

Abstract
Dual endothelin ETA and ETB receptor antagonists are approved therapy for pulmonary artery hypertension (PAH). We hypothesized that ETB receptor‐mediated clearance of endothelin‐1 at specific vascular sites may compromise this targeted therapy. Concentration‐response curves (CRC) to endothelin‐1 or the ETB agonist sarafotoxin S6c were constructed, with endothelin receptor antagonists, in various rat and mouse isolated arteries using wire myography or in rat isolated trachea. In rat small mesenteric arteries, bosentan displaced endothelin‐1 CRC competitively indicative of ETA receptor antagonism. In rat small pulmonary arteries, bosentan 10 μmol L−1 left‐shifted the endothelin‐1 CRC, demonstrating potentiation consistent with antagonism of an ETB receptor‐mediated endothelin‐1 clearance mechanism. Removal of endothelium or L‐NAME did not alter the EC 50 or Emax of endothelin‐1 nor increase the antagonism by BQ788. In the presence of BQ788 and L‐NAME, bosentan displayed ETA receptor antagonism. In rat trachea (ETB), bosentan was a competitive ETB antagonist against endothelin‐1 or sarafotoxin S6c. Modeling showed the importance of dual receptor antagonism where the potency ratio of ETA to ETB antagonism is close to unity. In conclusion, the rat pulmonary artery is an example of a special vascular bed where the resistance to antagonism of endothelin‐1 constriction by ET dual antagonists, such as bosentan or the ETB antagonist BQ788, is possibly due to the competition of potentiation of endothelin‐1 by blockade of ETB‐mediated endothelin‐1 clearance located on smooth muscle and antagonism of ETA‐ and ETB‐mediated contraction. This conclusion may have direct application for the efficacy of endothelin‐1 antagonists for treating PAH.
Keywords: ambrisentan, bosentan, BQ788, endothelin‐1, ETA receptors, ETB receptor‐mediated clearance mechanism, ETB receptors, macitentan, sarafotoxin S6c
Abbreviations
- D100
artery (mesenteric or tail) internal diameter (µm) at an equivalent transmural pressure of 100 mm Hg
- D20
artery (pulmonary) internal diameter (µm) at an equivalent transmural pressure of 20 mm Hg
- Emax
maximum possible effect for the agonist
- KPSS
isotonic potassium physiological salt solution (K+ 124 mmol L−1 for arteries or K+ 62 mmol L−1 for trachea)
- pEC50
negative log10 M concentration of agonist that evokes the half‐maximal response
- pKB
negative log10 M concentration of antagonist that shifted the agonist endothelin‐1 EC50 twofold to the right
- PSS
physiological salt solution
1. INTRODUCTION
In rats,1 rabbits,2 and nonhuman primates,3 dual ETA and ETB receptor antagonists or ETB‐selective endothelin‐1 antagonists increased the immunoreactive endothelin‐1 plasma level acutely by 3‐ to 10‐fold. After chronic oral dosing in rats with A‐182086, a dual ETA and ETB antagonist, the endothelin‐1 plasma levels rose by more than 24‐fold after 9 days.4 Micro positron emission tomography using 18F‐labeled endothelin‐1 in anesthetized rats confirmed that endothelin‐1 rapidly binds to rat lung and is cleared from the circulation (t 0.5 0.43 minutes).5 Pretreatment with the ETB‐selective antagonist BQ788 decreased the endothelin‐1 clearance by 85%.
While this intriguing mechanism of endothelin‐1 clearance by ETB receptors was first determined in vivo, we asked, could this mechanism affect the pharmacodynamics of endothelin‐1 interactions with ETA and ETB receptors mediating smooth muscle contraction in isolated tissue assays when determining the pKB of endothelin‐1 receptor antagonists? The impact of sites of loss of agonist or antagonist concentrations on pKB estimations has been observed in the acid‐secreting mouse stomach (figure 1 in Angus and Black6) and further developed by Kenakin.7 Indeed, we have previously reported that endothelin‐1 concentration‐contraction curves in rat small interlobar pulmonary arteries were surprisingly LEFT‐shifted; ie, endothelin‐1 contractions were “potentiated” in the presence of the dual ETA and ETB antagonist bosentan 10 μmol L−1,8 an observation that is consistent with blockade of a site of loss of endothelin‐1.
Here, we report the pharmacodynamic interactions and analyses of endothelin‐1 receptor antagonists in a range of isolated arteries and tracheal smooth muscle preparations with endothelin‐1 and the selective ETB receptor agonist venom peptide sarafotoxin S6c. Some arteries were treated with L‐NAME or had the endothelial cell layer removed. Our results show that the localized ETB clearance mechanism for endothelin‐1 on smooth muscle cells could explain the dramatic effect on the estimation of the dissociation constant for ETA and ETB antagonists when endothelin‐1 is used as the agonist and the endothelin‐1 clearance mechanism is present.
The conclusions provide a theoretical framework to test for the “ideal” dual ETA and ETB receptor antagonist if significant antagonism is to occur at ETA or ETB constrictor receptors and the ETB receptor‐mediated clearance of endothelin‐1 is blocked which potentiates the potency of endothelin‐1. This clearance mechanism, thus, joins other well‐known mechanisms of ETB‐mediated endothelin‐1 release of thromboxane A2, prostacyclin, and nitric oxide that would either enhance or functionally antagonize ETA‐ or ETB‐mediated vasoconstriction.9, 10, 11, 12
2. MATERIALS AND METHODS
The ethics committee of the University of Melbourne approved the experiments in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes (8th edition, 2013; National Health & Medical Research Council, Canberra, Australia). Animal studies are reported in compliance with the ARRIVE guidelines.13, 14 Male Sprague‐Dawley rats (280‐320 g; Biomedical Sciences Animal Facility, University of Melbourne, Australia) and male Swiss mice (30‐40 g; Animal Resources Centre, Murdoch, WA, Australia) were used in this study. Animals were housed (3‐4 per high‐topped cage with shredded paper bedding) at 22°C on a 12‐hour light/dark cycle with access to food and water ad libitum. Rats and mice were individually placed in a secure chamber and deeply anesthetized by inhalation of 5% isoflurane in oxygen, then killed by rapid excision of the heart. The rat and mouse tissues were rapidly excised and placed in cold physiological salt solution (PSS) with the following composition (mM): NaCI 119; KCl 4.69; MgSO4.7H2O 1.17; KH2PO4 1.18; glucose 5.5; NaHCO3 25; CaCl2.6H2O 2.5; EDTA 0.026 and saturated with carbogen (O2 95%; CO2 5%) at pH 7.4. Tissues were pinned down on a Silastic‐covered petri dish filled with cold PSS. A minimum of 5 rats or mice was used for each experimental group, with exact n values shown in the figure legends or Results section. Group sizes were equal by design; however, variations due to predetermined criteria (described in the methodology) are explained in the figure legends. Animal tissues were randomized to treatment groups.
2.1. Arteries
As previously described,15 third‐order rat and mouse mesenteric arteries, rat and mouse pulmonary arteries, and mouse tail arteries were dissected clear of their connective tissue and prepared as 2‐mm‐long segments for mounting on 40‐μm diameter wires for isometric force measurement in Mulvany and Halpern style myographs (model 620M, Danish Myo Technology, Aarhus, Denmark). Responses were captured by a Powerlab 4/30 A/D converter (ADInstruments, Sydney, Australia) and measured on a computer running LabChart 7 data acquisition software (ADInstruments).
After 30‐minute equilibration in PSS at 37°C, the arteries were passively stretched under micrometer (Mitutoyo, Kawasaki, Japan) control according to the normalization protocol to determine the internal diameter at equivalent transmural pressure of 100 mm Hg (D100) for all arteries, except for the pulmonary artery where 20 mm Hg was used (D20). The micrometer was then adjusted to decrease the passive stretch to an equivalent diameter of 90% of D100 (or 90% of D20, as applicable) and the artery remained at that setting of passive stretch for the remainder of the experiment.15, 16 Thirty minutes later the arteries were exposed for 2 minutes to potassium depolarizing solution (K+ replacing Na+ in PSS, ie, 124 mmol L−1; termed KPSS), before replacing with PSS. Subsequent responses were expressed as a % of this KPSS reference contraction in each artery. Rat or mouse mesenteric arteries that contracted to KPSS with <3 mN force, mouse tail arteries that contracted to KPSS with <20 mN force, or rat and mouse pulmonary arteries that contracted to KPSS with <1 mN force were considered as violations of predetermined criteria. As a further test of viability of the artery, a single 2‐minute exposure to 10 μmol L−1 noradrenaline was performed and then replaced with drug‐free PSS. To test the integrity of the endothelium, arteries were precontracted with noradrenaline 1 μmol L−1 (which contracts to about 80% of KPSS) and acetylcholine 1 μmol L−1 was added which would normally completely relax the artery in <30 seconds to the baseline force. Some arteries were equilibrated for 30 minutes with L‐NAME (Nω‐nitro‐L‐arginine methyl ester; 100 μmol L−1) and one concentration of an endothelin antagonist (bosentan 1, 10, or 100 μmol L−1; BQ788 0.3, 1, or 3 μmol L−1). In one study, BQ788 3 μmol L−1 and 0, 1, or 10 μmol L−1 bosentan were equilibrated before the concentration‐response curve was constructed to endothelin‐1. In each study, the artery was then exposed to a single cumulative concentration‐contraction curve (0.1 nmol L−1 to 3 μmol L−1, depending on agonist, tissue, and treatment) to either sarafotoxin S6c or endothelin‐1, added in half‐log10 M increments allowing time for the contraction to reach a plateau before raising the concentration.
2.2. Endothelium removal
In rat small pulmonary artery, the normalization procedure was completed before testing the relaxation to acetylcholine 1 μmol L−1 in arteries contracted by U46619 (0.1 μmol L−1). The artery passive force was then relaxed, and a human black hair was inserted into the artery lumen. Lateral movement of the hair and careful rotation of the artery loosely suspended on the 2 wires removed the endothelial cells. The passive force was reapplied to the level prior to the endothelial cell removal and the acetylcholine (1 μmol L−1) test repeated in the presence of U46619 (0.1 μmol L−1). Failure to relax to acetylcholine was considered the functional test of endothelial cell removal. The endothelium‐denuded arteries can still deliver a full relaxation response to sodium nitroprusside 1 μmol L−1.
2.3. Trachea
The main trachea (10 mm long) was dissected free from the rat, cut into 2‐ to 3‐mm‐long ring segments, and mounted on wires in 15‐mL organ baths (see figure 1 in Angus and Wright),15 used for large diameter ring segments. In some trachea ring segments, the epithelial cell layer was removed by using a splinter of wood and gently rubbing the lumen for 1 minute. The rings were stretched to a passive force of 1 g and equilibrated in PSS at 37°C for 60 minutes. A reference contraction to KPSS (62 mmol L−1 for trachea, see Clozel et al17) was obtained before washing the tissue with drug‐free PSS. Subsequent responses were expressed as a % of this KPSS reference contraction in each tracheal ring. Tracheae that contracted to KPSS with <1 g force were considered as violations of predetermined criteria. The resting force was readjusted to 1 g and the trachea left to equilibrate for 30 minutes in the absence or presence of bosentan (3, 10, or 30 μmol L−1). A single concentration‐contraction curve to sarafotoxin S6c or endothelin‐1 was constructed up to a maximum concentration of 0.3 μmol L−1 for sarafotoxin S6c or 3 μmol L−1 for endothelin‐1.
Figure 1.
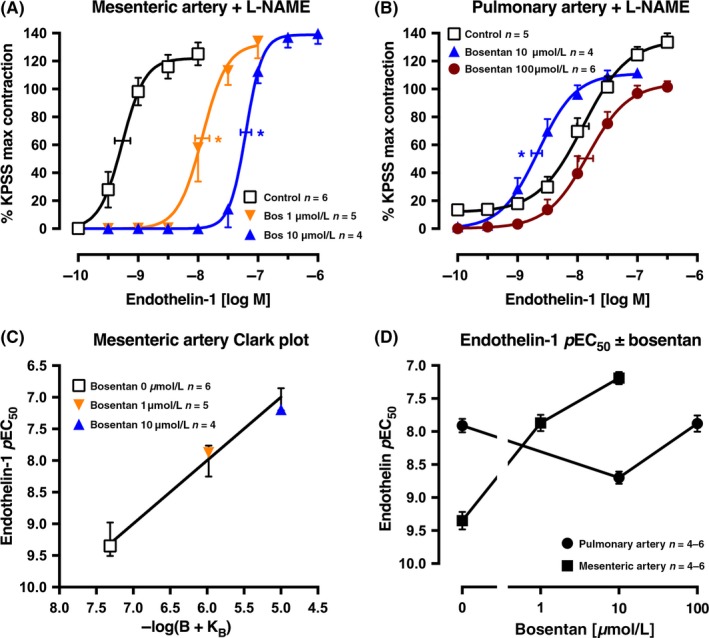
Average single exposure concentration‐contraction curves to endothelin‐1 in rat (A) mesenteric artery (n = 15) and (B) pulmonary artery (n = 15), pretreated with L‐NAME 100 μmol L−1, in the absence Control, (0 μmol L−1) or presence of bosentan 1, 10 or 100 μmol L−1. Data are expressed as % KPSS maximum contraction (y axis). (C) Clark plot display for the relationship in the rat mesenteric artery between the endothelin‐1 pEC 50 values (y axis; −log M) and −log(B + KB) where B is concentration of bosentan (0, 1, or 10 μmol L−1) and KB is the global‐fitted dissociation constant. The error bars are ± 2 SEM of the difference between the nonlinear regression‐fitted pEC 50 values for endothelin‐1 and the pEC 50 values fitted for the individual artery for each concentration of bosentan (B). (D) The pEC 50 values for the endothelin‐1 curves in (A) and (B) are plotted on the y axis against the bosentan concentration (x axis) for each artery type. Vertical error bars in (A, B, and D) are ± 1 SEM (those not shown are contained within the symbol). Horizontal error bars (A‐B) represent the EC 50 ± 1 SEM. n, number of arteries isolated from separate animals. *P < .05, pEC 50 values compared with respective control (0 μmol L−1) pEC 50 values. Variations in n are due to violation of predetermined criteria: mesenteric arteries that contracted to KPSS with <3 mN force or pulmonary arteries that contracted to KPSS with <1 mN force
2.4. Drugs
Drugs used were acetylcholine bromide (Sigma, St Louis, MO, USA); ambrisentan (Selleckchem, Houston, TX, USA); bosentan sodium salt (Selleckchem); BQ788 sodium salt (Peptides International, Louisville, KY, USA); endothelin‐1 (Genscript, Piscataway, NJ, USA); macitentan (Selleckchem); Nω‐nitro‐L‐arginine methyl ester hydrochloride (Sigma); (‐)‐noradrenaline bitartrate (Sigma); and sarafotoxin S6c (Auspep, Parkville, Victoria, Australia). All drugs were dissolved in MilliQ water except for endothelin‐1 which was dissolved in 10% dimethylformamide to 10−4 mol L−1, then diluted in MilliQ water, macitentan which was dissolved in DMSO to 10−3 mol L−1, then diluted in MilliQ water, and BQ788 which was dissolved in DMSO to 10−4 mol L−1.
2.5. Statistics and analyses
All data are expressed as mean ± SEM from n experiments. The data and analyses comply with the recommendations on experimental design and analysis in pharmacology.18 All contraction responses to endothelin‐1 or sarafotoxin S6c were measured as a % of the Emax (maximum response to agonist) to KPSS within each artery or tracheal ring. Each individual sigmoidal concentration‐contraction curve to endothelin‐1 or sarafotoxin S6c in the absence or presence of an endothelin receptor antagonist was fitted using Prism 7 (GraphPad Software, La Jolla, CA, USA). The pEC50 ± SEM values (−log10 M EC50) were determined for each treatment group. In endothelin‐1 experiments in rat trachea, the concentration‐contraction curves were not fitted as Emax values were not defined; instead, endothelin‐1 pEC50 values were calculated at responses of 50% KPSS maximum contraction. pEC50 values in treatment groups were compared to the respective control group with a one‐way ANOVA and Dunnett's post hoc test (Prism 7). Blinding was not performed for this study as all experiments yielded strict quantitative data.
2.6. Clark plot and analyses
Endothelin‐1 rapidly activates the respective ETA or ETB receptors before being internalized for recycling (ETA) or destruction (ETB) (see Bremnes et al19 and Paasche et al19, 20). This phenomenon makes it particularly difficult to establish multiple concentration‐response curves within a particular artery. In practical terms, the ETA or ETB receptor may be rapidly activated, but the resultant calcium mobilization and contraction takes a considerable time to develop even in small arteries <200 μm diameter. Thus, we routinely designed our experiments around a single cumulative concentration‐response curve in the presence or absence of an antagonist concentration.
Our chosen experimental design of only one concentration‐response curve per tissue does not allow for Schild plot analyses or determination of concentration ratios within tissue. By preference, we used the Clark plot and global fit analysis with its robust advantages.21 To determine the antagonist dissociation constant (KB) for each endothelin antagonist, we applied the global regression method22 that was simplified from that developed originally by Stone and Angus.21 A computer‐based nonlinear regression was performed to solve for KB (pKB = −log KB) by iterative approximation for ALL the endothelin‐1 (or sarafotoxin S6c) pEC50 values in the absence or presence of antagonist (B) concentrations thus:
| (1) |
where n is a “power departure” equivalent to allowing the slope of a Schild plot to vary from unity (see Lew and Angus22).
Having solved pKB, the relationship between the mean pEC50 values of the actual data were plotted against the antagonist concentration −log(B + KB) at concentrations of bosentan (0, 1, 3, 10, 30, or 100 μmol L−1), ambrisentan (0, 1, 3, or 10 μmol L−1), or macitentan (0, 0.3, 1, or 10 μmol L−1). This graphical display was named the Clark plot by Stone and Angus21 as it was similar to the plot developed by Clark23 of log(agonist) vs log(antagonist) at equal level of response. There are 2 important ways to test whether the concentration‐response curves are displaced to the right of the control pEC50 according to simple competitive antagonism. First, whether the 95% confidence limits for n contains 1; if so, the equation (1) is fitted where n = 1. Second, the error bars on the Clark plot are ± 2 times the standard error of the differences between the observed endothelin‐1 (or sarafotoxin S6c) pEC50 values and the predicted pEC50 values from the fitted equation (1). This provides an estimate of the confidence band around the line. If the point showing the average of the observed pEC50 values at a level of −log(B + KB) fell outside the error bar, this would indicate a departure from the simple competitivity model.
For comparison of pKB values between each antagonist in different settings, an unpaired Student's t test (Prism 7) was performed. Statistical significance was taken when P < .05.
3. RESULTS
3.1. Rat mesenteric and pulmonary small arteries
In rat small mesenteric arteries (i.d. 352 ± 6 μm), single endothelin‐1 concentration‐response curves had a pEC50 of 8.12 ± 0.02 and an Emax of 108 ± 5% KPSS (n = 4; data not shown). In the presence of L‐NAME (100 μmol L−1), the pEC50 for endothelin‐1 was 9.35 ± 0.13 (n = 6), significantly higher (17‐fold more potent) than in the absence of L‐NAME, and the Emax was 123 ± 9% KPSS (Figure 1A). In rat small second‐order pulmonary arteries (524 ± 20 μm i.d.), the pEC50 for endothelin‐1 was 7.55 ± 0.20 with an Emax of 124 ± 4% KPSS (n = 5; data not shown). In the presence of L‐NAME (100 μmol L−1), the pEC50 was 7.91 ± 0.10, not significantly different from control, and the Emax was 135 ± 7% KPSS (n = 5; Figure 1B). In the presence of L‐NAME and bosentan 1 and 10 μmol L−1, the endothelin‐1 concentration‐response curves were right‐shifted in a competitive manner in the rat mesenteric artery (Figure 1A), but significantly left‐shifted with bosentan 10 μmol L−1 in the rat pulmonary artery (Figure 1B). In the presence of bosentan 100 μmol L−1, the endothelin‐1 curve was located not significantly different to the control in the presence of L‐NAME (Figure 1B). For the mesenteric artery, the Clark plot and analyses indicate a pKB of 7.31 ± 0.16 (n = 15 points), congruent with the model of competitive antagonism (Figure 1C). A display of the pEC50 values for endothelin‐1 concentration‐response curves shows the significantly different control pEC50 for endothelin‐1 in the 2 artery types and the opposite effect on the pEC50 by bosentan 10 μmol L−1, all in the presence of L‐NAME (Figure 1D). Clearly, the presence of 100 μmol L−1 bosentan had very little effect in antagonizing the endothelin‐1 contraction when compared with control (0 μmol L−1 bosentan) in the pulmonary artery.
The failure to obtain an estimate of the pKB for bosentan in rat small pulmonary artery, possibly due to the removal of the agonist endothelin‐1, prompted the use of the nonendogenous ETB‐selective ligand sarafotoxin S6c in the absence or presence of L‐NAME. The control (0 μmol L−1 bosentan) curve was more potent (4.1‐fold) in the presence of L‐NAME (pEC50 with L‐NAME 9.31 ± 0.09, n = 6, and without L‐NAME 8.70 ± 0.19, n = 5; Figure 2A,B), suggesting a small effect of NO release in functionally antagonizing the contraction. The Clark plots and analyses show that bosentan is a competitive antagonist with similar pKB values in the absence or presence of L‐NAME of 5.52 ± 0.17 (n = 15 points) and 5.91 ± 0.24 (n = 17 points), respectively (Figure 2C,D).
Figure 2.
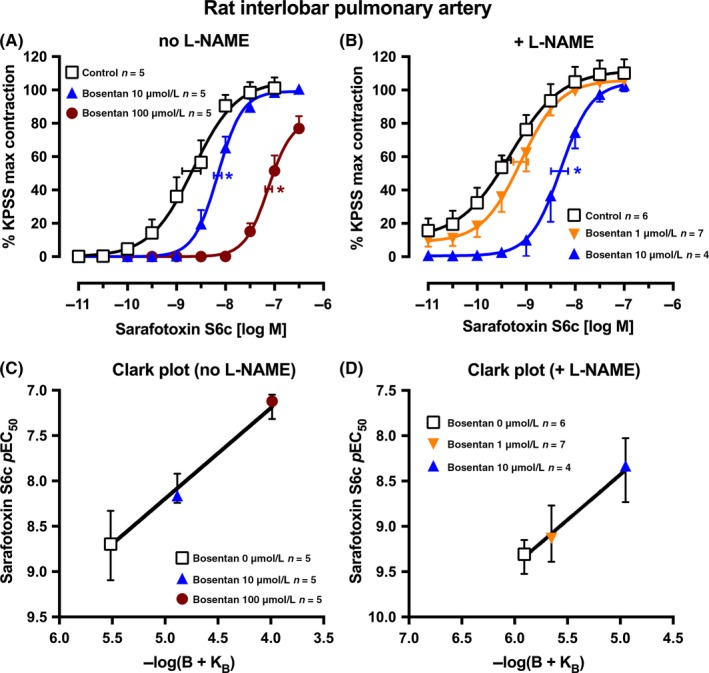
Average single exposure concentration‐contraction curves to sarafotoxin S6c in rat pulmonary artery in the (A) absence or (B) presence of L‐NAME 100 μmol L−1 and of bosentan 0 (Control), 1, 10, or 100 μmol L−1. Data are expressed as % KPSS maximum contraction (y axis). Vertical error bars are ± 1 SEM (those not shown are contained within the symbol). Horizontal error bars represent the EC 50 ± 1 SEM. (C‐D) Clark plot displays for the corresponding figure panel above for the relationship between the sarafotoxin S6c pEC 50 values (y axis; −log M) and −log(B + KB) values (see legend for Figure 1C) in the absence (C) or presence (D) of L‐NAME. n, number of arteries isolated from separate animals. *P < .05, pEC 50 values compared with respective control (0 μmol L−1) pEC 50 values. Variations in n are due to violation of a predetermined criterion: arteries that contracted to KPSS with <1 mN force
The more direct test for the role of ETB receptors in the endothelial clearance of endothelin‐1 was concluded from the interaction of the highly selective ETB receptor antagonist BQ788 and endothelin‐1 in the absence of the endothelium (Figure 3A). Again BQ788 (0.3‐3 μmol L−1) appeared to slightly left‐shift (potentiate) the endothelin‐1 concentration‐response curve pEC50. Moreover, this family of curves was similar in pattern to the curves in the presence of L‐NAME and endothelium (Figure 3B,E‐F), suggesting that ETB‐mediated clearance of endothelin‐1 may well be dependent on the smooth muscle cells in this artery rather than on the endothelium. To calibrate the ETB receptor on smooth muscle in the absence of NO release or clearance, we tested individual concentration‐response curves for sarafotoxin S6c with increasing concentrations of BQ788 in the presence of L‐NAME (100 μmol L−1) (Figure 3D). The global fit and Clark plot gave a pKB of 7.20 ± 0.21 (n = 20 points; Figure 3H). In the presence of BQ788 3 μmol L−1 to antagonize the ETB‐mediated clearance of endothelin‐1 and antagonize the ETB‐mediated contraction, endothelin‐1 still contracted the pulmonary artery with a pEC50 of 7.05 ± 0.16 indicating that ETA receptors were now operating (Figure 3C). To test this, equilibration with bosentan 0, 1, and 10 μmol L−1 gave right‐shifted concentration‐response curves and a pKB of 6.26 ± 0.23 (n = 13 points; Clark plot, Figure 3G). Note that the Clark plot display indicates that competitivity was not achieved as points lie outside the error bars for bosentan 1 and 10 μmol L−1.
Figure 3.
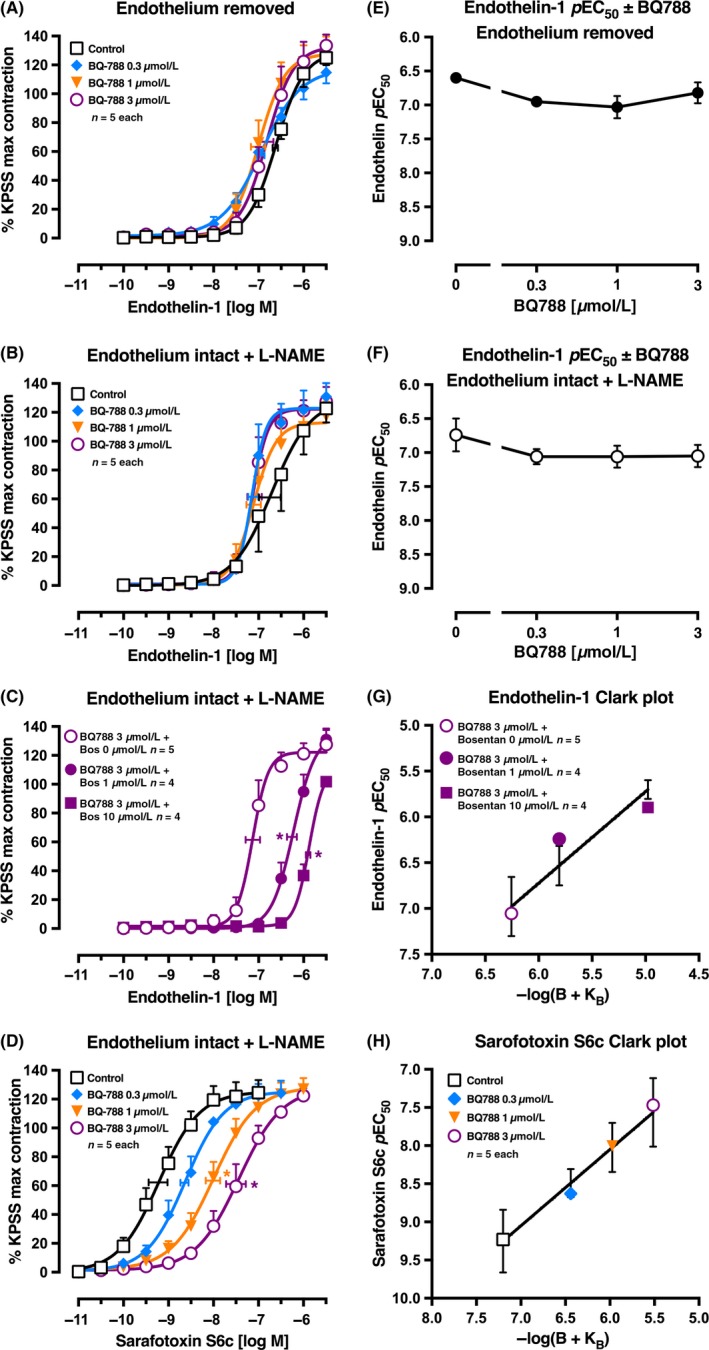
Average single exposure concentration‐contraction curves to (A‐C) endothelin‐1 or (D) sarafotoxin S6c in rat pulmonary artery in the (A) absence of endothelium or (B‐D) presence of endothelium plus L‐NAME 100 μmol L−1. (A, B, and D) Curves were completed in the absence (Control, 0 μmol L−1) or presence of BQ788 0.3, 1, or 3 μmol L−1. (C) Curves were completed in the presence of BQ788 3 μmol L−1 plus bosentan 0, 1, or 10 μmol L−1. Data are expressed as % KPSS maximum contraction (y axis). (E‐F) The pEC 50 values from (A‐B) are plotted on the y axis against the BQ788 concentration (x axis). Vertical error bars (A‐F) are ± 1 SEM (those not shown are contained within the symbol). Horizontal error bars (A‐D) represent the EC 50 ± 1 SEM. (G‐H) Clark plot displays for the corresponding left figure panel for the relationship between the (G) endothelin‐1 or (H) sarafotoxin S6c pEC 50 values (y axis; −log M) and −log(B + KB) values (see legend for Figure 1C) in the presence of L‐NAME. *P < .05, pEC 50 values compared with respective control (0 μmol L−1) pEC 50 values. n, number of arteries isolated from separate animals. Variations in n are due to violation of a predetermined criterion: arteries that contracted to KPSS with <1 mN force
Evidence that L‐NAME or endothelial cell removal had abolished the relaxation to acetylcholine 1 μmol L−1 was shown by the result that before treatment with L‐NAME or endothelial removal the relaxation to acetylcholine 1 μmol L−1 as a % of the precontractile tone was −54 ± 4% (n = 19) or −56 ± 2% (n = 18), respectively, and after treatment was −2 ± 2% or −1 ± 2%, respectively (data not shown).
3.2. Other arteries
In the mouse, we examined 3 different arteries to determine if the responses to bosentan and endothelin‐1 in the small pulmonary artery of the rat could be replicated. In the main pulmonary artery (i.d. 648 ± 20 μm), mesenteric artery (i.d. 275 ± 15 μm), and tail artery (i.d. 370 ± 6 μm), the patterns of endothelin‐1 concentration‐response curves and antagonism by bosentan were similar (Figure 4A‐C). The Clark plots and analyses showed pKB values for bosentan of 7.16 ± 0.13 (n = 17 points), 6.24 ± 0.16 (n = 14 points), and 6.52 ± 0.18 (n = 17 points) in the pulmonary, mesenteric, and tail arteries, respectively, and complied with the model of simple competitivity (Figure 4D‐F).
Figure 4.
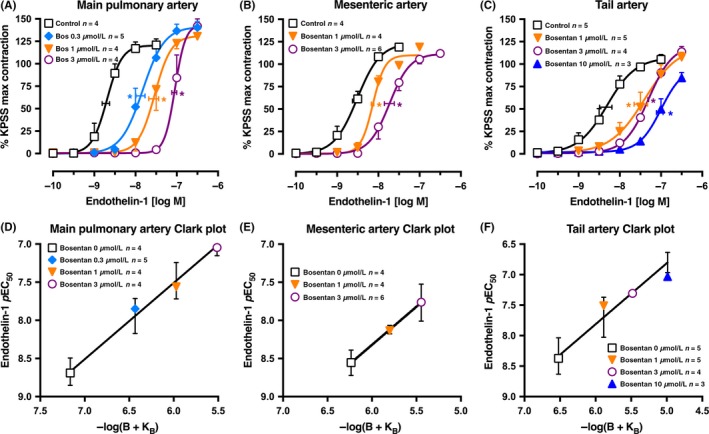
Average single exposure concentration‐contraction curves to endothelin‐1 in Swiss mouse isolated (A) main pulmonary, (B) mesenteric, and (C) tail arteries in the absence and presence of bosentan (0 (Control), 0.3, 1, 3, or 10 μmol L−1). Data are expressed as % KPSS maximum contraction (y axis). Vertical error bars are ± 1 SEM (those not shown are contained within the symbol). Horizontal error bars represent the EC 50 ± 1 SEM. (D‐F) Clark plot displays for the corresponding figure panel above for the relationship between the endothelin‐1 pEC 50 values (y axis; −log M) and −log(B + KB) values (see legend for Figure 1C). n, number of arteries isolated from separate animals. *P < .05, pEC 50 values compared with respective control (0 μmol L−1) pEC 50 values. Variations in n are due to violation of predetermined criteria: pulmonary arteries that contracted to KPSS with <1 mN force; mesenteric arteries that contracted to KPSS with <3 mN force; or tail arteries that contracted to KPSS with <20 mN force
3.3. Macitentan
In the rat small mesenteric artery, macitentan (0.3 and 1 μmol L−1) was a potent competitive endothelin‐1 receptor antagonist (Figure 5A). The Clark plot and analyses gave a pKB of 7.05 ± 0.10 (n = 15 points) and fitted the competitive model. The endothelin‐1 concentration‐contraction curves in the rat small pulmonary artery were completely unaffected by 1 and 10 μmol L−1 macitentan, as shown in Figure 5B.
Figure 5.
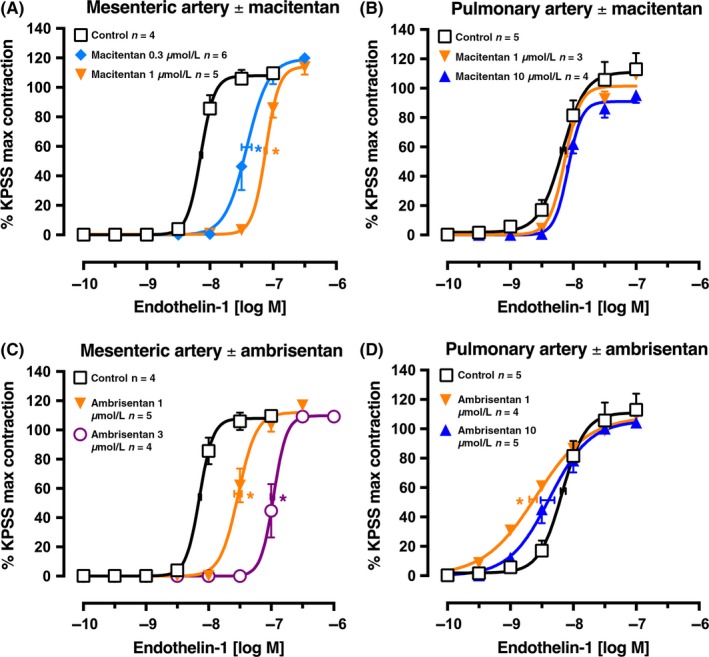
Average single exposure concentration‐contraction curves to endothelin‐1 in rat (A) and (C) mesenteric artery (n = 13‐15) or (B) and (D) pulmonary artery (n = 12‐14) in the absence (Control, 0 μmol L−1) or presence of (A‐B) macitentan 0.3, 1, or 10 μmol L−1 or (C‐D) ambrisentan 1, 3, or 10 μmol L−1. Data are expressed as % KPSS maximum contraction (y axis). Vertical error bars are ± 1 SEM (those not shown are contained within the symbol). Horizontal error bars represent the EC 50 ± 1 SEM. *P < .05, pEC 50 values compared with respective control (0 μmol L−1) pEC 50 values. Variations in n are due to violation of predetermined criteria: mesenteric arteries that contracted to KPSS with <3 mN force or pulmonary arteries that contracted to KPSS with <1 mN force
3.4. Ambrisentan
In the rat small mesenteric artery, ambrisentan (1 and 3 μmol L−1) was a potent competitive endothelin‐1 receptor antagonist (Figure 5C). The Clark plot and analyses gave a pKB of 6.60 ± 0.07 (n = 13 points) and fitted the competitive model. In contrast, endothelin‐1 concentration‐contraction curves in the rat small pulmonary artery were slightly left‐shifted from control by ambrisentan 1 μmol L−1 before showing a small right‐shift at 10 μmol L−1 (Figure 5D).
3.5. Rat trachea
As was observed in rat small pulmonary artery, sarafotoxin S6c was significantly more potent than endothelin‐1 in contracting the rat isolated trachea with the epithelium intact in the absence of any antagonist (pEC50 values: sarafotoxin S6c 8.72 ± 0.22, n = 4; endothelin‐1 6.82 ± 0.09, n = 7; Figure 6A,B). In the absence of epithelium, the pEC50 for sarafotoxin S6c was not changed (8.63 ± 0.13, n = 5) and similarly for endothelin‐1 (6.61 ± 0.17, n = 4) (Figure 6C,D). The low potency of endothelin‐1 in rat trachea prevented exploration of the full concentration‐response curve, and therefore, we calculated pEC50 values at responses of 50% KPSS maximum contraction. In contrast, the full sarafotoxin S6c concentration‐response curves were obtained to allow pEC50 values to be calculated from logistic curve analysis, often when the Emax was more than 100% KPSS.
Figure 6.
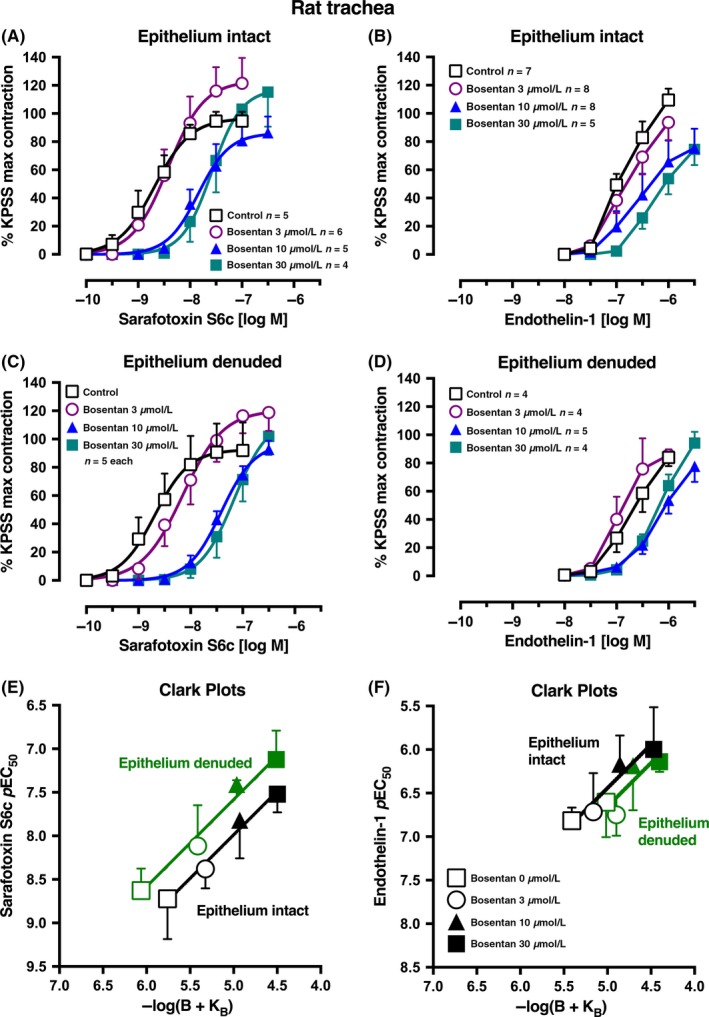
Average single concentration‐contraction curves to (A) sarafotoxin S6c or (B) endothelin‐1 in rat isolated trachea with intact epithelium in the absence (Control, 0 μmol L−1) or presence of bosentan 3, 10, or 30 μmol L−1. (C‐D) Corresponding agonist curves in trachea with epithelium denuded. Data are expressed as % KPSS maximum contraction (y axis). Error bars in (A‐D) are ± 1 SEM (those not shown are contained within the symbol). (E‐F) Clark plot displays for the relationship in the rat trachea between the (E) sarafotoxin S6c or (F) endothelin‐1 pEC 50 values (y axis; −log M) and −log(B + KB) where B is concentration of bosentan (0, 3, 10, or 30 μmol L−1) and KB is the global‐fitted dissociation constant (see legend for Figure 1C). n, number of tracheal rings isolated from separate animals. Variations in n are due to violation of a predetermined criterion: tracheae that contracted to KPSS with <1 g force
Bosentan (3‐30 μmol L−1) right‐shifted the endothelin‐1 and sarafotoxin S6c concentration‐contraction curves. The Clark plot and analysis show that with sarafotoxin S6c and epithelium intact, bosentan's pKB was 5.76 ± 0.23 (n = 20 points), not significantly different from endothelin‐1 as agonist with a pKB of 5.41 ± 0.28 (n = 28 points; Figure 6E,F). In epithelium‐denuded trachea, with sarafotoxin S6c, the bosentan pKB was 6.06 ± 0.18 (n = 20 points), significantly higher than for endothelin‐1 as agonist (pKB 5.02 ± 0.31, n = 17 points).
3.6. Modeling
To model the interaction between endothelin‐1 clearance and ETB and ETA receptor antagonism of the contraction response in small pulmonary arteries, we set the following criteria:
The ETB receptor‐sensitive endothelin‐1 clearance mechanism (CETB) can decrease the endothelin‐1 concentration at the ETA or ETB receptor environment by a maximum of 10‐fold (1 pEC50 unit).
The theoretical dual ETA and ETB receptor antagonist has the same pKB value (8.5) at the “clearance ETB receptor” as at the ETB and ETA receptor modulating contraction.
The efficiency of endothelin‐1 at ETA and ETB constrictor receptors is the same.
In Figure 7A, we set the control pEC50 for the sarafotoxin S6c concentration‐response curve at 8.8 so that a twofold shift (log 0.3) would result in a pEC50 of 8.5 in the presence of an ETA and ETB antagonist with a pKB of 8.5 (3 nmol L−1). Similarly, we set the control pEC50 for endothelin‐1 at 7.8. Assuming that the ETB (sarafotoxin S6c) assay was not compromised by clearance nor the endothelin‐1 assay (like mesenteric artery or rat aorta), then the pEC50 in the presence of the dual ET receptor antagonist (8.5‐6.5 −log M) would rise as shown in Figure 7A and the Schild plot would show competitive antagonism (slope = 1) and pKB 8.5 (Figure 7B). If the assay is of predominantly ETB receptors and the clearance mechanism is active, as we hypothesize in the small pulmonary artery, the control pEC50 for endothelin‐1 would lower to 6.8 as endothelin‐1 is removed from the receptor locus by the clearance mechanism (Figure 7A; ●). In the presence of the dual ETA and ETB antagonist, endothelin‐1 would be both potentiated in concentration available to ETB receptors as the clearance mechanism is antagonized, but in addition, the endothelin‐1 concentration would be inhibited at the ETB receptor mediating contraction. This is shown graphically in Figure 7A where at 3 concentrations of the dual antagonist, the endothelin‐1 pEC50 is both enhanced (●) and antagonized (▲) with 8.5, 8, 7.5, 7, and 6.5 −log M. To the experimenter of course, only the resultant of the potentiation and antagonism might be observed as shown by the ▲ flat line until surmountable antagonism is observed at 7 (–log M).
Figure 7.
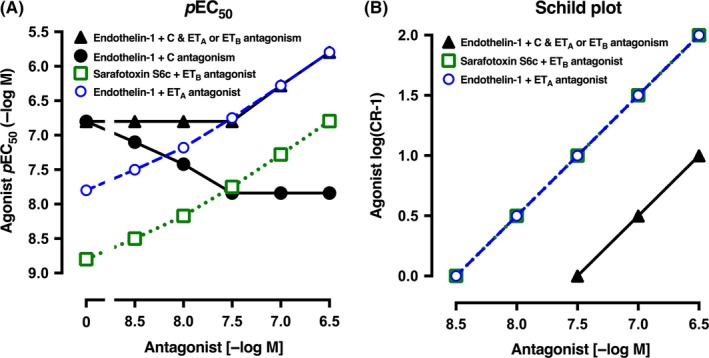
(A) Hypothetical relationship between the pEC
50 values for endothelin‐1 or sarafotoxin S6c concentration‐contraction curves and the concentration of a theoretical dual ETA and ETB receptor antagonist with a pKB of 8.5 at both receptors. For simplicity, the control (0 antagonist) pEC
50 for sarafotoxin S6c ( ) was set 1 log unit higher (10‐fold more potent) than for endothelin‐1 (
) was set 1 log unit higher (10‐fold more potent) than for endothelin‐1 ( ). In the presence of ETB receptors and the clearance (C) mechanism for endothelin‐1, the maximum clearance was set at 10‐fold (1 log unit) so that the pEC
50 in the presence of no antagonist (0) rises 1 log unit (● or ▲). As the ETB antagonism starts to block the endothelin‐1 clearance, so the pEC
50 rises (●) but just as does the ETB and ETA antagonism so that the resultant shows the actual pEC
50 is not altered. (B) The Schild plot for endothelin‐1 (or sarafotoxin S6c) as the agonist and the dual ETA and ETB antagonist with pKB of 8.5 is shown. Separate theoretical lines are shown for ETA (
). In the presence of ETB receptors and the clearance (C) mechanism for endothelin‐1, the maximum clearance was set at 10‐fold (1 log unit) so that the pEC
50 in the presence of no antagonist (0) rises 1 log unit (● or ▲). As the ETB antagonism starts to block the endothelin‐1 clearance, so the pEC
50 rises (●) but just as does the ETB and ETA antagonism so that the resultant shows the actual pEC
50 is not altered. (B) The Schild plot for endothelin‐1 (or sarafotoxin S6c) as the agonist and the dual ETA and ETB antagonist with pKB of 8.5 is shown. Separate theoretical lines are shown for ETA ( ; eg, rat aorta) and ETB (
; eg, rat aorta) and ETB ( ; eg, trachea). In the presence of ETB‐mediated clearance (C) that removes endothelin‐1, as in pulmonary artery, the Schild plot points (▲) move parallel 1 log unit to decrease the potency of the dual antagonist by 10‐fold (ie, the pKB of 8.5 becomes 7.5). The y axis is the agonist log(concentration ratio–1) and the x axis shows the concentration of dual ETA and ETB antagonist (−log M)
; eg, trachea). In the presence of ETB‐mediated clearance (C) that removes endothelin‐1, as in pulmonary artery, the Schild plot points (▲) move parallel 1 log unit to decrease the potency of the dual antagonist by 10‐fold (ie, the pKB of 8.5 becomes 7.5). The y axis is the agonist log(concentration ratio–1) and the x axis shows the concentration of dual ETA and ETB antagonist (−log M)
The relationship between the endothelin‐1 pEC50 and the ETA and ETB receptor antagonist concentrations is best illustrated in a Schild plot (Figure 7B). The dual ETA and ETB antagonist in the absence of the ETB clearance mechanism shows slope 1 and pKB 8.5 for the agonists endothelin‐1 at an ETA receptor‐only assay and pKB 8.5 for sarafotoxin S6c at an ETB receptor assay. Note that at pAntagonist 6.5, the ETA and ETB shift is 100‐fold (log(concentration ratio − 1) = 2). But if the endothelin‐1 clearance mechanism is active, as in small pulmonary arteries, the shift at constrictor ETB receptors is now only 10‐fold at pAntagonist 6.5 as the pKB has moved to 7.5. Clinically, this scenario would demand at least a plasma level of endothelin antagonist of p6.5 (ie, 0.3 μmol L−1) if the pKB at ETA and ETB receptors was 8.5.
Two further scenarios have been modeled. First, if the ETA to ETB selectivity ratio was ETA‐selective by 30‐fold, ie, pKB for the ET antagonist at ETA receptors was 8.5 and 7.0 for ETB receptors, the Schild plot with endothelin‐1 clearance active shows that to reach a 10‐fold antagonism at ETB constrictor receptors, then the plasma concentration would need to reach 10 μmol L−1 (p5 mol L−1), and at this concentration, the antagonism of ETA receptors would be 3000‐fold (Figure 8A). The second scenario is when an endothelin antagonist has a 10‐fold selectivity for ETB over ETA receptors; thus, pKB at ETB receptors is set at 8.5 and for ETA receptors at 7.5. The Schild plot (Figure 8B) shows that endothelin‐1 clearance will effectively decrease the pKB of antagonism at ETB constrictor receptors to 7.5, the same as ETA. Thus, a concentration of 0.3 μmol L−1 would result in a 10‐fold shift in both ETA and ETB receptors in the presence of endothelin‐1 clearance.
Figure 8.
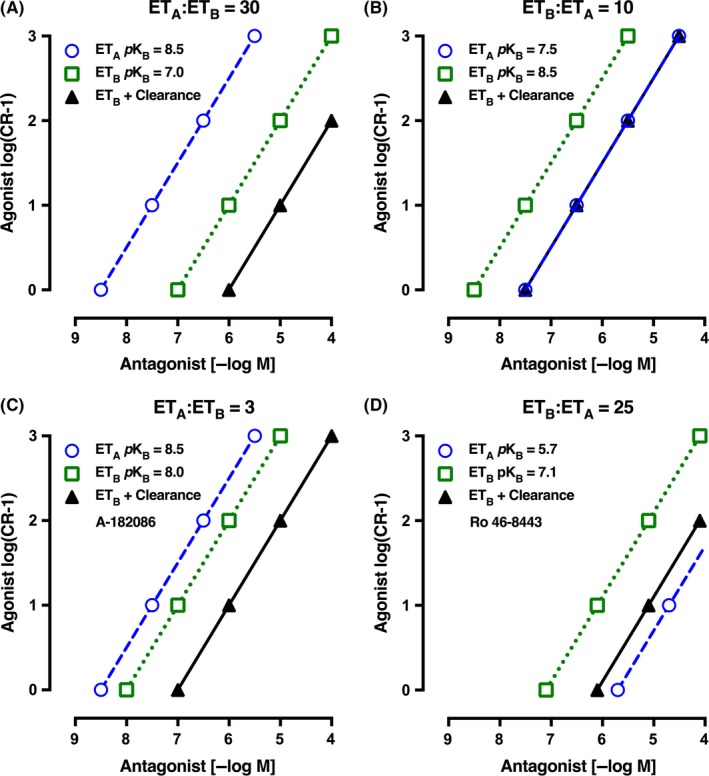
(A) Schild plot for a theoretical endothelin‐1 antagonist that is 30‐fold more selective at ETA vs ETB receptors (pKB: ETA 8.5 and ETB 7.0). Note that in the presence of ETB‐mediated clearance, the plasma concentration of the dual antagonist must rise to 10 μmol L−1 to give a 10‐fold antagonism at ETB constrictor receptors and 3000‐fold antagonism at ETA constrictor receptors. (B) Schild plot for a theoretical endothelin‐1 antagonist that is 10‐fold more selective at ETB vs ETA receptors (pKB: ETB 8.5 and ETA 7.5). In the presence of ETB‐mediated clearance, the plasma concentration of the dual antagonist must rise to 0.3 μmol L−1 to give a 10‐fold antagonism at both ETB and ETA receptors. (C) Schild plot for endothelin‐1 antagonist A‐182086 that is threefold more selective for ETA vs ETB receptors (pKB: ETA 8.5 and ETB 8.0; see Table 1). In the presence of ETB‐mediated clearance, the plasma concentration of the dual antagonist must rise to 1 μmol L−1 to give a 10‐fold antagonism at ETB constrictor receptors and 300‐fold antagonism at ETA constrictor receptors. (D) Schild plot for endothelin‐1 antagonist Ro 46‐8443 that is 25‐fold more selective at ETB vs ETA receptors (pKB: ETB 7.1 and ETA 5.7; see Table 1). In the presence of ETB‐mediated clearance, the plasma concentration of the dual antagonist must rise to 7.9 μmol L−1 to give a 10‐fold antagonism at ETB receptors, with a fivefold antagonism at ETA receptors. The y axis is the agonist log(concentration ratio–1) and the x axis shows the concentration of dual ETA and ETB antagonist (−log M)
We also present the Schild plot for compound A‐182086 which was developed with just threefold ETA to ETB selectivity (pKB at ETA 8.5 and at ETB 8; Figure 8C). Thus, in the presence of clearance, the plasma levels would need to be about 1 μmol L−1 (ie, 6 −log M) for a 10‐fold shift in ETB receptor constrictor activity, while there would be at least 300‐fold shift for ETA receptors. Indeed, these peak plasma levels of 4.3 μmol L−1 were achieved in rats given A‐182086 10 mg·kg−1 oral or even greater in dogs (34.5 μmol L−1), but significantly less in monkeys (0.16 μmol L−1) as the bioavailability varied from 54%, 71%, and 11%, respectively.24
Finally, we present the theoretical Schild plot for a selective ETB antagonist Ro 46‐8443 where the pKB at ETB is 7.1 and 5.7 at ETA receptors (Figure 8D). This ETB to ETA selectivity of 25 shows an important effect that when even with clearance in operation there is still more ETB constrictor antagonism than ETA.
4. DISCUSSION
Our work supports the hypothesis that in very specific vascular beds, the local clearance of endothelin‐1 lowers the endothelin‐1 concentration that would activate ETA or ETB endothelin receptors. The pKB estimate for ETA, ETB, or mixed ETA and ETB receptor antagonists will be confounded by 2 competing processes: one to potentiate the agonist endothelin‐1 and the second to antagonize its action at ETA and/or ETB receptors.
The tissue assays reported here confirm that there are special defined locations in some vascular beds and tracheal tissue that have a major population of ETB receptors on smooth muscle. Functional ETB receptors were defined by the substantial contraction up to the tissue maximum by the potent ETB‐selective agonist sarafotoxin S6c. This agonist is not a substrate for the ETB receptor‐sensitive clearance mechanism specifically shown for endothelin‐1 and blocked by ETB antagonists. Thus, the rat tracheal ring with agonist sarafotoxin S6c and epithelium intact proved to be a robust assay to define the pKB for ETB antagonists. We calculated the pKB for bosentan as 5.76 ± 0.23 for ETB receptors with sarafotoxin S6c and similarly 5.41 ± 0.28 with endothelin‐1. Importantly, the pKB for bosentan and sarafotoxin S6c was the same whether the epithelium was present or absent (pKB 5.76 ± 0.23 and 6.06 ± 0.18, respectively). In the original bosentan report, in rat tracheal rings, the pA2 was reported as 5.94 ± 0.04 with Schild slope 0.90 ± 0.18.17 Thus, tracheal smooth muscle ETB receptors mediate contraction, but there is no evidence of clearance of endothelin‐1 in this assay.
For the ETA receptor, the analysis is less certain as there is no selective ETA receptor agonist.25 The main assay used to determine the pKB (7.28) for bosentan at ETA receptors was the contraction to endothelin‐1 of rat aortic rings, with endothelium removed.26 Our competitive pKB values for bosentan and endothelin‐1 in human large diameter arteries such as pulmonary (i.d. 5.5 mm) and radial (i.d. 3.23 mm) 27 and in rat mesenteric small artery (i.d. 0.25 mm) or mouse main pulmonary (i.d. 0.65 mm), mesenteric (i.d. 0.28 mm), and tail (i.d. 0.37 mm) arteries all fall in the range 6.04‐7.31, consistent with ETA receptor antagonism. The one outstanding artery, of those we tested, where the dual ETA and ETB antagonist bosentan was apparently very weak was the rat small pulmonary artery.
There are 3 possible factors that could affect the pKB estimation: (i) endothelin‐1 could activate endothelial ETB receptors to release nitric oxide to functionally antagonize the contraction through smooth muscle cell ETA or ETB receptors; (ii) in some arteries, there may be a mix of ETA and ETB receptors; and (iii) the ETB receptor‐mediated clearance mechanism has an important action to decrease endothelin‐1 local concentrations by as much as 10‐fold.
First to the role of nitric oxide, L‐NAME made no significant difference to the pKB estimation for bosentan in rat small pulmonary artery (Figure 2). L‐NAME (100 μmol L−1) was effective in eliminating the release of NO as demonstrated by the complete abolition of the relaxation to acetylcholine (1 μmol L−1) in U46619‐precontracted arteries. Second, despite L‐NAME being present, endothelin‐1 was much less potent (lower pEC50) in rat pulmonary small artery than in rat mesenteric artery. Third, in the presence of bosentan 10 μmol L−1, the pEC50 for endothelin‐1 was right‐shifted and lowered to 7.2 (−log M) in the mesenteric artery, while in contrast, it was left‐shifted and raised to a pEC50 of 8.7 compared with control in the pulmonary artery (Figure 1A,B). We suggest that this anomalous result and inability to determine a pKB with bosentan in rat pulmonary artery is explained by the continuous removal of endothelin‐1 by the ETB receptor‐sensitive clearance mechanism found in this particular artery, but not in the rat mesenteric artery or aorta, nor human large pulmonary or radial artery.27
Further, direct functional evidence of the clearance of endothelin‐1 in rat pulmonary artery comes from the selective ETB antagonist BQ788 assay. With the agonist sarafotoxin S6c, and L‐NAME present, the pattern of BQ788 competitive antagonism shows right‐shifted concentration‐response curves with a pKB of 7.2 ± 0.21 from Clark plot analysis (Figure 3D,H). In stark contrast, when endothelin‐1 was the agonist, BQ788 up to 3 μmol L−1 caused no significant rightward shift, if anything a small left‐shift indicative of blockade of endothelin‐1 ETB clearance (Figure 3B,F). Removal of the endothelium failed to change the pattern of the endothelin‐1 and BQ788 interaction (Figure 3A,E). But bosentan was close to being a competitive ETA antagonist in the pulmonary artery in the presence of L‐NAME AND BQ788 to antagonize the clearance of endothelin‐1 (Figure 3C,G). Similarly, in human pulmonary resistance arteries, the ETA receptor antagonist BMS182874 was ineffective against low concentrations of endothelin‐1.28 The finding that endothelium removal did not affect the EC50 nor Emax to endothelin‐1 or change the action of BQ788 in the rat small pulmonary artery compared to endothelium‐intact tissues suggests that the arterial smooth muscle cells are the primary location of the proposed clearance mechanism (Figure 9).
Figure 9.
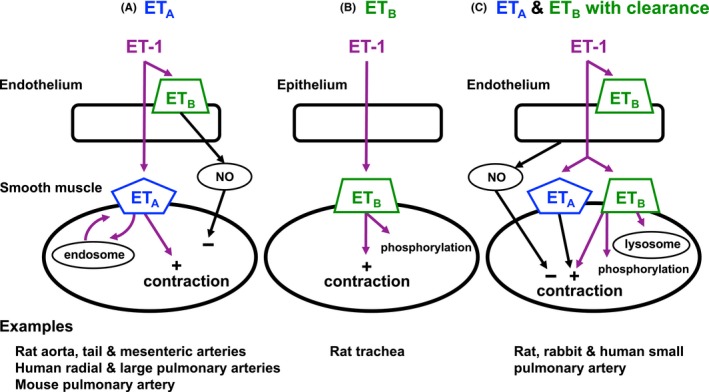
Schematic diagram of the location and function of ETA and ETB receptors in 3 tissue assays. (A) ETA receptors located on vascular smooth muscle mediate contraction. ETA receptors are internalized and recycled slowly through endosomes.36 (B) ETB receptors located on smooth muscle cells mediate contraction and are rapidly removed by phosphorylation.36 (C) ETB receptors located on smooth muscle cells bind endothelin‐1 and clear endothelin‐1 from the environment through lysosomal metabolism. The remaining endothelin‐1 binds to ETA and ETB receptors on smooth muscle to mediate contraction before being recycled by endosomes or destroyed by phosphorylation, respectively. In (A) and (C), ETB receptors on the endothelium mediate release of NO that transiently relaxes smooth muscle. Examples of the species and tissues assumed to have these particular receptor profiles are given below each panel. ET‐1, endothelin‐1. NO, nitric oxide
In earlier work, Hay et al29 reported that in rabbit pulmonary artery, sarafotoxin S6c gave a pKB of 7.7 for the mixed ETA and ETB receptor antagonist SB209670, but 6.7 when endothelin‐1 was the agonist. In human small pulmonary arteries (150‐200 μm i.d.) sarafotoxin S6c was more than 100‐fold more potent than endothelin‐1 and the authors concluded that both ETA and ETB receptor antagonists are required to antagonize endothelin‐1.28 We also found that the apparently weak antagonism of endothelin‐1 by bosentan in rat pulmonary arteries is shared with ambrisentan and macitentan (Figure 5). Indeed, these latter 2 endothelin‐1 antagonists are more ETA than ETB receptor selective (Table 1). However, it is important to note that the active metabolite of macitentan ACT‐132577 may additionally play a significant role as a dual ETA/ETB antagonist in vivo. Other factors such as pharmacokinetic differences will also affect the translation of these isolated tissue assay results into the clinic. Thus, there are a number of species including man, rabbit, and rat where small interlobar pulmonary arteries potentially have the ETB‐sensitive endothelin‐1 clearance mechanism and significant populations of ETB and ETA receptors on the smooth muscle mediating contraction.
Table 1.
Estimates of pKB from functional isolated tissue assays and selectivity ratios for dual ETA and ETB receptor antagonists in the absence or presence of endothelin‐1 clearance
| Receptor assay pKB | Ratio | ||||
|---|---|---|---|---|---|
| Endothelin antagonist | ETA Aortag , h | ETB Tracheag , i | ETB + Cj | ETA:ETB k | ETA:ETB + Cl |
| Bosentana | 7.3 | 5.9 | 4.9 | 25 | 250 |
| Ambrisentanb | 7.1 | 5.6 | 4.6 | 32 | 320 |
| Macitentanc | 7.6 | 5.9 | 4.9 | 50 | 500 |
| A‐182086d | 8.5m | 8.0m | 7.0 | 3 | 30 |
| Ro 46‐8443e | 5.7 | 7.1 | 6.1 | 0.04 | 0.4 |
| “Ideal”f | 8.5 | 8.5 | 7.5 | 1 | 10 |
Clozel et al.17
Bolli et al.33
Iglarz et al.34
Wessale et al.24
Breu et al.35
“Theoretical dualist.”
pKB from Schild plots.
Rat aorta (without endothelium) and agonist endothelin‐1.
Rat trachea (without epithelium) and agonist sarafotoxin S6c.
pKB for ETB receptors under the influence of endothelin‐1 clearance theoretically taken to be 10‐fold (ie, 1 log unit).
ETA to ETB selectivity ratio calculated as antilog (pKB ETA − pKB ETB).
ETA to ETB + C selectivity ratio calculated as antilog (pKB ETA − pKB ETB + C).
With endothelium.
Rabbit pulmonary artery (without endothelium).
4.1. Potential clinical implications
We have analyzed the endothelin receptor pharmacology in a wide range of arteries and the small pulmonary artery appears to be unique with its mix of ETA and ETB receptors and clearance mechanism. If this finding can be extrapolated to the clinic, there are a number of caveats that must be considered. (i) There is evidence in rats with monocrotaline‐induced pulmonary hypertension that the ETB receptor mRNA and protein expression in small pulmonary arteries are down‐regulated.30 However, in patients with severe pulmonary artery hypertension, the ETB receptor mRNA and protein expression were upregulated in the media of pulmonary arteries, while the ETA receptor gene expression was unaffected.31 (ii) That ETB receptors on endothelial cells are protective in limiting vascular remodeling and development of pulmonary hypertension, so that ETB antagonism may well be deleterious.32 (iii) That pharmacokinetic actions and active metabolites together with protein binding will significantly alter the resultant activity of endothelin receptor antagonists.
Noting the above, there are 3 dual endothelin antagonists approved for pulmonary artery hypertension. On isolated tissue assay data, all have a significant ETA to ETB receptor selectivity ratio of 25‐50 (Table 1). Our theoretical modeling which reflects our in vitro experimental data (Figures 1 and 8A) suggests that to obtain a 10‐fold antagonism of the ETB constrictor receptor in the presence of clearance, then a plasma concentration of 3000 times higher than the pKB at ETA receptors would be required. For bosentan, for example, plasma levels would need to be in the range of 200 μmol L−1! If these levels are not obtained, the antagonist would generally behave only as an effective ETA antagonist in the clinic.
Our modeling suggests that a 10‐fold selective ETB vs ETA antagonist might be ideal in antagonizing the pulmonary artery ETB receptors in the presence of CLEARANCE (Figure 8B). Ro 46‐844335 is 25‐fold selective for ETB vs ETA, and modeling would suggest that with a pKB of 7.1 at ETB receptors (Figure 8D), a plasma level would be required of nearly 10 μmol L−1 to give a 10‐fold antagonism of ETB receptors, but ETA antagonism would not be sufficient if inhibition of clearance presented a higher level of endothelin‐1. Another nonselective and potent ETA and ETB antagonist with selectivity ratio of just 3, A‐182086 (Table 1), has been used in animals and shows that effective ETA and ETB receptor antagonism was achieved after oral dosing.24
Given that any antagonism of clearance will raise plasma endothelin‐1 levels, there must be sufficient ETA receptor antagonism present to obviate vasoconstriction from this raised endothelin‐1 concentration. Theoretically then, an ETA vs ETB selectivity of 10‐fold would be sufficient, provided a high plasma concentration is achieved for ETB antagonism. From Table 1, we predict that given clearance of endothelin‐1 in important tissues such as pulmonary artery, the effective ETB antagonism is 10‐fold weaker so that the ETA to ETB + clearance selectivity ratio increases by 10‐fold. In effect, this suggests that the 3 antagonists in the clinic for pulmonary artery hypertension are principally ETA‐selective agents. The “ideal” antagonist would have identical pKB values at ETA and ETB receptors.
5. CONCLUSION
This experimental work in isolated tissue assays offers an explanation for the mechanism of the failure of “dual” ETA and ETB antagonists to competitively antagonize endothelin‐1 in some important arteries such as the small pulmonary artery where ETA and ETB receptors predominate to cause contraction. The experimental results and theoretical modeling are consistent with an endothelin‐1 clearance mechanism through internalization of endothelin‐1 bound to ETB receptors on smooth muscle of some blood vessels that can lower the endothelin‐1 concentration by 10‐fold. When this mechanism is blocked by ETB antagonists, the endothelin‐1 concentration will rise. The combination of a possible endothelin‐1 clearance and contraction mediated by ETA and ETB receptors provides an environment that would prevent effective endothelin‐1 receptor antagonism. This conclusion may have important implications for the effective use of endothelin antagonists in the treatment of pulmonary artery hypertension.
AUTHORS’ CONTRIBUTIONS
J.A.A. and C.E.W. conceived the study and designed the protocol. R.J.A.H. performed wire myography studies. J.A.A., C.E.W., and R.J.A.H. collected and analyzed data. J.A.A. and C.E.W. wrote the manuscript. R.J.A.H. critically reviewed the manuscript. All authors approved the final version of the manuscript.
DISCLOSURES
None declared.
ACKNOWLEDGEMENTS
We thank Mr Mark Ross‐Smith for expert technical support in isolated artery wire myography and isolated trachea studies. We thank the reviewers for their helpful suggestions which improved our work.
Angus JA, Hughes RJA, Wright CE. Distortion of KB estimates of endothelin‐1 ETA and ETB receptor antagonists in pulmonary arteries: Possible role of an endothelin‐1 clearance mechanism. Pharmacol Res Perspect. 2017;e00374 https://doi.org/10.1002/prp2.374
REFERENCES
- 1. Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, Nishikibe M. Clearance of circulating endothelin‐1 by ETB receptors in rats. Biochem Biophys Res Commun. 1994;199:1461‐1465. [DOI] [PubMed] [Google Scholar]
- 2. Gratton JP, Cournoyer G, Loffler BM, Sirois P, D'Orleans‐Juste P. ETB receptor and nitric oxide synthase blockade induce BQ‐123‐sensitive pressor effects in the rabbit. Hypertension. 1997;30:1204‐1209. [DOI] [PubMed] [Google Scholar]
- 3. Reinhart GA, Preusser LC, Burke SE, et al. Hypertension induced by blockade of ETB receptors in conscious nonhuman primates: role of ETA receptors. Am J Physiol Heart Circ Physiol. 2002;283:H1555‐H1561. [DOI] [PubMed] [Google Scholar]
- 4. Opgenorth TJ, Wessale JL, Dixon DB, et al. Effects of endothelin receptor antagonists on the plasma immunoreactive endothelin‐1 level. J Cardiovasc Pharmacol. 2000;36:S292‐S296. [DOI] [PubMed] [Google Scholar]
- 5. Johnstrom P, Fryer TD, Richards HK, et al. Positron emission tomography using 18F‐labelled endothelin‐1 reveals prevention of binding to cardiac receptors owing to tissue‐specific clearance by ET B receptors in vivo. Br J Pharmacol. 2005;144:115‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Angus JA, Black JW. Analysis of anomalous pK B values for metiamide and atropine in the isolated stomach of the mouse. Br J Pharmacol. 1979;67:59‐65. [PMC free article] [PubMed] [Google Scholar]
- 7. Kenakin TP. Pharmacologic analysis of drug‐receptor interaction, 2nd edn New York: Raven Press; 1993:305‐310. [Google Scholar]
- 8. Angus JA, Wright CE. Endothelin and the sympathetic nervous system In: Clozel M, Rubin LJ, eds. The Endothelin System in Cardiopulmonary Diseases. Basel: Friedrich Reinhardt Verlag; 2004:97‐125. [Google Scholar]
- 9. Allcock GH, Warner TD, Vane JR. Roles of endothelin receptors in the regional and systemic vascular responses to ET‐1 in the anaesthetized ganglion‐blocked rat: use of selective antagonists. Br J Pharmacol. 1995;116:2482‐2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. D'Orléans‐Juste P, Claing A, Télémaque S, Maurice MC, Yano M, Gratton JP. Block of endothelin‐1‐induced release of thromboxane A2 from the guinea pig lung and nitric oxide from the rabbit kidney by a selective ETB receptor antagonist, BQ‐788. Br J Pharmacol. 1994;113:1257‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Nucci G, Thomas R, D'Orléans‐Juste P, et al. Pressor effects of circulating endothelin are limited by its removal in the pulmonary circulation and by the release of prostacyclin and endothelium‐derived relaxing factor. Proc Natl Acad Sci U S A. 1988;85:9797‐9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maurice MC, Gratton JP, D'Orléans‐Juste P. Pharmacology of two novel mixed ETA/ETB receptor antagonists, BQ‐928 and 238, in the carotid and pulmonary arteries and the perfused kidney of the rabbit. Br J Pharmacol. 1997;120:319‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW . Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 2010;160:1577‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McGrath JC, Lilley E. Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol. 2015;172:3189‐3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Angus JA, Wright CE. Techniques to study the pharmacodynamics of isolated large and small blood vessels. J Pharmacol Toxicol Meth. 2000;44:395‐407. [DOI] [PubMed] [Google Scholar]
- 16. Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 1977;41:19‐26. [DOI] [PubMed] [Google Scholar]
- 17. Clozel M, Breu V, Gray GA, et al. Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J Pharmacol Exp Ther. 1994;270:228‐235. [PubMed] [Google Scholar]
- 18. Curtis MJ, Bond RA, Spina D, et al. Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol. 2015;172:3461‐3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bremnes T, Paasche JD, Mehlum A, Sandberg C, Bremnes B, Attramadal H. Regulation and intracellular trafficking pathways of the endothelin receptors. J Biol Chem. 2000;275:17596‐17604. [DOI] [PubMed] [Google Scholar]
- 20. Paasche JD, Attramadal T, Sandberg C, Johansen HK, Attramadal H. Mechanisms of endothelin receptor subtype‐specific targeting to distinct intracellular trafficking pathways. J Biol Chem. 2001;276:34041‐34050. [DOI] [PubMed] [Google Scholar]
- 21. Stone M, Angus JA. Developments of computer‐based estimation of pA2 values and associated analysis. J Pharmacol Exp Ther. 1978;207:705‐718. [PubMed] [Google Scholar]
- 22. Lew MJ, Angus JA. Analysis of competitive agonist‐antagonist interactions by nonlinear regression. Trends Pharmacol Sci. 1995;16:328‐337. [DOI] [PubMed] [Google Scholar]
- 23. Clark AJ. The antagonism of acetyl choline by atropine. J Physiol (Lond). 1926;61:547‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wessale JL, Adler AL, Novosad EI, et al. Pharmacology of endothelin receptor antagonists ABT‐627, ABT‐546, A‐182086 and A‐192621: ex vivo and in vivo studies. Clin Sci (Lond). 2002;103(Suppl 48):112S‐117S. [DOI] [PubMed] [Google Scholar]
- 25. Davenport AP, Hyndman KA, Dhaun N, et al. Endothelin. Pharmacol Rev. 2016;68:357‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clozel M, Breu V, Burri K, et al. Pathophysiological role of endothelin revealed by the first orally active endothelin receptor antagonist. Nature. 1993;365:759‐761. [DOI] [PubMed] [Google Scholar]
- 27. Angus JA, Soeding PF, Hughes RJA, Wright CE. Functional estimation of endothelin‐1 receptor antagonism by bosentan, macitentan and ambrisentan in human pulmonary and radial arteries in vitro. Eur J Pharmacol. 2017;804:111‐116. [DOI] [PubMed] [Google Scholar]
- 28. McCulloch KM, Docherty CC, Morecroft I, MacLean MR. EndothelinB receptor‐mediated contraction in human pulmonary resistance arteries. Br J Pharmacol. 1996;119:1125‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hay DW, Luttmann MA, Beck G, Ohlstein EH. Comparison of endothelin B (ETB) receptors in rabbit isolated pulmonary artery and bronchus. Br J Pharmacol. 1996;118:1209‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sauvageau S, Thorin E, Villeneuve L, Dupuis J. Change in pharmacological effect of endothelin receptor antagonists in rats with pulmonary hypertension: role of ETB‐receptor expression levels. Pulm Pharmacol Ther. 2009;22:311‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bauer M, Wilkens H, Langer F, Schneider SO, Lausberg H, Schäfers HJ. Selective upregulation of endothelin B receptor gene expression in severe pulmonary hypertension. Circulation. 2002;105:1034‐1036. [DOI] [PubMed] [Google Scholar]
- 32. Kelland NF, Kuc RE, McLean DL, et al. Endothelial cell‐specific ETB receptor knockout: autoradiographic and histological characterisation and crucial role in the clearance of endothelin‐1. Can J Physiol Pharmacol. 2010;88:644‐651. [DOI] [PubMed] [Google Scholar]
- 33. Bolli MH, Marfurt J, Grisostomi C, et al. Novel benzo[1,4]diazepin‐2‐one derivatives as endothelin receptor antagonists. J Med Chem. 2004;47:2776‐2795. [DOI] [PubMed] [Google Scholar]
- 34. Iglarz M, Binkert C, Morrison K, et al. Pharmacology of macitentan, an orally active tissue‐targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327:736‐745. [DOI] [PubMed] [Google Scholar]
- 35. Breu V, Clozel M, Burri K, Hirth G, Neidhart W, Ramuz H. In vitro characterisation of Ro 46‐8443, the first non‐peptide antagonist selective for the endothelin ETB receptor. FEBS Lett. 1996;383:37‐41. [DOI] [PubMed] [Google Scholar]
- 36. Cramer H, Müller‐Esterl W, Schroeder C. Subtype‐specific desensitization of human endothelin ETA and ETB receptors reflects differential receptor phosphorylation. Biochemistry. 1997;36:13325‐13332. [DOI] [PubMed] [Google Scholar]