

Abstract
Immune molecules such as cytokines and chemokines and the cells that produce them within the brain, notably microglia, are critical for normal brain development. This recognition has in recent years led to the working hypothesis that inflammatory events during pregnancy, e.g. in response to infection, may disrupt the normal expression of immune molecules during critical stages of neural development and thereby contribute to the risk for neurodevelopmental disorders such as autism spectrum disorder (ASD). This hypothesis has in large part been shepherded by the work of Dr. Paul Patterson and colleagues, which has elegantly demonstrated that a single viral infection or injection of a viral mimetic to pregnant mice significantly and persistently impacts offspring immune and nervous system function, changes that underlie ASD-like behavioral dysfunction including social and communication deficits. Subsequent studies by many labs – in humans and in non-human animal models - have supported the hypothesis that ongoing disrupted immune molecule expression and/or neuroinflammation contributes to at least a significant subset of ASD. The heterogeneous clinical and biological phenotypes observed in ASD strongly suggest that in genetically susceptible individuals, environmental risk factors combine or synergize to create a tipping or threshold point for dysfunction. Importantly, animal studies showing a link between maternal immune activation (MIA) and ASD-like outcomes in offspring involve different species and diverse environmental factors associated with ASD in humans, beyond infection, including toxin exposures, maternal stress, and maternal obesity, all of which impact inflammatory or immune pathways. The goal of this review is to highlight the broader implications of Dr. Patterson's work for the field of autism, with a focus on the impact that MIA by diverse environmental factors has on fetal brain development, immune system development, and the pathophysiology of ASD.
Introduction
Autism spectrum disorder (ASD) is a complex continuum of neurodevelopmental disorders with early childhood-onset. These disorders, for which there is presently no cure and only limited treatments, are typically associated with significant lifelong cognitive, social, communication and behavioral impairments for the individual (Birtwell et al., 2016; Magiati et al., 2016). The prevalence of ASD has been progressively increasing, and is more common than previously thought (Centers for Disease Control and Prevention, 2014). Though a component is certainly genetic, hundreds of diverse genes are now linked to ASD, each of which contributes to only a very small percentage of the affected population. The heterogeneous clinical and biological phenotypes observed in ASD strongly suggest that in genetically susceptible individuals, environmental risk factors also combine or synergize to create a tipping or threshold point for dysfunction. Multiple prenatal/maternal exposures, most notably infection, have been linked to an increased risk of ASD in offspring (Ashwood and Van de Water, 2004; Becker, 2007; Blaylock and Strunecka, 2009; Lauritsen et al., 2005; Volk et al., 2014). A large body of work by the highly influential Dr. Paul Patterson, and colleagues, has illustrated some of the mechanisms by which maternal immune activation (MIA) with viral infection or viral mimetics can persistently alter offspring immune function, disrupt fetal brain development, and induce the onset of ASD-like behaviors in animal models. The immune system is our interface with the environment, and a role for immunological involvement in at least a subtype of ASD has been hypothesized for some time (Money et al., 1971). Family studies document a significant relationship between familial autoimmune disorders and ASD (Comi et al., 1999; Molloy et al., 2006a; Molloy et al., 2006b; Sweeten et al., 2003a; Sweeten et al., 2003b). Results from post-mortem and neuroimaging studies utilizing PET/MRI have identified stable and persistent inflammation in the brains of some subjects with ASD compared to control subjects (Pardo et al., 2005; Suzuki et al., 2013; Vargas et al., 2005). Findings from multiple animal models have demonstrated marked immune abnormalities that correlate with abnormalities in behavior (Bauman et al., 2013; Bauman et al., 2014; Borrell et al., 2002; Braunschweig et al., 2012; Brimberg et al., 2016; Dalton et al., 2003; Malkova et al., 2012; Martin et al., 2008; Shi et al., 2003; Singer et al., 2009; Smith et al., 2007a). These studies involve different species and diverse environmental factors associated with ASD in humans, beyond infection, including toxin exposures, maternal stress, and maternal obesity, all of which impact inflammatory or immune pathways (Krakowiak et al., 2012; Volk et al., 2014; Zerbo et al., 2013). The goal of this review is to highlight the broader implications of Dr. Patterson's work for the field of autism, with a focus on the impact that MIA by diverse environmental factors may have on fetal brain development, immune system development, and the risk of ASD.
The Immune System is a Regulator of Normal Brain Development
Immune molecules are critical for normal brain development (Bilbo and Schwarz, 2012; Deverman and Patterson, 2009; Schwarz and Bilbo, 2012). A novel role for major histocompatibility (MHC) class I proteins in activity-dependent synapse formation within the visual cortex was identified over two decades ago (Corriveau et al., 1998), and a role for complement proteins in synapse elimination was described several years later (Stevens et al., 2007), two pivotal findings that fundamentally changed our concept of “immune privilege” within the healthy brain. Several chemokines and cytokines have now been identified for their critical roles in neuronal and glial cell migration, differentiation, synaptic maturation, and many other processes (Deverman and Patterson, 2009; Nawa and Takei, 2006). Time-dependence and regional specificity has been demonstrated for cytokines during brain development (Pousset, 1994), suggesting distinct roles for individual cytokines in the development of specific brain circuits. For instance, interleukin (IL)-1β is expressed at high levels throughout the late prenatal/early postnatal hippocampus and cortex, but at very low (constitutive) levels in the adult (Giulian et al., 1988; Schmitz and Chew, 2008). Microglia, the primary immune and cytokine-producing cells of the central nervous system (CNS), are also important for normal brain development via the phagocytosis of extraneous synapses (Paolicelli et al., 2011; Schafer et al., 2012) and apoptotic cells (Sierra et al., 2010), and for aspects of axonal growth and angiogenesis (Rakic and Zecevic, 2000; Streit, 2001). Microglia colonize the fetal brain as primitive myeloid precursor cells from the yolk sac, starting around embryonic (E) day 9 in the rodent and late 1st trimester in humans (Chan et al., 2007; Ginhoux et al., 2010). They enter the parenchyma via the blood stream and ventricles, and are initially clustered around subcortical regions such as the hippocampus and corpus callosum (Cuadros and Navascues, 1998; Wang et al., 2002; Xu et al., 1993). From there, microglia migrate throughout the brain where they continue to proliferate throughout the first postnatal weeks in humans and rodents. They are long-lived cells that are now accepted to be self-renewing, e.g. without contribution from the hematopoietic cells in the periphery under normal conditions (Elmore et al., 2014; Schulz et al., 2012). Microglia largely appear thick, reactive or amoeboid during the early perinatal period, in contrast to the ramified morphology found in adults (Fujita et al., 1981; Ling and Wong, 1993; Rezaie and Male, 2002), though little is known about what this means for function. Interestingly, the developmental peak in cytokine concentrations within distinct brain regions depends on the presence of amoeboid microglia in the rat (Giulian et al., 1988). Due to the critical role that immune molecules play in normal brain development, many, notably Dr. Patterson (Deverman and Patterson, 2009), have hypothesized that aberrant expression of these molecules in response to immune activation is harmful for neural development. That is, it is because immune moleculesand glia are critical for normal development that they are also implicated in abnormal development (Bilbo and Schwarz, 2009, 2012).
Immune Activation and Neural Dysfunction
We now recognize that immune activation or abnormalities within the brain may play a pivotal role in the etiology and/or progression of neuropsychiatric conditions as diverse as Alzheimer's disease, schizophrenia, ASD, and depression. The link between influenza virus in pregnancy and increased risk for ASD and other neurodevelopmental disorders such as schizophrenia has been documented for many years (Brown, 2012; Canetta and Brown, 2012). Severe bacterial infections in pregnancy, in particular those associated with fever, are similarly associated with increased risk of ASD in children (Atladottir et al., 2010). Very recent data show the association extends to parasites, as low circulating maternal immunoglobulin (Ig) levels against Toxoplasma gondii, a common parasite, is linked to increased odds of ASD in offspring, whereas high levels of Ig predict the opposite, suggesting a protective effect of adequate maternal Ig (Spann et al., 2017). Conversely, fetal brain-specific antibodies have been identified in a subset of mothers of autistic children in several different studies (Brimberg et al., 2016; Careaga et al., 2013; Fox-Edmiston and Van de Water, 2015; Piras et al., 2014; Rossi et al., 2014). These same isolated antibodies produce ASD-like symptoms in naïve rhesus monkey offspring following injection of the mothers during pregnancy (Bauman et al., 2013). These data strengthen the intriguing evidence that autoimmune disorders are more common in individuals and first degree relatives with ASD, broadly suggesting a critical role of immune dysregulation.
The developing CNS may be especially vulnerable to inflammatory disruption (Rodier, 1980), in large part due to its remarkable plasticity within discrete critical windows, perturbations of which may produce effects on brain and behavior that manifest much later in life, or persist throughout an organism's life span (Cai et al., 2000; Pang et al., 2003; Richardson-Burns and Tyler, 2004; Urakubo et al., 2001; Yu et al., 2004). For instance, the neural circuitry underlying sensory systems such as vision and audition require environmental input during discrete developmental windows for adult function to emerge intact – disruption of this input (e.g. via sensory deprivation) results in severe functional deficiencies throughout the remainder of the lifespan. Though less well characterized, there is emerging evidence that immune molecules such as complement proteins and cytokines guide the normal development of neural circuits (Stevens et al., 2007), and thus their abnormal expression in response to immune challenge – during a critical period – is likely to have devastating consequences.
Indeed, altered cytokine expression early in life is increasingly associated with the risk of neurodevelopmental disorders including ASD and severe developmental delay. Concentrations of IL-1β, IL-6, and tumor necrosis factor (TNF) are elevated in infants with severe perinatal complications (Miller et al., 1990), and children with bacterial meningitis have elevated levels of IL-1β that strongly correlate with the occurrence of neurological disorders (Mustafa et al., 1989). Increased levels of IL-6 in amniotic fluid have also been a clinically useful marker of increased risk for neurological disorders and morbidity (Yoon et al., 1995). Lipopolysaccharide (LPS), the cell wall component of gram-negative bacteria, has been used to mimic bacterial infection in many pre-clinical studies because it initiates a rapid and well-characterized immune response, notably robust cytokine expression (see (Wang et al., 2006) for review). Prenatal LPS injections in mice or rats induce long-term changes in immune function, synaptic plasticity, white matter development, and in social, cognitive, and motor behaviors of offspring (Batinic et al., 2016; Fernandez de Cossio et al., 2017; Fidel et al., 1994; Foley et al., 2015; Gao et al., 2015; Golan et al., 2005; Urakubo et al., 2001). Similarly, influenza virus administered to pregnant mice increases cytokine levels in placental tissue and amniotic fluid, in particular IL-6, and results in abnormal development of the offspring (Shi et al., 2003), as does the viral mimic polyinosinic:polycytidylic acid (poly IC) (Shi et al., 2009). Importantly, the infectious agents themselves most often do not enter the fetal circulation, and experiments demonstrating that specific cytokine antagonists in the presence of infection protect the fetal brain from adverse outcomes have implicated the critical role of the maternal immune response itself (Smith et al., 2007b). Moreover, the strong association between infection and developmental disorders spans a diverse number of bacterial, viral, and mimetic agents, suggesting a common mechanism such as cytokine release.
Evidence of immune system abnormalities in autism
Immune system abnormalities have been reported throughout the body and brain of many autistic individuals. As described briefly above, these include evidence of brain specific auto-antibodies, altered lymphocyte responses to antigen, altered cytokine production, and an increased incidence of allergies and other autoimmune disorders (Ashwood and Wakefield, 2006; Ashwood et al., 2006; Blaylock, 2009; Blaylock and Strunecka, 2009; Derecki et al., 2010; Enstrom et al., 2010; Maezawa and Jin, 2010; Pardo et al., 2005). Recent epidemiological studies have shown statistical correlations between risk for ASD and either maternal or infantile atopic diseases, such as asthma, eczema, food allergies and food intolerance (Theoharides et al., 2016), and mouse models of allergic asthma priming in the mother results in ASD-like behavior in the offspring (Schwartzer et al., 2015). Microglia appear to be persistently activated or altered in a significant subset of individuals with ASD (Morgan et al., 2010; Pardo et al., 2005; Vargas et al., 2005). These cells are critical for host defense in response to any perturbation of body or brain homeostasis, including infection, trauma, or hypoxia, in part via their production of cytokines, chemokines, and reactive oxygen species (e.g., nitric oxide) (Graeber and Streit, 2010). In response to injury or immune stimulation, microglia up-regulate multiple surface receptors, including those for complement proteins, MHC II (important for antigen presentation), and cytokines, which in turn initiate both repair and cytotoxic processes via interactions with numerous other CNS cell types (e.g., astrocytes, neurons, pericytes) (Rostene et al., 2011). Microglia are a good candidate for long-term or persistent changes and/or pathology within the brain, because these cells are long-lived and can become and remain activated chronically (Town et al., 2005). The molecular mechanisms of “activation” are not well defined, though they have been characterized in situ by changes in morphology consistent with injury, e.g. enlarged cell bodies and short, thick processes, a phenotypic switch that has been observed in post-mortem ASD brains in multiple studies (Morgan et al., 2010; Pardo et al., 2005; Vargas et al., 2005). Note that there is also evidence that microglia become dystrophic with disease progression, in contrast to sensitized or reactive, which exhibit stripped or disembodied processes and impairments in their normal homeostatic functions such as phagocytosis (Streit et al., 2009; Streit and Xue, 2009). In either case, because microglia are believed to be long-living cells, glial pathology has the capacity to significantly alter neural function and behavior, perhaps over the entire lifespan.
Because the prenatal period is a time of such intense microglial proliferation and activity, we have hypothesized that it is particularly vulnerable to the induction of long-term changes in microglial cell number or function (Bilbo and Schwarz, 2009, 2012; Bilbo et al., 2011). Indeed, our work has demonstrated significant and persistent impacts of perinatal infection or inflammation on microglial development and function in rodents, which produce sensitized inflammatory responses to subsequent insults later in life that are directly implicated in behavioral abnormalities (Bilbo et al., 2005a; Bilbo and Tsang, 2010; Williamson et al., 2011). For the remainder of this review, we focus on the working hypothesis that microglial activation by non-infectious environmental factors in pregnancy, resulting in MIA, is a critical mechanism in the pathophysiology of a significant subtype of ASD, which if true has implications for a wide segment of the population, but also for treatment and prevention strategies. Air pollution, one of the most relevant and pervasive environmental toxins in the modern world, may be a particularly important threat to child health, and increasing evidence links exposure early in life to increased risk of ASD (Carter and Blizard, 2016; Roberts et al., 2013; Volk et al., 2013), a topic we expand upon in the sections that follow.
Environmental Factors and ASD
Diverse exposures to environmental factors, beyond infection, are increasingly recognized as a risk factor for ASD (reviewed in (Carter and Blizard, 2016)). These factors include pesticides, various components of air pollution (diesel exhaust, NO2, heavy metals), phthalates, polychlorinated biphenyls (e.g., BPA), and even dietary components such as common food allergens (e.g., gluten, casein) (Lin et al., 2016). All of these chemicals can impact immune or inflammatory pathways. Though the evidence is primarily derived from large scale epidemiological studies with their inherent caveats regarding causation, the animal literature in support of these associations at a mechanistic level is steadily increasing. Roadway exposures account for the majority of air pollution in the environment, of which diesel exhaust is a primary toxic component (Block and Levesque, 2009; Hartz et al., 2008; Inoue et al., 2006a; Inoue et al., 2006b; Inoue et al., 2006c). Air levels of diesel exhaust at the time and place of birth is one of the strongest and most consistent environmental links to ASD (Roberts et al., 2013; Rossignol et al., 2014). In animal studies, maternal air pollutant chemical exposures during pregnancy have been linked with asthma in offspring, even independent of postnatal exposures (Gilliland et al., 2000; Jedrychowski et al., 2004), suggesting similar impacts are present for the brain. Mechanistic studies have revealed that diesel exhaust particles markedly activate microglia in vitro and in vivo, in adult rodents and canines (Block and Levesque, 2009; Block and Calderon-Garciduenas, 2009; Block et al., 2012; Block et al., 2007; Costa et al., 2017) though less is known about its impact during the prenatal period.
We have recently determined the impact of maternal exposure to diesel exhaust on microglial development in the offspring mouse brain. Pregnant mice received aspirations of vehicle (VEH) or diesel exhaust particles (DEP) intermittently (every 2-3 days) throughout gestation, a regimen which induces significant lung inflammation in the mother similar to that observed in humans (Ghio et al., 2012). We tested the hypothesis that DEP-induced MIA would impact fetal microglial development, with significant consequences for brain development and long-term function. Microglial morphology is dynamic throughout development, and exhibits a stereotypical pattern of maturation in most brain regions, with a transition from a predominantly round or “amoeboid” morphology early in development (E10-14) to one with small cell bodies and long, thin processes by around postnatal day (P) 30 (Figure 1A). In the E18 cortex, maternal DEP exposure increased the number of round microglia compared to offspring of VEH-exposed dams, without any changes in the total number of cells, but notably only in males (Figure 1B; (Bolton et al., 2017)). By P30, when the majority of microglia in normal cortex exhibit small cell bodies with long, thin processes, the number of microglia with thick processes was increased by DEP, once again independent of changes in total number, and again only in males. These data suggest the maturation of microglia within the cortex was delayed by DEP in males. We saw the same effect in other brain regions including the hippocampus and hypothalamus (not shown), suggesting a global effect on the CNS. Notably, all of the DEP-induced changes in microglial morphology in males were dependent on the presence of the innate immune receptor toll-like-receptor (TLR) 4, as TLR4-/- mice did not exhibit DEP-induced effects in any brain region (Figure 1C). The impact of diesel particles on the airway inflammatory response is dependent on TLR4 (Inoue et al., 2006b), which is classically defined for its recognition of pathogen-associated molecular patterns (PAMPs, e.g. LPS). Increasing evidence shows the function of some TLRs extend beyond that of pathogen recognition to the discrimination of “danger” associated molecules (DAMPs) (Bianchi and Manfredi, 2009; Matzinger, 2002; Seong and Matzinger, 2004), including a class of endogenous molecules known as “alarmins” (e.g., hyaluronic acid, high mobility box group 1 protein (HMGB1), heat shock proteins, fibrinogen) which are released systemically in response to sterile inflammation or in response to multiple chemicals or toxins, e.g. as part of an inflammatory cascade or in response to cell death or tissue distress (Bianchi, 2007) (Figure 2). Interestingly, sensitized monocyte responses to TLR ligands are widely reported in individuals with ASD (Enstrom et al., 2010; Nadeem et al., 2017; Nazeen et al., 2016). These combined data suggest a model in which prenatal inflammation by air pollution delays the maturation of microglia particularly within the male brain, which we hypothesize leads to long-term alterations in their function, ultimately leading to abnormal neural circuit development and ASD-like behaviors.
Figure 1.

(A) Microglia transition from a round/amoeboid morphology early in development (∼E10, when they first begin to colonize the CNS) to a ramified morphology with small cell bodies and thin processes by P30. (B) DEP increases the number of round microglia in cortex at E18, and the number of thick microglia at P30, in males only, suggesting DEP delays their maturation and/or leads to their activation. There are similar effects in other brain regions including hippocampus (not shown) (C) All of the effects of DEP in males are dependent on TLR4. *p<0.05 for all; NS = not significantly different.
Figure 2.
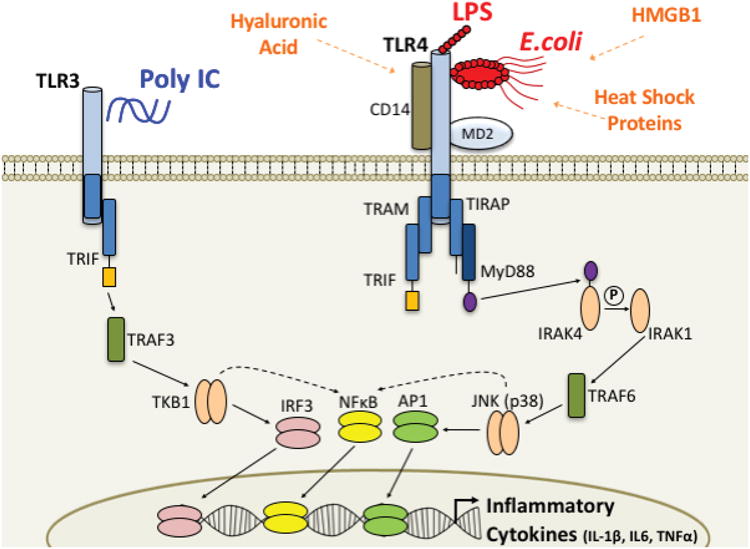
Toll-like receptors (TLR)s are the primary innate immune receptors which recognize pathogens in the body and brain. TLR4 recognizes bacteria, such as E. coli, and lipopolysaccharide (LPS), which is the cell wall component. TLR3 recognizes double-stranded RNA, the genetic code for viruses, as well as viral mimetics, such as Poly IC. Though TLR4 and TLR3 activation induces very different intracellular signaling pathways, activation of either receptor results in the transcription and translation of many similar inflammatory cytokines. The role of these receptors has recently expanded to include the recognition of more general “danger” associated molecular patterns (DAMPs). TLRs, including both TLR4 and TLR2, can identify and respond to DAMPs that are produced and released by nearby cells undergoing cell death or distress. To date, proteins that have been identified as DAMPs include hyaluronic acid, Heat Shock Proteins, and high mobility group box (HMGB) 1. CD14 = cluster of differentiation 14, TRIF = Toll-like receptor adaptor molecule, TIRAP = Toll-interleukin 1 receptor (TIR) domain containing adaptor protein, MyD88 = Myeloid differentiation primary response gene (88), IRAK = interleukin-1 receptor-associated kinase, TRAF = TNF receptor-associated factor, JNK = c-Jun N-terminal kinase, TKB1 = a tyrosine kinase, AP1 = activator protein 1 transcription factor, NFκB = nuclear factor kappa-light-chain-enhancer of activated B cells transcription factor, IRF3 = interferon regulatory factor 3 transcription factor. Adapted from (Schwarz and Bilbo, 2011).
Based on the hypothesis that perturbations of normal microglial development have significant consequences for neural development and thus behavioral function throughout the lifespan, we assessed learning and memory using a fear conditioning test, and anxiety using an elevated zero maze test, in young adult mice exposed prenatally to DEP or VEH; however, there was no impact of DEP on either measure (Bolton et al., 2013). These data suggest that maternal exposure to air pollution, even at a relatively high dose that induces inflammation in the mother and alters microglial development in the fetal (male) brain, is insufficient to cause changes in behavior. Notably, the effect sizes of even the largest single environmental factor associations with ASD are quite small. This strongly suggests that it is the combination of multiple perinatal exposures that increases vulnerability in the offspring. Most exposure models from both the pre-clinical and epidemiological literatures suffer from oversimplification, with single factors in isolation, which simplifies models but lends little insight into mechanisms by which diverse environmental exposures combine or synergize to cause pathology. A growing body of research suggests that maternal well-being during pregnancy is a crucial determinant of lifelong physical and mental health of the offspring (Case et al., 2005; Hackman et al., 2010; Susser et al., 1999). Chemical toxins such as lead exposure are well known to adversely affect brain development (Needleman et al. 1990; Weiss and Landrigan 2000). However, “social toxins”, such as violence, poverty, and other factors that generate psychological stress in mothers and children, have in recent years gained recognition as risk factors that can also alter the trajectory of brain development (Tamayo et al., 2017; Wright et al., 2010). For example, one study noted an association between air pollution and asthma only in children that were also living with a chronic stressor (e.g. domestic violence) (Clougherty et al. 2007). Similarly, parental stress can increase the effect of in utero toxin exposures (i.e., tobacco smoke) on childhood asthma risk (Shankardass et al. 2009). In such cases, stress may increase vulnerability, permitting a toxin to initiate significant injury to physiological systems when it would have been insufficient to do so in isolation. We hypothesize these synergistic effects of stress and pollutants are possible because they likely act on common biological systems, such as innate immune pathways (Frank et al. 2007; Levesque et al. 2011) within the developing nervous system. Notably, glucocorticoids may upregulate TLRs on microglia, augmenting subsequent neuroinflammatory responses (Frank et al. 2010; Gárate et al. 2012). These findings could explain why a single exposure or risk factor in isolation is a modest predictor of ASD risk (Kinney et al., 2008a; Kinney et al., 2008b; Magnusson et al., 2012; Roberts et al., 2014).
Based on this intriguing literature, we developed a mouse model that combines prenatal exposure to DEP, as described previously, with a novel model of maternal stress (MS), in which the nesting material within the cage is restricted during the final week of pregnancy, a psychological stressor designed to model poor housing conditions (Rice et al., 2008). We predicted the combination of prenatal exposures would activate the maternal immune system, and thereby impact the developing fetal brain (Figure 3). We found significant impacts of combined maternal DEP+MS on offspring communication and social behavior early in life, two of the hallmark behavioral deficits observed in ASD (Towbin et al., 2005; Truitt et al., 2007; White et al., 2009). We assessed ultrasonic vocalizations (USVs) at P5, during a 1 min task in which pups were separated from dam and littermates, and found a significant impact of DEP+MS only in males (Figure 3B). We next assessed social exploratory behavior at P15 (in a behaviorally naïve cohort), a time period when pups become more independent, in a task that assesses time spent in home cage bedding vs. unfamiliar bedding (collected from a conspecific dam and pups). Control pups explored both areas of the arena equally, whereas DEP+MS males and females spent time almost exclusively in home cage bedding. Moreover, male DEP+MS pups took significantly longer to enter the unfamiliar bedding, and overall activity was higher in DEP+MS pups compared to controls (Figure 3C). Thus, DEP+MS males immediately entered the home nest bedding, and were hyperactive in that area once they arrived, with virtually no exploration of the unfamiliar bedding, likely indicative of profound anxiety. Taken together, there are clear social and communication alterations in DEP+MS pups, especially males, at ages similar to the emergence of ASD in humans.
Figure 3.

(A) Experimental timeline, treatments, and outcomes assessed. (B) USVs were collected during a 1 min separation from dam and littermates. (C) P15 pups were placed into the center of a novel arena with equal portions of home cage vs. unfamiliar nest material, and total activity, latency to enter and time spent in each portion were assessed. (D) Cognitive and anxiety testing in adulthood, see text for details. (E) Inflammatory gene expression and (F) cytokine protein in whole brain at P30; *p<0.05. VEH = vehicle control, DEP = diesel exhaust particles, MS = maternal stress.
We also assessed learning and memory in young adult (P60) mice from each prenatal treatment group using a fear-conditioning paradigm, as before. Cognitive deficits are a core component of many neurodevelopmental disorders, including ASD (DeLong, 1992; Pellicano et al., 2006; Piven and Palmer, 1997; Rutter, 1983; Sheinkopf, 2005; Solomon et al., 2009). In this task, mice receive a series of foot shocks in a novel context, and increased freezing in that context on a later test day is indicative of intact memory (more freezing = better memory). This type of contextual memory (i.e., memory for the conditioned context) depends on the hippocampus, which is densely populated with glucocorticoid and innate immune receptors (e.g., TLRs), and is often vulnerable to environmental perturbation (stress, trauma) (Williamson and Bilbo, 2013). Connectivity between the hippocampus and cortex is impaired in an animal model of ASD (Zhan et al., 2014). Males but not females exposed prenatally to combined exposures (DEP+MS) showed contextual memory deficits on the test day. We tested the same animals for anxiety using an elevated zero maze, in which increased time in the dark arms is indicative of greater anxiety. Again, anxiety-like behavior is significantly increased only in DEP+MS males (Bolton et al., 2013) (Figure 3D).
Mechanisms Underlying Long-term Changes in Brain and Behavior
What are the potential mechanisms underlying these persistent behavioral deficits in combined exposure mice? DEP exposure alone impacts the maturation of microglia within the male brain, as described previously, but this is insufficient to impact behavior. We have demonstrated in a different animal model that bacterial infection in newborn rats similarly induces changes in microglial activation and development, and results in profound cognitive deficits in adulthood. However, the deficit is only observed if the rats infected as newborns receive a systemic injection of LPS around the time of learning (Bilbo et al., 2005a; Bilbo et al., 2005b). This low dose of LPS is insufficient to impact memory in control rats, as is the neonatal infection in isolation, without the “second hit”. Subsequent experiments demonstrated the systemic LPS causes the microglia of previously infected rats to respond more vigorously, even months later, with an overproduction of the inflammatory cytokine IL-1β, which impairs memory (Williamson et al., 2011), potentially via the downregulation of brain-derived-neurotrophic-factor (Bilbo et al., 2008), which is important for memory consolidation. These data bear a striking similarity to the phenomenon of immunological or glial “priming” that is well described in the neurodegeneration and aging literatures (Cunningham et al., 2009; Godbout and Johnson, 2006; Hennessy et al., 2017; Perry, 2004; Perry et al., 2007; Perry et al., 2010), and increasingly within the delirium literature as well (Maclullich et al., 2013; Sanders et al., 2014). A central characteristic of glial priming is the transition to and maintenance of an “activated” morphology (e.g. amoeboid or reactive) in response to an initial stimulus, e.g. infection, trauma, or cumulative aging alone. However, primed glia do not chronically produce cytokines and other pro-inflammatory mediators typical of cells in an acutely activated state. Upon challenge, however, such as infection or injury in the periphery, these primed cells will over-produce cytokines within the brain compared to cells that were not previously primed or sensitized (Perry et al., 2002). Notably, we have already demonstrated that adult mice born to DEP-exposed dams gain more weight when placed on a high fat “inflammatory” diet as adults, and exhibit greater central and peripheral measures of inflammation; and these effects are once again greater in males than females (Bolton et al., 2014; Bolton et al., 2012).
These collective data suggested to us that microglia may be sensitized or “primed” by DEP, and subsequently over respond to the secondary challenge of maternal stress, which would have persistent consequences for neural development and function. To broadly assess neuroinflammatory changes within the brains of mice in each condition, we measured inflammatory gene expression in whole forebrain of offspring from each group at P30 using a pathway-focused qPCR Array. We analyzed candidate genes implicated within the peripheral immune system in autoimmune disorders such as asthma, which are known to be impacted by DEP and which we suspected may also be altered in the brain. Only 2 genes were significantly altered, and only in the male offspring of DEP+MS dams: toll-like-receptor (TLR) 4 and caspase 1, the enzyme which converts the pro-inflammatory cytokine IL-1β into its active form downstream of TLR4 ligation (Figure 3E). As introduced previously, TLRs are a family of highly conserved pattern recognition receptor proteins that transduce signals through cytosolic adapter proteins (e.g. myeloid differentiation factor 88 (MyD88)) following ligation by PAMPS (e.g. LPS) or DAMPS (e.g. HMGB1), that link to downstream signaling cascades, notably the activation of the transcription factor NF-kB, which is key to subsequent cytokine release. TLRs 2, 3, and 4 are the best characterized within the mammalian CNS, and growing evidence demonstrates their recognition of putative DAMPS, including a wide array of environmental factors that are hypothesized to directly or indirectly activate TLRs (Seong and Matzinger, 2004); Figure 2; notably TLR2 and 4 are primarily expressed by microglia within the CNS (Zhang et al., 2014).
The “double hit” of DEP plus maternal stress exposure required to lend significant, persistent gene changes in male mice is intriguing given the critical function of TLRs within the inflammasome pathway. IL-1β is transcribed by immune cells including microglia as a larger pro-hormone, pro-IL-1β, which must be cleaved by caspase-1 to form the biologically active, mature form of the protein. The process of cleaving pro-IL-1β often requires the formation of an inflammasome, an intracellular multiprotein complex that mediates processing and maturation of IL-1β via activation of caspase-1 (Martinon et al., 2009). Importantly, signaling at TLR4 initiates the intracellular signaling cascade to activate NF-kB and the subsequent production of Nod-Like-Receptor family Pyrin domain containing 3 (NLRP3) (Kawai and Akira, 2010), which is a critical step in the formation of the inflammasome (Figure 4). The inflammasome is of particular interest for our model because it requires a priming step and an activation step (Hornung and Latz, 2010), and there is evidence that glucocorticoids can act as a priming stimulus (Busillo and Cidlowski, 2013; Frank et al., 2016). Our data suggest the opposite may also occur; i.e. that DEP exposure (or, presumably, a broad array of environmental exposures) is the priming stimulus, and maternal stress-induced DAMP production is the activation step. Both DEP and stress are likely activating the inflammasome within microglia indirectly via the release of an endogenous DAMP(s), which remain to be characterized. Notably, a recent report shows multiple inflammasome complexes are activated in peripheral blood cells of individuals with ASD, along with increased IL-1β (Saresella et al., 2016b), suggesting that regardless of initial priming/activation stimuli, the inflammasome pathway may represent a novel target for treatment.
Figure 4.
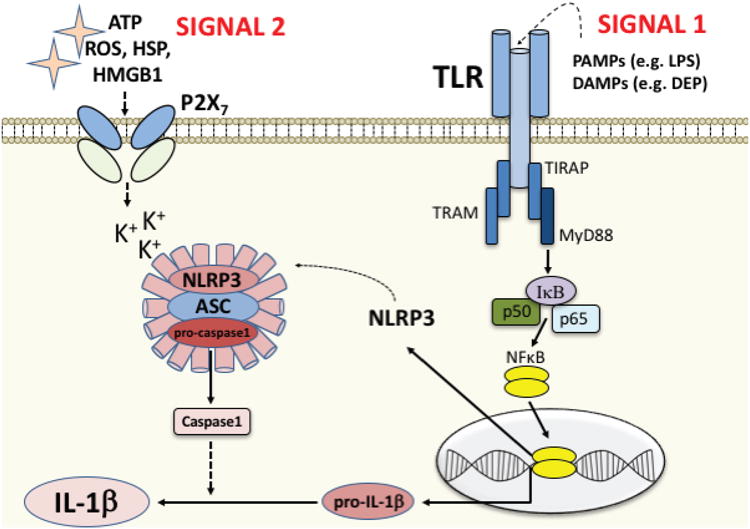
The process of cleaving pro-IL-1β into the mature, active protein often requires the formation of an inflammasome, an intracellular multiprotein complex that mediates processing and maturation of IL-1β via activation of caspase-1. This signaling cascade requires both a priming step (SIGNAL 1, acting on the TLR), and an activation step (SIGNAL 2, acting on, e.g. a purinergic receptor such as P2X7 or a variety of other scavenger receptors, see Hornung and Latz, 2010). SIGNAL 1 candidates include both exogenous ligands/PAMPs such as LPS, and endogenous “danger” signals/DAMPs that are released in response to toxins such as DEP (or cellular damage induced by DEP). Ligation of the TLR by SIGNAL 1 initiates the intracellular signaling cascade to activate the transcription factor NFkB and the subsequent production of pro-IL-1β, as well as Nod-Like-Receptor family Pyrin domain containing 3 (NLRP3). NLRP3 then translocates to the cytosol to form a complex with the proteins ASC and pro-caspase1, a critical step in the formation of the inflammasome. SIGNAL 2 candidates include ATP, reactive oxygen species (ROS), heat shock proteins (HSP), HMGB1, and many others, released in response to cellular distress or tissue damage. This second signal activates the inflammasome complex for cleavage of pro-IL-1β into IL-1β and release.
Consistent with the hypothesis of inflammasome activation, male offspring exposed to DEP+MS exhibit consistently increased levels of IL-1β protein along with increased caspase-1 mRNA, and decreased levels of anti-inflammatory IL-10 protein within the hypothalamus, hippocampus, and cortex, whereas the same brain regions in females showed the opposite pattern. Overall, DEP+MS males exhibit a greater pro-inflammatory bias (IL-1β/IL-10 ratio) than all other groups (Figure 3F), while females exhibit an intriguing anti-inflammatory bias driven by high IL-10 (Bolton et al., 2013). This constitutive change in cytokine protein was nonetheless quite surprising, as cytokine expression is exquisitely and tightly regulated within the CNS (Yirmiya and Goshen, 2011); however, a persistent neuroinflammatory process would agree with the stable pathophysiology thought to underlie ASD and associated behavioral dysfunction. Notably, elevated circulating levels of IL-1β have often been reported in ASD, though it is unclear if this is true for the brain (Masi et al., 2015; Saresella et al., 2016a). Subsequent analyses in our experiments revealed striking correlations between the behavioral deficits in male mice, and their brain levels of IL-1β. Specifically, high brain levels of IL-1β in DEP+MS males were significantly correlated with impaired memory and increased anxiety (Bolton et al., 2013). Moreover, these same mice produce exaggerated levels of peripheral IL-1β in response to a systemic LPS injection (Bolton et al., 2014), similar to that reported in ASD patients (Masi et al., 2015; Saresella et al., 2016a).
In sum, these data suggest a persistent priming or sensitization of the TLR/IL1/inflammasome pathway in this multiple exposure MIA model, especially in males, which lends enduring ASD-relevant behavioral deficits. These data are compelling given a recent transcriptomic meta-analysis of control vs. ASD genetic data sets showing a significant convergence onto innate immune genes - and TLR genes in particular - between ASD and co-morbid conditions such as asthma, inflammatory bowel disease, and others (Nazeen et al., 2016), and lend further strength to the hypothesis that there is a meaningful, neuroinflammation-linked subtype of ASD.
Conclusions
Taken together, both the associations between immune system dysfunction and ASD, and the list of environmental contributors to ASD, appear to be increasing. Existing epidemiology and association studies in humans are intriguing but difficult to ascribe causality; thus there is an urgent need for animal models that incorporate multiple factors in a rigorous and mechanistic way. Dr. Paul Patterson laid the critical groundwork for the hypothesis that MIA by infection leads to altered neural development and thus behavioral dysfunction throughout life, with relevance to ASD and other neurodevelopmental disorders. The important work of many labs has just begun to build upon this foundation, both to unravel the mysteries of how and why ASD rates continue to rise, and to determine if and how immune system (dys) function in response to diverse environmental factors and gene × environment interactions during neurodevelopment may hold an important key. We believe this knowledge will be critical for prevention and treatment going forward.
Acknowledgments
This work has been supported in part by NIH (NIMH) R01 MH101183 and NIH (EHS) R01 ES025549 to SDB.
References
- Ashwood P, Van de Water J. A review of autism and the immune response. Clinical & developmental immunology. 2004;11:165–174. doi: 10.1080/10446670410001722096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Wakefield AJ. Immune activation of peripheral blood and mucosal CD3+ lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms. Journal of neuroimmunology. 2006;173:126–134. doi: 10.1016/j.jneuroim.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Wills S, Van de Water J. The immune response in autism: a new frontier for autism research. J Leukoc Biol. 2006;80:1–15. doi: 10.1189/jlb.1205707. [DOI] [PubMed] [Google Scholar]
- Atladottir HO, Thorsen P, Schendel DE, Ostergaard L, Lemcke S, Parner ET. Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders: a Danish cohort study. Archives of pediatrics & adolescent medicine. 2010;164:470–477. doi: 10.1001/archpediatrics.2010.9. [DOI] [PubMed] [Google Scholar]
- Batinic B, Santrac A, Divovic B, Timic T, Stankovic T, Obradovic A, Joksimovic S, Savic MM. Lipopolysaccharide exposure during late embryogenesis results in diminished locomotor activity and amphetamine response in females and spatial cognition impairment in males in adult, but not adolescent rat offspring. Behavioural brain research. 2016;299:72–80. doi: 10.1016/j.bbr.2015.11.025. [DOI] [PubMed] [Google Scholar]
- Bauman MD, Iosif AM, Ashwood P, Braunschweig D, Lee A, Schumann CM, Van de Water J, Amaral DG. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Translational Psychiatry. 2013;3:e278. doi: 10.1038/tp.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman MD, Iosif AM, Smith SE, Bregere C, Amaral DG, Patterson PH. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biological psychiatry. 2014;75:332–341. doi: 10.1016/j.biopsych.2013.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KG. Autism, asthma, inflammation, and the hygiene hypothesis. Medical hypotheses. 2007;69:731–740. doi: 10.1016/j.mehy.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of leukocyte biology. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Bianchi ME, Manfredi AA. Immunology. Dangers in and out. Science (New York, N Y) 2009;323:1683–1684. doi: 10.1126/science.1172794. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Barrientos RM, Eads AS, Northcutt A, Watkins LR, Rudy JW, Maier SF. Early-life infection leads to altered BDNF and IL-1beta mRNA expression in rat hippocampus following learning in adulthood. Brain, behavior, and immunity. 2008;22:451–455. doi: 10.1016/j.bbi.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. Journal of Neuroscience. 2005a;25:8000–8009. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behavioral neuroscience. 2005b;119:293–301. doi: 10.1037/0735-7044.119.1.293. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Frontiers in behavioral neuroscience. 2009;3:1–14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Frontiers in neuroendocrinology. 2012;33:267–286. doi: 10.1016/j.yfrne.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Smith SH, Schwarz JM. A Lifespan Approach to Neuroinflammatory and Cognitive Disorders: A Critical Role for Glia. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2011 doi: 10.1007/s11481-011-9299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Tsang V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:2104–2115. doi: 10.1096/fj.09-144014. [DOI] [PubMed] [Google Scholar]
- Birtwell KB, Willoughby B, Nowinski L. Social, cognitive, and behavioral development of children and adolescents with autism spectrum disorder (Chapter 2) In: Mcdougle CJ, editor. Primer on Autism Spectrum Disorder. Oxford University Press; New York, NY: 2016. pp. 19–30. [Google Scholar]
- Blaylock RL. A possible central mechanism in autism spectrum disorders, part 2: immunoexcitotoxicity. Alternative Therapeutics and Health Medicine. 2009;15:60–67. [PubMed] [Google Scholar]
- Blaylock RL, Strunecka A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Current Medicinal Chemistry. 2009;16:157–170. doi: 10.2174/092986709787002745. [DOI] [PubMed] [Google Scholar]
- Block M, Levesque S. Diesel exhaust particles cause neuroinflammation and prime microglia. Society for Neuroscience Abstracts; Chicago, IL: 2009. [Google Scholar]
- Block ML, Calderon-Garciduenas L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends in neurosciences. 2009;32:506–516. doi: 10.1016/j.tins.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Elder A, Auten RL, Bilbo SD, Chen H, Chen JC, Cory-Slechta DA, Costa D, Diaz-Sanchez D, Dorman DC, Gold DR, Gray K, Jeng HA, Kaufman JD, Kleinman MT, Kirshner A, Lawler C, Miller DS, Nadadur SS, Ritz B, Semmens EO, Tonelli LH, Veronesi B, Wright RO, Wright RJ. The outdoor air pollution and brain health workshop. Neurotoxicology. 2012;33:972–984. doi: 10.1016/j.neuro.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews Neuroscience. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bolton JL, Auten RL, Bilbo SD. Prenatal air pollution exposure induces sexually dimorphic fetal programming of metabolic and neuroinflammatory outcomes in adult offspring. Brain, behavior, and immunity. 2014;37:30–44. doi: 10.1016/j.bbi.2013.10.029. [DOI] [PubMed] [Google Scholar]
- Bolton JL, Huff NC, Smith SH, Mason SN, Foster WM, Auten RL, Bilbo SD. Maternal stress and effects of prenatal air pollution on offspring mental health outcomes in mice. Environmental health perspectives. 2013;121:1075–1082. doi: 10.1289/ehp.1306560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Marinero S, Hassanzadeh T, Natesan D, Le D, Belliveau C, Mason SN, Auten RL, Bilbo SD. Gestational Exposure to Air Pollution Alters Cortical Volume, Microglial Morphology, and Microglia-Neuron Interactions in a Sex-Specific Manner. Frontiers in Synaptic Neuroscience. 2017;9:10. doi: 10.3389/fnsyn.2017.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Smith SH, Huff NC, Gilmour MI, Foster WM, Auten RL, Bilbo SD. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J. 2012;26:4743–4754. doi: 10.1096/fj.12-210989. [DOI] [PubMed] [Google Scholar]
- Borrell J, Vela JM, Arevalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats. Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2002;26:204–215. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- Braunschweig D, Golub MS, Koenig CM, Qi L, Pessah IN, Van de Water J, Berman RF. Maternal autism-associated IgG antibodies delay development and produce anxiety in a mouse gestational transfer model. Journal of neuroimmunology. 2012;252:56–65. doi: 10.1016/j.jneuroim.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimberg L, Mader S, Jeganathan V, Berlin R, Coleman TR, Gregersen PK, Huerta PT, Volpe BT, Diamond B. Caspr2-reactive antibody cloned from a mother of an ASD child mediates an ASD-like phenotype in mice. Molecular psychiatry. 2016;21:1663–1671. doi: 10.1038/mp.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Developmental Neurobiology. 2012;72:1272–1276. doi: 10.1002/dneu.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends in Endocrinology and Metabolism. 2013;24:109–119. doi: 10.1016/j.tem.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatric research. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Canetta SE, Brown AS. Prenatal Infection, Maternal Immune Activation, and Risk for Schizophrenia. Translational Neuroscience. 2012;3:320–327. doi: 10.2478/s13380-012-0045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga M, Hansen RL, Hertz-Piccotto I, Van de Water J, Ashwood P. Increased anti-phospholipid antibodies in autism spectrum disorders. Mediators of Inflammation. 2013;2013:935608. doi: 10.1155/2013/935608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CJ, Blizard RA. Autism genes are selectively targeted by environmental pollutants including pesticides, heavy metals, bisphenol A, phthalates and many others in food, cosmetics or household products. Neurochemistry International. 2016 doi: 10.1016/j.neuint.2016.10.011. [DOI] [PubMed] [Google Scholar]
- Case A, Fertig A, Paxson C. The lasting impact of childhood health and circumstance. Journal of Health Economics. 2005;24:365–389. doi: 10.1016/j.jhealeco.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Chan WY, Kohsaka S, Rezaie P. The origin and cell lineage of microglia: new concepts. Brain Research Reviews. 2007;53:344–354. doi: 10.1016/j.brainresrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Comi AM, Zimmerman AW, Frye VH, Law PA, Peeden JN. Familial clustering of autoimmune disorders and evaluation of medical risk factors in autism. Journal of Child Neurology. 1999;14:388–394. doi: 10.1177/088307389901400608. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- Costa LG, Cole TB, Coburn J, Chang YC, Dao K, Roque PJ. Neurotoxicity of traffic-related air pollution. Neurotoxicology. 2017;59:133–139. doi: 10.1016/j.neuro.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadros MA, Navascues J. The origin and differentiation of microglial cells during development. Progress in neurobiology. 1998;56:173–189. doi: 10.1016/s0301-0082(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, Rawlins JN, Perry VH. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biological psychiatry. 2009;65:304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton P, Deacon R, Blamire A, Pike M, McKinlay I, Stein J, Styles P, Vincent A. Maternal neuronal antibodies associated with autism and a language disorder. Annals of Neurology. 2003;53:533–537. doi: 10.1002/ana.10557. [DOI] [PubMed] [Google Scholar]
- DeLong GR. Autism, amnesia, hippocampus, and learning. Neuroscienceand Biobehavioral Reviews. 1992;16:63–70. doi: 10.1016/s0149-7634(05)80052-1. [DOI] [PubMed] [Google Scholar]
- Derecki NC, Privman E, Kipnis J. Rett syndrome and other autism spectrum disorders-brain diseases of immune malfunction? Molecular psychiatry. 2010;15:355–363. doi: 10.1038/mp.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enstrom AM, Onore CE, Van de Water JA, Ashwood P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain, behavior, and immunity. 2010;24:64–71. doi: 10.1016/j.bbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez de Cossio L, Guzman A, van der Veldt S, Luheshi GN. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain, behavior, and immunity. 2017;63:88–98. doi: 10.1016/j.bbi.2016.09.028. [DOI] [PubMed] [Google Scholar]
- Fidel PL, Jr, Romero R, Wolf N, Cutright J, Ramirez M, Araneda H, Cotton DB. Systemic and local cytokine profiles in endotoxin-induced preterm parturition in mice. American Journal of Obstetrics and Gynecology. 1994;170:1467–1475. doi: 10.1016/s0002-9378(94)70180-6. [DOI] [PubMed] [Google Scholar]
- Foley KA, MacFabe DF, Kavaliers M, Ossenkopp KP. Sexually dimorphic effects of prenatal exposure to lipopolysaccharide, and prenatal and postnatal exposure to propionic acid, on acoustic startle response and prepulse inhibition in adolescent rats: relevance to autism spectrum disorders. Behavioural brain research. 2015;278:244–256. doi: 10.1016/j.bbr.2014.09.032. [DOI] [PubMed] [Google Scholar]
- Fox-Edmiston E, Van de Water J. Maternal Anti-Fetal Brain IgG Autoantibodies and Autism Spectrum Disorder: Current Knowledge and its Implications for Potential Therapeutics. CNS Drugs. 2015;29:715–724. doi: 10.1007/s40263-015-0279-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Weber MD, Watkins LR, Maier SF. Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders. Neurobiology of Stress. 2016;4:62–70. doi: 10.1016/j.ynstr.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Tsuchihashi Y, Kitamura T. Origin, morphology and function of the microglia. Progress in Clinical Biology Research. 1981;59A:141–169. [PubMed] [Google Scholar]
- Gao Y, Liu L, Li Q, Wang Y. Differential alterations in the morphology and electrophysiology of layer II pyramidal cells in the primary visual cortex of a mouse model prenatally exposed to LPS. Neuroscience letters. 2015;591:138–143. doi: 10.1016/j.neulet.2015.02.043. [DOI] [PubMed] [Google Scholar]
- Ghio AJ, Smith CB, Madden MC. Diesel exhaust particles and airway inflammation. Current Opinion in Pulmonary Medicine. 2012;18:144–150. doi: 10.1097/MCP.0b013e32834f0e2a. [DOI] [PubMed] [Google Scholar]
- Gilliland FD, Berhane K, McConnell R, Gauderman WJ, Vora H, Rappaport EB, Avol E, Peters JM. Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax. 2000;55:271–276. doi: 10.1136/thorax.55.4.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (New York, N Y) 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Young DG, Woodward J, Brown DC, Lachman LB. Interleukin-1 is an astroglial growth factor in the developing brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1988;8:709–714. doi: 10.1523/JNEUROSCI.08-02-00709.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Neurologic clinics. 2006;24:521–538. doi: 10.1016/j.ncl.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology. 2005;48:903–917. doi: 10.1016/j.neuropharm.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ. Microglia: biology and pathology. Acta neuropathologica. 2010;119:89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- Hackman DA, Farah MJ, Meaney MJ. Socioeconomic status and the brain: mechanistic insights from human and animal research. Nature Reviews Neuroscience. 2010;11:651–659. doi: 10.1038/nrn2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Block ML, Hong JS, Miller DS. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J. 2008;22:2723–2733. doi: 10.1096/fj.08-106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy E, Gormley S, Lopez-Rodriguez AB, Murray C, Murray C, Cunningham C. Systemic TNF-alpha produces acute cognitive dysfunction and exaggerated sickness behavior when superimposed upon progressive neurodegeneration. Brain, behavior, and immunity. 2017;59:233–244. doi: 10.1016/j.bbi.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. European Journal of Immunology. 2010;40:620–623. doi: 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Takano H, Sakurai M, Oda T, Tamura H, Yanagisawa R, Shimada A, Yoshikawa T. Pulmonary exposure to diesel exhaust particles enhances coagulatory disturbance with endothelial damage and systemic inflammation related to lung inflammation. Experimental biology and medicine (Maywood, N J) 2006a;231:1626–1632. doi: 10.1177/153537020623101007. [DOI] [PubMed] [Google Scholar]
- Inoue K, Takano H, Yanagisawa R, Hirano S, Ichinose T, Shimada A, Yoshikawa T. The role of toll-like receptor 4 in airway inflammation induced by diesel exhaust particles. Archives of Toxicology. 2006b;80:275–279. doi: 10.1007/s00204-005-0040-6. [DOI] [PubMed] [Google Scholar]
- Inoue K, Takano H, Yanagisawa R, Hirano S, Kobayashi T, Ichinose T, Yoshikawa T. Effects of organic chemicals derived from ambient particulate matter on lung inflammation related to lipopolysaccharide. Archives of Toxicology. 2006c;80:833–838. doi: 10.1007/s00204-006-0105-1. [DOI] [PubMed] [Google Scholar]
- Jedrychowski W, Bendkowska I, Flak E, Penar A, Jacek R, Kaim I, Spengler JD, Camann D, Perera FP. Estimated risk for altered fetal growth resulting from exposure to fine particles during pregnancy: an epidemiologic prospective cohort study in Poland. Environmental health perspectives. 2004;112:1398–1402. doi: 10.1289/ehp.7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunology. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kinney DK, Miller AM, Crowley DJ, Huang E, Gerber E. Autism prevalence following prenatal exposure to hurricanes and tropical storms in Louisiana. Journal of autism and developmental disorders. 2008a;38:481–488. doi: 10.1007/s10803-007-0414-0. [DOI] [PubMed] [Google Scholar]
- Kinney DK, Munir KM, Crowley DJ, Miller AM. Prenatal stress and risk for autism. Neuroscience and Biobehavioral Reviews. 2008b;32:1519–1532. doi: 10.1016/j.neubiorev.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowiak P, Walker CK, Bremer AA, Baker AS, Ozonoff S, Hansen RL, Hertz-Picciotto I. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics. 2012;129:e1121–1128. doi: 10.1542/peds.2011-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritsen MB, Pedersen CB, Mortensen PB. Effects of familial risk factors and place of birth on the risk of autism: a nationwide register-based study. Journal of child psychology and psychiatry, and allied disciplines. 2005;46:963–971. doi: 10.1111/j.1469-7610.2004.00391.x. [DOI] [PubMed] [Google Scholar]
- Lin X, Cheng C, Terry P, Chen J, Cui H, Wu J. Rapid and sensitive detection of bisphenol a from serum matrix. Biosensors and Bioelectronics. 2016;91:104–109. doi: 10.1016/j.bios.2016.12.024. [DOI] [PubMed] [Google Scholar]
- Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia. 1993;7:9–18. doi: 10.1002/glia.440070105. [DOI] [PubMed] [Google Scholar]
- Maclullich AM, Anand A, Davis DH, Jackson T, Barugh AJ, Hall RJ, Ferguson KJ, Meagher DJ, Cunningham C. New horizons in the pathogenesis, assessment and management of delirium. Age and Ageing. 2013;42:667–674. doi: 10.1093/ageing/aft148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. Journal of Neuroscience. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiati I, Ong C, Lim XY, Tan JW, Ong AY, Patrycia F, Fung DS, Sung M, Poon KK, Howlin P. Anxiety symptoms in young people with autism spectrum disorder attending special schools: Associations with gender, adaptive functioning and autism symptomatology. Autism : the international journal of research and practice. 2016;20:306–320. doi: 10.1177/1362361315577519. [DOI] [PubMed] [Google Scholar]
- Magnusson C, Rai D, Goodman A, Lundberg M, Idring S, Svensson A, Koupil I, Serlachius E, Dalman C. Migration and autism spectrum disorder: population-based study. The British journal of psychiatry : the journal of mental science. 2012;201:109–115. doi: 10.1192/bjp.bp.111.095125. [DOI] [PubMed] [Google Scholar]
- Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain, behavior, and immunity. 2012;26:607–616. doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LA, Ashwood P, Braunschweig D, Cabanlit M, Van de Water J, Amaral DG. Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain, behavior, and immunity. 2008;22:806–816. doi: 10.1016/j.bbi.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annual Review of Immunology. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. Molecular psychiatry. 2015;20:440–446. doi: 10.1038/mp.2014.59. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science (New York, N Y) 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Miller LC, Isa S, LoPreste G, Schaller JG, Dinarello CA. Neonatal interleukin-1 beta, interleukin-6, and tumor necrosis factor: cord blood levels and cellular production. Journal of Pediatrics. 1990;117:961–965. doi: 10.1016/s0022-3476(05)80145-3. [DOI] [PubMed] [Google Scholar]
- Molloy CA, Morrow AL, Meinzen-Derr J, Dawson G, Bernier R, Dunn M, Hyman SL, McMahon WM, Goudie-Nice J, Hepburn S, Minshew N, Rogers S, Sigman M, Spence MA, Tager-Flusberg H, Volkmar FR, Lord C. Familial autoimmune thyroid disease as a risk factor for regression in children with Autism Spectrum Disorder: a CPEA Study. Journal of autism and developmental disorders. 2006a;36:317–324. doi: 10.1007/s10803-005-0071-0. [DOI] [PubMed] [Google Scholar]
- Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P, Altaye M, Wills-Karp M. Elevated cytokine levels in children with autism spectrum disorder. Journal of neuroimmunology. 2006b;172:198–205. doi: 10.1016/j.jneuroim.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Money J, Bobrow NA, Clarke FC. Autism and autoimmune disease: a family study. Journal of Autism and Childhood Schizophrenia. 1971;1:146–160. doi: 10.1007/BF01537954. [DOI] [PubMed] [Google Scholar]
- Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, Courchesne E, Everall IP. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biological psychiatry. 2010;68:368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- Mustafa MM, Lebel MH, Ramilo O, Olsen KD, Reisch JS, Beutler B, McCracken GH., Jr Correlation of interleukin-1 beta and cachectin concentrations in cerebrospinal fluid and outcome from bacterial meningitis. The Journal of pediatrics. 1989;115:208–213. doi: 10.1016/s0022-3476(89)80067-8. [DOI] [PubMed] [Google Scholar]
- Nadeem A, Ahmad SF, Bakheet SA, Al-Harbi NO, Al-Ayadhi LY, Attia SM, Zoheir KM. Toll-like receptor 4 signaling is associated with upregulated NADPH oxidase expression in peripheral T cells of children with autism. Brain, behavior, and immunity. 2017;61:146–154. doi: 10.1016/j.bbi.2016.12.024. [DOI] [PubMed] [Google Scholar]
- Nawa H, Takei N. Recent progress in animal modeling of immune inflammatory processes in schizophrenia: implication of specific cytokines. Neuroscience research. 2006;56:2–13. doi: 10.1016/j.neures.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Nazeen S, Palmer NP, Berger B, Kohane IS. Integrative analysis of genetic data sets reveals a shared innate immune component in autism spectrum disorder and its co-morbidities. Genome Biology. 2016;17:228. doi: 10.1186/s13059-016-1084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y, Cai Z, Rhodes PG. Disturbance of oligodendrocyte development, hypomyelination and white matter injury in the neonatal rat brain after intracerebral injection of lipopolysaccharide. Developmental Brain Research. 2003;140:205–214. doi: 10.1016/s0165-3806(02)00606-5. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science (New York, N Y) 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Vargas DL, Zimmerman AW. Immunity, neuroglia and neuroinflammation in autism. International Review of Psychiatry. 2005;17:485–495. doi: 10.1080/02646830500381930. [DOI] [PubMed] [Google Scholar]
- Pellicano E, Maybery M, Durkin K, Maley A. Multiple cognitive capabilities/deficits in children with an autism spectrum disorder: “weak” central coherence and its relationship to theory of mind and executive control. Development and psychopathology. 2006;18:77–98. doi: 10.1017/S0954579406060056. [DOI] [PubMed] [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain, behavior, and immunity. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Boche D. Atypical inflammation in the central nervous system in prion disease. Current opinion in neurology. 2002;15:349–354. doi: 10.1097/00019052-200206000-00020. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nature reviews immunology. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nature reviews neurology. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Piras IS, Haapanen L, Napolioni V, Sacco R, Van de Water J, Persico AM. Anti-brain antibodies are associated with more severe cognitive and behavioral profiles in Italian children with Autism Spectrum Disorder. Brain, behavior, and immunity. 2014;38:91–99. doi: 10.1016/j.bbi.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piven J, Palmer P. Cognitive deficits in parents from multiple-incidence autism families. Journal of child psychology and psychiatry, and allied disciplines. 1997;38:1011–1021. doi: 10.1111/j.1469-7610.1997.tb01618.x. [DOI] [PubMed] [Google Scholar]
- Pousset F. Developmental expression of cytokine genes in the cortex and hippocampus of the rat central nervous system. Brain research. Developmental brain research. 1994;81:143–146. doi: 10.1016/0165-3806(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Rakic S, Zecevic N. Programmed cell death in the developing human telencephalon. The European journal of neuroscience. 2000;12:2721–2734. doi: 10.1046/j.1460-9568.2000.00153.x. [DOI] [PubMed] [Google Scholar]
- Rezaie P, Male D. Mesoglia & microglia--a historical review of the concept of mononuclear phagocytes within the central nervous system. Journal of the history of the neurosciences. 2002;11:325–374. doi: 10.1076/jhin.11.4.325.8531. [DOI] [PubMed] [Google Scholar]
- Rice CJ, Sandman CA, Lenjavi MR, Baram TZ. A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology. 2008;149:4892–4900. doi: 10.1210/en.2008-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson-Burns SM, Tyler KL. Regional differences in viral growth and central nervous system injury correlate with apoptosis. Journal of Virology. 2004;78:5466–5475. doi: 10.1128/JVI.78.10.5466-5475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AL, Koenen KC, Lyall K, Ascherio A, Weisskopf MG. Women's posttraumatic stress symptoms and autism spectrum disorder in their children. Research in autism spectrum disorders. 2014;8:608–616. doi: 10.1016/j.rasd.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AL, Lyall K, Hart JE, Laden F, Just AC, Bobb JF, Koenen KC, Ascherio A, Weisskopf MG. Perinatal air pollutant exposures and autism spectrum disorder in the children of Nurses' Health Study II participants. Environmental health perspectives. 2013;121:978–984. doi: 10.1289/ehp.1206187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier PM. Chronology of neuron development: animal studies and their clinical implications. Developmental Medicine and Child Neurology. 1980;22:525–545. doi: 10.1111/j.1469-8749.1980.tb04363.x. [DOI] [PubMed] [Google Scholar]
- Rossi CC, Fuentes J, Van de Water J, Amaral DG. Brief Report: Antibodies reacting to brain tissue in Basque Spanish children with Autism Spectrum Disorder and their mothers. Journal of autism and developmental disorders. 2014;44:459–465. doi: 10.1007/s10803-013-1859-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol DA, Genuis SJ, Frye RE. Environmental toxicants and autism spectrum disorders: a systematic review. Translational Psychiatry. 2014;4:e360. doi: 10.1038/tp.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostene W, Guyon A, Kular L, Godefroy D, Barbieri F, Bajetto A, Banisadr G, Callewaere C, Conductier G, Rovere C, Melik-Parsadaniantz S, Florio T. Chemokines and chemokine receptors: new actors in neuroendocrine regulations. Frontiers in neuroendocrinology. 2011;32:10–24. doi: 10.1016/j.yfrne.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Rutter M. Cognitive deficits in the pathogenesis of autism. Journal of child psychology and psychiatry, and allied disciplines. 1983;24:513–531. doi: 10.1111/j.1469-7610.1983.tb00129.x. [DOI] [PubMed] [Google Scholar]
- Sanders RD, Coburn M, Cunningham C, Pandharipande P. Risk factors for postoperative delirium. Lancet Psychiatry. 2014;1:404–406. doi: 10.1016/S2215-0366(14)00012-1. [DOI] [PubMed] [Google Scholar]
- Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, Rainone V, Nemni R, Mancuso R, Clerici M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Molecular Neurodegeneration. 2016a;11:23. doi: 10.1186/s13024-016-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saresella M, Piancone F, Marventano I, Zoppis M, Hernis A, Zanette M, Trabattoni D, Chiappedi M, Ghezzo A, Canevini MP, la Rosa F, Esposito S, Clerici M. Multiple inflammasome complexes are activated in autistic spectrum disorders. Brain, behavior, and immunity. 2016b;57:125–133. doi: 10.1016/j.bbi.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz T, Chew LJ. Cytokines and myelination in the central nervous system. TheScientificWorldJournal. 2008;8:1119–1147. doi: 10.1100/tsw.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Schwartzer JJ, Careaga M, Chang C, Onore CE, Ashwood P. Allergic fetal priming leads to developmental, behavioral and neurobiological changes in mice. Translational Psychiatry. 2015;5:e543. doi: 10.1038/tp.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD. The Immune System and the Developing Brain. Morgan & Claypool Life Sciences 2011 [Google Scholar]
- Schwarz JM, Bilbo SD. Sex, glia, and development: interactions in health and disease. Hormones and behavior. 2012;62:243–253. doi: 10.1016/j.yhbeh.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nature reviews immunology. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- Sheinkopf SJ. Hot topics in autism: cognitive deficits, cognitive style, and joint attention dysfunction. Medicine and health, Rhode Island. 2005;88:152–153. 157–158. [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain, behavior, and immunity. 2009;23:116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer HS, Morris C, Gause C, Pollard M, Zimmerman AW, Pletnikov M. Prenatal exposure to antibodies from mothers of children with autism produces neurobehavioral alterations: A pregnant dam mouse model. Journal of neuroimmunology. 2009;211:39–48. doi: 10.1016/j.jneuroim.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007a;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007b;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon M, Ozonoff SJ, Ursu S, Ravizza S, Cummings N, Ly S, Carter CS. The neural substrates of cognitive control deficits in autism spectrum disorders. Neuropsychologia. 2009;47:2515–2526. doi: 10.1016/j.neuropsychologia.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann MN, Sourander A, Surcel HM, Hinkka-Yli-Salomaki S, Brown AS. Prenatal toxoplasmosis antibody and childhood autism. Autism research : official journal of the International Society for Autism Research. 2017;10:769–777. doi: 10.1002/aur.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and macrophages in the developing CNS. Neurotoxicology. 2001;22:619–624. doi: 10.1016/s0161-813x(01)00033-x. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta neuropathologica. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Xue QS. Life and death of microglia. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2009;4:371–379. doi: 10.1007/s11481-009-9163-5. [DOI] [PubMed] [Google Scholar]
- Susser EB, Brown A, Matte TD. Prenatal factors and adult mental and physical health. Canadian Journal of Psychiatry. 1999;44:326–334. doi: 10.1177/070674379904400402. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Sugihara G, Ouchi Y, Nakamura K, Futatsubashi M, Takebayashi K, Yoshihara Y, Omata K, Matsumoto K, Tsuchiya KJ, Iwata Y, Tsujii M, Sugiyama T, Mori N. Microglial activation in young adults with autism spectrum disorder. JAMA psychiatry. 2013;70:49–58. doi: 10.1001/jamapsychiatry.2013.272. [DOI] [PubMed] [Google Scholar]
- Sweeten TL, Bowyer SL, Posey DJ, Halberstadt GM, McDougle CJ. Increased prevalence of familial autoimmunity in probands with pervasive developmental disorders. Pediatrics. 2003a;112:e420. doi: 10.1542/peds.112.5.e420. [DOI] [PubMed] [Google Scholar]
- Sweeten TL, Posey DJ, McDougle CJ. High blood monocyte counts and neopterin levels in children with autistic disorder. American Journal of Psychiatry. 2003b;160:1691–1693. doi: 10.1176/appi.ajp.160.9.1691. [DOI] [PubMed] [Google Scholar]
- Tamayo YOM, Tellez-Rojo MM, Trejo-Valdivia B, Schnaas L, Osorio-Valencia E, Coull B, Bellinger D, Wright RJ, Wright RO. Maternal stress modifies the effect of exposure to lead during pregnancy and 24-month old children's neurodevelopment. Environment International. 2017;98:191–197. doi: 10.1016/j.envint.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theoharides TC, Tsilioni I, Patel AB, Doyle R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Translational Psychiatry. 2016;6:e844. doi: 10.1038/tp.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin KE, Pradella A, Gorrindo T, Pine DS, Leibenluft E. Autism spectrum traits in children with mood and anxiety disorders. Journal of child and adolescent psychopharmacology. 2005;15:452–464. doi: 10.1089/cap.2005.15.452. [DOI] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. Journal of Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt WA, Sajdyk TJ, Dietrich AD, Oberlin B, McDougle CJ, Shekhar A. From anxiety to autism: spectrum of abnormal social behaviors modeled by progressive disruption of inhibitory neuronal function in the basolateral amygdala in Wistar rats. Psychopharmacology. 2007;191:107–118. doi: 10.1007/s00213-006-0674-y. [DOI] [PubMed] [Google Scholar]
- Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophrenia research. 2001;47:27–36. doi: 10.1016/s0920-9964(00)00032-3. [DOI] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of Neurology. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Volk HE, Kerin T, Lurmann F, Hertz-Picciotto I, McConnell R, Campbell DB. Autism spectrum disorder: interaction of air pollution with the MET receptor tyrosine kinase gene. Epidemiology. 2014;25:44–47. doi: 10.1097/EDE.0000000000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk HE, Lurmann F, Penfold B, Hertz-Picciotto I, McConnell R. Traffic-related air pollution, particulate matter, and autism. JAMA psychiatry. 2013;70:71–77. doi: 10.1001/jamapsychiatry.2013.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Wu CH, Shieh JY, Wen CY. Microglial distribution and apoptosis in fetal rat brain. Developmental Brain Research. 2002;139:337–342. doi: 10.1016/s0165-3806(02)00584-9. [DOI] [PubMed] [Google Scholar]
- Wang X, Rousset CI, Hagberg H, Mallard C. Lipopolysaccharide-induced inflammation and perinatal brain injury. Seminars in Fetal and Neonatal Medicine. 2006;11:343–353. doi: 10.1016/j.siny.2006.04.002. [DOI] [PubMed] [Google Scholar]
- White SW, Oswald D, Ollendick T, Scahill L. Anxiety in children and adolescents with autism spectrum disorders. Clinical psychology review. 2009;29:216–229. doi: 10.1016/j.cpr.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson LL, Bilbo SD. Chemokines and the hippocampus: a new perspective on hippocampal plasticity and vulnerability. Brain, behavior, and immunity. 2013;30:186–194. doi: 10.1016/j.bbi.2013.01.077. [DOI] [PubMed] [Google Scholar]
- Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and memory: modulation by early-life infection. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:15511–15521. doi: 10.1523/JNEUROSCI.3688-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RJ, Visness CM, Calatroni A, Grayson MH, Gold DR, Sandel MT, Lee-Parritz A, Wood RA, Kattan M, Bloomberg GR, Burger M, Togias A, Witter FR, Sperling RS, Sadovsky Y, Gern JE. Prenatal maternal stress and cord blood innate and adaptive cytokine responses in an inner-city cohort. American journal of respiratory and critical care medicine. 2010;182:25–33. doi: 10.1164/rccm.200904-0637OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kaur C, Ling EA. Variation with age in the labelling of amoeboid microglial cells in rats following intraperitoneal or intravenous injection of a fluorescent dye. Journal of anatomy. 1993;182(Pt 1):55–63. [PMC free article] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain, behavior, and immunity. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Yoon BH, Romero R, Kim CJ, Jun JK, Gomez R, Choi JH, Syn HC. Amniotic fluid interleukin-6: a sensitive test for antenatal diagnosis of acute inflammatory lesions of preterm placenta and prediction of perinatal morbidity. American Journal of Obstetrics and Gynecology. 1995;172:960–970. doi: 10.1016/0002-9378(95)90028-4. [DOI] [PubMed] [Google Scholar]
- Yu HM, Yuan TM, Gu WZ, Li JP. Expression of glial fibrillary acidic protein in developing rat brain after intrauterine infection. Neuropathology. 2004;24:136–143. doi: 10.1111/j.1440-1789.2003.00539.x. [DOI] [PubMed] [Google Scholar]
- Zerbo O, Qian Y, Yoshida C, Grether JK, Van de Water J, Croen LA. Maternal Infection During Pregnancy and Autism Spectrum Disorders. Journal of autism and developmental disorders. 2013 doi: 10.1007/s10803-013-2016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nature neuroscience. 2014;17:400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]