

Abstract
Current therapies for autoimmune diseases focus on treating the symptoms rather than the underlying disease cause. A major setback in improving current therapeutics for autoimmunity is the lack of antigen specificity. Successful antigen-specific immunotherapy (ASIT) would allow for improved treatment of autoimmune diseases. In this work, dexamethasone was co-delivered with autoantigen (PLP) in vivo to create effective ASIT for the treatment of experimental autoimmune encephalomyelitis (EAE). Using an emulsion of incomplete Freund’s adjuvant (IFA) as a co-delivery vehicle, it was discovered that the controlled release of autoantigen was important for the suppression of clinical disease symptoms. Analysis of the immune response via cytokines revealed that dexamethasone was important for shifting the immune response away from inflammation. Co-delivery of both autoantigen and dexamethasone increased B-cell populations and antibody production, signifying an increased humoral immune response. Overall, this data indicated that the co-delivery of PLP and dexamethasone with a water-in-oil emulsion is effective in treating a murine autoimmune model.
Keywords: experimental autoimmune encephalomyelitis (EAE), incomplete Freund’s adjuvant (IFA), dexamethasone, proteolipid protein (PLP), co-delivery, antigen-specific immunotherapy
Graphical abstract
INTRODUCTION
The current therapies for autoimmune diseases, including multiple sclerosis (MS), are often unsuccessful in stopping or reversing disease progression and may lead to non-specific immunosuppression [1, 2]. Effective antigen-specific immunotherapy (ASIT) has the potential to suppress the immune response in regard to a specific autoantigen, and therefore would not hinder the ability of the patient’s immune system to fight off foreign pathogens. Unfortunately, attempts to create ASIT for autoimmunity using only autoantigen have largely been unsuccessful [3-5]. In recent years, the idea of combining autoantigen and immunomodulators has emerged as a way to improve upon ASIT for autoimmunity [6]. Promising research has indicated that co-delivery of a small molecule immunomodulator with an autoantigen can lead to the creation of antigen-specific treatment of autoimmune disease [7-11].
Co-delivery of autoantigen and immunomodulator has been successful in animal models of autoimmunity [7-14]. These promising results may be attributed to the spatial and temporal combination of the components in order to elicit the appropriate antigen-specific immune response. In fact, traditional vaccines have utilized co-delivery of antigen and immunomodulator (i.e., adjuvant) to successfully induce a protective antigen-specific response [15]. One hypothesized mechanism for the success of co-delivery is the ability to mimic the natural two-signal paradigm of antigen-specific immunity, wherein concurrent antigen and secondary signal (co-stimulatory or co-inhibitory) are needed to elicit an antigen-specific immune response [15, 16]. In this regard, several studies have explored co-delivery of autoantigen and immunomodulator that allow for the controlled release of one or both components, increasing the exposure of immune cells to antigen and/or immunomodulator, which is known to contribute to efficacy of antigen-specific responses in autoimmune models [7-11, 15].
The potential to mimic vaccine mechanisms, not only through antigen and adjuvant co-delivery but also through induction of humoral and/or T-helper type-2 (Th2) immunity, has shown efficacy in the treatment of T-helper type-1 (Th1) related autoimmune diseases. Both MS and the murine model of the disease, experimental autoimmune encephalomyelitis (EAE), are believed to be primarily Th1 and T-helper type-17 (Th17)-mediated [17, 18]. Utilizing an adjuvant initially developed for protective vaccination against foreign pathogens, incomplete Freund’s adjuvant (IFA), studies have shown a shift from an autoantigen-specific Th1/Th17 towards a Th2/humoral response resulting in the suppression of autoimmune disease in animals [19, 20]. IFA is composed of 85% mineral oil (paraffin oil) and 15% emulsifier (mannide monooleate), and forms a water-in-oil emulsion when combined with an aqueous phase [21] A 1:1 emulsion of IFA with an aqueous solution containing antigen has been shown to enhance the antigen-specific immune response in animals [16, 21-23]. Additionally, IFA emulsions both alone and in combination with autoantigen have demonstrated the capacity to mitigate symptoms of autoimmune disease [19, 20, 24-29]. A recent clinical trial has also shown the ability of IFA to deliver autoantigen for the treatment of type 1 diabetes in humans [30].
Recently our lab has found that co-delivery of dexamethasone (DEX) and a disease-specific autoantigen was able to suppress inflammation in EAE splenocytes ex vivo [31]. Although traditionally thought of as a general immunosuppressant, DEX has been shown to be an effective immunomodulator with the ability to decrease the Th1 response, enhance the Th2 response, and even skew the immune system towards a regulatory response [32-36]. Our lab has verified these results in a screen of eleven immunomodulators in antigen-specific splenocytes obtained from EAE mice, and determined that DEX was capable of suppressing pro-inflammatory Th1-related cytokines, increasing regulatory cytokines, and decreasing the overall T-cell response [31]. Other groups have tested the co-administration or co-delivery of DEX and autoantigen in animal models of autoimmunity with successful outcomes [9, 37].
In this manuscript, we investigated the co-delivery of DEX and proteolipid protein epitope (PLP139-151), the antigen used to induce EAE, via an IFA emulsion. Additionally, all individual components of the co-delivery system and their possible combinations were investigated for the treatment of EAE. To our knowledge, this is the first study demonstrating the use of IFA for co-delivery of an immunomodulator and autoantigen for autoimmune therapy. Our approach seeks to expand upon current knowledge of co-delivery in ASIT immunotherapy, the unique immune effects of IFA, and the immunomodulatory mechanisms of DEX in order to create an antigen-specific therapy for autoimmunity.
MATERIALS AND METHODS
Materials
The peptide antigen, PLP139-151 (PLP), was obtained from PolyPeptide Laboratories (Torrance, CA). For EAE induction incomplete Freund’s adjuvant (IFA) and killed Mycobacterium tuberculosis strain H37RA were purchased from Difco (Sparks, MD), and pertussis toxin was purchased from List Biological Laboratories (Campbell, CA). Dexamethasone (DEX) was purchased from Sigma-Aldrich (St. Louis, MO). For flow cytometry studies, Alexa Fluor® 488-conjugated anti-mouse CD3, Alexa Fluor® 647-conjugated anti-mouse CD19, and Pacific Blue™-conjugated anti-mouse CD11c, and their respective isotype controls were purchased from Biolegend (San Diego, CA). All other chemicals and reagents were analytical grade and used as received.
Preparation of IFA emulsion
All IFA emulsions were created with a 1:1 volumetric ratio of IFA to phosphate buffered saline (PBS), unless otherwise noted. Before emulsification of peptide-containing emulsions, PLP was solubilized in PBS at 2x the final concentration of 2000 μM. For formulations including DEX, DEX was first dissolved in DMSO and added to the PBS phase before emulsification such that the final amount of DMSO was less than 0.5% (the maximum concentration recommended by ICH Guideline, Impurities: Guideline for Residual Solvents, Q3C (R5), 2011) and the final concentration of DEX was 20 μg/mL. The IFA and PBS phases were then emulsified using a 20 gauge micro-emulsifying needle (stainless steel, 20G X 2-7/8” with reinforcing bar, Cadence Science, Inc.) using two 6 cc plastic syringes, and the emulsion was passed through the needle 15 times. For in vivo treatments, IFA emulsions were vortexed (speed 10, Mini Vortexer, Fisher Scientific) for 3 minutes immediately prior to injection.
Characterization of IFA Emulsions
To characterize properties of the IFA emulsions, Nile Blue A (Sigma Aldrich) was dissolved in ethanol and introduced to IFA oil (10 μM final dye concentration, less than 0.5% residual ethanol). The dye-containing oil was sonicated for five minutes to disintegrate Nile Blue particles, then emulsified with PBS and vortexed as described above. 5 μL of the IFA emulsion was inserted to an uncoated μ-Slide Chemotaxis (ibidi), and sample insertion ports were blocked to immobilize the emulsion. The slide was imaged using an Olympus IX-81 with ZDC (Olympus Scientific Solutions Americas, Waltham, MA) scope configured with a Prior H117 with Proscan III stage (Prior Scientific Inc, Rockland, MA), Hamamatsu ORCA-R2 CCD camera (Hamamatsu Corporation, Bridgewater, NJ), Sutter Lambda 10-3 filter wheel and Lambda LS Xenon light source (Sutter Instrument, Novato, CA), and Olympus Objectives – 10X 0.3NA, 20X 0.5NA, 40X 0.6NA, 60X 0.7NA (Olympus Scientific Solutions Americas, Waltham, MA). Nile Blue A fluorescence (excitation 631 nm/emission 660 nm) was captured with filters 560 nm ± 25, 607 nm ± 36, 650 nm ± 13, and 684 nm ± 24. Images were acquired and analyzed using SlideBook 6.0 (Intelligent Imaging Innovations, Denver, CO). Stability was also investigated for IFA emulsions by monitoring the formulations hourly for phase separation and photographically documenting the emulsion integrity.
Characterization of PLP and DEX Release
For the release characterization, 4 mL of the IFA emulsion was placed into regenerated cellulose dialysis bags with 6,000-8,000 MWCO (30 μm wall thickness, Fisherbrand Dialysis Tubing) which was then placed into 100 mL of PBS in a glass beaker covered using parafilm. Alternatively, PLP and DEX were also placed in PBS at the same concentration as in the final emulsion, and release was characterized via the same procedure. All release studies were kept at 37°C on an incubator shaker (79 rpm, Excella E24 Incubator Shaker, New Brunswick Scientific), and 1 mL samples were taken from the outermost PBS phase at specific time points; with 1 mL of PBS added for media replacement at each time point. Characterization of the PLP and DEX concentrations released from the dialysis bags were performed using gradient reverse-phase HPLC (Waters 2796 Bioseparations Module, Waters Corp) on a C4 analytical column (Waters XBridge Protein BEH column, 300 Å, 3.5 μm, 4.6 mm × 150 mm, 10-500 K). Samples were eluted using mobile phases A (100% water with 0.1% trifluoroacetic acid (TFA)) and B (100% acetonitrile with 0.1% TFA) with a linear gradient of 100% A to 20% A over 25 minutes at a constant flow of 1 mL/min. PLP was detected at 220 nm and DEX was detected at 240 nm with a dual wavelength absorbance detector (Waters 2487 Dual λ Absorbance Detector, Waters Corp). Data was collected and processed using Empower 3 Software (Waters Corp).
Induction of EAE and Therapeutic Study
SJL/J female mice, 4 – 5 weeks old, were purchased from Envigo Laboratories and housed under specified, pathogen-free conditions at The University of Kansas. All protocols involving live mice were approved by The University of Kansas Institutional Animal Care and Use Committee. Mice were immunized subcutaneously with 200 μg of PLP in a 0.2 mL emulsion of Complete Freund’s Adjuvant (CFA), composed of equal volumes of PBS and IFA also containing killed Mycobacterium tuberculosis strain H37RA at a final concentration of 4 mg/mL. The CFA containing PLP was administered to regions above the shoulders and the flanks (total of four sites; 50 μL at each injection site). In addition, 100 μL of pertussis toxin (200 ng) was injected intraperitoneally on the day of immunization (day 0) and 2 days post-immunization. Each treatment group contained six mice, except for the group with PLP treatment, which only contained five mice due to an unexpected death before disease induction. Mice received 100 μL subcutaneous injections of treatment at the nape of the neck on days 4, 7, and 10 of the study. All DEX-containing treatment groups were given at 20 μg DEX per injection and all PLP-containing treatments groups were given on a 200 nmol PLP per injection basis. All IFA emulsions were administered within 30 minutes of fabrication. Disease progression was evaluated using clinical scoring as follows: 0, no clinical signs of the disease; 1, tail weakness or limp tail; 2, paraparesis (weakness or incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with forelimb weakness or paralysis; and 5, moribund. Body weight was also measured daily.
Splenocyte Isolation and ex vivo treatment
Mouse spleens were resected from EAE mice on day 25 following disease-induction and cultured as previously described [13]. Briefly, the spleens were first passed through a wire mesh using the rubber end of a sterile 1 mL syringe plunger, and collected in 5 mL of RPMI 1640 media. The crude cellular extract was then centrifuged and the resulting cell pellet was resuspended in 3.5 mL of 1x Gey’s lysis solution and placed on ice for 3.5 minutes to lyse splenic red blood cells. The lysis reaction was stopped by the addition of 10 mL RPMI 1640 media containing 10% FBS and centrifuged at 1,100 × g for 5 minutes. The remaining cell pellet was resuspended in fresh media (RPMI 1640 media containing 10% FBS and 1% Penicillin-Streptomycin) and seeded in 96-well cell culture plates at a cell density of 1×106 cells/well in a final volume of 200 μL. The cells were immediately rechallenged with and without 25 μM PLP antigen. Stimulated cell cultures were incubated for 120 hours at 37°C in a CO2 (5%) incubator.
Measurement of Cytokines
Supernatants of cell cultures were collected after a120 hour incubation period following the day 25 spleen harvest. Secreted cytokines; GM-CSF, TNF-α, IL-2, IFN-γ, IL-10, IL-17, IL-15, and IL-23 were detected using a U-Plex assay kit according to manufacturer instructions (Meso Scale Discovery). Briefly, each U-Plex was coated with 50 μL of the multiplex coating solution containing linker and biotinylated capture antibody combinations for each cytokine and incubated on a shaker at 700 rpm for 1 hour at room temperature. Following washing each well 3 times with 150 μL PBS containing 0.05% Tween 20, 25 μL of diluent and 25 μL of sample was added to each well and incubated again for 1 hour on a shaker at room temperature. Detection antibody was added at 50 μL/well and incubated for 1 hour. Finally, each assay plate was read using the QuickPlex multiplex plate reader (Meso Scale Discovery).
Measurement of Cellular Metabolism
Cell metabolism was determined by a resazurin (7-hydroxy- 3H-phenoxazin-3-one 10-oxide) assay. Briefly, resazurin (75 μmol/L final) was added to splenocyte cultures and incubated for 3 hours. Metabolic reductive capacity was determined by a change in fluorescence (excitation 560 nm/emission 590 nm). Background fluorescence was determined in RPMI media and was subtracted from each experimental read. All fluorescent readings were performed using a Spectramax M5 (Molecular Devices) plate reader.
Fluorescent staining and Flow Cytometry
Immediately after isolation, splenocytes were seeded in 12-well cell culture plates at a cell density of 5×106 cells/well in a final volume of 1.5 mL. Treatments were then given at the concentration of interest, both with and without 25 μM PLP. Cell cultures were then incubated for 120 hours at 37°C in a CO2 (5%) incubator. At 120 hrs, splenocytes were stained with the desired antibodies. Briefly, 1×106 cells were washed with 1 mL of wash buffer (RPMI 1640 media containing 5% FBS) before centrifugation and resuspension in 50 μL block buffer containing 20 μg/mL TruStain fcX (anti-mouse CD16/32 antibody, Biolegend) in wash buffer. Cells were incubated on ice for 25 minutes before adding the desired antibodies or isotype controls in 50 μL at 2x the manufacturer recommended concentration for 1 hour. For flow cytometry analysis, 40,000 cells per sample were detected using a BD FACSFusion cytometer. Data was analyzed using FlowJo Software.
Detection of PLP-Specific Antibody-secreting Cells by ELISpot
96-well plates (Immunlon 2HB) were coated with 50 μg/mL PLP in 100 μL of PBS and incubated overnight at 4°C. The PLP coating solution was discarded and the plate was blocked with PBS containing 0.5% FBS for 1 hour prior to addition of splenocytes. Immediately after their isolation, splenocytes were seeded onto the coated 96-well plate at a cell density of 1×106 cells per well in a final volume of 100 μL. The splenocyte cultures were incubated for 48 hours at 37°C in a CO2 (5%) incubator. Following incubation, each plate was washed 4x with PBS containing 0.05% Tween 20 for 2 minutes, followed by washing 2x with PBS for 2 minutes. 100 μL of horseradish peroxidase (HRP) conjugated anti-mouse IgG at a concentration of 1 μg/mL was added to each well and incubated for 1 hour at 37°C. Following a second wash step, identical to previously described, a buffer containing TrueBlue Peroxidase Substrate (Kirkegaard & Perry Laboratories, Inc) and agarose (1:1 ratio) was heated in a water bath to 56°C and 100 μL was added to each well using reverse pipetting to avoid bubbles. Plates were incubated overnight at 4°C before reading on the CTL Ultimate S6 ImmunoSpot Analyzer (Cellular Technology Limited). Analysis of spots was done using CTL ImmunoSpot software (Cellular Technology Limited).
Statistical analysis
Statistical evaluation of data was performed using a one-way or two-way analysis of variance (ANOVA) as experimentally appropriate, followed by Tukey and Sidak multiple comparison tests. Trends were described following individual t-tests and linear regression analysis. A single outlier was removed from the cytokine analysis of the IFA with DEX and PLP group (Figure 5) following outlier confirmation via the Grubb’ Outlier Test. The criteria for statistical significance for all analyses was set at p<0.05. The majority of analyses were performed using GraphPad Software (GraphPad Software Inc.) and linear regression analysis to determine data trends was performed using MATLAB (The MathWorks, Inc).
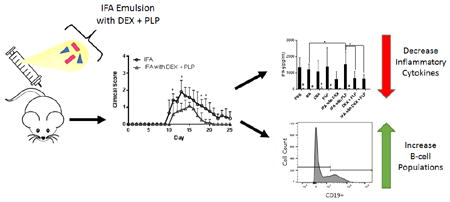
Figure 5.
Splenocytes were harvested on day 25 from EAE mice treated in vivo with components of the co-delivery treatment, IFA with DEX + PLP. Splenocytes were incubated for 120 hours with or without 25 μM PLP rechallenge. A.) Cell metabolism and supernatant cytokine levels of B.) GM-CSF, C.) IFN-γ, D.) IL-10, E.) TNF-α, F.) IL-6, G.) IL-17, H.) IL-2, I.) IL-4, J.) IL-23, and K.) IL-15 were determined. (n= 5-6 per group, * p< 0.05 comparing different treatment groups, # p<0.05 for 25 μM PLP versus no PLP for the same treatment, ND indicates cytokine levels were not detectable, Black bars indicate 25 μM PLP and Gray bars indicate no PLP rechallenge)
RESULTS
Characterization of IFA Emulsion containing PLP and DEX
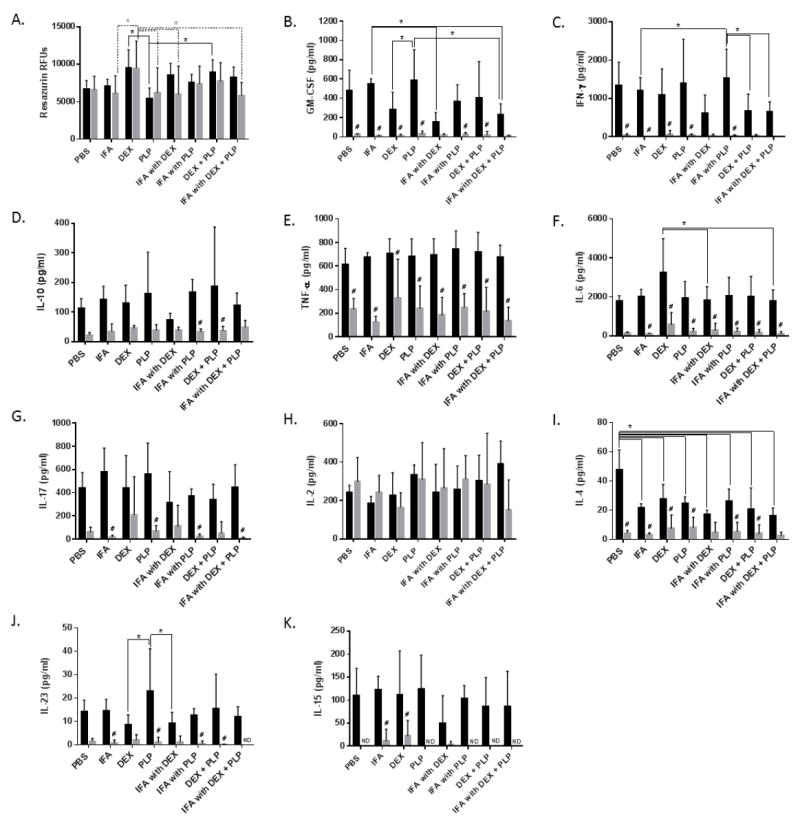
IFA oil was combined at a 1:1 ratio with phosphate buffered saline (PBS) using a micro-emulsifying needle to create an emulsion that was observed to be physically stable for upwards of 9 hours (Supplemental Figure 1). The emulsion was imaged using Nile Blue A as an oil-partitioning dye (Figure 1). The resulting Bright Field (Figure 1A), fluorescent (Figure 1B) and composite (Figure 1C) images depict aqueous droplets of PBS contained by an oil phase medium; confirming the water-in-oil structure of the emulsion. Droplet sizes reached diameters of up to 50 μm.
Figure 1.
Representative images of the IFA emulsion via A.) Bright Field, B.) fluorescence of Nile Blue A (excitation 631 nm/ emission 660 nm) in the oil phase, and C.) both images overlaid. (40x magnification)
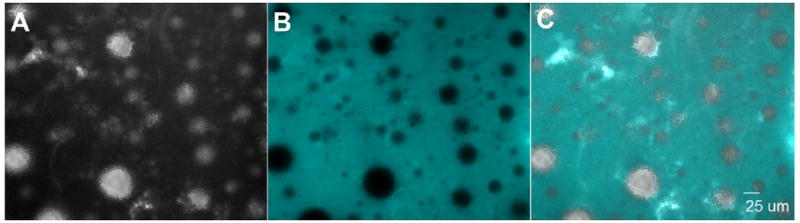
A 1:1 IFA oil: PBS emulsion was created containing PLP and DEX for the co-delivery of autoantigen and immunomodulator. The release of both PLP and DEX from either the IFA emulsion or PBS was measured by placing each formulation into a dialysis membrane (6,000-8,000 Da MWCO) and tracking the release into sink conditions of PBS at 37°C. PLP and DEX were found to be fully released from PBS within the membrane after 24 hours and 10 hours, respectively (Figure 2). DEX within the IFA emulsion was not fully released until approximately 120 hours (Figure 2A). PLP was much slower to release from the IFA emulsion, with less than 10% of the total PLP in the formulation being released within 192 hours (Figure 2B). Several additional emulsions with varying IFA oil to PBS ratios were tested in order to verify the presence and eventual release of PLP, revealing that decreasing oil content increased the release of PLP within 48 hours (Supplemental Figure 2).
Figure 2.
Release at 37°C of A.) DEX and B.) PLP from IFA emulsion (open circle) and PBS (black square) within the dialysis bag in sink conditions of PBS. Concentrations determined at each time point via RP-HPLC. Data shown as a percentage of total DEX or PLP in the formulation. (n=3 per formulation)
Treatments containing PLP in IFA emulsion Suppress EAE Symptoms
IFA emulsion containing PLP and DEX, along with each combination of the components and a PBS control, were tested in mice induced with EAE. Treatments groups include PBS, IFA, DEX, PLP, IFA with DEX, IFA with PLP, DEX + PLP, and IFA with DEX + PLP. Treatments were given subcutaneously between the shoulder blades on days 4, 7, and 10 of the study (as shown on Figure 3A-B and Figure 4A-B). By day 12, half of the mice in the PBS control group had died. Due to these deaths, and the corresponding disease score of 5, the PBS was statistically significant from all other treatments, including the IFA vehicle control, on at least one day of the study (Figure 3A-B). Additionally, due to the high error in clinical score associated with the days on which the PBS mice died (days 11-12), the IFA group is shown as an alternative control compared to all other treatments (Figure 3). Weight changes were also monitored throughout the study and are shown as a percent change as compared to day 8, the last day with no disease symptoms (Figure 4). Although the death of the three PBS treated mice contributed to a significant increase in clinical score, the weight change was unaffected. In fact, there was no statistical difference between IFA and PBS treatment groups in terms of weight change (Figure 4A).
Figure 3.
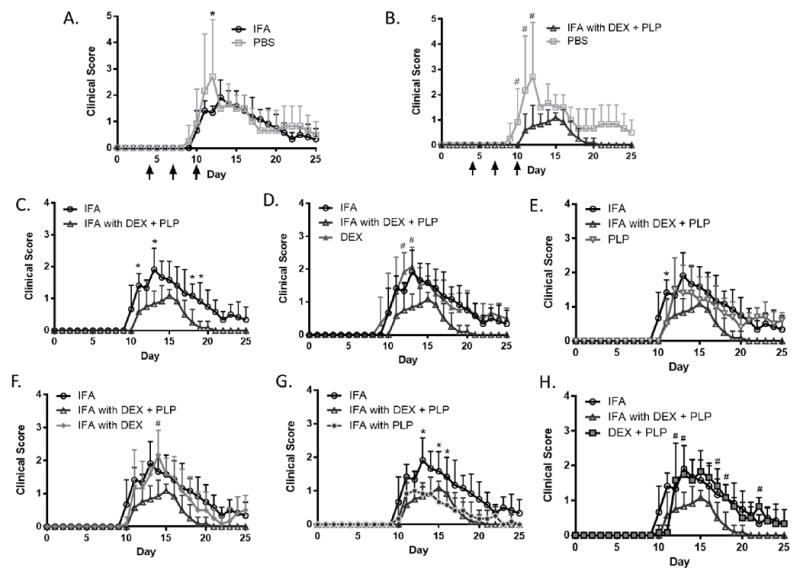
Clinical disease scores in EAE mice given components of co-delivery treatment, IFA with DEX + PLP. Disease score with PBS control treatment compared to A.) IFA and B.) IFA with DEX + PLP. IFA and IFA with DEX + PLP treatments were compared to C.) each other, D.) DEX treatment, E.) PLP treatment, F.) IFA with DEX, G.) IFA with PLP and H.) DEX + PLP. All treatments were given on days 4, 7, and 10 (black arrows). (n = 5-6 mice per group, * p< 0.05 as compared to IFA, # p<0.05 as compared to IFA with DEX + PLP)
Figure 4.
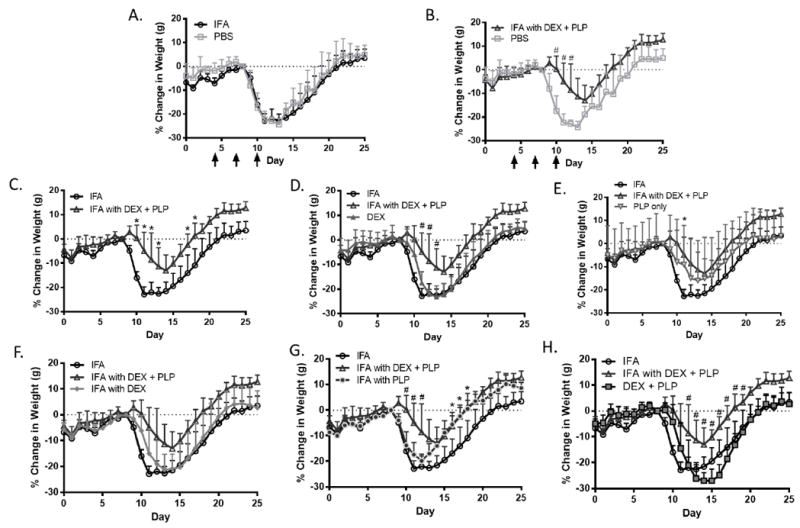
Percent change in weight as compared to day 8 (last day before symptoms) in EAE mice given components of co-delivery treatment, IFA with DEX + PLP. Percent weight change with PBS control treatment compared to A.) IFA and B.) IFA with DEX + PLP. IFA and IFA with DEX + PLP treatments were compared to C.) each other, D.) DEX treatment, E.) PLP treatment, F.) IFA with DEX, G.) IFA with PLP and H.) DEX + PLP. All treatments were given on days 4, 7, and 10 (black arrows). (n = 5-6 mice per group, * p< 0.05 as compared to IFA, # p<0.05 as compared to IFA with DEX + PLP)
In determining the potential benefits offered by co-delivery of autoantigen and immunomodulator, IFA with DEX + PLP was also compared to the clinical score and change in weight from each treatment group (Figures 3 & 4). IFA with DEX + PLP significantly decreased clinical score and weight change as compared to PBS on three days of the study (Figure 3B & Figure 4B). Additionally, as compared to IFA alone, IFA with DEX + PLP decreased both the clinical score and weight change during days before, during, and after peak disease (as determined by peak clinical scores and maximum weight change, see Supplemental Table 1) (Figure 3C & Figure 4C). IFA with DEX + PLP also significantly suppressed disease symptoms on at least one day during the study as compared to DEX only, IFA with DEX, and DEX + PLP (Figure 3D, F, H). Treatments that were not statistically different than IFA with DEX + PLP; both PLP and IFA with PLP, did significantly decrease clinical scores as compared to IFA (Figure 3 E, G). Several PLP-containing treatments (PLP, IFA with PLP, and IFA with DEX + PLP) also significantly decreased the peak clinical score as compared to the PBS control. Interestingly, both IFA with DEX + PLP and IFA with PLP suppressed EAE symptoms to a similar degree. The suppression by both treatments containing PLP in IFA is most clearly observed by examining the area under the curve for clinical score and peak clinical disease score (Supplemental Figure 3). Surprisingly, this same trend was not seen in weight change data, as IFA with PLP actually had significantly more weight loss as compared to IFA with DEX + PLP around the peak of disease (Figure 4G).
Dexamethasone Shifts Cytokine Profiles Away from Inflammation
Following disease remission, on day 25 of the EAE study, splenocytes were collected from the mice and were rechallenged in vitro with and without PLP antigen. Cellular metabolic activity and cytokine responses were measured at 120 hours post antigen rechallenge (Figure 5). Metabolic activity as measured by resazurin was found to significantly increase with DEX treatment as compared to IFA, IFA with DEX, and IFA with DEX + PLP without rechallenge (Figure 5A). DEX treatment also increased metabolic activity both with and without rechallenge as compared to treatment with PLP (Figure 5A). Additionally, DEX + PLP was found to increase metabolic activity as compared to PLP with antigen rechallenge (Figure 5A).
Rechallenge with PLP was found to significantly increase the cytokine levels of GM-CSF, IFN-γ, TNF-α, IL-6, and IL-4 for the majority of treatment groups and increased levels of IL-10, IL-17, IL-23, and IL-15 in some of the treatment groups (Figure 5). Significant differences between the treatment groups were found in the cytokine response of GM-CSF, IFN-γ, IL-6, IL-4, and IL-23; however, differences were only found for samples including PLP rechallenge. Treatments containing DEX tended to decrease pro-inflammatory cytokines including GM-CSF, IFN-γ, and IL-23 as compared to other treatment groups upon rechallenge with PLP (Figure 5B, C, J). Both treatments of IFA and PLP alone were found to significantly increase the pro-inflammatory GM-CSF as compared to IFA with DEX and IFA with DEX + PLP (Figure 5B). IL-23 also significantly increased due to treatment with PLP, this time compared to DEX, and IFA with DEX (Figure 5J). Surprisingly, the general pro-inflammatory cytokine IFN-γ was shown to increase with IFA with PLP following rechallenge, a treatment that was effective at suppressing clinical score in vivo. IFA with PLP significantly increased levels of IFN-γ as compared to treatments of IFA only, DEX + PLP, and IFA with DEX + PLP (Figure 5C). IL-6 was shown to be significantly increased with DEX treatment as compared to IFA with DEX and IFA with DEX + PLP (Figure 5F). Unlike the majority of cytokines analyzed, IL-4 significantly increased with PBS treatment as compared to all other treatment groups (Figure 5I).
Effective in vivo Therapies Increase B-cell-related Responses in Splenocytes
In addition to cytokine responses, changes in cell populations in the splenocytes obtained from the different in vivo treatment groups were examined both with and without PLP antigen rechallenge (Figure 6). The population of T-cells, as identified by CD3, was found to be statistically similar for all treatment groups, both with and without rechallenge (Figure 6A). CD11c, which is a common marker for dendritic cells (DCs), was found to be increased both with and without antigen rechallenge for IFA with DEX + PLP and with antigen for IFA with PLP (Figure 6B). B-cells, as identified by CD19, were found to be significantly increased in the IFA with DEX+ PLP treatment group (Figure 6C, F). CD19+CD11c+ cells were also analyzed, as these cells have been shown to act as autoimmune associated B-cells (ABCs) [38, 39]. The CD19+CD11c+ population was significantly increased for the IFA with DEX + PLP treatment group without antigen rechallenge (Figure 6D). The ABC population in the IFA with DEX + PLP group was significantly higher than all other treatment groups as a percent of the total splenocytes; however, the ABCs were not increased as a percentage of the B-cell (CD19+) population (Figure 6E).
Figure 6.
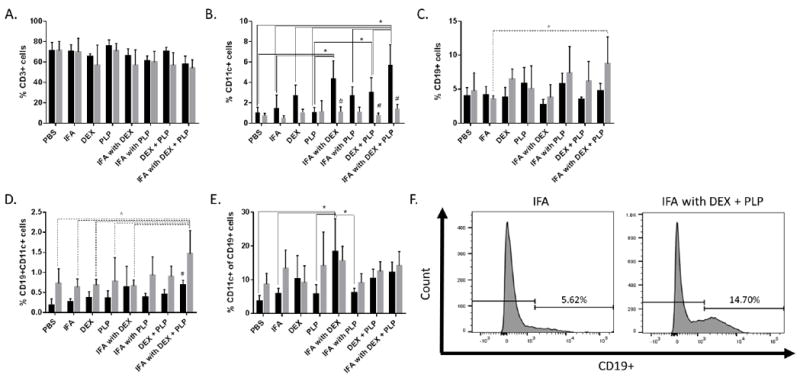
Splenocytes were harvested on day 25 from EAE mice treated in vivo with components of co-delivery treatment, IFA with DEX + PLP. Splenocytes were incubated for 120 hours with or without 25 μM PLP rechallenge. The cells were then stained with antibodies for CD3 (Alexa Fluor 488), CD19 (Alexa Fluor 647), and CD11c (Pacific Blue) and analyzed by flow cytometry. Cell populations were determined for A.) T-cells (CD3+), B.) Dendritic Cells (CD11c+), C.) B-cells (CD19+) and D.) autoimmune associated B-cells (ABCs, CD19+CD11c+) as a percentage of the total cell population. E.) The percentage of CD11c+ cells out of the CD19+ cell population was also determined. F.) Gating for CD19+ B-cells is shown for both IFA and IFA with DEX + PLP, without antigen rechallenge. (n= 3-5 per group, * p< 0.05 comparing different treatment groups, # p<0.05 for 25 μM PLP versus no PLP for the same treatment, Black bars indicate 25 μM PLP and Gray bars indicate no PLP rechallenge)
Autoantibody production was also measured using ELISpot to determine the number of cells producing PLP-specific antibodies in the spleen following each in vivo treatment. Similar to the increased presence of B-cells demonstrated by flow cytometry, ELISpot showed significantly higher anti-PLP antibody producing cells in splenocytes from mice treated by IFA with DEX + PLP as compared to other treatment groups (Figure 7).
Figure 7.
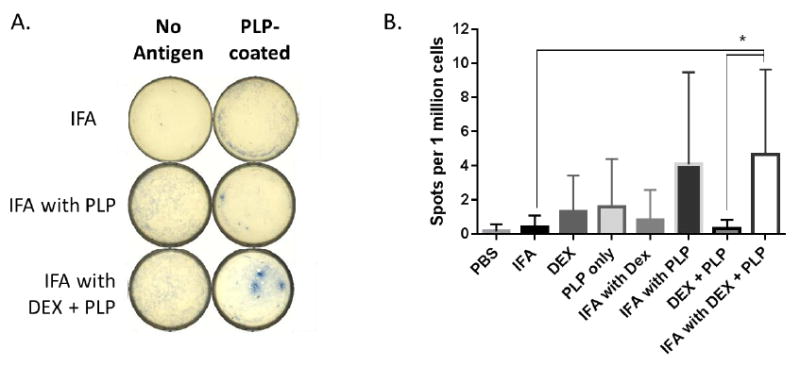
Splenocytes were harvested on day 25 from EAE mice treated in vivo with components of co-delivery treatment, IFA with DEX + PLP, and were plated on wells coated with or without PLP antigen for 48 hours. Spots associated with B-cells producing PLP-specific antibodies were detected using HRP anti-IgG and TrueBlue stain, and imaged with a CTL ImmunoSpot Analyzer. A.) Representative wells both with and without antigen coating are shown. B.) The number of spots per 1 million cells were compared for different treatment groups. (n= 5-6 per group in duplicate wells, * p< 0.05 comparing different treatment groups)
DISCUSSION
The combination of autoantigen and immunomodulators has recently emerged as a potentially effective ASIT strategy for the treatment of autoimmunity [6]. In this study, IFA was investigated as a co-delivery vehicle in order to treat EAE with a combination of DEX and PLP. Our studies demonstrate that controlled release of DEX and PLP from IFA may assist in suppressing EAE. IFA has been previously indicated to have significant immunosuppressive properties alone [27]; however, we did not observe this phenomenon. IFA suppressed disease symptoms as compared to the PBS control on only one day, which was the same as was seen for all other treatment groups (Figure 3A). When used as a delivery vehicle for autoantigen or immunomodulator, IFA containing PLP or DEX + PLP resulted in the most pronounced disease suppression (Figure 3 & Supplemental Figure 3).
The delivery of PLP appeared to play a major role in EAE disease suppression, such that co-delivery of DEX and PLP in IFA did not improve the suppression of disease clinical scores to any noticeable extent as compared to IFA with only PLP (Figure 3G). The importance of PLP in symptom suppression by an IFA formulation may be due to controlled release from aqueous droplets within the emulsion. In particular, the release of PLP into solution was greatly slowed when it was incorporated into IFA emulsions containing 1:1 ratios of oil to PBS (Figure 2B & Supplemental Figure 2). The slow release of PLP from the IFA emulsion may be extending the exposure of autoantigen to antigen-presenting cells (APCs), allowing the therapy to influence the immune system long after the injection is administered. Previous studies in animal models of autoimmunity have seen similar phenomena, where the controlled release of autoantigen increases disease suppression both with and without an immunomodulatory signal [7, 8, 11, 40-42]. Another reason for the ability of PLP in IFA to suppress disease is the entrapment of the aqueous phase, containing the hydrophilic PLP, within the IFA oil matrix as visually demonstrated in Figure 1. Due to the low hydrophile- lipophile balance of mannide monooleate, the surfactant used in IFA, the propensity to create emulsions in which the water phase is entrapped in the oil is favored. The water-in-oil IFA emulsion structure with the entrapment of a hydrophilic antigen has been found to both protect the antigen from degradation and allow for a prolonged immune response [21]. The entrapment of PLP within water droplets of our IFA emulsion may contribute to antigen persistence at the injection site, and consequentially, improved uptake by APCs [43, 44]. The emulsion stability of up to 9 hours (Supplemental Figure 1) may also help prolong antigen presence at for enhanced APC uptake.
We also observed that treatments containing PLP, even without controlled release, significantly decreased peak clinical score and appeared to delay disease onset (Figure 3 & Supplemental Figure 3). Treatment with an appropriate dose of autoantigen has often been reported to decrease autoimmune symptoms in animals, but has not been successfully translated to the clinic [3, 45]. The EAE in vivo efficacy of this treatment was also contradictory to the cytokine response observed in splenocytes obtained on day 25 of the study. Treating EAE mice with PLP alone was shown to promote pro-inflammatory cytokine production by significantly increasing GM-CSF and IL-23, and slightly increasing IFN-γ and IL-2 although not to a significant degree compared to other treatments (Figure 5). The correlation of poor efficacy of autoantigen alone in humans [3, 45] and the increased pro-inflammatory cytokine production by PLP treatment in EAE, as demonstrated in this manuscript, emphasizes the importance of analyzing the immune response in addition to clinical symptoms.
The in vivo data indicated the importance of controlled release of autoantigen in the creation of effective ASIT, while the addition of an immunomodulator such as DEX had little effect on clinical symptoms (Figure 3 & 4). When analyzing the immune response more closely, it was found that DEX plays an important role in shifting the cytokine profile away from pro-inflammatory Th1/Th17 responses (Figure 5). Decreases in pro-inflammatory cytokine levels are in direct contrast with an increase in cellular metabolic activity upon DEX treatment, therefore the suppression of pro-inflammatory cytokine by DEX may be even more significant when considering the results on a per metabolizing cell basis (Figure 5A). DEX was also shown to increase IL-6 (Figure 5F); however, this cytokine can be indicative of both pro-inflammatory (Th17) and regulatory (Treg) immune responses depending on the presence of other cytokine responses [46, 47]. The increase in IL-6 coupled with a decrease in IL-23 and GM-CSF, along with no change in IL-17, suggests that DEX is not producing a Th17 response and therefore IL-6 production may not be indicative of inflammation [46, 47]. Another cytokine demonstrating unexpected results was IL-4, which was shown to decrease with all therapies, even IFA alone (Figure 5I); however, the role of IL-4 in the treatment of EAE has been debated [48].
Interesting immunological trends due to DEX treatment were also observed in the CD11c+ dendritic cell (DC) population upon rechallenge with PLP (Figure 6B). IFA with DEX and IFA with DEX + PLP significantly increased the population of DCs compared to the majority of treatments, and DEX + PLP increased DCs as compared to PLP only treatment. The increase of DCs with treatments containing DEX is directly reverse of the trend for DEX to decrease GM-CSF (Figure 5B). GM-CSF has been known to be essential for the generation of inflammatory DCs from monocytes in the spleen, and plays a crucial role in EAE pathogenesis [49]. The inversely proportional trend of GM-CSF and CD11c+ DCs following treatments with DEX may indicate that the DCs being produced are not pro-inflammatory, and may actually be immature or even tolerogenic [50, 51]. CD11c+ DCs have been shown to act as tolerogenic DCs in the spleen [50]. Also, the treatment of DCs with DEX has previously been demonstrated to produce higher levels of tolerogenic DCs [49, 52-54]. Additionally, although not statistically significant, a slight trend in the decrease of CD3+ T-cells with the addition of DEX was also noted (Figure 6A).
Previous studies co-administering IFA and autoantigen have shown that a shift towards a Th2/humoral immune response allows for the suppression of Th1/Th17-mediated autoimmune diseases [19, 20]. Our study indicates an increased humoral response with both increased autoantibody production and B-cell populations in splenocytes from mice treated with either IFA with PLP or IFA with DEX + PLP (Figures 6 & 7). Although IFA with DEX + PLP increases the percent of autoimmune associated B-cells (ABCs) in the entire splenocyte population, this trend seems to be associated with an increase in B-cells overall rather than an increase in the proportion of B-cells that are ABCs (Figure 6D, E). Both B-cell and ABC populations are found to significantly decrease upon rechallenge with antigen. Unresponsiveness to antigen rechallenge may indicate that many of these cells are anergic and therefore not contributing to EAE progression [55, 56].
In measuring PLP-specific autoantibody producing B-cells via ELISpot, we discovered that the treatments that suppressed clinical symptoms of EAE had much higher levels of PLP-specific antibodies (Figure 7). Increased production of antibody following effective treatment with IFA and autoantigen has been observed before in both human autoimmunity and animal models [20, 30]. An increase in antibody production against autoantigen has been shown to correspond to skewing the immune response towards a Th2/humoral response resulting in suppression of Th1/Th17-mediated autoimmunity [20]. Our results demonstrating both an increase in the B-cell population and PLP-specific antibody production with IFA with DEX + PLP treatment of EAE support the hypothesis that the clinical disease suppression may be due to an increased Th2/humoral immune response.
CONCLUSIONS
As the combination of autoantigen and immunomodulator increases in popularity as an effective autoimmune therapeutic strategy, it is important to understand how each component contributes to the desired outcome. By investigating all possible treatment combinations of DEX and PLP using IFA as a delivery vehicle, we were able to identify important aspects of EAE treatment efficacy. Although co-delivery of DEX and PLP was hypothesized to be most effective, it was found that IFA with PLP was just as efficacious at ameliorating disease symptoms in vivo, indicating the importance of controlled delivery of autoantigen. Upon closer analysis of the immunological responses, it was found that DEX plays an important role in decreasing pro-inflammatory cytokines and possibly increasing tolerogenic DCs. Additionally, the co-delivery treatment of IFA with PLP and DEX was shown to increase B-cell populations and antibody production; demonstrating an increased humoral immune response as compared to controls. Overall, this data indicates that co-delivery of PLP autoantigen and DEX in an IFA emulsion is effective in the treatment of a murine autoimmune model.
Supplementary Material
Acknowledgments
LN was supported by the American Association of Pharmaceutical Scientists (AAPS) Foundation Graduate Fellowship. JDG and BLH were supported by the Madison and Lila Self Graduate Fellowship at the University of Kansas. MAC was supported by the Biotechnology Predoctoral Training Program at the University of Kansas. CJP was supported by NIH training grant T32 GM008545 and the Howard Rytting pre-doctoral fellowship from the Department of Pharmaceutical Chemistry at the University of Kansas. The authors would like to acknowledge the Kansas Vaccine Institute at the University of Kansas, specifically Francisco J. Martinez-Becerra, for his assistance with flow cytometry, ELISpot, and cytokine measurements with MSD Multiplex. Additionally, the authors thank Joseph St. Amand for his assistance with data analysis to determine trends. Lastly, the authors thank Heather Shinogle of the Microscopy and Analytical Imaging Core Laboratory at the University of Kansas for her assistance in collecting and analyzing IFA emulsion images.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luo X, Miller SD, Shea LD. Immune Tolerance for Autoimmune Disease and Cell Transplantation. Annu Rev Biomed Eng. 2016;18:181–205. doi: 10.1146/annurev-bioeng-110315-020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65:239–248. doi: 10.1002/ana.21640. [DOI] [PubMed] [Google Scholar]
- 3.Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, Gran B, Eaton J, Antel J, Frank JA, McFarland HF, Martin R. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–1175. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 4.Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, Steinman L. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. 2000;6:1176–1182. doi: 10.1038/80525. [DOI] [PubMed] [Google Scholar]
- 5.Freedman MS, Bar-Or A, Oger J, Traboulsee A, Patry D, Young C, Olsson T, Li D, Hartung HP, Krantz M, Ferenczi L, Verco T M.-. Investigators. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology. 2011;77:1551–1560. doi: 10.1212/WNL.0b013e318233b240. [DOI] [PubMed] [Google Scholar]
- 6.Northrup L, Christopher MA, Sullivan BP, Berkland C. Combining antigen and immunomodulators: Emerging trends in antigen-specific immunotherapy for autoimmunity. Adv Drug Deliv Rev. 2015 doi: 10.1016/j.addr.2015.10.020. [DOI] [PubMed] [Google Scholar]
- 7.Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM. Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell reports. 2016;16:2940–2952. doi: 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, Kishimoto TK. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci U S A. 2015;112:E156–165. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, Peng H, Yang Y, Vangundy Z, Papenfuss TL, Lovett-Racke AE, Bachelder EM, Ainslie KM. Treatment of experimental autoimmune encephalomyelitis by codelivery of disease associated Peptide and dexamethasone in acetalated dextran microparticles. Molecular pharmaceutics. 2014;11:828–835. doi: 10.1021/mp4005172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle- mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109:11270–11275. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capini C, Jaturanpinyo M, Chang HI, Mutalik S, McNally A, Street S, Steptoe R, O’Sullivan B, Davies N, Thomas R. Antigen-specific suppression of inflammatory arthritis using liposomes. Journal of immunology. 2009;182:3556–3565. doi: 10.4049/jimmunol.0802972. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. The Journal of pharmacology and experimental therapeutics. 2007;322:879–886. doi: 10.1124/jpet.107.123257. [DOI] [PubMed] [Google Scholar]
- 13.Sestak JO, Sullivan BP, Thati S, Northrup L, Hartwell B, Antunez L, Forrest ML, Vines CM, Siahaan TJ, Berkland C. Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Molecular Therapy — Methods & Clinical Development. 2014;1 doi: 10.1038/mtm.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northrup L, Sestak JO, Sullivan BP, Thati S, Hartwell BL, Siahaan TJ, Vines CM, Berkland C. Co-delivery of autoantigen and b7 pathway modulators suppresses experimental autoimmune encephalomyelitis. The AAPS journal. 2014;16:1204–1213. doi: 10.1208/s12248-014-9671-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kindt TJ, Goldsby RA, Osborne BA, Kuby J. Kuby immunology. 6. W.H. Freeman and Company; New York: 2007. [Google Scholar]
- 17.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clinical and experimental immunology. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Legroux L, Arbour N. Multiple Sclerosis and T Lymphocytes: An Entangled Story. J Neuroimmune Pharmacol. 2015;10:528–546. doi: 10.1007/s11481-015-9614-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heeger PS, Forsthuber T, Shive C, Biekert E, Genain C, Hofstetter HH, Karulin A, Lehmann PV. Revisiting tolerance induced by autoantigen in incomplete Freund’s adjuvant. Journal of immunology. 2000;164:5771–5781. doi: 10.4049/jimmunol.164.11.5771. [DOI] [PubMed] [Google Scholar]
- 20.Lin MS, Tse HM, Delmastro MM, Bertera S, Wong CT, Lakomy R, He J, Sklavos MM, Coudriet GM, Pietropaolo M, Trucco MM, Piganelli JD. A multivalent vaccine for type 1 diabetes skews T cell subsets to Th2 phenotype in NOD mice. Immunol Res. 2011;50:213–220. doi: 10.1007/s12026-011-8215-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koh YT, Higgins SA, Weber JS, Kast WM. Immunological consequences of using three different clinical/laboratory techniques of emulsifying peptide-based vaccines in incomplete Freund’s adjuvant. J Transl Med. 2006;4:42. doi: 10.1186/1479-5876-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powell MF, Newman MJ. Vaccine design: the subunit and adjuvant approach. Plenum Press; New York: 1995. [Google Scholar]
- 23.Dietrich FM. The Immune Response of C57bl-6 Mice to Protein Antigens Incorporated into Freund’s Incomplete Adjuvant. Pathol Microbiol (Basel) 1964;27:1025–1034. doi: 10.1159/000161596. [DOI] [PubMed] [Google Scholar]
- 24.Zheng H, Zhang H, Liu F, Qi Y, Jiang H. T cell-depleted splenocytes from mice pre-immunized with neuroantigen in incomplete Freund’s adjuvant involved in protection from experimental autoimmune encephalomyelitis. Immunol Lett. 2014;157:38–44. doi: 10.1016/j.imlet.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Liddi R, Beales PE, Rosignoli G, Pozzilli P. Incomplete Freund’s adjuvant reduces diabetes in the non-obese diabetic mouse. Horm Metab Res. 2000;32:201–206. doi: 10.1055/s-2007-978622. [DOI] [PubMed] [Google Scholar]
- 26.Cannon GW, Griffiths MM, Woods ML. Suppression of adjuvant-induced arthritis in DA rats by incomplete Freund’s adjuvant. Arthritis Rheum. 1993;36:126–131. doi: 10.1002/art.1780360120. [DOI] [PubMed] [Google Scholar]
- 27.Zamora A, Matejuk A, Silverman M, Vandenbark AA, Offner H. Inhibitory effects of incomplete Freund’s adjuvant on experimental autoimmune encephalomyelitis. Autoimmunity. 2002;35:21–28. doi: 10.1080/08916930290005873. [DOI] [PubMed] [Google Scholar]
- 28.Luo J, Lindstrom J. Antigen-specific immunotherapeutic vaccine for experimental autoimmune myasthenia gravis. Journal of immunology. 2014;193:5044–5055. doi: 10.4049/jimmunol.1401392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fousteri G, Dave A, Bot A, Juntti T, Omid S, von Herrath M. Subcutaneous insulin B:9-23/IFA immunisation induces Tregs that control late-stage prediabetes in NOD mice through IL-10 and IFNgamma. Diabetologia. 2010;53:1958–1970. doi: 10.1007/s00125-010-1777-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orban T, Farkas K, Jalahej H, Kis J, Treszl A, Falk B, Reijonen H, Wolfsdorf J, Ricker A, Matthews JB, Tchao N, Sayre P, Bianchine P. Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin B-chain immunotherapy. Journal of autoimmunity. 2010;34:408–415. doi: 10.1016/j.jaut.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Northrup L, Sullivan BP, Hartwell BL, Garza A, Berkland C. Screening Immunomodulators To Skew the Antigen-Specific Autoimmune Response. Molecular pharmaceutics. 2017;14:66–80. doi: 10.1021/acs.molpharmaceut.6b00725. [DOI] [PubMed] [Google Scholar]
- 32.Franchimont D, Galon J, Gadina M, Visconti R, Zhou Y, Aringer M, Frucht DM, Chrousos GP, O’Shea JJ. Inhibition of Th1 immune response by glucocorticoids: dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. Journal of immunology. 2000;164:1768–1774. doi: 10.4049/jimmunol.164.4.1768. [DOI] [PubMed] [Google Scholar]
- 33.Zheng G, Zhong S, Geng Y, Munirathinam G, Cha I, Reardon C, Getz GS, van Rooijen N, Kang Y, Wang B, Chen A. Dexamethasone promotes tolerance in vivo by enriching CD11clo CD40lo tolerogenic macrophages. Eur J Immunol. 2013;43:219–227. doi: 10.1002/eji.201242468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Oppenheim JJ, Winkler-Pickett RT, Ortaldo JR, Howard OM. Glucocorticoid amplifies IL-2-dependent expansion of functional FoxP3(+)CD4(+)CD25(+) T regulatory cells in vivo and enhances their capacity to suppress EAE. Eur J Immunol. 2006;36:2139–2149. doi: 10.1002/eji.200635873. [DOI] [PubMed] [Google Scholar]
- 35.Elenkov IJ. Glucocorticoids and the Th1/Th2 balance. Ann N Y Acad Sci. 2004;1024:138–146. doi: 10.1196/annals.1321.010. [DOI] [PubMed] [Google Scholar]
- 36.Wust S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, Luhder F, Reichardt HM. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. Journal of immunology. 2008;180:8434–8443. doi: 10.4049/jimmunol.180.12.8434. [DOI] [PubMed] [Google Scholar]
- 37.Kang Y, Xu L, Wang B, Chen A, Zheng G. Cutting edge: Immunosuppressant as adjuvant for tolerogenic immunization. Journal of immunology. 2008;180:5172–5176. doi: 10.4049/jimmunol.180.8.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubtsov AV, Rubtsova K, Kappler JW, Jacobelli J, Friedman RS, Marrack P. CD11c-Expressing B Cells Are Located at the T Cell/B Cell Border in Spleen and Are Potent APCs. Journal of immunology. 2015;195:71–79. doi: 10.4049/jimmunol.1500055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, Marrack P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. 2011;118:1305–1315. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, Gigliotti CL, Boggio E, Clemente N, Comi C, Dianzani C, Boldorini R, Chiocchetti A, Reno F, Dianzani U. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine. 2014;32:5681–5689. doi: 10.1016/j.vaccine.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 41.Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG. A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin Immunol. 2015 doi: 10.1016/j.clim.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belogurov AA, Jr, Stepanov AV, Smirnov IV, Melamed D, Bacon A, Mamedov AE, Boitsov VM, Sashchenko LP, Ponomarenko NA, Sharanova SN, Boyko AN, Dubina MV, Friboulet A, Genkin DD, Gabibov AG. Liposome-encapsulated peptides protect against experimental allergic encephalitis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27:222–231. doi: 10.1096/fj.12-213975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, Liu C, Lou Y, Wang Z, Ma W, Rabinovich B, Sowell RT, Schluns KS, Davis RE, Hwu P, Overwijk WW. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465–472. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reinhardt RL, Bullard DC, Weaver CT, Jenkins MK. Preferential accumulation of antigen-specific effector CD4 T cells at an antigen injection site involves CD62E-dependent migration but not local proliferation. J Exp Med. 2003;197:751–762. doi: 10.1084/jem.20021690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Current opinion in immunology. 2010;22:609–615. doi: 10.1016/j.coi.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganeshan K, Bryce PJ. Regulatory T cells enhance mast cell production of IL-6 via surface-bound TGF-beta. Journal of immunology. 2012;188:594–603. doi: 10.4049/jimmunol.1102389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 48.Liblau R, Steinman L, Brocke S. Experimental autoimmune encephalomyelitis in IL-4-deficient mice. Int Immunol. 1997;9:799–803. doi: 10.1093/intimm/9.5.799. [DOI] [PubMed] [Google Scholar]
- 49.Moreau A, Varey E, Beriou G, Hill M, Bouchet-Delbos L, Segovia M, Cuturi MC. Tolerogenic dendritic cells and negative vaccination in transplantation: from rodents to clinical trials. Front Immunol. 2012;3:218. doi: 10.3389/fimmu.2012.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J, Cao X. Regulatory dendritic cells in autoimmunity: A comprehensive review. Journal of autoimmunity. 2015;63:1–12. doi: 10.1016/j.jaut.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 51.Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H. Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol Cell Biol. 2002;80:477–483. doi: 10.1046/j.1440-1711.2002.01115.x. [DOI] [PubMed] [Google Scholar]
- 52.Garcia-Gonzalez P, Morales R, Hoyos L, Maggi J, Campos J, Pesce B, Garate D, Larrondo M, Gonzalez R, Soto L, Ramos V, Tobar P, Molina MC, Pino-Lagos K, Catalan D, Aguillon JC. A short protocol using dexamethasone and monophosphoryl lipid A generates tolerogenic dendritic cells that display a potent migratory capacity to lymphoid chemokines. J Transl Med. 2013;11:128. doi: 10.1186/1479-5876-11-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleijwegt FS, Jansen DT, Teeler J, Joosten AM, Laban S, Nikolic T, Roep BO. Tolerogenic dendritic cells impede priming of naive CD8(+) T cells and deplete memory CD8(+) T cells. Eur J Immunol. 2013;43:85–92. doi: 10.1002/eji.201242879. [DOI] [PubMed] [Google Scholar]
- 54.Emmer PM, van der Vlag J, Adema GJ, Hilbrands LB. Dendritic cells activated by lipopolysaccharide after dexamethasone treatment induce donor-specific allograft hyporesponsiveness. Transplantation. 2006;81:1451–1459. doi: 10.1097/01.tp.0000208801.51222.bd. [DOI] [PubMed] [Google Scholar]
- 55.Cambier JC, Getahun A. B cell activation versus anergy; the antigen receptor as a molecular switch. Immunol Lett. 2010;128:6–7. doi: 10.1016/j.imlet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol. 2005;6:1160–1167. doi: 10.1038/ni1256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.