

Abstract
The leading cause of synthetic graft failure includes thrombotic occlusion and intimal hyperplasia at the site of vascular anastomosis. Herein, we report a co- immobilization strategy of heparin and potent anti-neointimal drug (Mitogen Activated Protein Kinase II inhibitory peptide; MK2i) by using a tyrosinase-catalyzed oxidative reaction for preventing thrombotic occlusion and neointimal formation of synthetic vascular grafts. The binding of heparin–tyramine polymer (HT) onto the polycarprolactone (PCL) surface enhanced blood compatibility with significantly reduced protein absorption (64.7% decrease) and platelet adhesion (82.2% decrease) compared to bare PCL surface. When loading MK2i, 1) the HT depot surface gained high MK2i- loading efficiency through charge-charge interaction, and 2) this depot platform enabled long-term, controlled release over 4 weeks (92–272 μg/mL of MK2i). The released MK2i showed significant inhibitory effects on VSMC migration through down-regulated phosphorylation of target proteins (HSP27 and CREB) associated with intimal hyperplasia. In addition, it was found that the released MK2i infiltrated into the tissue with a cumulative manner in ex vivo human saphenous vein (HSV) model. This present study demonstrates that enzymatically HT-coated surface modification is an effective strategy to induce long-term MK2i release as well as hemocompatibility, thereby improving anti- neointimal activity of synthetic vascular grafts.
Keywords: Tyrosinase-triggered surface functionalization, Heparin, MK2 inhibitory peptide, Anti-thrombosis, Intimal hyperplasia, Synthetic vascular grafts
Graphical Abstract
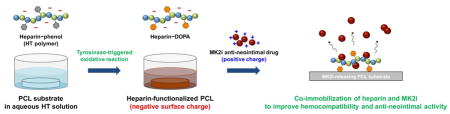
1. Introduction
Cardiovascular and peripheral vascular diseases are a leading cause of morbidity and mortality worldwide [1, 2]. Vascular grafting serves as a major treatment by inserting between the arterial and venous circulation for bypassing occlusions. Along with this clinical need, prosthetic vascular grafts composed of synthetic polymer (e.g., polyethylene terephthalate; Dacron, expanded polytetrafluoroethylene; ePTFE, polyurethane; PU, and polycarprolactone; PCL) have been developed [3, 4]. Although significant progress has been made in their design and application, two major issues resulting in synthetic graft failure are: 1) thrombus formation by non-specific protein/platelet adhesion, and 2) restenosis development by intimal hyperplasia. To address these clinical unmet needs, surface modification with bioactive and/or bioinert compounds would be an effective option to improve hemocompatibility and patency of synthetic vascular grafts.
At the early stage of synthetic vascular access, a foreign material exerts an unfavorable reaction with blood, leading to initiation of coagulation cascade with platelet aggregation [5]. Hence, incorporating antithrombotic and antiplatelet agents (e.g., heparin, hirudin, clopidogrel, aspirin, warfarin, and etc.) to the surface of blood-contacting biomaterials has been tested so far [6, 7]. Up to date, bioactive heparin coating on the polymer surfaces is still recognized as a gold-standard treatment for the enhanced blood compatibility. The interaction between heparin and antithrombin III (AT III) inactivates thrombin, factor Xa and other proteases involved in blood clotting pathway, thereby preventing thrombus formation [8]. Moreover, hydrophilic nature and negative charge of heparin compound were proved to have non- fouling effects against protein and platelet adhesion [9]. However, although heparin-bound materials have shown desirable anti-thrombogenic activity at blood–material interfaces, the restenosis occurrence proceeded by intimal hyperplasia at the late postsurgical stage still remain challenging for successful vascular grafting.
Intimal hyperplasia refers to excessive proliferation and migration of vascular smooth muscle cells (VSMCs) primarily in the arterial wall [10]. When a vascular graft is implanted, the mechanical and biochemical stresses (e.g., transforming growth factor-beta; TGF-β, platelet-derived growth factor; PDGF) triggered by vascular injury result in pathologic remodeling including intimal thickening and graft stenosis/occlusion, ultimately leading to graft failure [11]. Various drugs that can inhibit abnormal activities of VSMCs were employed to prevent development of intimal hyperplasia [12]. In particular, we have been studying potent inhibitory effects of a cell-penetrating peptide inhibitor of Mitogen Activated Protein Kinase II (MK2i) on neointimal hyperplasia, fibrosis and inflammation [7, 13]. MK2 is a downstream regulator of the TGF-β/p38 stress-activated protein kinase pathway that is implicated in intimal hyperplasia. Our previous studies have demonstrated that this MK2i peptide inhibits VSMC proliferation and migration as well as synthetic phenotypic modulation. In addition, the anti- neointimal effects of MK2i were also demonstrated in ex vivo and in vivo mouse/rabbit vein interposition models [14, 15].
While vein wall thickening continues over 12 weeks into arterial exposure, a 2–4 week sustained release profile of MK2i is ideal because the majority of VSMC proliferation and migration occur within this window, and MK2i inhibits the VSMC actions by its anti- hyperplasia effects [14–16]. However, there is no clear evidence for current drug release approaches to meet this expectation, and burst release at the early stage of drug elusion is considered as a major drawback. Hence, instead of blending or simple coating to base materials, strong interactions such as charge-charge interaction between cationic MK2i and depot material surface need to be investigated to achieve the goal. To design drug-eluting materials enabling sustained and controlled delivery of target drug, various positively or negatively charged polymers have been incorporated into biomaterials using surface modification techniques [17, 18]. In particular, a negatively charged heparin material has been extensively explored for drug- or gene-delivery applications because it provides attractive properties such as high-affinity with growth factors or positively charged therapeutics, excellent biocompatibility and favorable biological functions [19]. The effort continues to develop facile surface heparin modification method that enables long-term stability of the immobilized heparin, easy manipulation of drug release profile and high drug-loading efficacy for effective bioactivity of the target drug.
For the past few decades, various surface modification methods have been employed to immobilize bioactive heparin compound on the surface of biomaterials: typically, physical absorption, covalent chemical conjugation and photochemical attachment [20]. However, these approaches have shown some drawbacks such as short-term stability of the immobilized molecules, and complicated and time-consuming procedures. In addition, pre-treatment is sometimes required to introduce functional groups on the surface. Recently, bioinspired-catecholic reaction using 3,4-dihydroxyphenylalanine (DOPA) molecules derived from adhesive mussel proteins has shown great promise as a biomimetic surface modification method [21, 22]. DOPA- functionalized molecules can be immobilized on the versatile surfaces (e.g. metals, ceramics, and polymers) by a simple dipping method. During the reaction, DOPA is oxidized to active o-quinone (DOPA-quinone) that has surface adhesive properties. However, this DOPA molecule can easily lose their adhesiveness before treatment because of the molecular oxidation when exposed to air. Moreover, surface binding of DOPA requires long reaction time (> 12 h), which means that this process is uncontrollable and time-consuming. We previously reported a simple surface modification method using a tyrosinase-triggered oxidative reaction that can overcome the limitations of DOPA system [23, 24]. The tyrosinase catalyzes the molecular conversion from highly stable phenol moiety to adhesive DOPA/DOPA-quinone within a short time (< 1 h) while maintaining the benefits of DOPA system. Accordingly, we were able to control biological interactions at interfaces of implantable biomaterials by surface immobilization of phenol-functionalized bioactive and/or bioinert compounds (e.g., RGD-phenol and mPEG-phenol) [23, 24].
Herein, we report a co- immobilization strategy of heparin and MK2i peptide by using the tyrosinase-catalyzed oxidative reaction for sustained release of MK2i up to 4 weeks while preventing thrombotic occlusion and neointimal formation of synthetic vascular grafts. In this study, the heparin polymer was employed with two goals: 1) to prevent thrombus formation through their non- fouling properties, and 2) to load and induce the sustained release of MK2i via electrostatic interaction between drug and heparin. To prepare heparin- immobilized polycaprolactone (PCL–HT) surfaces, we first synthesized phenol-grafted heparin polymer (heparin–tyramine; HT), followed by surface immobilization of heparin with the tyrosinase treatment. We characterized the surface properties (i.e., heparin density, wettability, atomic composition) to verify heparin coating on the PCL surfaces, and investigated in vitro non- fouling properties of PCL–HT against protein/platelet adhesion for the antithrombotic effect. After loading cationic MK2i peptides on the heparinized polymer graft surface, we determined cumulative MK2i release kinetics over 28 days, and evaluated bioactivities of the released MK2i from the PCL substrates including its inhibitory effects on VSMC migration and phosphorylation of target proteins associated with intimal hyperplasia. Furthermore, we also confirmed if the released MK2i peptides can be delivered into human saphenous vein (HSV) tissue in an ex vivo model of adventitial delivery from external polymer wrapping.
2. Materials and Methods
2.1. Materials
Heparin sodium (molecular weight = 12,000–15,000 g/mol) was supplied from Acros Organics (Geel, Belgium). Polycaprolactone (average molecular weight = 80,000 g/mol; PCL), tyramine hydrocholoride (TA), 1-ethyl-3-(3-dimethylamino-propyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS), toluidine blue O (TBO) and tyrosinase (from mushroom, 3610 units/mg solid) were obtained from Sigma-Aldrich (St. Louis, MO, USA). MK2i peptide (YARAAARQARAKALARQLGVAA) was supplied from EZBiolab (Carmel, IN, USA). Dulbecco’s phosphate-buffered saline (DPBS) was purchased from Gibco BRL (Grand Island, NY, USA), and Tris-HCl buffer (10 mM, pH 8.5) was purchased from bioWORLD (Dublin, OH, USA).
For in vitro cell studies, rat vascular smooth muscle cells (VSMCs) were purchased from ATCC (Manassas, VA, USA), and were cultured at 37 °C and 5% CO2 in Dulbecco’s modified Eagle medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (PS). For western blot analysis, the specific antibodies for total HSP27 and phosphor-HSP27 (serine 15) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA) and Thermo Fisher Scientific (Waltham, MA, USA), respectively. The antibodies for CREB and phosphor-CREB were obtained from Cell signaling Technology (Danvers, MA, USA).
2.2. Surface immobilization of HT polymer through tyrosinase-triggered oxidative reaction
Prior to the heparin- functionalization step, PCL substrates were first prepared by a solvent casting method [25, 26]. The PCL solution (10 wt%) dissolved in 150 mL of THF was poured onto a glass Petri dish, and the solvent was evaporated slowly at room temperature. The round-shape PCL substrates (13 mm in diameter) were collected by punching, and they were washed three times with DW to remove the residual THF.
To prepare HT- immobilized PCL substrate (PCL–HT), the HT polymer solution was treated to the PCL surface by a simple dipping method as previously described [23]. Briefly, the PCL substrates were immersed in a solution of HT polymer (1 wt%) dissolved in Tris-HCl buffer (10 mM, pH 8.5), and then 0.4 kU/mL of tyrosinase was added. After 3 h of incubation at room temperature, they were washed thoroughly with DW, and dried under vacuum.
2.3. Characterizations of the HT-immobilized PCL surfaces
After tyrosinase treatment, the immobilized heparin amount on the surface was quantified using a TBO assay [2, 27, 28]. Briefly, the bare PCL, PCL/HT (without tyrosinase) and PCL–HT samples were immersed in 1 mL of PBS (pH 7.4) solution containing 0.002 wt% TBO, and were shaken for 30 min at room temperature. Then, 2 mL of hexane was added and vortexed for 5 min. The absorbance of aqueous phase was measured at 620 nm using a microplate reader (M1000, Tecan, Switzerland), and heparin amount was determined by a calibration curve of heparin solution.
To characterize HT- immobilized PCL substrates, changes in the surface wettability were analyzed by measuring static water contact angle using a contact angle analyzer (SEO, Phoenix 150, South Korea). A droplet of DW on the surface of pure PCL and PCL–HT samples was visualized using an equipped camera. Changes in the surface atomic composition after HT immobilization on PCL surface were also analyzed by X-ray photoelectron spectroscopy (Thermo Electron, K-Alpha, USA) at Center for Research Facilities, Kyunghee University, Korea.
2.4. Characterizations of MK2i peptide-loaded PCL–HT substrates
To confirm the electrostatic interaction between MK2i and HT polymer, changes in zeta potential of HT/MK2i nanoparticles were analyzed by increasing MK2i feed amounts, as previously reported [2]. The nanoparticles formed with 1 mg/mL of HT (500 μL) and 0.1–50 mg/mL MK2i (500 μL) were prepared in PBS medium, and their zeta potential was measured using a DLS Zetasizer (Nano ZS, Malvern, USA). The concentration of HT polymer was fixed at 1 mg/mL, and mixing ratio (w/w) was varied from [MK2i]/[HT] = 0.1 to 50 to investigate the molecular interactions.
For in vitro MK2i release study, the MK2i peptides with different concentrations (100, 500 and 1000 μg/mL) were first loaded on the PCL–HT substrates by a simple dipping method. Briefly, the substrates with 13 mm in diameter were immersed in 1 mL of Tris-HCl buffer (10 mM, pH 8.5) containing MK2i peptide, and the reaction was allowed at room temperature for 24 h. Then, the dehydrated PCL–HT/MK2i discs were placed in 24-well plate, and PBS (1 mL) was added to each well. The release experiment was carried out at 37 °C for 28 days. At predetermined time intervals, the supernatant was collected and replaced with the same volume of fresh buffer media for the next time point. The amounts of the released MK2i peptide in the collected media were measured at 562 nm using a Micro BCA assay kit (Pierce, USA), and the cumulative MK2i release kinetics were drawn as a function of time. As a negative control for heparin functionalization, the bare PCL incubated in MK2i solution was used. In addition, FITC-conjugated MK2i peptides (EZBiolab, USA; 100, 500 and 1000 μg/mL) were also visualized by fluorescence microscopy (Axio Observer, Zeiss, Germany) to confirm the distribution and loading amount of MK2i on the surface. In follow-up studies, 1000 μg/mL of MK2i solution was used to prepare PCL–HT/MK2i substrates.
2.5. In vitro protein/platelet adhesion tests on the HT-immobilized PCL surface
For in vitro protein absorption test, the substrates (bare PCL and PCL–HT) were immersed and incubated in PBS medium containing fibrinogen (100 μg/mL) at 37 °C. After 3 h of incubation, the surface of each sample was gently rinsed three times with PBS, and then treated with 1% SDS to collect the absorbed protein. The amount of absorbed fibrinogen on the surface was quantified using a Micro BCA assay kit (Pierce, USA)
For in vitro platelet adhesion test, the platelet was obtained from fresh rat whole blood (Sprague-Dawley (SD) rat; weights: 250–300 g) in accordance with the guidelines of Yonsei Laboratory Animal Research Center (YLARC; Seoul, Korea). The isolated platelet pellets were diluted with buffer solution, and added to bare PCL, PCL–HT and PCL–HT/MK2i substrates (13 mm in diameter). Following incubation in platelet solution at 37 °C for 1 h, each sample was thoroughly washed three times, and fixed with 2% glutaraldehyde in PBS (0.1 M, pH 7.4) for 6 h. They were then fixed with 1% osmium tetroxide (OsO4) for 90 min, and dehydrated using a Critical Point Dryer (EM CPD300, Leica, Austria). The adhered platelets on the surface were observed using the SEM (FE-SEM, Carl ZEISS, Germany) after platinum sputter coating (EM ACE600, Leica, Austria), and were quantified from the SEM images on random positions.
2.6. In vitro studies of VSMC migration and proliferation
To investigate the inhibition effect of the released MK2i on cellular migration, in vitro scratch-wound assay was performed using a Culture-Insert 2 well migration assay kit (Ibidi, USA) according to the manufacturer’s instructions. VSMCs were seed within Insert 2 well at a seeding density of 1 × 104 cells/well, and cultured to be reached at confluency (90–95%) for 1–2 day in DMEM-supplemented with 10% FBS and 1% PS, under standard culture condition. After gently removing the Culture-Insert 2 well to create the scratch wound, the culture medium was replaced with low serum growth medium containing 1% FBS to minimize cellular growth, and incubated overnight [7, 14]. Then, the cells were covered with samples (bare PCL, PCL–HT and PCL–HT/MK2i), and incubated in the medium containing 50 ng/mL of PDGF (R&D systems, USA). To prevent from direct physical contact of sample discs with the adhered cells on the plat e, a rubber O-ring with 13 mm in diameter (Parker, USA) was placed as a spacer between the bottom plate and disc-type sample. As a positive and negative control, the medium with and without 200 μg/mL of MK2i peptide was used, respectively. After 24 h of treatment with each sample, the wound areas were imaged under optical microscopy (Axio Observer, Zeiss, Germany), and quantified using an ImageJ software (NIH, USA). The wound closure rate (%) was determined by measuring the wound area (Wt) at 24 h post sample treatment with respect to the initial scratch-wound area (Wi) using the following equation: (Wt − Wi)/Wi × 100. Each migration assay was repeated three times.
In vitro VSMC proliferation assay was also performed as a control for cell migration test. As described above, the same experiment condition with the scratch-wound assay was used. VSMCs were incubated with the culture medium containing 1% FBS in 24-well plate for 24 h, and then treated with 50 ng/mL of PDGF and samples (culture medium, bare PCL, PCL–HT, PCL–HT/MK2i and 200 μg/mL of MK2i). After 24 h of treatment, the WST-1 reagents (100 μL) were added to each well, and their optical density (O.D.) was measured at 450 nm to confirm VSMCs proliferation in each test condition.
2.7. Western blot analysis of target protein phosphorylation in VSMCs
VSMCs were grown to 90–95% confluence in a cell culture Petri dish (60 mm in diameter) with normal culture media (10% FBS and 1% PS). Separately, bare PCL and PCL–HT/MK2i were incubated in low serum growth medium (1% FBS), and the extracts were collected at predetermined time intervals (6 h, 1, 3 and 7 days). Adhered cells were serum-starved for 24 h to minimize cell growth, and then treated with pre-extracted MK2i solutions for 2 h. As positive and negative control, the medium with and without 200 μg/mL of MK2i peptide were used, respectively. Following 2 h of treatment with the extracts, 30 μM lysophosphatidic acid (LPA; Tocris Bioscience, UK) was treated to each well. As a control for LPA treatment, the medium without LPA was also treated in the same condition. After 30 min of incubation with LPA at 37 °C, the cells were lysed, and then centrifuged at 14,000 rpm for 15 min at 4 °C. The total protein amount in the collected supernatant was measured using a Bio-Rad Protein assay kit. Equal amounts of protein (50 μg/lane) were loaded into 10% TGX Precast Gels (Bio-Rad, USA); the proteins were separated by electrophoresis, and they were transferred to a nitrocellulose membrane. For analysis of heat shock protein 27 (HSP27) phosphorylation, the membrane was stained overnight at 4 °C with specific antibodies for total HSP27 (1:500 dilution) and phosphor-HSP27 (1:1000 dilution). For analysis of cAMP-response element binding protein (CREB) phosphorylation, the membrane was stained overnight at 4 °C with specific antibodies for total CREB (1:500 dilution) and phosphor-CREB (1:1000 dilution). After washing three times, the blots were treated with appropriate secondary antibodies for 1 h at room temperature, and imaged using Odyssey infrared scanner (Li-COR Bioscience, USA). Signal intensity of each band was quantified with Li-COR Odyssey software at 800 and 680 nm wavelengths. Each western blot experiment was repeated three times.
2.8. Ex vivo MK2i delivery into human saphenous vein (HSV)
For ex vivo study using MK2i-releasing PCL–HT sheath, the discarded human saphenous vein (HSV) was obtained from the patients undergoing peripheral or coronary vascular bypass surgery in accordance with approved IRB guidelines provided by Vanderbilt University Medical Center (VUMC). The HSV segment was cut into consecutive rings (2–3 mm in width), and PEG-based hydrogel was then filled inside HSV rings to prevent shrinkage. After that, the HSV rings were wrapped with each sample sheath (bare PCL or PCL–HT/Alexa 568–MK2i), and then they were incubated in RPMI 1640 medium containing 30% FBS, 1% L-glutamine and 1% PS under standard culture condition (37 °C and 5% CO2). To visualize the delivered MK2i in the HSV tissue, Alexa 568 (red fluorescence) was conjugated to MK2i peptide before loading on the surface of PCL–HT sheath (3/3.5 mm in internal/external diameter and 3 mm in width). After ex vivo organ culture for 6 h and 3 days, the HSV tissues were frozen in NEG-50 medium (Thermo Scientific, USA), and sectioned (10 μm in thickness). The cross-sectioned tissues were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, USA), and then imaged using a fluorescent digital scanner (Aperio VERSA 200, Leica, Germany).
2.9. Statistical analysis
Quantitative data is expressed as mean ± standard deviation (S.D.). Statistical analysis was performed using a Student’s t-test, and the significance of the results was set to P-value < 0.05.
3. Results and Discussion
3.1. Preparation of tyrosinase-reactive HT polymer and HT-immobilized PCL substrates
The leading cause of synthetic graft failure includes thrombotic occlusion and intimal hyperplasia at the site of vascular anastomosis [3, 4]. To prevent these adverse clinical outcomes, various approaches combining with potent therapeutic agents (e.g., simple drug-coating/blending, hydrogel coating, nanoparticles) have been explored so far [12]. However, there is still an unmet need in terms of long-term drug delivery because burst release at the early stage of drug elusion limits their endovascular delivery and anti-hyperplasia effects. Hence, we developed the heparin- functionalized polymer graft surface that can elute a potent anti- neointimal hyperplasia drug (i.e. MK2i peptide) up to 4 weeks to improve hemocompatiblity and anti-neointimal activity.
As the first step to achieve this goal, heparin was selected as both an anticoagulant agent and a surface depot layer, and then was immobilized on the polycaprolactone (PCL) surface using enzymatic oxidative reaction. We previously reported a facile surface modification method using a tyrosinase enzyme to immobilize bioactive and/or bioinert molecules on the surfaces for biomedical applications: phenol-functionalized molecules were bound to the versatile surfaces by tyrosinase-mediated oxidative reaction, which was performed via a simple dipping procedure [23, 24]. This approach provides several benefits such as high reaction selectivity toward phenol moiety and rapid reaction rate (< 1 h) under mild condition as well as easy control of surface density [3]. Therefore, we employed this surface immobilization system to make the heparin- modified surface for producing MK2i drug depot layer as well as improving hemocompatibility.
To prepare an active heparin derivative for the tyrosinase-catalyzed reaction, an amine-reactive tyramine containing phenol moiety was grafted to the carboxyl groups of heparin backbone through an EDC/NHS coupling reaction (Figure S1a). Then, the chemical structure of heparin–TA (HT) polymer was characterized by 1H NMR and UV analysis, as previously reported [29, 30]. As shown in Figure S1b, 1H NMR spectrum of the HT polymer dissolved in D2O clearly showed the characteristic peaks of phenol group (aromatic protons of TA) at 6.8 and 7.1 ppm, indicating successful grafting of TA to heparin polymer. In addition, unlike heparin polymer, the specific UV absorbance (275 nm) for this phenol group was also observed in HT polymer: their phenolic content quantitatively measured by a calibration curve of tyramine hydrochloride solutions was approximately 350 μmol/g of HT polymer (Figure S1c). In terms of anticoagulant activity, the anti- factor Xa assay revealed that binding affinity of HT polymer to AT III was slightly decreased to 85% compared to the pure heparin (100%). This result indicates that grafting tyramine to carboxylic groups of the heparin did not result in a significant loss of anticoagulant activity in heparin [31].
Next, the HT- immobilized PCL (PCL–HT) surface was prepared by simply immersing the PCL substrates into an aqueous HT polymer (1 wt%) solution, followed by addition of tyrosinase solution (0.4 KU/mL) (Figure 1a). As an oxidizing agent, tyrosinase has an ability to catalyze hydroxylation of phenol moiety into o-quinone which is highly adhesive onto the versatile surfaces including polymers [22, 24, 32]. Therefore, the tyrosinase treatment during this process enables surface binding of phenol-containing HT polymer. Figure 1b shows the time-dependent color changes of HT solution by tyrosinase-catalyzed oxidative reaction. The transparent HT solution turned into dark black (like oxidized DOPA solution) after 90 min of incubation, which indirectly proves that HT immobilization onto the PCL surface is achieved during this process.
Figure 1. Schematic illustration of enzymatically catalyzed heparin-immobilization and MK2 inhibitory (MK2i) loading on the surface of polycaprolactone (PCL) for anti-thrombogenic effect and sustained release of therapeutics.

(a) PCL substrate (13 mm in diameter) was immersed in aqueous polymer solution containing heparin-tyramine (HT; 1 wt%), and heparin was then immobilized on the PCL surface by the tyrosinase-triggered oxidative reaction. After that, MK2i was loaded on the heparinized PCL layer through an electrostatic interaction between MK2i peptide and HT polymer. (b) Photographs of the HT solution, and PCL–HT substrate after the tyrosinase (0.4 kU/mL) treatment for 3 h.
As for a surface model, we chose PCL substrate because it is one of representative polymers used for developing synthetic vascular grafts due to excellent biocompatibility, slow biodegradability and mechanical compliance [4, 33]. Especially, PCL has demonstrated to be suitable for the design of implantable material in vascular tissue engineering because their appropriate biodegradability (> 1 year) proved to minimize infection risk/inflammatory response, and promoted favorable endothelialization for blood vessel reconstruction. [34, 35].
3.2. Surface characterizations of HT-immobilized PCL substrates
To verify that the tyrosinase treatment induces immobilization of the synthesized HT polymer onto the PCL surface, we first analyzed the surface heparin density using a TBO assay depending on the reaction time (1–12 h). As the reaction time increased, the heparin amount increased on the surface from 5.8 to 18.0 μg/cm2, and this amount was almost saturated after 3 h (Figure 2a). Hence, we fixed the reaction time to 3 h for preparation of PCL–HT substrates in the follow- up experiments. In previous studies, the immobilization reaction of phenol-containing molecules (e.g. mPEG-phenol and RGD-phenol) was completed within 0.5–1 h [23, 24]. However, when applying this HT polymer, there was a time-delay in reaching the equilibrium state of surface density. According to some researches, the chemical structures such as polysaccharides and carboxyl group in heparin have been proved to have an antioxidant property [36, 37]. Therefore, this diminished surface-binding efficiency of phenol molecules in HT polymer was caused most likely due to an antioxidant property of heparin that counteracts the oxidative reaction catalyzed by tyrosinase. When compared to bare PCL (control), there also remained a small amount of heparin (1.1 μg/cm2) on the surface of PCL/HT incubated in HT solution without tyrosinase, which may be due to physical absorption (Figure 2b).
Figure 2. Characterizations of heparin-immobilized PCL (PCL–HT) substrate prepared by tyrosinase-catalyzed reaction.

Surface density of heparin: (a) PCL–HT substrates with different tyrosinase-reaction times (1–12 h), and (b) bare PCL, PCL/HT (without tyrosinase treatment) and PCL–HT (with tyrosinase treatment) for 3 h. (c) Surface wettability and water contract angles before and after HT functionalization. XPS analysis: (d) wide scan and (e) narrow scan spectra for S2p/N1s in bare PCL and PCL–HT surfaces. Results in (a) (c) are shown as the average values ± S.D. (n = 3).
To characterize the surface properties of PCL–HT substrate, changes in both surface wettability and surface atomic composition were determined before and after heparin modification. Figure 2c shows the images of water droplets on bare PCL and PCL–HT surfaces. While the water contact angle of bare PCL surface was 81 ± 4°, it was significantly decreased to 19 ± 4° after 3 h of heparin functionalization. Heparin has many hydrophilic groups including hydroxyl, sulfate, amine and carboxyl groups [9]. Accordingly, this decrease in water contact angle can be explained by the exposure of hydrophilic HT polymer on the surface consisting of hydrophobic PCL. In XPS wide and narrow scan spectra of the modified PCL–HT, the corresponding peaks of nitrogen (N1s, 399 eV) and sulfur (S2p, 168 eV) were observed as compared to bare PCL spectra. As expected, the S2p and N1s peaks appeared due to the sulfate groups in heparin backbone and amide bonding formed by TA conjugation, respectively [38]. Together, these results demonstrate that the synthesized HT polymer was successfully immobilized onto the PCL surfaces through the tyrosinase-catalyzed oxidative reaction.
3.3. Characterizations of MK2i peptide-loaded PCL–HT surface
Before loading MK2i peptide on the heparinized PCL–HT surface, we investigated the molecular interaction between MK2i peptide and HT polymer. The changes in zeta-potential of HT/MK2i complex were analyzed by varying mixing ratio (w/w; [MK2i]/[HT] = 0.1 to 50) [2, 39]. Figure 3a shows zeta-potential values with increasing the MK2i concentration (0.1–50 mg/mL), while maintaining the consistent HT concentration (1 mg/mL). The HT/MK2i complex consisting of mostly HT ([MK2i]/[HT] = 0.1) had strong negative charge (ζ = −19.1 mV), and the absolute value gradually decreased from 19.1 to 2.1 mV. The inversely-correlated relationship between the MK2i amount and the surface net charge of HT/MK2i complex indicates that negatively charged groups of HT polymer interact with positively charged MK2i peptide via electrostatic interaction. In addition, we identified that MK2i peptide could be loaded at approximately 50 times higher amount compared to the immobilized HT amount on the PCL surface (HT density = 18.0 μg/cm2) because the net charge of HT/MK2i complex ([MK2i]/[HT] = 50) was still negative. Thus, three different concentrations (100, 500 and 1000 μg/mL) of MK2i peptide in an available range were used to load them on the PCL–HT surface in this study.
Figure 3. Characterizations of MK2i peptide-loaded PCL–HT substrate.

(a) Zeta potential values of HT/MK2i complex by varying the MK2i feed amount at fixed concentration of HT polymer (1 mg/mL). The mixing ratio (w/w) was varied from [MK2i]/[HT] = 0.1 to 50. (b) In vitro release kinetics of MK2i peptide loaded on bare PCL and PCL–HT substrates. The MK2i peptides (100, 500 and 1000 μg/mL) were treated to the PCL–HT surfaces, and then the released MK2i amounts in the collected media were measured over 28 days. The cumulative MK2i release kinetics were drawn as a function of time. (c) Fluorescence images on the surfaces of PCL–HT after treating FITC–MK2i peptides (100, 500 and 1000 μg/mL). Results in (a) and (b) are shown as the average values ± S.D. (n = 3).
Next, to estimate the release kinetics of MK2i peptide loaded on bare PCL and PCL–HT surfaces, we measured cumulative MK2i release amounts as a function of time over a 28-day period (Figure 3b) The released amount of MK2i was also shown as the percentage of the total Mk2i amount loaded (Figure S2). In PCL/MK2i_1000 sample without HT functionalization, there was the initial burst of MK2i, and then no further release. On the other hand, although the heparin modified PCL–HT/MK2i_1000 also had the initial burst (< 1 day, 130 μg/mL), they showed the continuous and sustained MK2i release behavior with a higher amount over 28 days. For example, when 1000 μg/mL of MK2i peptide was treated to each of bare PCL and PCL–HT, the total released MK2i amount from the PCL–HT surface (272 μg/mL; 27.2%) was much greater (6.6-time) than that from bare PCL (41 μg/mL; 4.1%). This result validates the importance of heparin modified surface for efficient loading and eluting MK2i peptide. Additionally, the MK2i release kinetic from the PCL–HT surface could be varied by controlling their loading amount. Figure 3c shows the fluorescence microscopy images on the surface of PCL–HT treated with FITC–MK2i peptide at different concentrations (100, 500 and 1000 μg/mL). It was clearly observed that FITC–MK2i peptides with green fluorescence were homogeneously distributed throughout the surface, and the relative intensity was higher along with an increase in the feed amount of FITC–MK2i peptide. Thus, this observation can support the feed amount-dependent release kinetics of MK2i as seen in Figure 3b. Taken together, we demonstrated that HT immobilization 1) led to higher loading efficiency of MK2i peptide on the PCL surface through the electrostatic interaction, and 2) enabled long-term sustained delivery (over 28 days) with a control release behavior.
3.4. In vitro cytotoxicity evaluation of PCL–HT/MK2i substrate
To assess in vitro cytotoxicity of MK2i-releasing PCL substrate for vascular graft application, the solution extracted from PCL–HT/MK2i substrate was treated to the adhered NIH3T3 cells as described previously [40]. Following incubation of extracts with NIH3T3 cells for 24 h, cell viability was evaluated using a WST-1 assay and a live/dead staining assay. In comparison to TCPS control (normal culture medium without sample incubation), all samples (bare PCL, PCL–HT and PCL–HT/MK2i) showed > 90% mitochondrial activities of NIH3T3 cells (Figure S3a). In addition, as seen in Figure S3b, there was no significant morphological difference between cells cultured in culture medium or extracts. Most cells also appeared to be highly viable in live/dead staining images, showing > 95% green color-stained live cells. Therefore, we confirmed excellent biocompatibility of the MK2i-releasing PCL–HT to apply for vascular grafting.
3.5. Evaluation of hemocompatibility of the PCL–HT substrate
In general, thrombus formation on the surface of blood-contacting implants or devices is activated by non-specific protein absorption and platelet adhesion [41, 42]. Once foreign materials are exposed to blood, plasma proteins (e.g., albumin, fibrinogen and von Willebrand factor) are firstly absorbed on their surfaces, thereby inducing platelet adhesion and activation. Subsequently, the activated platelet aggregates and forms a fibrin network via the blood coagulation cascade, ultimately leading to thrombus formation. Therefore, a highly repellent surface of vascular graft material against protein/platelet absorption is crucial in the graft design.
For in vitro protein absorption test, we selected fibrinogen protein, and treated them to each sample. Among plasma proteins, it is well known that fibrinogen is of particularly importance in thrombus formation process as it is highly abundant in blood, and has a high affinity to platelet [43]. When samples (bare PCL and PCL–HT) were incubated in culture media containing fibrinogen, PCL–HT exhibited a significantly decreased protein absorption level (0.6 μg/cm2) compared to bare PCL (1.7 μg/cm2), indicating that surface density of the immobilized HT is sufficient to repel non-specific protein absorption (Figure 4a). This result is mostly like due to both negative charge and hydrophilic characters of HT polymer grafted on the PCL surface. The previous studies demonstrated that the negatively charged groups (SO3- and COO−) in heparin reduced absorption of albumin (isoelectric point; PI = 4.8) and fibrinogen (PI = 5.5) proteins by electrostatic repulsion effect, and the non- fouling degree against protein absorption was dependent on the surface heparin density [44]. In addition, the hydrophilic polymer-bound surface can also serve as a physical barrier against protein/platelet absorption through hydrogen bond-induced surface hydration [45, 46].
Figure 4. In vitro evaluation of hemocompatibility of the PCL–HT substrate.

(a) Absorbed fibrinogen amounts on bare PCL and PCL–HT surfaces. The substrates were incubated in protein solution (100 μg/mL), and then the absorbed fibrinogen amount was measured by a Micro BCA assay. (b) Adhered platelet amounts on bare PCL, PCL–HT and PCL–HT/MK2i surfaces. (c) SEM images of the adhered platelets treated on the substrates for 1 h. All results are shown as the average values ± S.D. (n = 3; *P < 0.05 compared to bare PCL, #P < 0.05 compared to PCL–HT).
The protein adsorption generally affects the platelet adhesion and activation. The mechanism of platelet adhesion on the surface of blood-contacting materials involves plasma protein absorption at the initial stage which provides cell-binding sites. Therefore, reducing protein adsorption decreases cell attachment [5, 47]. In order to investigate the effect of HT coating on the platelet adhesion, the platelets isolated from rat whole blood were treated onto bare PCL, PCL–HT and PCL–HT/MK2i surfaces, and then the number of adhered platelet was quantified from the SEM images observed at randomly chosen positions. It was confirmed that the platelet adhesion was not significant on the PCL–HT surface, as opposed to the bare PCL (Figure 4b). The average number of platelets adhered on bare PCL and PCL–HT surfaces was 2.6 × 106 and 3.7 × 105 cells/cm2, respectively, which means 85.6% decrease in the adhered platelet density after HT-immobilization. In addition, MK2i- loaded PCL–HT substrate showed most reduced platelet adhesion (96.2%; 1.0 × 105 cells/cm2) compared to bare PCL. Although the surface negative charge was relatively diminished by loading positively charged MK2i peptide on the heparin- immobilized substrate, it did not cause a visible decrease in anti-platelet adhesion. We speculate that our MK2i peptide also has a potential to prevent the platelet adhesion.
A reduction in platelet adhesion on both PCL–HT and PCL–HT/MK2i might be caused by the bioactivity of immobilized heparin as well as the electrostatic repelling effect. Indeed, many studies have demonstrated that heparin has a strong ability to inhibit the platelet activation and aggregation via molecular interactions such as AT III or p-selectin binding [2, 48]. As shown in Figure 4c, while the platelets on the bare PCL without heparin treatment exhibited the adhered and aggregated morphologies, the sphere- like shape of the platelet was observed on the heparinized surfaces (PCL–HT and PCL–HT/MK2i). Taken together, we demonstrated that HT and MK2i immobilization on the PCL surface by tyrosinase treatment remarkably enhanced blood compatibility with significantly reduced protein absorption and platelet adhesion.
3.6. Inhibitory effect of the released MK2i on VSMC migration
It is well known that graft restenosis is primarily due to excessive migration and proliferation of VSMCs triggered by venous response to surgical injury [11, 49]. During vascular remodeling, chemotactic molecules (e.g., TGF-β and PDGF) are known to stimulate VSMC recruitment and proliferation [50, 51]. Thus, inhibiting abnormal activities of VSMCs has become an attractive strategy to prevent graft stenosis/occlusion by intimal hyperplasia. In this study, a promising anti- neointimal therapeutic drug, the MK2i peptide was loaded on the heparinized PCL surface, and then the bioactivity of the release MK2i was investigated.
To evaluate inhibitory effect of the released MK2i from PCL–HT surface on VSMC migration, we performed in vitro wound-healing assay using a Culture-Insert 2 well. The scratch-wounds were created, and then cells were incubated with samples (bare PCL, PCL–HT and PCL–HT/MK2i) in the medium containing PDGF (50 ng/mL) for 24 h. As a negative and positive control, the medium with or without MK2i peptide (200 μg/mL) was used [7]. As shown in Figure 5a, VSMCs migrated into the scratched gap between the confluent cell areas. Based on these images, wound closures (%) were calculated by quantitatively measuring the wound area at 24 h with respect to the initial scratch-wound area (Figure 5b). Firstly, PDGF treatment accelerated wound closure rate compared to NT (no treatment) without PDGF. In addition, we confirmed that treatment of MK2i solution (49.0%) clearly decreased PDGF-induced cell migration compared with the cells stimulated with PDGF but not treated with MK2i (NT with PDGF; 75.6%). Among the PCL substrate-treated groups, PCL–HT/MK2i (26.7%) resulted in significantly reduced VSMC migration as compared to bare PCL (59.7%) and PCL–HT (60.8%), exhibiting a comparable inhibitory effect on VSMC migration. This result validates that the bioactivity and stability of the released MK2i peptide are well maintained even after loading on the modified PCL–HT surface, and the released amount is enough to effectively inhibit PDGF-stimulated VSMC migration. The PCL substrates without MK2i loading (bare PCL and PCL–HT) also had inhibitory effect compared to NT control with PDGF. We speculate that it was affected from the experimental condition where the cells were covered with PCL disk on the top of culture medium.
Figure 5. In vitro migration and proliferation of VSMC.

(a) Representative images and (b) quantified wound closure of the migrated VSMCs treated with MK2i-loaded PCL–HT. The cells were incubated in the medium containing PDGF (50 ng/mL) for 24 h. The wound closure (%) was determined by measuring the wound area at 24 h compared to the initial scratch wound area. As positive and negative control, the medium with or without 200 μg/mL of MK2i was used, respectively. Scale bars represent 200 μm. (c) Effect of the released MK2i on proliferation of VSMCs. To prove that wound closure is not affected by cell proliferation, the proliferation assay was performed in the same experimental condition with wound scratch assay. Results in (b) and (c) are shown as the average values ± S.D. (n = 3; *P < 0.05 compared to NT, #P < 0.05 compared to bare PCL and PCL–HT).
To validate that the above- inhibited VSMC migration results were not influenced by cellular growth rate, we also performed in vitro cell proliferation assay under the same experimental condition with scratch-wound assay. As shown in Figure 5c, among the sample groups, no statistical differences in cellular growth of VSMCs were identified. Therefore, this result supports that the released MK2i from PCL–HT surface certainly inhibited VSMC migration.
3.7. Inhibited phosphorylation of HSP27 and CREB in LPA-induced VSMCs by PCL–HT/MK2i
HSP27 and CREB are major proteins involving development of intimal hyperplasia [14, 52]. They are produced by VSMCs in response to exposure of the mechanical and biochemical stresses induced by vascular grafting procedure. In addition, these proteins have been identified as a substrate of MAPK-activated MK2 that is an intracellular kinase regulator activated by p38 MAPK. During vascular pathologic remodeling, the expressions of HSP27 and CREB are up-regulated by the MK2 kinase pathway regulating VSMC migration/proliferation and inflammation [53]. Thus, the down-regulated expression and phosphorylation of these target proteins (HSP27 and CREB) is critical to prevent the neo-intimal formation.
To assess the bioactivity of the released MK2i toward phosphorylation of HSP27 and CREB in VSMCs, changes in phosphorylation of target proteins were analyzed by a western blot assay using MK2i-extracted solutions. Each extract was separately collected by incubation of PCL–HT/MK2i substrate in the medium for predetermined time periods (6 h, 1, 3 and 7 days), and then treated to the adhered VSMCs. After that, 30 μM LPA was treated to stimulate the phosphorylation of target proteins in VSMCs [14]. When exposed to LPA alone, phosphorylation of both HSP27 and CREB was stimulated when compared to that in VSMCs not treated with LPA: phosphor-HSP27 (54.8% increase) and phosphor-CREB (131.7% increase) vs. NT without LPA (data not shown). Figure 6a shows the western blots of phosphor-target proteins and total target proteins. Based on their signal intensities, we quantified the relative ratios (phosphor-HSP27/total HSP27 and phosphor-CREB/total CREB). As a positive control, pretreatment with MK2i solution (200 μg/mL) resulted in a decrease in both phosphor-HSP27 (35.7% decrease) and phosphor-CREB (31.6% decrease). In addition, PCL–HT/MK2i group showed a gradual decrease in phosphor-HSP27 and phosphor-CREB depending on the MK2i releasing period, whereas bare PCL had a similar level to negative control (NT with LPA alone) (Figure 6b). Concretely, the released MK2i (121–210 μg/mL) from the PCL–HT/MK2i for 1–7 days period significantly inhibited phosphorylation of HSP27 (38.0–41.4% decrease, *P < 0.05 vs. NT) and CREB (20.6–31.7% decrease, *P < 0.05 vs. NT) in LPA-stimulated VSMCs compared to NT control. When considering MK2i concentrations in each collected medium, the inhibitory bioactivity of MK2i toward phosphorylation of target proteins appears to be dependent on the released MK2i amount. The results demonstrate that the released MK2i shown ability to suppress VMSC migration can also inhibit phosphorylation of target proteins (HSP27 and CREB) associated with neointimal development following vascular access surgery.
Figure 6. Effect of the released MK2i from PCL–HT/MK2i substrate on phosphorylation of HSP27 and CREB in VSMCs.

(a) Representative images of western blots showing the phosphorylation of target proteins (pHSP27 and pCREB) in VSMCs treated with the released MK2i. The PCL–HT/MK2i samples were incubated in low serum growth medium (1% FBS), and the extract solutions were collected after 6 h, 1, 3 and 7 days. Following treatment of each extract to VSMCs for 2 h, LPA (30 μM) was exposed for 30 min. As a positive and negative control, the medium with or without 200 μg/mL of MK2i was used. (b) Quantification of the relative ratios (phosphor-HSP27/total HSP27 and phosphor-CREB/total CREB). Results are shown as the average values ± S.D. (n = 3; *P < 0.05 compared to NT).
3.8. Ex vivo MK2i delivery of PCL–HT/MK2i to human saphenous vein (HSV)
To examine the bio-availability resulting from endovascular delivery of the released MK2i from PCL–HT surface, the MK2i peptide-releasing PCL–HT sheath wrapped around a HSV segment, and then the presence of MK2i in the HSV tissue was confirmed after ex vivo organ culture for 6 h and 3 days. To visualize the delivered MK2i peptide, Alexa 568 (red fluorescence)- labeled MK2i was loaded on the surface of PCL–HT sheath. Figure 7 shows the fluorescence images of the cross-sectioned HSVs treated with bare PCL and PCL–HT/Alexa 568–MK2i. Compared to control group (bare PCL), it was clearly observed that Alexa 568–MK2i was released from the PCL–HT, and delivered into the vessel tissue. Especially, as the treatment time increased, improved infiltration and abundant distribution of the Alexa 568–MK2i were observed in the HSV tissue. While the initially released MK2i for 6 h was only detected in the tunica adventitia, the continuous release of MK2i over 3 days resulted in overall diffusion up to the tunica media and subintima layer, as indicated by relatively more distributed red color in the entire vascular tissue. Thus, these results suggest that the continuously released MK2i from PCL–HT surface enables perivascular delivery into the vessel tissue in a cumulative fashion.
Figure 7. MK2i peptide delivery of PCL–HT/MK2i to human vein tissue.

Representative fluorescence images of the HSVs wrapped with bare PCL and PCL–HT/MK2i sheaths for 6 h and 3 days. MK2i peptide was labelled with Alexa 568 (red) before loading on the surface of PCL–HT sheath. The cross-sectioned HSVs were stained with DAPI (blue). Scale bars indicate 200 μm.
4. Conclusions
The present study reports a co- immobilization strategy of heparin and anti- neointimal MK2i drug to prevent thrombotic occlusion and neotinitmal formation of synthetic vascular grafts. The heparin was employed with two goals: 1) to prevent thrombus formation through their non- fouling properties, and 2) to load and induce the sustained release of MK2i via electrostatic interaction between cationic drug and anionic heparin. The surface binding of phenol-containing HT polymer on the PCL surface was successfully achieved by a tyrosinase- mediated simple dipping method within 3 h. After heparin modification, hydrophilic and negatively charged surface was created on the PCL substrate, and the HT immobilization resulted in remarkably enhanced blood compatibility with significantly decreased protein adsorption and platelet adhesion compared to the bare PCL surface. The characterization results of MK2i-loaded surface demonstrated that the enzymatically HT- immobilized surface 1) led to high loading efficiency of cationic peptide such as MK2i through electrostatic interaction, and 2) enabled sustained MK2i release up to 4 weeks with a control release behavior. In addition, the bioactivity and stability released MK2i were maintained, exhibiting significant inhibitory effects on VSMC migration and phosphorylation of target proteins (HSP27 and CREB) associated with intimal hyperplasia. When MK2i peptide-releasing PCL–HT sheath was applied to ex vivo HSV model, they showed perivascular delivery into the vessel tissue in a cumulative fashion. These findings indicate that enzymatically modified HT surface eluting MK2i peptide is a promising strategy to enhance hemocompatibility and anti-neointimal activity for vascular grafting.
Supplementary Material
Figure S1. Synthesis and characterizations of phenol-conjugated he parin (HT) derivatives. (a) Synthetic route of HT polymer by an EDC/NHS coupling reaction, (b) 1H NMR spectrum of HT polymer dissolved in D2O, and (c) UV absorbance spectra of heparin and HT polymers.
Figure S2. In vitro release profiles of MK2i peptide from PCL and PCL–HT substrates over 28 days. The cumulative released amounts of MK2i were drawn as a percentage with respect to the total MK2i amount loaded.
Figure S3. In vitro cytotoxicity evaluation of PCL–HT/MK2i substrate using NIH3T3 cells. (a) Cell cytotoxicity of bare PCL, PCL–HT and MK2i- loaded PCL–HT. The adhered NIH3T3 cells were treated with extracted culture media for 24 h, and then their mitochondrial activities were quantified by a WST-1 assay. As a positive control, fresh medium without sample extraction was used. Results are shown as the average values ± S.D. (n = 3) (b) Photographed images of the NIH3T3 cells cultured in each extracted medium for 24 h: phase contrast and live/dead staining images (green: live cells, red: dead cells). Scale bars indicate 100 μm.
Acknowledgments
This research was funded and supported by AHA GRANT25890018, NSF CBET BME 1056046, NSF DMR BMAT 1506717, and NIH EB 019509. This study was also financially supported by the Faculty Research Assistance Program of Yonsei University College of Medicine for 2016 (6- 2016- 0031) and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2016M3A9E9941743, 2015M3A9E2028578, and 2015M3A9E2028643). Frozen sectioning was performed by Vanderbilt Translational Pathology Shared Resource. Fluorescent whole slide imaging of HSV segments was conducted by Digital Histology Shared Resource at Vanderbilt University Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Vries MR, Simons KH, Jukema JW, Braun J, Quax PH. Vein graft failure: from pathophysiology to clinical outcomes. Nat Rev Cardiol. 2016;13:451–470. doi: 10.1038/nrcardio.2016.76. [DOI] [PubMed] [Google Scholar]
- 2.Liu T, Liu Y, Chen Y, Liu S, Maitz MF, Wang X, Zhang K, Wang J, Wang Y, Chen J, Huang N. Immobilization of heparin/poly-(L)- lysine nanoparticles on dopamine-coated surface to create a heparin density gradient for selective direction of platelet and vascular cells behavior. Acta Biomater. 2014;10:1940–1954. doi: 10.1016/j.actbio.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 3.Adipurnama I, Yang MC, Ciach T, Butruk-Raszeja B. Surface modification and endothelialization of polyurethane for vascular tissue engineering applications: a review. Biomater Sci. 2016;5:22–37. doi: 10.1039/c6bm00618c. [DOI] [PubMed] [Google Scholar]
- 4.Ren X, Feng Y, Guo J, Wang H, Li Q, Yang J, Hao X, Lv J, Ma N, Li W. Surface modification and endothelialization of biomaterials as potential scaffolds for vascular tissue engineering applications. Chem Soc Rev. 2015;44:5680–5742. doi: 10.1039/c4cs00483c. [DOI] [PubMed] [Google Scholar]
- 5.Qi P, Maitz MF, Huang N. Surface modification of card iovascular materials and implants. Surf Coat Technol. 2013;233:80–90. [Google Scholar]
- 6.Kent KC, Liu B. Intimal hyperplasia--still here after all these years! Ann Vasc Surg. 2004;18:135–137. doi: 10.1007/s10016-004-0019-4. [DOI] [PubMed] [Google Scholar]
- 7.Evans BC, Hocking KM, Osgood MJ, Voskresensky I, Dmowska J, Kilchrist KV, Brophy CM, Duvall CL. MK2 inhibitory peptide delivered in nanopolyplexes prevents vascular graft intimal hyperplasia. Sci Transl Med. 2015;7:291ra295. doi: 10.1126/scitranslmed.aaa4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 9.Sarkar S, Sales KM, Hamilton G, Seifalian AM. Addressing thrombogenicity in vascular graft construction. J Biomed Mater Res, Part B. 2007;82:100–108. doi: 10.1002/jbm.b.30710. [DOI] [PubMed] [Google Scholar]
- 10.Sugiura T, Agarwal R, Tara S, Yi T, Lee YU, Breuer CK, Weiss AS, Shinoka T. Tropoelastin inhibits intimal hyperplasia of mouse bioresorbable arterial vascular grafts. Acta Biomater. 2017;52:74–80. doi: 10.1016/j.actbio.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleinedler JJ, Foley JD, Orchard EA, Dugas TR. Novel nanocomposite stent coating releasing resveratrol and quercetin reduces neointimal hyperplasia and promotes re-endothelialization. J Controlled Release. 2012;159:27–33. doi: 10.1016/j.jconrel.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Chaudhary MA, Guo LW, Shi X, Chen G, Gong S, Liu B, Kent KC. Periadventitial drug delivery for the prevention of intimal hyperplasia following open surgery. J Controlled Release. 2016;233:174–180. doi: 10.1016/j.jconrel.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopes LB, Flynn C, Komalavilas P, Panitch A, Brophy CM, Seal BL. Inhibition of HSP27 phosphorylation by a cell-permeant MAPKAP Kinase 2 inhibitor. Biochem Biophys Res Commun. 2009;382:535–539. doi: 10.1016/j.bbrc.2009.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopes LB, Brophy CM, Flynn CR, Yi Z, Bowen BP, Smoke C, Seal B, Panitch A, Komalavilas P. A novel cell permeant peptide inhibitor of MAPKAP kinase II inhibits intimal hyperplasia in a human saphenous vein organ culture model. J Vasc Surg. 2010;52:1596–1607. doi: 10.1016/j.jvs.2010.06.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muto A, Panitch A, Kim N, Park K, Komalavilas P, Brophy CM, Dardik A. Inhibition of Mitogen Activated Protein Kinase Activated Protein Kinase II with MMI-0100 reduces intimal hyperplasia ex vivo and in vivo. Vasc Pharmacol. 2012;56:47–55. doi: 10.1016/j.vph.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zwolak RM, Adams MC, Clowes AW. Kinetics of vein graft hyperplasia: association with tangential stress. J Vasc Surg. 1987;5:126–136. [PubMed] [Google Scholar]
- 17.Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomed. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakiyama-Elbert SE. Incorporation of heparin into biomaterials. Acta Biomater. 2014;10:1581–1587. doi: 10.1016/j.actbio.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang Y, Kiick KL. Heparin- functionalized polymeric biomaterials in tissue engineering and drug delivery applications. Acta Biomater. 2014;10:1588–1600. doi: 10.1016/j.actbio.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murugesan S, Xie J, Linhardt RJ. Immobilization of heparin: approaches and applications. Curr Top Med Chem. 2008;8:80–100. doi: 10.2174/156802608783378891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye Q, Zhou F, Liu W. Bioinspired catecholic chemistry for surface modification. Chem Soc Rev. 2011;40:4244–4258. doi: 10.1039/c1cs15026j. [DOI] [PubMed] [Google Scholar]
- 22.Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel- inspired surface chemistry for multifunctional coatings. Science. 2007;318:426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee Y, Park KM, Bae JW, Park KD. Facile surface PEGylation via tyrosinase-catalyzed oxidative reaction for the preparation of non-fouling surfaces. Colloids Surf, B. 2013;102:585–589. doi: 10.1016/j.colsurfb.2012.08.059. [DOI] [PubMed] [Google Scholar]
- 24.Park KM, Park KD. Facile surface immobilization of cell adhesive peptide onto TiO2 substrate via tyrosinase-catalyzed oxidative reaction. J Mater Chem. 2011;21:15906–15908. [Google Scholar]
- 25.Li S, Garreau H, Pauvert B, McGrath J, Toniolo A, Vert M. Enzymatic degradation of block copolymers prepared from epsilon-caprolactone and poly(ethylene glycol) Biomacromolecules. 2002;3:525–530. doi: 10.1021/bm010168s. [DOI] [PubMed] [Google Scholar]
- 26.Tang ZG, Black RA, Curran JM, Hunt JA, Rhodes NP, Williams DF. Surface properties and biocompatibility of solvent-cast poly[-caprolactone] films. Biomaterials. 2004;25:4741–4748. doi: 10.1016/j.biomaterials.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Kim SE, Song SH, Yun YP, Choi BJ, Kwon IK, Bae MS, Moon HJ, Kwon YD. The effect of immobilization of heparin and bone morphogenic protein-2 (BMP-2) to titanium surfaces on inflammation and osteoblast function. Biomaterials. 2011;32:366–373. doi: 10.1016/j.biomaterials.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Kim SE, Yun YP, Shim KS, Park K, Choi SW, Shin DH, Suh DH. Fabrication of a BMP -2-immobilized porous microsphere modified by heparin for bone tissue engineering. Colloids Surf, B. 2015;134:453–460. doi: 10.1016/j.colsurfb.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Lee Y, Bae JW, Oh DH, Park KM, Chun YW, Sung HJ, Park KD. In situ forming gelatin-based tissue adhesives and their phenolic content-driven properties. J Mater Chem B. 2013;1:2407–2414. doi: 10.1039/c3tb00578j. [DOI] [PubMed] [Google Scholar]
- 30.Lee SH, Lee Y, Chun YW, Crowder SW, Young PP, Park KD, Sung HJ. In Situ Crosslinkable Gelatin Hydrogels for Vasculogenic Induction and Delivery of Mesenchymal Stem Cells. Adv Funct Mater. 2014;24:6771–6781. doi: 10.1002/adfm.201401110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tae G, Kim YJ, Choi WI, Kim M, Stayton PS, Hoffman AS. Formation of a novel heparin-based hydrogel in the presence of heparin-binding biomolecules. Biomacromolecules. 2007;8:1979–1986. doi: 10.1021/bm0701189. [DOI] [PubMed] [Google Scholar]
- 32.Lee H, Lee BP, Messersmith PB. A reversible wet/dry adhesive inspired by mussels and geckos. Nature. 2007;448:338–341. doi: 10.1038/nature05968. [DOI] [PubMed] [Google Scholar]
- 33.Boire TC, Gupta MK, Zachman AL, Lee SH, Balikov DA, Kim K, Bellan LM, Sung HJ. Pendantallyl crosslinking as a tunable shape memory actuator for vascular applications. Acta Biomater. 2015;24:53–63. doi: 10.1016/j.actbio.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Valence S, Tille JC, Mugnai D, Mrowczynski W, Gurny R, Moller M, Walpoth BH. Long term performance of polycaprolactone vascular grafts in a rat abdominal aorta replacement model. Biomaterials. 2012;33:38–47. doi: 10.1016/j.biomaterials.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 35.Gong W, Lei D, Li S, Huang P, Qi Q, Sun Y, Zhang Y, Wang Z, You Z, Ye X, Zhao Q. Hybrid small-diameter vascular grafts: Anti-expansion effect of electrospun poly epsilon-caprolactone on heparin-coated decellularized matrices. Biomaterials. 2016;76:359–370. doi: 10.1016/j.biomaterials.2015.10.066. [DOI] [PubMed] [Google Scholar]
- 36.Lopalco A, Stella VJ. Effect of Molecular Structure on the Relative Hydrogen Peroxide Scavenging Ability of Some alpha-Keto Carboxylic Acids. J Pharm Sci. 2016;105:2879–2885. doi: 10.1016/j.xphs.2016.03.041. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Liu YM, Qi ZM, Wang SY, Liu SX, Li X, Wang HJ, Xia XC. An overview on natural polysaccharides with antioxidant properties. Curr Med Chem. 2013;20:2899–2913. doi: 10.2174/0929867311320230006. [DOI] [PubMed] [Google Scholar]
- 38.Lee M, Kim Y, Ryu JH, Kim K, Han YM, Lee H. Long-term, feeder- free maintenance of human embryonic stem cells by mussel- inspired adhesive heparin and collagen type I. Acta Biomater. 2016;32:138–148. doi: 10.1016/j.actbio.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 39.Evans BC, Hocking KM, Kilchrist KV, Wise ES, Brophy CM, Duvall CL. Endosomolytic Nano-Polyplex Platform Technology for Cytosolic Peptide Delivery To Inhibit Pathological Vasoconstriction. ACS Nano. 2015;9:5893–5907. doi: 10.1021/acsnano.5b00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryu JH, Lee Y, Do MJ, Jo SD, Kim JS, Kim BS, Im GI, Park TG, Lee H. Chitosan- g-hematin: enzyme- mimicking polymeric catalyst for adhesive hydrogels. Acta Biomater. 2014;10:224–233. doi: 10.1016/j.actbio.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Feng W, Gao X, McClung G, Zhu SP, Ishihara K, Brash JL. Methacrylate polymer layers bearing poly(ethylene oxide) and phosphorylcholine side chains as non- fouling surfaces: In vitro interactions with plasma proteins and platelets. Acta Biomater. 2011;7:3692–3699. doi: 10.1016/j.actbio.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 42.Keuren JFW, Wielders SJH, Willems GM, Morra M, Lindhout T. Fibrinogen adsorption, platelet adhesion and thrombin generation at heparinized surfaces exposed to flowing blood. Thromb Haemostasis. 2002;87:742–747. [PubMed] [Google Scholar]
- 43.Yang Z, Wang J, Luo R, Maitz MF, Jing F, Sun H, Huang N. The covalent immobilization of heparin to pulsed-plasma polymeric allylamine films on 316L stainless steel and the resulting effects on hemocompatibility. Biomaterials. 2010;31:2072–2083. doi: 10.1016/j.biomaterials.2009.11.091. [DOI] [PubMed] [Google Scholar]
- 44.Lin WC, Liu TY, Yang MC. Hemocompatibility of polyacrylonitrile dialysis membrane immobilized with chitosan and heparin conjugate. Biomaterials. 2004;25:1947–1957. doi: 10.1016/j.biomaterials.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 45.Leng C, Hung HC, Sun S, Wang D, Li Y, Jiang S, Chen Z. Probing the Surface Hydration of Nonfouling Zwitterionic and PEG Materials in Contact with Proteins. ACS Appl Mater Interfaces. 2015;7:16881–16888. doi: 10.1021/acsami.5b05627. [DOI] [PubMed] [Google Scholar]
- 46.Chen SF, Li LY, Zhao C, Zheng J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer. 2010;51:5283–5293. [Google Scholar]
- 47.Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451:914–918. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye X, Hu X, Wang H, Liu J, Zhao Q. Polyelectrolyte multilayer film on decellularized porcine aortic valve can reduce the adhesion of blood cells without affecting the growth of human circulating progenitor cells. Acta Biomater. 2012;8:1057–1067. doi: 10.1016/j.actbio.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 49.Conte MS. Technical factors in lower-extremity vein bypass surgery: how can we improve outcomes? Semin Vasc Surg. 2009;22:227–233. doi: 10.1053/j.semvascsurg.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19:116–127. doi: 10.1038/cr.2008.326. [DOI] [PubMed] [Google Scholar]
- 51.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chava KR, Karpurapu M, Wang D, Bhanoori M, Kundumani-Sridharan V, Zhang Q, Ichiki T, Glasgow WC, Rao GN. CREB- mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler, Thromb Vasc Biol. 2009;29:809–815. doi: 10.1161/ATVBAHA.109.185777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapopara PR, von Felden J, Soehnlein O, Wang Y, Napp LC, Sonnenschein K, Wollert KC, Schieffer B, Gaestel M, Bauersachs J, Bavendiek U. Deficiency of MAPK-activated protein kinase 2 (MK2) prevents adverse remodelling and promotes endothelial healing after arterial injury. Thromb Haemostasis. 2014;112:1264–1276. doi: 10.1160/TH14-02-0174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Synthesis and characterizations of phenol-conjugated he parin (HT) derivatives. (a) Synthetic route of HT polymer by an EDC/NHS coupling reaction, (b) 1H NMR spectrum of HT polymer dissolved in D2O, and (c) UV absorbance spectra of heparin and HT polymers.
Figure S2. In vitro release profiles of MK2i peptide from PCL and PCL–HT substrates over 28 days. The cumulative released amounts of MK2i were drawn as a percentage with respect to the total MK2i amount loaded.
Figure S3. In vitro cytotoxicity evaluation of PCL–HT/MK2i substrate using NIH3T3 cells. (a) Cell cytotoxicity of bare PCL, PCL–HT and MK2i- loaded PCL–HT. The adhered NIH3T3 cells were treated with extracted culture media for 24 h, and then their mitochondrial activities were quantified by a WST-1 assay. As a positive control, fresh medium without sample extraction was used. Results are shown as the average values ± S.D. (n = 3) (b) Photographed images of the NIH3T3 cells cultured in each extracted medium for 24 h: phase contrast and live/dead staining images (green: live cells, red: dead cells). Scale bars indicate 100 μm.