WT littermates were obtained along with PV::Cre homozygous/CNTNAP2 heterozygous mice, which were bred for subsequent generations (Fig. 1A). We then injected AAV5-EF1α::DIO-SSFO-eYFP to specifically express the microbial opsin SSFO in mouse PV neurons (Fig. 1B).
Fig. 1. Generation of PV::Cre/CNTNAP2 mice and characterization of SSFO properties in mPFC PV neurons.
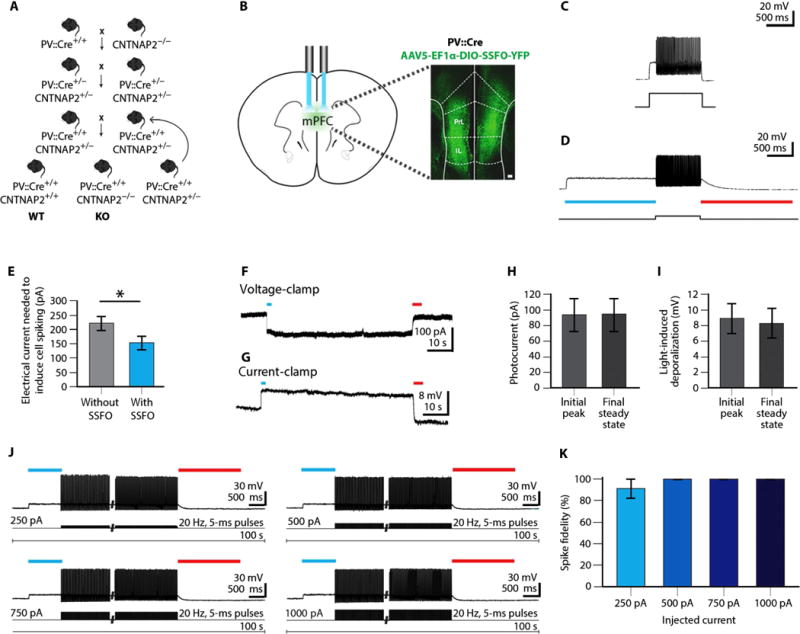
(A) Breeding strategy to generate PV::Cre/CNTNAP2 mice is shown. Only the combination of genotypes that were chosen for the next breeding step is shown. PV::Cre+/+ CNTNAP2+/− mice were bred to generate WT and CNTNAP2 KO mice. (B) AAV5-EF1α::DIO-SSFO-YFP expression in PV neurons in WT mice. Bilateral optical fiber implantation above the mPFC in WT mice is also shown. Scale bar, 100 μm. (C) Representative short–time scale current-clamp trace of a PV neuron [identified by yellow fluorescent protein (YFP) expression] in a brain slice from the mPFC of a PV::Cre mouse, prepared 2 to 3 weeks after injection of a Cre-dependent adeno-associated virus (AAV) encoding the SSFO. Trace shows the minimum amount of electrical current injection (200 pA) required for spike generation. Resting potential, −72 mV. (D) Representative trace of the same PV neuron showing the much lower minimum current injection (50 pA) required for spike generation during SSFO activation by blue light of wavelength 475 nm (28-nm bandpass) (light blue bar). Light power density was 8 mW/mm2. SSFO was deactivated using red light of wavelength 632/622 nm (red bar). Light power density was 5 mW/mm2. (E) Bar graph summarizing the electrical current needed to induce spiking without or with SSFO activation (n = 8 cells) (table S1). (F) Representative extended–time scale voltage-clamp trace of an SSFO-expressing PV neuron after blue light and red light application. Cell was held at −77 mV. (G) Representative current-clamp trace of same cell after blue and red light application. Note the lack of directly elicited spiking. (H) Voltage-clamp studies showing SSFO-induced initial peak and final steady-state photocurrent amplitude (n = 8 cells). Resting potential of cells held in voltage clamp at −80 mV is shown (table S2). (I) Current-clamp studies showing SSFO-induced initial peak and final steady-state depolarization (n = 8 cells) (table S3). (J) Sample traces showing stable, high-fidelity action potential generation in a PV neuron expressing SSFO after blue light delivery, combined with increasing amounts of electrical current injection (5-ms pulses, 20-Hz frequency), followed by deactivation with red light. Current amplitude was applied in 250-pA steps, beginning at 250 pA. In all cases, spiking persisted for 60 s until experimental termination of the costimulation. (K) Bar graph summary of spike fidelity (spikes per 5-ms current pulse) throughout the entire SSFO activation protocol, in combination with the varying electrical current injection steps, as shown in (J). n = 7 cells from WT mice; P = 0.101 by Kruskal-Wallis H test (table S4). Bar graphs in (E), (H), (I), and (K) are presented as mean ± SEM. *P < 0.05 by paired t test.
SSFO is a double-mutant excitatory channelrhodopsin that was engineered to have slow off-kinetics (22). Upon activation with a pulse of blue light, SSFO induces stable subthreshold depolarization of neurons without directly driving action potentials, which elevates the excitability and strengthens the influence of targeted cells (22). Because of a channel closure time constant of ~30 min, the higher probability of spiking is stable after a single light flash over the period required for complex behavioral testing, unless the channel is switched off by yellow light (22). We found that activation of SSFO expressed by mouse PV neurons, when compared to no-SSFO activation (Fig. 1C), resulted in the neurons requiring lower inward-current amplitudes to generate action potentials (Fig. 1D) (with SSFO, 153.1 ± 22.4 pA; without SSFO, 221.9 ± 20.8 pA; n = 8; Fig. 1E). This indicated the utility of SSFO for favoring excitability of the inhibitory neuronal population.
To further validate this approach, we characterized SSFO activity in greater detail. Blue light delivery reliably generated prolonged photocurrents (initial peak: 93.7 ± 19.9 pA; final steady state: 94.7 ± 20.0 pA; n = 8), which corresponded to stable subthreshold depolarization in current-clamp recordings (peak: 8.9 ± 1.8 mV; steady state: 8.23 ± 1.9 mV), and was immediately reversible by red light (Fig. 1, F to I). Because SSFOs have been optimized for extended excitability, we sought to ensure that mouse PV neurons would be able to sustain action potential generation over long time scales. We provided electrical stimulation by intracellular current injection through the patch-clamp pipette during light-induced depolarization to examine prolonged time scale spiking, spanning a range from moderate (250 pA) to large (1000 pA) injected currents. We found that all tested cells were able to generate stable spike trains even when large-amplitude (1000 pA) pulses were applied (no depolarization block was seen; spike probability, 91.3 ± 8.5% with 250-pA current injection, 100% with 500 pA, 100% with 750 pA, and 100% with 1000 pA) (Fig. 1, J and K).
Rescue of autism-like behaviors in CNTNAP2-deficient mice
Given that an increase in E:I balance in genetic mouse models of autism has been suggested to underlie social behavior deficits (23, 25, 26), we hypothesized that reducing cortical E:I balance in CNTNAP2 KO mice could improve social deficits. We selectively expressed SSFO in PV neurons and implanted bilateral optical fibers into the mPFC of WT and CNTNAP2 KO mice. To bias excitability of PV neurons, we applied a 2-s light pulse (473 nm); immediately after light stimulation, we initiated a 10-min social exploration test (Fig. 2A).
Fig. 2. SSFO or SwiChR++ modulation of mPFC neurons and rescue of social behavior deficits in CNTNAP2 KO mice.
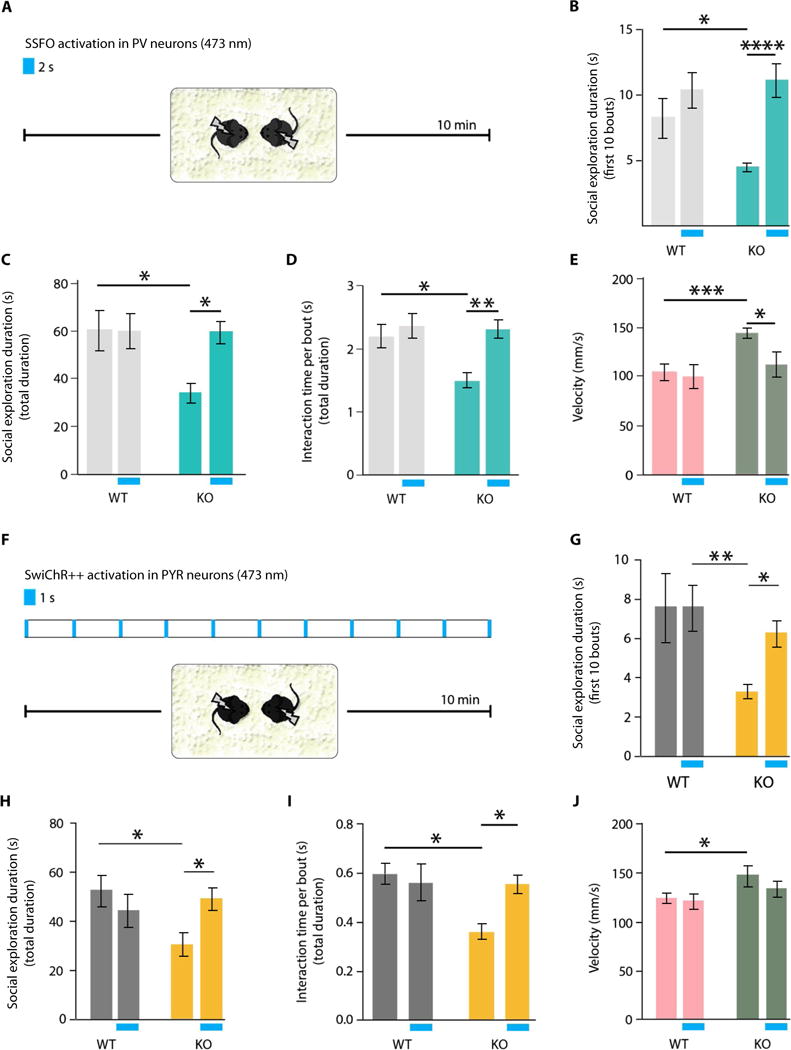
(A) Testing of social interaction behavior for 10 min in mouse pairs matched by genotype and sex is shown. A single 2-s, 473-nm pulse of blue light was administered immediately before the behavioral test in the stimulation condition. (B) The duration of social explorations during the first 10 interaction bouts with or without SSFO stimulation is shown (WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P < 0.0001) (table S5). In (B) to (J), two-way ANOVA was used, followed by Dunnett’s pairwise comparisons; parameter interaction values and n values (at least eight pairs per condition) for each arm are shown in the Supplementary Materials. (C) Duration of social explorations during the total 10 min of the test with and without SSFO stimulation (WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P < 0.05) (table S6). (D) Mean social exploration duration per interaction bout with and without SSFO stimulation (WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P < 0.01) (table S7). (E) Mean social approach velocity with and without SSFO stimulation (WTbaseline versus KObaseline, P < 0.001; KObaseline versus KOstimulation, P < 0.05) (table S8). (F) Social interaction test after a 1-s intermittent 473-nm pulse of blue light to stimulate the inhibitory channelrhodopsin SwiChR++. (G) Social exploration duration during the first 10 interaction bouts with and without inhibitory SwiChR++ stimulation (WTstimulation ver sus KObaseline, P < 0.01; KObaseline versus KOstimulation, P < 0.05) (table S9). (H) Social exploration duration during the total length of the test (10 min) with and without inhibitory SwiChR++ stimulation (WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P < 0.05) (table S10). (I) Mean social exploration duration per interaction bout with and without inhibitory SwiChR++ stimulation (WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P < 0.05) (table S11). (J) Mean social approach velocity with and without inhibitory SwiChR++ stimulation (WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P > 0.05) (table S12). Two separate cohorts were tested and included in the analyses. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***, ***P < 0.001, and ****P < 0.0001.
CNTNAP2 KO mice exhibited a profound deficit in social interactions at baseline [one-way analysis of variance (ANOVA), Dunnett’s pairwise comparison to control; WTbaseline versus KObaseline, P < 0.05; over the first 10 interaction bouts; Fig. 2B]. Strikingly, reducing mPFC cellular E:I balance with SSFO rescued this social deficit (KObaseline versus KOstimulation, P < 0.0001; Fig. 2B). Similar effects were seen over the entire 10-min interaction period (one-way ANOVA, Dunnett’s pairwise comparison to control; WTbaseline versus KObaseline, P < 0.05; Fig. 2C). The significant differences in social exploration between WT and CNTNAP2 KO mice (23) were rescued through SSFO activation (KObaseline versus KOstimulation, P < 0.05; Fig. 2C). In addition to total social interaction time differences, we also noted that mean social-bout duration was significantly shorter in KO mice compared to WT mice in the absence of SSFO stimulation (one-way ANOVA, Dunnett’s pairwise comparison to control; WTbaseline versus KObaseline, P < 0.05); after SSFO activation, social exploration per bout was fully restored in CNTNAP2 KO mice (one-way ANOVA, Dunnett’s pairwise comparison to control; KObaseline versus KOstimulation, P < 0.01; Fig. 2D).
CNTNAP2 KO mice have been previously reported to show greater locomotor activity than WT littermates in the open-field test (23); hyperactivity symptoms can be up to 85% comorbid with autism in clinical populations (27). We observed that CNTNAP2 KO mice were hyperactive compared to WT littermates during social behavior bouts (one-way ANOVA, Dunnett’s pairwise comparison; WTbaseline versus KObaseline, P < 0.001; Fig. 2E). We found that reducing E:I balance in the mPFC through SSFO activation in PV neurons corrected this key behavioral phenotype (one-way ANOVA, Dunnett’s pairwise comparison; KObaseline versus KOstimulation, P < 0.05; Fig. 2E).
Next, to test whether manipulation of E:I balance was essential for reversal of the social behavior deficits, we sought to reduce cortical E:I balance by an independent, but equally temporally precise, optogenetic method. In this case, we decreased the excitability of excitatory PYR neurons in the mPFC of WT or CNTNAP2 KO mice. We selectively expressed SwiChR++, a blue light–activated bistable chloride channel, in PYR neurons by injection of AAV8-CKIIα-SwiChR++-eYFP into the mPFC (targeted as shown in fig. S1). Taking into account the channel closure rate of ~115 s, we delivered 1-s pulses of 473-nm wavelength light at 1-min intervals during the 10-min social exploration test to achieve sustained inhibition of PYR cells (Fig. 2F).
Notably, the behavioral results after PYR neuron inhibition were concordant with the effects of PV neuron activation in the mPFC of CNTNAP2 KO mice. Upon SwiChR++ activation, CNTNAP2 KO mice interacted longer with littermates during the first 10 bouts of the social exploration test (one-way ANOVA, Dunnett’s pairwise comparison; WTstimulation versus KObaseline, P < 0.01; KObaseline versus KOstimulation, P < 0.05; Fig. 2G). The effects of increased inhibition of PYR neurons became more evident over the 10-min duration of the test (one-way ANOVA, Dunnett’s pairwise comparison; WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P < 0.05; Fig. 2H). Mean social-bout duration (length of individual interaction periods) was also rescued upon SwiChR++ activation (WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P < 0.05; Fig. 2I). Differences in locomotor activity in the observation arena between CNTNAP2 KO and WT mice were eliminated upon inhibition of PYR neurons by SwiChR++ activation (WTbaseline versus KObaseline, P < 0.05; KObaseline versus KOstimulation, P > 0.05; Fig. 2J).
To test for the specificity of the two optogenetic manipulations on cellular E:I balance and social behavior, we performed object interaction tests (fig. S2, A to D). After optogenetic manipulation, we observed specificity for an increase in social interactions compared to interactions with inanimate objects with no difference seen in object interaction times. Optogenetic manipulations of E:I balance trended toward reduced object exploration behavior in the CNTNAP2 KO animals (two-way ANOVA treatment-genotype interaction; SSFO cohort, F1,32 = 1.348, P = 0.2542; fig. S2B; SwiChR++ cohort, F1,27 = 0.0025, P = 0.9599; fig. S2D). As a further test, we assessed hyperactivity independent of social behaviors in the open-field chamber test (fig. S3). CNTNAP2 KO animals were hyperactive in the open-field test compared to WT litter-mates (one-way ANOVA, Dunnett’s pairwise comparison; WTbaseline versus KObaseline, P < 0.001 for both SSFO and SwiChR++ cohorts; fig. S3, A and B, respectively). This phenotypic difference was no longer detectable after optogenetic modulation of cellular E:I balance (one-way ANOVA, Dunnett’s pairwise comparison to control; WTstimulation versus KOstimulation, P > 0.05 for both SSFO and SwiChR++ cohorts).
Neurophysiological activity in freely moving mice
To explore underlying neural activity signatures, we sought to track and quantify local neural circuit activity in freely moving mice using fiber photometry, a technique that allows recording of cell population Ca2+ signals linked to neural activity (28, 29). A genetically encoded Ca2+ indicator designed to change fluorescence properties during neuronal firing (30–32) was expressed in PV neurons of the mPFC. Fluorescence fluctuations were then measured using an optical fiber implanted in freely moving mice. Altered neuronal activity within the targeted population of neurons was then plotted as a transient change in fluorescence normalized to baseline (dF/F) and could be time-locked to the behavior of mice on fast time scales (hundreds of milliseconds) (29, 33). We tested the method’s capability to track physiological effects of optogenetic E:I balance modulation by exploring whether SSFO activation in local mPFC PV neurons would modulate projection-neuron activity in freely moving mice. Mouse brain fiber photometry recordings of cortical projections to striatum were chosen as the activity readout because disruptions in corticostriatal circuitry have been implicated in the CNTNAP2 KO mouse phenotype (23). We found that upon SSFO activation in mPFC PV neurons, activity along the corticostriatal projections, defined by origin (mPFC) and target (striatum), was reduced (fig. S4) in a temporally precise and reversible fashion.
We then sought to use this method to define naturally occurring activity in mPFC PV neurons during social interactions with mice and novel objects. In freely moving mice (33) that expressed the genetically encoded Ca2+ indicator GCaMP6f in mPFC PV neurons (Fig. 3A), we continuously recorded Ca2+ signals (after acquisition of a baseline) while behaviorally testing novel social interactions with nonlittermates, familiar social interactions with littermates, and novel object interactions, each separated by rest periods (Fig. 3B and movies S1 and S2). Recordings were acquired over a continuous session to counteract variability in the baseline activity.
Fig. 3. Differential mPFC PV neuron activity during exploratory behaviors in CNTNAP2 KO and WT mice.
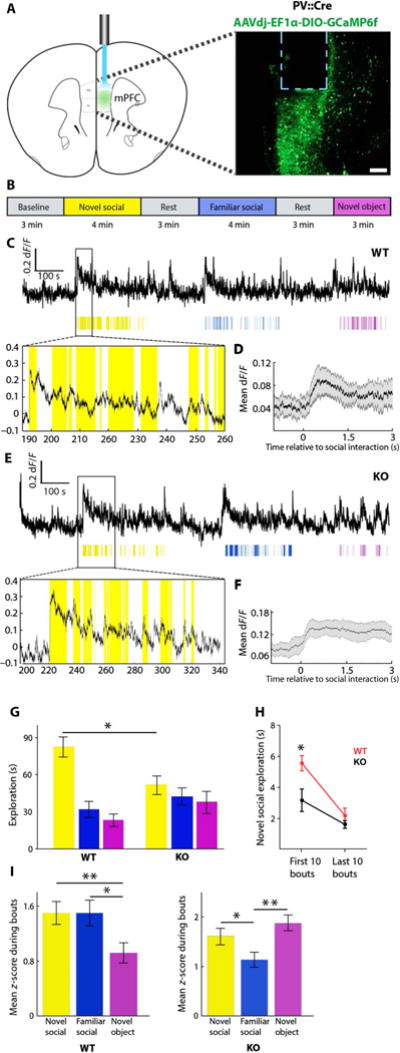
(A) Left: Schematic showing viral targeting of mPFC PV neurons for the purposes of fiber photometry recordings. Right: Neurons expressing the fluorescent activity reporter GCaMP6f adjacent to the optical fiber. Scale bar, 150 mm. (B) Behavioral test during 20 min of continuous fiber photometry recording. Animals were presented with a sex- and age-matched new (non-cagemate) mouse (yellow), a familiar (cagemate) mouse (blue), and a novel object (pink) for 3 or 4 min, with rest periods in between. (C) Photometry traces from WT animals during the behavioral test. Behavioral bouts are represented as colored lines with thickness reflecting duration of the bout. The yellow panel below is a zoomed-in image of the corresponding trace. (D) Representative plot showing the average photometry trace aligned to the start of the social interaction for a WT animal. Black line represents mean dF/F (change in fluorescence divided by baseline fluorescence) of the first 10 bouts. Gray lines correspond to SE. (E) Photometry traces from CNTNAP2 KO animals during the behavioral test. Behavioral bouts are represented as colored lines with thickness reflecting their duration. The panel below is the zoomed-in image of the corresponding trace above. (F) Representative plot showing the average photometry trace aligned to the start of the interaction for a CNTNAP2 KO animal. Black line represents average mean dF/F of the first 10 bouts. Gray lines correspond to SEM. (G) Mean total exploration time during each condition [novel social interaction (yellow), Student’s t test, P = 0.01; WT, n = 6; KO, n = 12; familiar social interaction (blue), P = 0.55; WT, n = 6; KO, n = 8; novel object interaction (pink), P = 0.20; WT, n = 10; KO, n = 11] (table S13). (H) Mean novel social exploration duration during the initial and final 10 bouts of the social interaction behavior test (initial 10 bouts, Student’s t test, P = 0.03; final 10 bouts, P = 0.27; WT, n = 6; KO, n = 9) (table S14). (I) Mean z-scored dF/F during the first 10 bouts. WT mice (left, n = 5; novel social interaction versus novel object interaction, P = 0.003; novel social interaction versus familiar social interaction, P = 0.86; familiar social interaction versus novel object interaction, P = 0.04) and CNTNAP2 KO mice (right, n = 5; novel social interaction versus novel object interaction, P = 0.21; novel social interaction versus familiar social interaction, P = 0.0240; familiar social interaction versus novel object interaction, P = 0.0012) (movies S1 and S2; note social PV recruitment in both cases, but to a greater extent than novel-object PV recruitment only for WT genotype as shown in figure). Wilcoxon rank-sum test was used to compare combined bouts across all mice for each condition. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01.
In freely moving WT and CNTNAP2 KO animals, activity of PV neurons increased in response to all three types of interactions (Fig. 3, C and E). Upon introduction of a nonlittermate mouse to a freely moving WT or CNTNAP2 KO mouse, there was a consistent increase in PV neuron activity (Fig. 3, D and F). Parallel trends were observed with familiar social interactions and novel object interactions in both genotypes (fig. S5). As expected, the total novel social interaction time in CNTNAP2 KO mice was lower than in WT mice, whereas mean duration of the interaction time with a novel object or familiar litter-mates did not significantly differ (novel social interaction, P = 0.0110; WT, n = 6; KO, n = 10; familiar social interaction, P = 0.5506; WT, n = 6; KO, n = 7; novel object interaction, P = 0.1965; WT, n = 10; KO, n = 11) (Fig. 3G). The difference in social interaction times was apparent within the first 10 interaction bouts (initial 10 bouts, P = 0.0301; final 10 bouts, P = 0.2741; WT, n = 6; KO, n = 9) (Fig. 3H).
In WT mice, PV neuron activity was higher during novel (non-familiar) social interactions compared to novel object interactions; in CNTNAP2 KO mice, PV neuronal activity was similar under both conditions (WT, n = 5; novel social interaction versus novel object interaction, P = 0.0033; KO, n = 5; novel social interaction versus novel object interaction, P = 0.2059) (Fig. 3I). Moreover, when novel social interactions were compared to familiar social interactions, no significant difference in PV neuron activity was observed in WT mice (WT, n = 5; novel social interaction versus familiar social interaction, P = 0.8605) (Fig. 3I, left). However, PV neurons in CNTNAP2 KO mice showed increased activity upon introduction to a new mouse compared to a familiar mouse (KO, n = 5; novel social interaction versus familiar social interaction, P = 0.0240) (Fig. 3I, right). Neither WT nor CNTNAP2 KO PV neurons showed changes in activity associated with altered locomotion (WT, n = 5; prelocomotion versus postlocomotion, P = 0.9974; KO, n = 5; prelocomotion versus postlocomotion, P = 0.5202) (fig. S6). Together, these data demonstrate distinct underlying neural dynamics of mPFC PV neurons in CNTNAP2 KO mice compared to WT mice, concordant with a change in the E:I balance of mPFC PV neurons.
DISCUSSION
Demonstration of a causal link between targetable pathophysiological pathways and clinically relevant symptoms is critical for testing translational ideas, but this process has been particularly challenging for neuropsychiatric diseases such as autism. Several previous studies have pointed to structural or functional abnormalities in inhibitory neurons in genetic models of cognitive dysfunction; in some cases, behavioral improvements were achieved through systemic pharmacological modulation of GABA transmission (19, 26, 34), but GABA modulation has not yet been found to be clinically effective for treating the core social interaction/communication and stereotyped behavioral symptoms of autism (35).
Here, we report that acute and reversible reduction in cellular E:I balance in the mPFC region of adult mouse brain, either by increasing PV neuron excitability or reducing PYR neuron excitability, rescued social behavior deficits in the CNTNAP2 KO autism-like mouse model. We also observed, using fiber photometry in freely moving mice, differential mPFC PV neuron activity in CNTNAP2 KO mice compared to WT animals. The observation that the activity of WT (but not CNTNAP2 KO) PV neurons increased more during social interactions compared to novel-object interactions, together with previous observations that PV neuron activity enhances the information-carrying capacity of mPFC circuitry (14), may be consistent with a hypothesis that the complexity of social interactions poses unique informational challenges for mPFC circuitry. Novel social interactions may therefore normally require robust and responsive E:I balance modulation by PV neurons, which may fail to occur properly in autism-related conditions.
Fast-spiking PV neurons form synapses at and near the cell bodies of PYR neurons, providing (among other circuit roles) feedforward and feedback inhibition (36), gamma rhythm modulation (13, 14), gain control over sensory responses (37–39), and output control over sub-cortically projecting PYR neurons (40). Recent optogenetic elucidation of these diverse circuit roles has led to the suggestion of PV neuron involvement in multiple psychiatric disorders (41, 42), including not only autism but also schizophrenia and anxiety, and broad potential utility as a therapeutic target. Anxiety responses toward new social stimuli in CNTNAP2 KO mice could have taken place in our study, consistent with the ~16% comorbidity between autism and social anxiety disorder (43). CNTNAP2 KO mice have not been found to be more anxious than WT littermates in a nonsocial light/dark box study (23), but selective anxiety in response to social stimuli was not ruled out.
From a translational perspective, the ability to restore social behavior in CNTNAP2 KO animals through PYR neuron inhibition or PV neuron activation is notable because the PYR neuron intervention did not require a transgenic (for example, Cre driver) line. The only requirement for expression of the transgene (the microbial opsin SwiChR++ in this case) by PYR neurons was a viral vector and promoter fragment [calcium/calmodulin-dependent protein kinase IIa (CaMKIIα)], both known to be effective for PYR neuron targeting in primates. Targeting cell types in a well-defined area of the brain may thus represent a potentially new strategy, contrasting in specificity with systemic pharmacological strategies (35). Pharmacological interventions that enhance GABA receptor action directly affect inhibition at the neurotransmitter-receptor level, but GABA receptors are present on both excitatory and inhibitory neurons. In contrast, modulation of the cellular E:I balance, which affects the relative number of active excitatory or inhibitory neurons, may exert a more refined influence on cortical processing. Future studies are required to reveal the mechanistic interrelationships between these two E:I concepts (excitatory/inhibitory synaptic inputs onto an individual neuron versus excitable excitatory/inhibitory cells among a population of neurons) and how various interventions may alter neural circuit dynamics in autism and other disease states.
In-depth behavioral studies in nonhuman primates are needed to demonstrate that interventions such as the optogenetic tools used in this study would provide benefits for the wider range of social interactions encountered by patients with autism and to address any potential adverse effects on nonsocial behaviors. People with autism spectrum disorders have difficulties in forming relationships with other people, both with and without autism. Thus, future animal studies will be required where rodents interact with animals of diverse genotypes under a broad range of conditions. In addition, to examine any potential adverse effects of increased PV neuron activity or dampened PYR neuron activity, a broad range of nonsocial behavioral assays should be conducted in nonhuman primates.
Together, the results presented here support the notion that cortical E:I imbalance may contribute to core behavioral disturbances in autism. Future studies are required to explore whether E:I imbalance in other brain regions, such as sensory associative areas, striatum, and hippocampus (44), may be involved in the emergence of behavioral phenotypes in the CNTNAP2 KO mice and whether similar pathophysiological mechanisms occur in other genetic models of autism. These studies will be critical to form a unified mechanistic and translational understanding of the neurons and circuits that underlie autism spectrum disorders.
MATERIALS AND METHODS
Study design
A sample size of at least 16 mice (eight pairs) per group was used for all optogenetic experiments; 5 mice per group were used for fiber photometry studies. Animals were randomly assigned to treatment with optogenetic stimulation. Both behavioral and photometry data were analyzed entirely blinded to the condition, and we used consistent blinded and readout-specific criteria to exclude animals from analysis. First (for social or novel object), animals in which at least 10 bouts of social interaction could not be acquired (or exhibiting seizure, escape, or aggressive behaviors) were excluded (validated by follow-up analysis revealing that the appearance of such behaviors led to scores at least 3 SD away from the mean). Second, for photometry recordings, exclusions were made blinded to any knowledge of group or intervention, only on the basis of looking at the entire trace before generation of averaged plots aligned to behavioral epochs and eliminating those cases where there was a clear baseline shift/motion artifact or loss of signal (caused by patch cord twisting or loosening during the recording). To prevent experimenter bias, behavioral data were automatically analyzed where possible. Velocity of social approach was analyzed with TopScan software (CleverSys Inc.); movement velocity and duration of time spent in each zone were recorded and analyzed using a video tracking system (Biobserve). Configuration settings for automated scoring were kept the same for all subjects. Social and object interactions were analyzed manually; all analyses were performed by the same researcher blinded to genotype and condition. For social and object interaction, MATLAB scripts were written and used to reliably score, allowing the researcher to push a key when the interaction began, hold onto the key as the physical interaction continued, and release the key as soon as the physical interaction ended. All graphs and numerical values in the figures are presented as mean ± SEM. Each experiment was scored twice, and means were used for statistical analysis.
Animals
Mice lacking the CNTNAP2 gene were obtained from D. Geschwind (23). Mutant mice were bred with transgenic PV::Cre C57BL/6J mice. The three obtained genotypes (Fig. 1A) were housed together, with two to five same-sex littermates per cage. All mice were housed on a reverse 12-hour light/dark cycle and were given food and water ad libitum. All experimental protocols were approved by the Stanford University Institutional Animal Care and Use Committee (IACUC) following the National Institutes of Health guidelines for the care and use of laboratory animals.
Acute slice electrophysiology
Three hundred–micrometer acute coronal slices were obtained from virus-injected mice after intracardial perfusion with ice-cold, sucrose-containing artificial cerebrospinal fluid (ACSF) containing 85 mM NaCl, 75 mM sucrose, 2.5 mM KCl, 25 mM glucose, 1.25 mM NaH2PO4, 4 mM MgCl2, 0.5 mM CaCl2, and 24 mM NaHCO3. Slices were kept in sucrose-containing ACSF for 1 hour at 32° to 34°C and then transferred to room temperature to an oxygenated standard ACSF solution containing 123 mM NaCl, 3 mM KCl, 26 mM NaHCO3, 2 mM CaCl2, 1 mM MgCl2, 1.25 mM NaH2PO4, and 11 mM glucose. Electrophysiological recordings were performed at 32° to 34°C with synaptic transmission blockers D(−)-2-amino-5-phosphonovaleric acid (25 mM), 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline-2,3-dione (10 μM), and gabazine (10 μM) added to the ACSF. Neurons were visualized using an upright microscope (BX61WI, Olympus) with a 40× water-immersion objective and infrared differential interference contrast optics. Individual recordings were obtained from cells using fluorescent protein expression to assay opsin expression. A SPECTRA X light engine (Lumencor) was attached to the fluorescence port of the microscope and used both to detect fluorescence and to deliver light pulses for opsin activation. Light was filtered using a 475/28-nm filter for blue light and a 633/22-nm filter for red light. Power densities of the blue and red lights were 8 and 5 mW/mm2, respectively, measured with a power meter (Thorlabs). Recordings were conducted in a dark room to minimize opsin activation from ambient light. Whole-cell recordings were performed using patch-clamp pipettes pulled from borosilicate glass capillaries (Sutter Instrument) with a horizontal puller (P-2000, Sutter Instrument) and contained the following internal solution: 125 mM K-gluconate, 10 mM KCl, 10 mM Hepes, 4 mM Mg3ATP, 0.3 mM NaGTP, 10 mM phosphocreatine, and 1 mM EGTA. Values were corrected for a liquid junction potential of +11.5 mV. Recordings were made using a MultiClamp 700B amplifier (Molecular Devices), with signals filtered at 4 kHz and digitized at 10 kHz with a Digidata 1440A analog-digital interface (Molecular Devices). Data were recorded with pCLAMP 10.3 software (Molecular Devices) and analyzed with pCLAMP 10.3 and SigmaPlot (SPSS). The electrical current needed to induce spiking was obtained by injecting current in 25-pA steps and recorded as the lowest amplitude that induced action potentials. Peak photocurrent amplitude was measured within the beginning 3 s, and the steady-state photocurrent amplitude was measured within the last 3 s of a 1-min light pulse under voltage clamp. Electrical pulses for examining spike generation probability during SSFO activation were applied in 250-pA steps from 250 to 1000 pA. The electrical pulses were applied with a 5-ms duration (with a 20-Hz frequency). Spike probability for each cell, at each current step, was calculated as the percentage of action potentials generated following the blue light pulses combined with injected electrical pulses throughout the entire protocol length. Paired t tests were used for statistics.
Stereotaxic virus injection and cannula implantation
All experimental and surgical protocols were approved by the Stanford University’s IACUC. For all surgeries, stainless steel head plates and ferrules were fixed to the skull using Metabond (Parkell). Mice were anesthetized with 2.0% isoflurane and leveled using bregma and lambda skull landmarks in a stereotaxic apparatus (Kopf Instruments). To achieve cell type–specific opsin expression in the transgenic PV-2a-Cre line, we cloned the opsin gene (SSFO) into a loxP/lox2722-flanked (double-floxed), inverted open reading frame plasmid under control of the elongation factor 1α (EF1α) promoter (pAAV-EF1α-DIO-SSFO-eYFP). The virus was produced by the University of North Carolina Chapel Hill Vector Core (map: https://web.stanford.edu/group/dlab/optogenetics/sequence_info.html). For cell-specific expression in PYR neurons, we cloned SwiChR++ under CaMKIIα promoter (pAAV-CaMKIIα-SwiChR++-eYFP). mPFC (from bregma; 1.8 mm anterior, 0.35 mm lateral, and −2.4 mm ventral) was targeted using a 10-μl NanoFil Hamilton syringe and 33-g beveled needle (World Precision Instruments). One thousand nanoliters of the virus suspension was infused at a rate of 150 nl/min into both hemispheres. After infusion, the needle was kept at the injection site for 4 min and then slowly withdrawn. For mPFC PV neuron stimulation, mice were implanted with a 200-μm-diameter 0.22–numerical aperture (NA) dual fiber-optic cannula (Doric Lenses) directly above the mPFC at 1.8 mm [anterior-posterior (AP)], 0.35 mm [medial-lateral (ML)], and −2.0 mm [dorsal-ventral(DV)]. For fiber photometry recordings, PV neurons were targeted with AAVdj-EF1α-DIO-GCaMP6f that was produced at the Stanford Gene Vector and Virus Core (Stanford University). mPFC injections were performed as described above, but unilaterally. A low-fluorescence 400-μm-diameter 0.48-NA mono fiber-optic cannula (Doric Lenses) was implanted at 1.8 mm (AP), 0.35 mm (ML), and −2.4 mm (DV). Cannulas were secured to the skull using a base layer of adhesive dental cement (C&B-Metabond, Parkell), followed by a second layer of cranioplastic cement (Ortho-Jet, Lang). Behavioral experiments were conducted 2 weeks later for optogenetic stimulation and 3 weeks later for fiber photometry recordings to allow for sufficient viral expression and postsurgery recovery.
Immunohistochemistry and confocal microscopy
For all cohorts, animals were anesthetized and transcardially perfused with ice-cold 1× phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. Brains were postfixed for 48 hours in 4% PFA. Brains were sectioned into 100-μm-thick slices using a vibratome and stored in cryoprotectant at 4°C in the dark until antibody staining. Slices were washed three times for 10 min in PBS and mounted on microscope slides with PVA-DABCO. Images were acquired on a Leica TCS SP5 scanning laser microscope.
Behavioral tests
All tests were performed during the dark phase, and animals were allowed to acclimate to the behavior room for at least 2 days before the beginning of behavioral testing. Both females and males were included in behavioral tests. For optogenetic stimulation, a fiber-optic patch cord (Doric Lenses) was connected to the chronically implanted optical fiber on one side and a 473-nm solid-state laser diode on the other side. A Master-8 pulse stimulator (A.M.P.I.) controlled the laser output. Stimulation trials were conducted with the addition of a 2-s pulse of 8-mW light from the tip of the fiber; the fiber was then decoupled before experimentation beginning with a 1-min acclimation period (22). For fiber photometry recordings, a 3-m-long fiber-optic patch cord was connected to the chronically implanted optical fiber and suspended above the behavioral cage to allow animals to interact and move freely during the experiment. To habituate and minimize anxiety created through the environment, we placed each experimental mouse individually in a novel cage for 10 min the day before the test.
Reciprocal social interaction
Mice were placed in a new cage simultaneously with a novel mouse matched in age, genotype, and sex for 10 min (23). Both mice in the pair were treated under the same experimental condition, either with stimulation or without stimulation. The order of experimental condition was counterbalanced between pairs, and conditions were separated by 24 hours. The time engaged in social interaction (nose-to-nose sniffing, nose-to-anus sniffing, and following or crawling on/under each other) for the pair (combining the behavior of both animals) was manually scored by blind observation to genotype and condition. MATLAB scripts were written and used to reliably score social interactions, allowing the researcher to push a key when the interaction began, hold onto the key as the physical interaction continued, and release as soon as the physical interaction ended. Each experiment was scored twice and averaged to minimize human error, and the mean was used for statistical analysis. Velocity of social approach was analyzed with TopScan software (CleverSys Inc.). Mice that lost fiber-optic implants or manifested seizures upon stimulation were excluded from analysis.
Novel object interaction
The novel object test was performed in a novel, clean cage using the same general method with the social interaction assay. Instead of presenting a stranger mouse, either a plastic ball or wooden block (of equivalent size) was introduced to the animal’s home cage, and total time spent investigating the object over 3 min was scored manually. MATLAB scripts were used for more precise scoring, and the mean results from two independent scoring sessions were used for final analysis. The type of object was switched between experiments. We counterbalanced the condition and the object type between animals to prevent any potential inherent preferences for a given object influencing the results.
Open-field test
The open-field test was conducted on different cohorts of mice to measure the effect of optogenetic stimulation on anxiety-like responses and general locomotor ability. The mice were placed in the chamber (50 × 50 cm), and their movement was recorded and analyzed for 10 min using a video tracking system (Biobserve). The center zone of the open-field chamber was defined as the 23 × 23–cm central square area.
Fiber photometry recordings and analysis
As described previously (45, 46), we collected emitted fluorescence from targeted brain regions using a single optical fiber while delivering excitation light. We used multiple excitation wavelengths (488 and 405 nm) modulated at distinct carrier frequencies to allow for ratiometric measurements. Light collection, filtering, and demodulation were as previously described (45, 46). The ratiometric fluorescence signal was calculated for each continuous behavioral recording with custom-written MATLAB scripts. We fit a double exponential to a thresholded version of the fluorescence time series and subtracted the best fit from the unthresholded signal to account for slow bleaching artifacts (29). We then calculated a single baseline fluorescence value as the median of the entire trace (which robustly estimated the baseline fluorescence). We calculated the normalized change in fluorescence (dF/F) by subtracting the baseline fluorescence from the fiber fluorescence at each time point and dividing that value by the baseline fluorescence. Within-animal analyses were done by calculating z-scored dF/F and finding the mean dF/F during each interaction bout (that is, mean dF/F from the start of social interaction to the end). Next, we combined the first 10 bouts across all mice to measure mean z-scored dF/F for each genotype. To measure locomotion-induced activity changes, we calculated the mean dF/F during 1 s before locomotion initiation and during first 1 s of locomotion and ran a paired t test on the average pre- and postlocomotion values across animals.
Fiber photometry microscope setup, surgeries, and analysis
Frame-projected Independent-fiber Photometry (FIP) microscopy was as previously described (29). Ca2+ signals were acquired through a sCMOS (scientific complementary metal-oxide semiconductor) camera that is connected to a series of dichroic mirrors integrated into the microscope, which couples in various wavelengths of excitation light sources. We unilaterally injected EF1α-DIO-SSFO-eYFP (300 nl) and AAVdj-CKIIα-GCaMP6f (700 nl) into mPFC to specifically target PV interneurons and excitatory PYR neurons, respectively. With this design, SSFO was expressed in local PV interneurons, whereas GCaMP6f traveled along the PYR neuron processes and was expressed in striatal axon terminals. A 200-μm-diameter 0.22-NA mono fiber-optic cannula was implanted above mPFC (AP, 1.8 mm; ML, 0.35 mm; DV, −2.0 mm) for optogenetic stimulation, and a low-fluorescence 400-μm-diameter 0.48-NA mono fiber-optic cannula (Doric Lenses) was implanted into dorsal striatum (AP, 0.7 mm; ML, 1.00 mm; DV, −3.3 mm) for FIP recordings. We conducted FIP recordings 6 weeks after the surgeries to allow for sufficient expression in the axon terminals. On the day of recording, one patch cord was attached to the implanted mPFC fiber on one side and to a 470-nm laser source on the other side. A second patch cord connected the dorsal striatum fiber to the FIP microscope. After a 2-min baseline recording, a 2-s, 8-mW, 470-nm pulse was presented while the animals were freely moving. Recording continued for 3 min after the pulse. Analysis was performed with custom MATLAB scripts. Regions of interest were first manually drawn around the fiber on the basis of a mean image of the movie. The average fluorescence intensity was calculated for each fiber. dF/F was calculated as described (29). We found individual Ca2+ events by thresholding fluorescence changes and calculated event rate for each 20-s bin before and after the stimulation.
Statistical analysis
For each figure, a statistical test matching the structure of the experiment and the structure of the data was used. Two-way ANOVA, followed by Dunnett’s pairwise comparisons to controls, was used to compare pairs of experimental conditions and genotypes with each other. Nonparametric Wilcoxon rank-sum testing was used when normal variance was not assumed. Variances within each group of data are shown as SEM throughout. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data presentation and statistical analysis were performed with GraphPad Prism software.
Supplementary Material
Fig. S1. Bilateral expression of SwiChR++ in mPFC.
Fig. S2. CNTNAP2 KO and WT mice interact with objects similarly both in the presence and absence of SSFO recruitment.
Fig. S3. Both SSFO and SwiChR++ recruitments rescue CNTNAP2 KO hyperactivity in the open-field test.
Fig. S4. Modulation of outgoing corticostriatal activity traffic by SSFO recruitment in mPFC PV neurons.
Fig. S5. Temporal details of interactions with familiar mouse and novel object.
Fig. S6. Modulation of PV neuron activity with locomotion.
Table S1. Raw data of Fig. 1E.
Table S2. Raw data of Fig. 1H.
Table S3. Raw data of Fig. 1I.
Table S4. Raw data of Fig. 1K.
Table S5. Raw data of Fig. 2B.
Table S6. Raw data of Fig. 2C.
Table S7. Raw data of Fig. 2D.
Table S8. Raw data of Fig. 2E.
Table S9. Raw data of Fig. 2G.
Table S10. Raw data of Fig. 2H.
Table S11. Raw data of Fig. 2I.
Table S12. Raw data of Fig. 2J.
Table S13. Raw data of Fig. 3G.
Table S14. Raw data of Fig. 3H.
Movie S1. CNTNAP2 KO mPFC PV neuron activity during social behavior.
Movie S2. WT mPFC PV neuron activity during social behavior.
Acknowledgments
We thank D. Geschwind for sharing CNTNAP2 KO mice, O. Penagarikano for help with behavioral experimental design, E. Sylwestrak for helpful discussions, M. Lo for logistical support, and the Deisseroth Lab for their support.
Funding: K.D. was funded by NIH, Defense Advanced Research Projects Agency, the Simons Foundation, the Wiegers Foundation, and the Gatsby Foundation. A.S. was funded by a Howard Hughes Medical Institute International Student Fellowship. C.K.K. was supported by an NIH National Research Service Award F31 predoctoral fellowship (NIDA F31DA041795). M.I. was supported by a Japan Society for the Promotion of Science Overseas Research Fellowship. I.K. was funded by the NSF.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/9/401/eaah6733/DC1
Author contributions: A.S. and K.D. designed the experiments and wrote the paper. A.S. and M.I. carried out data collection and analysis. C.K.K. set up the fiber photometry rig and carried out data analysis. S.Y.L. conducted the electrophysiology experiments and analysis. A.S.O.H. performed animal breeding experiments and surgeries. I.K. generated supplementary movies. C.R. carried out opsin design and virus production. L.E.F. and M.W. participated in study design. T.J.D. participated in fiber photometry rig setup. K.D. supervised all aspects of the work.
Competing interests: K.D. is a founder, consultant, and scientific advisory board member of Circuit Therapeutics. The other authors declare that they have no competing interests.
Data and materials availability: The sequences of opsins can be found at http://web.stanford.edu/group/dlab/optogenetics/sequence_info.html. All reagents and methods are freely available for research purposes from the Deisseroth Lab at www.optogenetics.org.
REFERENCES AND NOTES
- 1.Chen JA, Peñagarikano O, Belgard TG, Swarup V, Geschwind DH. The emerging picture of autism spectrum disorder: Genetics and pathology. Annu Rev Pathol. 2015;10:111–144. doi: 10.1146/annurev-pathol-012414-040405. [DOI] [PubMed] [Google Scholar]
- 2.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. American Psychiatric Association Publishing; 2013. [Google Scholar]
- 3.Deisseroth K, Etkin A, Malenka RC. Optogenetics and the circuit dynamics of psychiatric disease. JAMA. 2015;313:2019–2020. doi: 10.1001/jama.2015.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubenstein JLR, Merzenich MM. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology. 2011;58:428–432. doi: 10.1212/wnl.58.3.428. [DOI] [PubMed] [Google Scholar]
- 6.Casanova MF, Buxhoeveden DP, Brown C. Clinical and macroscopic correlates of minicolumnar pathology in autism. J Child Neurol. 2002;17:692–695. doi: 10.1177/088307380201700908. [DOI] [PubMed] [Google Scholar]
- 7.Casanova MF, El-Baz AS, Kamat SS, Dombroski BA, Khalifa F, Elnakib A, Soliman A, Allison-McNutt A, Switala AE. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol Commun. 2013;1:67. doi: 10.1186/2051-5960-1-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashemi E, Ariza J, Rogers H, Noctor SC, Martínez-Cerdeño V. The number of parvalbumin-expressing interneurons is decreased in the medial prefrontal cortex in autism. Cereb Cortex. 2017;27:1931–1943. doi: 10.1093/cercor/bhw021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fatemi SH, Reutiman TJ, Folsom TD, Rooney RJ, Patel DH, Thuras PD. mRNA and protein levels for GABAAα4, α5, β1 and GABABR1 receptors are altered in brains from subjects with autism. J Autism Dev Disord. 2010;40:743–750. doi: 10.1007/s10803-009-0924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harada M, Taki MM, Nose A, Kubo H, Mori K, Nishitani H, Matsuda T. Non-invasive evaluation of the GABAergic/glutamatergic system in autistic patients observed by MEGA-editing proton MR spectroscopy using a clinical 3 Tesla instrument. J Autism Dev Disord. 2011;41:447–454. doi: 10.1007/s10803-010-1065-0. [DOI] [PubMed] [Google Scholar]
- 11.Dichter GS, Felder JN, Bodfish JW. Autism is characterized by dorsal anterior cingulate hyperactivation during social target detection. Soc Cogn Affect Neurosci. 2009;4:215–226. doi: 10.1093/scan/nsp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belmonte MK, Gomot M, Baron-Cohen S. Visual attention in autism families: “Unaffected” sibs share atypical frontal activation. J Child Psychol Psychiatry. 2010;51:259–276. doi: 10.1111/j.1469-7610.2009.02153.x. [DOI] [PubMed] [Google Scholar]
- 13.Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milne E, Scope A, Pascalis O, Buckley D, Makeig S. Independent component analysis reveals atypical electroencephalographic activity during visual perception in individuals with autism. Biol Psychiatry. 2009;65:22–30. doi: 10.1016/j.biopsych.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 16.Snijders TM, Milivojevic B, Kemner C. Atypical excitation–inhibition balance in autism captured by the gamma response to contextual modulation. NeuroImage Clin. 2013;3:65–72. doi: 10.1016/j.nicl.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Diessen E, Senders J, Jansen FE, Boersma M, Bruining H. Increased power of resting-state gamma oscillations in autism spectrum disorder detected by routine electroencephalography. Eur Arch Psychiatry Clin Neurosci. 2015;265:537–540. doi: 10.1007/s00406-014-0527-3. [DOI] [PubMed] [Google Scholar]
- 18.Dickinson A, Bruyns-Haylett M, Jones M, Milne E. Increased peak gamma frequency in individuals with higher levels of autistic traits. Eur J Neurosci. 2015;41:1095–1101. doi: 10.1111/ejn.12881. [DOI] [PubMed] [Google Scholar]
- 19.Gandal MJ, Sisti J, Klook K, Ortinski PI, Leitman V, Liang Y, Thieu T, Anderson R, Pierce RC, Jonak G, Gur RE, Carlson G, Siegel SJ. GABAB-mediated rescue of altered excitatory-inhibitory balance, gamma synchrony and behavioral deficits following constitutive NMDAR-hypofunction. Transl Psychiatry. 2012;2:e142. doi: 10.1038/tp.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar JC, Chen YH, Lanza M, Howell B, Chow VY, Heiken K, Liu S, Wootton C, Hunter MA, Huang M, Miller GA, Cañive JM. Cortical thickness as a contributor to abnormal oscillations in schizophrenia? NeuroImage Clin. 2014;4:122–129. doi: 10.1016/j.nicl.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berndt A, Lee SY, Wietek J, Ramakrishnan C, Steinberg EE, Rashid AJ, Kim H, Park S, Santoro A, Frankland PW, Iyer SM, Pak S, Ährlund-Richter S, Delp SL, Malenka RC, Josselyn SA, Carlén M, Hegemann P, Deisseroth K. Structural foundations of optogenetics: Determinants of channelrhodopsin ion selectivity. Proc Natl Acad Sci USA. 2016;113:822–829. doi: 10.1073/pnas.1523341113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JLR, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489:385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee DO, Ousley OY. Attention-deficit hyperactivity disorder symptoms in a clinic sample of children and adolescents with pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2006;16:737–746. doi: 10.1089/cap.2006.16.737. [DOI] [PubMed] [Google Scholar]
- 28.Schulz K, Sydekum E, Krueppel R, Engelbrecht CJ, Schlegel F, Schröter A, Rudin M, Helmchen F. Simultaneous BOLD fMRI and fiber-optic calcium recording in rat neocortex. Nat Methods. 2012;9:597–602. doi: 10.1038/nmeth.2013. [DOI] [PubMed] [Google Scholar]
- 29.Kim CK, Yang SJ, Pichamoorthy N, Young NP, Kauvar I, Jennings JH, Lerner TN, Berndt A, Lee SY, Ramakrishnan C, Davidson TJ, Inoue M, Bito H, Deisseroth K. Simultaneous fast measurement of circuit dynamics at multiple sites across the mammalian brain. Nat Methods. 2016;13:325–328. doi: 10.1038/nmeth.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Caldéron NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SSH, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, Tye KM, Anikeeva P, Malenka RC, Deisseroth K. Natural neural projection dynamics underlying social behavior. Cell. 2014;157:1535–1551. doi: 10.1016/j.cell.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, Khalilov I, Tsintsadze V, Brouchoud C, Chazal G, Lemonnier E, Lozovaya N, Burnashev N, Ben-Ari Y. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343:675–679. doi: 10.1126/science.1247190. [DOI] [PubMed] [Google Scholar]
- 35.Brondino N, Fusar-Poli L, Panisi C, Damiani S, Barale F, Politi P. Pharmacological modulation of GABA function in autism spectrum disorders: A systematic review of human studies. J Autism Dev Disord. 2016;46:825–839. doi: 10.1007/s10803-015-2619-y. [DOI] [PubMed] [Google Scholar]
- 36.Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- 37.Wilson NR, Runyan CA, Wang FL, Sur M. Division and subtraction by distinct cortical inhibitory networks in vivo. Nature. 2012;488:343–348. doi: 10.1038/nature11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. 2012;73:159–170. doi: 10.1016/j.neuron.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee SH, Kwan AC, Zhang S, Phoumthipphavong V, Flannery JG, Masmanidis SC, Taniguchi H, Huang ZJ, Zhang F, Boyden ES, Deisseroth K, Dan Y. Activation of specific interneurons improves V1 feature selectivity and visual perception. Nature. 2012;488:379–383. doi: 10.1038/nature11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karnani MM, Jackson J, Ayzenshtat I, Tucciarone J, Manoocheri K, Snider WG, Yuste R. Cooperative subnetworks of molecularly similar interneurons in mouse neocortex. Neuron. 2016;90:86–100. doi: 10.1016/j.neuron.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marín O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13:107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- 42.Hu H, Gan J, Jonas P. Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: From cellular design to microcircuit function. Science. 2014;345:1255263. doi: 10.1126/science.1255263. [DOI] [PubMed] [Google Scholar]
- 43.van Steensel FJA, Bögels SM, Perrin S. Anxiety disorders in children and adolescents with autistic spectrum disorders: A meta-analysis. Clin Child Fam Psychol Rev. 2011;14:302–317. doi: 10.1007/s10567-011-0097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jurgensen S, Castillo PE. Selective dysregulation of hippocampal inhibition in the mouse lacking autism candidate gene CNTNAP2. J Neurosci. 2015;35:14681–14687. doi: 10.1523/JNEUROSCI.1666-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lerner TN, Shilyansky C, Davidson TJ, Evans KE, Beier KT, Zalocusky KA, Crow AK, Malenka RC, Luo L, Tomer R, Deisseroth K. Intact-brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell. 2015;162:635–647. doi: 10.1016/j.cell.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zalocusky KA, Ramakrishnan C, Lerner TN, Davidson TJ, Knutson B, Deisseroth K. Nucleus accumbens D2R cells signal prior outcomes and control risky decision-making. Nature. 2016;531:642–646. doi: 10.1038/nature17400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Bilateral expression of SwiChR++ in mPFC.
Fig. S2. CNTNAP2 KO and WT mice interact with objects similarly both in the presence and absence of SSFO recruitment.
Fig. S3. Both SSFO and SwiChR++ recruitments rescue CNTNAP2 KO hyperactivity in the open-field test.
Fig. S4. Modulation of outgoing corticostriatal activity traffic by SSFO recruitment in mPFC PV neurons.
Fig. S5. Temporal details of interactions with familiar mouse and novel object.
Fig. S6. Modulation of PV neuron activity with locomotion.
Table S1. Raw data of Fig. 1E.
Table S2. Raw data of Fig. 1H.
Table S3. Raw data of Fig. 1I.
Table S4. Raw data of Fig. 1K.
Table S5. Raw data of Fig. 2B.
Table S6. Raw data of Fig. 2C.
Table S7. Raw data of Fig. 2D.
Table S8. Raw data of Fig. 2E.
Table S9. Raw data of Fig. 2G.
Table S10. Raw data of Fig. 2H.
Table S11. Raw data of Fig. 2I.
Table S12. Raw data of Fig. 2J.
Table S13. Raw data of Fig. 3G.
Table S14. Raw data of Fig. 3H.
Movie S1. CNTNAP2 KO mPFC PV neuron activity during social behavior.
Movie S2. WT mPFC PV neuron activity during social behavior.