

Abstract
Calcium (Cav1 and Cav2) and sodium channels possess homologous CaM-binding motifs, known as IQ motifs in their C termini, which associate with calmodulin (CaM), a universal calcium sensor. Cav3 T-type channels, which serve as pacemakers of the mammalian brain and heart, lack a C-terminal IQ motif. We illustrate that T-type channels associate with CaM using co-immunoprecipitation experiments and single particle cryo-electron microscopy. We demonstrate that protostome invertebrate (LCav3) and human Cav3.1, Cav3.2, and Cav3.3 T-type channels specifically associate with CaM at helix 2 of the gating brake in the I–II linker of the channels. Isothermal titration calorimetry results revealed that the gating brake and CaM bind each other with high-nanomolar affinity. We show that the gating brake assumes a helical conformation upon binding CaM, with associated conformational changes to both CaM lobes as indicated by amide chemical shifts of the amino acids of CaM in 1H-15N HSQC NMR spectra. Intact Ca2+-binding sites on CaM and an intact gating brake sequence (first 39 amino acids of the I–II linker) were required in Cav3.2 channels to prevent the runaway gating phenotype, a hyperpolarizing shift in voltage sensitivities and faster gating kinetics. We conclude that the presence of high-nanomolar affinity binding sites for CaM at its universal gating brake and its unique form of regulation via the tuning of the voltage range of activity could influence the participation of Cav3 T-type channels in heart and brain rhythms. Our findings may have implications for arrhythmia disorders arising from mutations in the gating brake or CaM.
Keywords: calcium channel, circular dichroism (CD), cryo-electron microscopy, gating, isothermal titration calorimetry (ITC), nuclear magnetic resonance (NMR), patch clamp, short hairpin RNA (shRNA)
Introduction
Calmodulin (CaM)3 is a universal resident calcium sensor that promotes a calcium-dependent regulation at a canonical IQ motif of the C-terminal tails of voltage-gated calcium channels (1–5) and sodium channels (6). CaM regulates a rapid and robust inactivation gating of L-type calcium channels (e.g. Cav1.2) (7) or skeletal muscle sodium channels (e.g. Nav1.4) (2) and a facilitation of channel currents in synaptic (e.g. Cav2.1) calcium channels (8). The conservation of CaM binding extends to basal Cav1 L-type channels in single-celled eukaryotes, such as ciliate Paramecium tetraurelia, which has an extended C terminus that includes a conserved IQ motif (9). CaM mutants of Paramecium (10) influence the calcium-dependent inactivation of L-type calcium currents (11, 12) and their control of ciliary beat frequency for swimming and turning behavior (13). CaM binding at the IQ motif and its conserved regulation are consistent with the observed calcium-dependent inactivation (known as CDI) even in expressed cnidarian L-type channel homolog (14). All vertebrate L-type channels display a calcium-dependent inactivation that ranges from minor to very robust, from Cav1.4 to Cav1.1 to Cav1.2 and Cav1.3 channels, respectively (15). A hallmark of the calcium-dependent inactivation in L-type calcium channels is its resilience even in the presence of high-calcium buffering in 10 mm EGTA or BAPTA. This resilience in calcium sensing in the presence of high-calcium buffering is conferred by a secondary CaM-binding site referred to as NSCaTE (N-terminal spatial Ca2+-transforming element) specifically contained within the N terminus of invertebrate Cav1 channels (9) and mammalian Cav1.2 and Cav1.3 channels (16, 17). NSCaTE is lacking outside of L-type calcium channels, but the highly conserved C-terminal IQ motif extends to members of the sodium channel family, from Nav2 channels in the early branching single-celled eukaryotes before the split of animals and fungi, such as apusozoan, Thecamonas trahens, and Nav1 channels (18), including cnidarian jellyfish, which are the extant relatives of the likely ancestors containing the first metazoan nervous systems (19, 20). The only member of the calcium and sodium channel superfamily to lack the reported CaM-binding elements (the C-terminal IQ motif and the N-terminal NSCaTE) are Cav3 T-type channels (1, 2, 21).
Here, we show that Cav3 T-type channels possess high-affinity calmodulin binding at the “gating brake,” a helix–loop–helix motif located in the proximal I–II linker of known Cav3 T-type channels in the analogous position where accessory Cavβ subunits regulate Cav1 and Cav2 channel complexes (22). This gating brake governs the low-voltage dependence of T-type channels, and its absence generates a “runaway gating” phenotype (23–26). Mutations that alter the function of the Cav3.2 gating brake have been found in human patients with absence epilepsy (27, 28).
CaM-binding sequences are variable, which can range from a striking likeness to the C-terminal IQ motif from L-type channels, as in the gating brake of the basal metazoan Trichoplax adherens, to diverse sequences that generate a nanomolar affinity for CaM binding in protostome invertebrates such as pond snail LCav3 and all human (Cav3.1, Cav3.2, and Cav3.3) channel isoforms. We demonstrate that CaM facilitates the formation of α-helices in gating brake sequences, can pre-associate with Cav3 T-type channels without calcium ions, and its binding involves structural conformational changes in both N- and C-terminal pairs of EF hands in CaM and the gating brake. Dialysis of CaM binding (CaMB) peptides, or co-expression of apo-CaM (CaM1234) generates a significant hyper-polarizing shift in voltage sensitivities and faster gating kinetics, consistent with the mutant phenotype of Cav3 T-type channels lacking a gating brake in the I–II linker.
Cav3 T-type channels contribute to pacemaker rhythms, such as the conducting system of the heart (29) and low-threshold calcium potentials (also known as low-threshold spikes), that trigger rhythmic burst firing classically associated with thalamic neurons during non-rapid eye movement sleep and to the spike wave discharge during absence seizures (30, 31). There is also significant evidence for T-type currents participating in “low threshold” neurotransmitter release (32) and in the maintenance of vascular tone (33). Cav3 T-type channels possess a significant “window current” that provides a small but continuous stream of calcium influx through a population of Cav3 T-type channels open at rest (34). This calcium, available through Cav3 T-type channels at rest, is modeled to contribute to cellular proliferation during organ development, to the aberrant proliferation in many cancers (35), and to the hypertrophied condition of the mammalian heart (36). Classically, Cav3 T-type channels are mostly inactivated at rest, and their participation rate is steeply voltage-dependent (37). CaM's regulation of the voltage sensitivities of Cav3 T-type channels at the gating brake thus has dramatic consequences to the participation of Cav3 T-type channels in normal functions, as well as during development and disease.
Results
CaM complexes with full-length Cav3 T-type channels
Full-length mammalian Cav3.1 channels were purified from Sf9 insect cells (38) and co-incubated with biotin–CaM complexed to a streptavidin–gold conjugate. Fig. 1A (left panel) shows a field of purified Cav3.1 (protein appears white) coupled to an electron dense (black) gold particle, indicating CaM is bound. Individual images illustrating different orientations of the single particles of Cav3.1 channels alone or calmodulin–gold alone and Cav3.1 channels in complex with calmodulin–gold are shown in montages of viewpoints on top, bottom, and middle panels, respectively, in Fig. 1A. The orientation of the channel in some images allows identification of the C terminus, a protruding, finger-like projection from the transmembrane domain of Cav3.1 as highlighted by an asterisk; a feature previously identified through single-particle electron microscopy 3-D reconstruction of Cav3.1 (38). We confirmed the complexing of calmodulin with Cav3 T-type channels as co-immunoprecipitants in Western blottings (see Fig. 1B). CaM–GFP fusion proteins transfected and purified from HEK-293T cells and isolated on anti-GFP Sepharose beads were identified in the complex with HA-tagged Cav3.2 channels in Western blottings, labeled with anti-HA antibody, as an ∼259-kDa band (Fig. 1B, middle lane) in the presence of calcium ions (33.3 μm CaCl2). The co-immunoprecipitation of CaM–Cav3.2 channels failed in the absence of co-expressed HA-tagged Cav3.2 channels (Fig. 1B, left lane) or in the absence of CaM (Fig. 1B, right lane).
Figure 1.

CaM associates with full-length Cav3 T-type channels illustrated by nano-particle cryoelectron microscopy (A) and co-immunoprecipitation (B). A, field of negatively stained (2% w/v uranyl acetate) full-length Cav3.1 in complex with CaM in the presence of 1.5 mm Ca2+. Scale bar is 50 nm. Left panel, CaM has a biotin label that binds streptavidin gold (5 nm). Protein appears white, and the gold that is electron-dense is black. Right panel, top row, montage of purified Cav3.1 particles (white density) presenting different views of the channel. Asterisk indicates “tail” domain that we have previously determined corresponds to the C terminus (38). Middle row, montage of Cav3.1 particles with CaM–biotin bond indicated by the presence of streptavidin gold (black sphere). Bottom row, control images of CaM–biotin–streptavidin gold particle without Cav3.1. Observed relative differences in Cav3.1 and streptavidin gold particle sizes relate to the orientation and contrast of each Cav3.1 channel particle adhered to the EM support film and the size variation in the streptavidin gold particles (3–6 nm with nominal size of 5 nm, according to the manufacturer). B, CaM–GFP bound to anti-GFP magnetic beads associates with hemagglutinin (HA)-tagged Cav3.2 channel (top panel, middle lane) as illustrated by HA antibody labeling (259-kDa band) of the Cav3.2 channel co-immunoprecipitant (IP) bound to beads. 259-kDa HA-tagged Cav3.2 channel band does not appear as a co-immunoprecipitant in the Western blot without co-expression of pCDNA3.1 plasmid inserts containing HA-tagged Cav3.2 channel (top panel, left lane) or without co-expression of CaM–GFP (top panel, right lane) in HEK-293T cells. Middle panel illustrates anti-HA antibody staining of the 259-kDa HA-tagged Cav3.2 channel of a replicate experiment of input proteins for the Western blot shown in the top panel without co-immunoprecipitation. Bottom panel illustrates anti-GFP antibody staining of the 44.2-kDa GFP-tagged CaM or GFP alone (27 kDa), in a replicate experiment of input proteins for the Western blot shown in the top panel without co-immunoprecipitation. C, CaM–GFP (left two lanes, 44.2 kDa) and GFP alone (right lane, 27 kDa) bound to anti-GFP magnetic beads as illustrated by anti-GFP antibody labeling (259-kDa band) of the Cav3.2 channel co-immunoprecipitant bound to beads. GFP alone generated two bands on the Western blot, which may result from differing post-translational modifications. Co-immunoprecipitation experiments were carried out in 33.3 μm CaCl2, pH 7.4. Vector for HEK-293T cell expressed inserts for Western blotting (Cav3.2–HA, EGFP, CaM–pGFP) were contained in pcDNA3.1. Membranes were stained with Ponceau red following protein transfer to evaluate the protein content in each lane. Co-immunoprecipitation experiments were carried out in 33.3 μm CaCl2, pH 7.4. Vector for HEK-293T cell expressed inserts for Western blotting (Cav3.2–HA, EGFP, CaM–pGFP) were contained in pcDNA3.1. Membranes were stained with Ponceau red following protein transfer to evaluate the protein content in each lane.
Cav3 T-type channels possess a predicted high-affinity CaM-binding site at their gating brake in the I–II linker
Cav3 T-type channels lack the C-terminal IQ motif shared among other calcium channels (Cav1 and Cav2) and sodium (Nav1 and Nav2) channels (1, 2, 21). Because both EM and co-immunoprecipitation experiments indicated that CaM binds to full-length mammalian Cav3.1 and Cav3.2 channels, we therefore next sought to delineate the CaM-binding domain. A unique but ubiquitous feature in Cav3 T-type channels is a helix–loop–helix, gating brake motif (similar in structure to a region in fumarase enzyme) in the proximal I–II linker that is in the analogous position of β subunit binding to Cav1 and Cav2 calcium channels (25). It is within the second helix of the gating brake of the I–II cytoplasmic linker where we have found an analogous region to the C-terminal IQ motif, with nanomolar affinity binding to CaM (Fig. 2A). Sequence alignments of Cav3 T-type channels (Fig. 2B) indicated a predicted CaM-binding site (illustrated by red color gradient) in helix-2 of the gating brake of representative metazoan species from cnidarians to the three human Cav3.1, Cav3.2, and Cav3.3 channels. A CaM-binding site is also predicted in Cav3 T-type channels by the on-line tool, CaM Target Database (39). Predictions suggest that the gating brake sequence is cytoplasmic, and a helical wheel analysis indicates its amphipathic nature.
Figure 2.

CaM is predicted to associate with Cav3 T-type channels in helix-2 of the gating brake in the proximal I–II linker. A, illustration of the four domain (DI, DII, DIII, and DIV) × 6 transmembrane helices structure common to sodium channels, calcium channels, and NALCN. Cav1, Cav2, and Nav channels contain a canonical proximal C-terminal IQ motif that possess high-affinity CaM-binding site. A proposed equivalence of the CaM-binding C-terminal IQ motif in Cav3 T-type channels is a helix–loop–helix gating brake motif in the proximal I–II linker that is in the analogous position of β-subunit binding to Cav1 and Cav2 calcium channels. B, sequence alignment of T-type channels illustrates a predicted CaM-binding site in helix-2 of the gating brake (illustrated by red color) in representative species from cnidarians to the three human genes. CaM-binding site prediction from CaM Target Database (39). Predictions suggested the putative CaMB peptide sequence is cytoplasmic, and helical wheel analysis indicates its amphipathic nature. In the most primitive metazoan with a T-type channel, Trichoplax (placozoan), the most basal extant T-type channel in multicellular organisms known possesses a gating brake motif resembling the C-terminal IQ motif shared with other calcium (Cav1 and Cav2) and sodium (Nav2 and Nav1) channels (illustrated by blue color). Yellow outlined residues are the sequences for the synthetic CaMB peptides. 39 and 111 amino acid sequence deleted in gating brake deletion mutant, Cav3.2(D453–491) and Cav3.2(D429–539) are indicated. C and D, protein similarities among CaMB peptide sequences in the I–II linker, illustrating the high similarity among invertebrate (snail) LCav3 and the human Cav3.x homologs (C) and the greater similarity of Trichoplax Cav3 CaMB peptide sequences to the C-terminal IQ motif of Cav1.2 channels (D), rather than other gating brake sequences (C).
Cav3 T-type channel from most basal multicellular animals resembles the C-terminal IQ motif of Cav1, Cav2, Nav2, and Nav1 channels
The most basal multicellular organism with a Cav3 T-type channel is T. adherens (a placozoan) (40), which has a region spanning the gating brake that is unlike other gating brake sequences (Fig. 2C, blue bar) with a core 17 amino acids that resembles the C-terminal CaM-binding IQ motif of Cav1, Cav2, and Nav2 channels (Fig. 2, C and D, blue bars). The Cav3 T-type channel in the basal multicellular placozoan may represent a structural intermediate involving the positioning of the C-terminal IQ motif in the common ancestor to calcium channels and sodium channels to the proximal I–II linker, before a divergence and appearance of a gating brake helix–loop–helix motif shared among all other metazoan Cav3 T-type channels. Comparatively, the IQ motif of Cav1 L-type channels is mostly identical from microbial eukaryotes to human homologs (9), where the CaM-binding gating brake motif in Cav3 T-type channels is more divergent. The protein similarity of gating brake sequences between protostome invertebrate (e.g. pond snail LCav3) and among the human Cav3 isoforms is 71–76% (Fig. 2C, red bars). Differences in gating brake sequences may reflect a local adaptability of the gating brake among Cav3 T-type channels in different animals.
Gating brake peptides from snail LCav3 and the three human Cav3.1, Cav3.2, and Cav3.3 T-type channels associate with Ca2+–CaM
26-mer peptide sequences spanning the gating brake region were synthesized for snail LCav3 and human Cav3.1, Cav3.2, and Cav3.3 channels dubbed “Cav3 CaMB” peptides. In addition, a PGPGPG substituted Cav3.2 channel peptide (Cav3.2mut) was synthesized, which served as a negative control peptide in experiments (see yellow highlighted sequence in Fig. 2B). CaMB peptides promoted a gel-mobility shift with CaM in the presence of 0.1 mm CaCl2-containing solution and increasing molar ratios (0.5 to 4×) of snail LCav3 or human Cav3.1, Cav3.2, and Cav3.3 channel CaMB peptides (Fig. 3). Mutated Cav3.2 CaMB peptide (Cav3.2mut) with the PGPGPG substitution did not promote a gel-mobility shift with CaM (Fig. 3). Cav3.3 peptide has the weakest apparent interaction compared with other Cav3.1 and Cav3.2 peptides, which correlates with its weaker predicted binding affinity (Fig. 2B). Presence of urea attenuated the weakest interacting human Cav3.3 channels with CaM without affecting the higher affinity LCav3 gating brake peptide with CaM.
Figure 3.
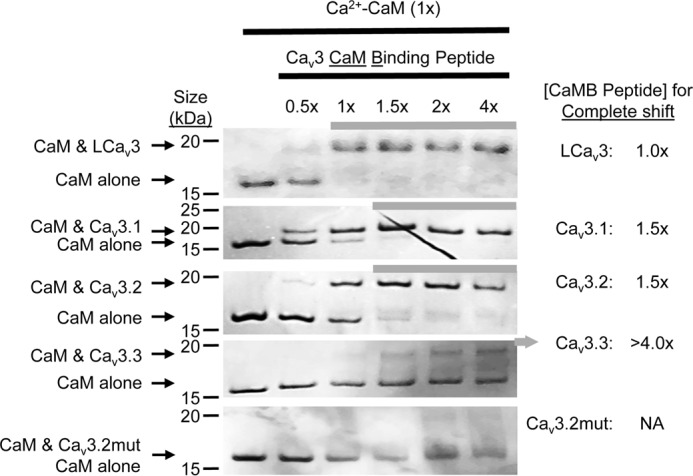
CaMB peptides from snail LCav3 and the three human Cav3.x T-type channels associate with Ca2+–CaM. Gel-mobility shifts with CaM are evident in the presence of 0.1 mm CaCl2-containing solution and increasing molar ratios (0.5–4×) of snail LCav3 or human Cav3.1, Cav3.2, and Cav3.3 channel CaMB peptides but not evident in the presence of mutated control Cav3.2 CaMB peptide (Cav3.2mut). 1st lane in each gel is Ca2+–CaM-only control. The rank order of weakest to strongest apparent interaction of Ca2+–CaM for peptides: Cav3.3 < Cav3.2 < Cav3.1 < LCav3 correlates with the rank order of calculated binding affinities using ITC (see Fig. 5). Note that Cav3.1 and Cav3.2 peptides both completely displace Ca2+–CaM at 1.5× peptide to CaM molar ratio, but Cav3.1 appears to possess a higher affinity based on sub-saturation levels of peptide. Urea attenuates the weaker interacting human Cav3.x channels but does not attenuate the higher affinity LCav3 CaMB peptide.
Gating brake peptides assume a helical conformation upon association with Ca2+–CaM
Differential circular dichroism (CD) spectroscopy of snail LCav3, human Cav3.2, and Cav3.3 gating brake peptides suggest that the peptides assume a more helical secondary structure upon co-incubation with CaM or with helix-stabilizing agent trifluoroethanol (TFE) (Fig. 4A). Helical propensity of peptides increases after titrating higher TFE concentrations from 10 to 50% (41, 42). The apparent α-helical formation of LCav3 and Cav3.2 resembles the CD spectral signature with 10% TFE, whereas Cav3.3 resembles more the spectral signature with 25% TFE. Human Cav3.1 is unique among the gating brake peptides in appearing to assume an α-helical conformation when free in solution (without CaM or TFE) (Fig. 4A), and this can be explained by its high-native alanine content (Fig. 4B). Although the circular dichroism results are suggestive of induction of helical secondary structure in the peptide upon binding CaM, some of the observed ellipticity changes may be induced by tertiary structural changes, not secondary structural ones (43). Mutated peptide (Cav3.2mut) has no α-helical propensity as predicted due to its proline substitutions (Fig. 4, A and B).
Figure 4.

CaMB peptides from snail Cav3 and human Cav3.x T-type channels assume an α-helical structure when bound to Ca2+–CaM. A, differential circular dichroism (CD) spectra of CaMB peptides co-incubated in the presence of helix-stabilizing agent, TFE (gray dashed line), 20 μm CaM (red line), or without CaM (blue line). 26-mer snail LCav3, human Cav3.2, and Cav3.3 CaMB peptides (sequence, bottom right) assume a more helical secondary structure upon addition of CaM and TFE. TFE has a much greater effect on the highest binding affinity peptide, snail LCav3, than the others. Cav3.1 peptide is the only isoform that is α-helical when free in solution, as evidenced by the characteristic negative peaks at 208 and 222 nm. Cav3.2-Gbmut peptide was incapable of adopting an α-helix even at 50% TFE, nor did it appear to interact with CaM. CaM-alone curve was subtracted from each spectra, converted to mean residue ellipticity (θ), and smoothed. For TFE experiments, 50 μm peptide in PBS was used, whereas the baseline was corrected against PBS as background. B, gating brake peptide sequences highlighting the alanine-rich Cav3.1 sequence in blue outline, and the “PGPGPG” substitution in red outline that serves as the Cav3.2 gating brake mutant. The 24 residues of the 26-mer peptides with a yellow-colored background are predicted to contain α-helices according to PSIPRED version 3.3 (Bioinformatics Group at University College London) (91).
Gating brake peptides bind with a high nanomolar affinity to Ca2+–CaM
CaM binds Cav3 CaMB peptides in 0.5 mm CaCl2 solution with a 1:1 stoichiometry, at nanomolar affinities that vary from 12, 43, 187, and 383 nm for snail LCav3, Cav3.1, Cav3.2, and Cav3.3 CaMB peptides, respectively (representative data in Fig. 5 and table of parameters in Fig. 6A). The rank order of measured binding affinities determined in the isothermal titration calorimetry (ITC) (Fig. 6A) is consistent with the rank order of concentration of CaMB peptide to CaM ratio required for a saturating gel-mobility shift, 1.0, 1.5, 1.5, and >4.0× for snail LCav3, Cav3.1, Cav3.2, and Cav3.3 CaMB peptides, respectively (Fig. 3). The Cav3.3 CaMB peptide possessed the weakest CaM-binding site of all isoforms as evidenced by the lowest measured affinity with ITC and incomplete gel shift of CaM in native PAGE (Fig. 3). Besides their variable sequence and binding affinity differences for CaM, the Cav3 CaMB peptides vary in their thermodynamic properties during their association with CaM. The association appears to be entropy-driven for LCav3, LCav3.1, and Cav3.3 peptides and enthalpy-driven for Cav3.2 peptide, as illustrated by the latter's negative enthalpy (exothermic) (Figs. 5 and 6A).
Figure 5.

Thermodynamic basis for CaM-binding peptide interactions with wild-type and mutant CaM. Raw data traces, isothermal titration calorimetry (above), and integrated heats of the measured interaction were fitted with a One Sites model using Malvern MicroCal (ITC200) add-on within Origin software (below). A, representative interactions with snail LCav3, and human Cav3.1 and Cav3.2 CaMB peptides and wild-type CaM used in the calculation for their 12.4, 42.7, and 187.1 average nanomolar affinities, respectively. B, sample interactions with human Cav3.2 and Cav3.1 CaMB peptides and mutant calmodulins, including N-lobe of CaM, and CaM1234 used in the calculation for their 31.1 and 543.3 average nanomolar affinities, respectively. Full table of ITC parameters (mean ± S.E., n = 3) is illustrated in Fig. 6A.
Figure 6.

ITC and NMR analyses indicate high-affinity, nanomolar binding, and associated conformation changes, respectively, upon Cav3 T-type channel CaM-binding peptides to N- and C-terminal lobes of CaM. A, tabulated data of ITC analysis indicates a 1:1 stoichiometry of CaM binding with a 10, 36, 196, and 383 nanomolar affinity for CaM-binding peptides for snail LCav3 and human Cav3.1, Cav3.2, and Cav3.3 channels respectively. All peptide and CaM interactions are endothermic, except Cav3.2 CaMB peptide, which is exothermic. LCav3 CaMB peptide in particular has a complex ITC curve and will bind to CaM with mutated N-terminal Ca2+-binding sites (CaM12), but not to CaM with mutated C-terminal Ca2+-binding sites (CaM34). N-CaM and C-CaM are 74-aa constructs (exactly half of the CaM molecule) representing each individual lobe. Both N- and C-CaM associate with Cav3.2 CaMB peptide, albeit a higher affinity for the N-lobe of CaM. CaM with all four Ca2+-binding sites mutated in its lobes (apo-CaM, CaM1234) still associates with CaM-binding peptides. Table headings: Binding stoichiometry of the interaction between CaM and CaM-binding peptide in solution (N), Binding affinity (Ka), enthalpy changes (ΔH), entropy changes (ΔS). ITC curves were fitted to a one-set-of-sites model with a high degree of fit for N, Ka, and ΔH values shown for representative experiments in Fig. 5A. The parameters for the all peptides were calculated from three replicate experiments. B, T-type channel GB peptides promote a conformational change involving both CaM lobes confirmed by amide chemical shifts of CaM's amino acids in 1H-15N HSQC NMR spectra. Overlay of 1H-15N HSQC spectra of CaM alone (red color) and CaM bound to gating brake peptides from Cav3.2 (green color), or snail LCav3 (cyan color). C, chemical shift differences between CaM and the CaM–Cav3.2 complex and CaM and the CaM–LCav3 complex. B and C illustrate representative data from one of three replicate experiments.
Gating brake peptides bind and induce structural changes to both N- and C-terminal lobes of Ca2+–CaM and will likely associate with CaM in the absence of Ca2+
Mutant CaM1234 possess critical Asp → Ala mutations in its EF hands that disable Ca2+ binding, creating a mutant that serves as a proxy for apo-CaM (44). The gating brake may serve as a pre-association site for binding CaM in the absence of calcium ions (apo-CaM), with binding of snail LCav3 and human Cav3.1 CaMB peptides with Ca2+-binding deficient CaM (CaM1234), albeit at an ∼12- and ∼349-fold lower affinity, respectively, compared with wild-type CaM (Figs. 5 and 6A). The interactions with CaMB peptides likely involve both lobes of EF hand pairs of CaM, because both N- and C-terminal CaM (74-aa constructs spanning exactly half of the CaM molecule) were able to bind Cav3.2 CaMB peptide, with the N-lobe having a significantly higher affinity (Figs. 5 and 6A). LCav3 CaMB peptide will bind to CaM with mutated N-terminal Ca2+-binding sites (CaM12) but not to CaM with mutated C-terminal Ca2+-binding sites (CaM34) (Fig. 6A). Chemical shift changes observed in the 1H-15N HSQC NMR spectra of CaM in the presence of both snail LCav3 (red to cyan color shift, Fig. 6B) and human Cav3.2 (red to green color shift, Fig. 6B) indicate a dramatic conformational rearrangement of both N- and C-terminal lobes of CaM in response to high-affinity binding to gating brake sequences. The widespread and dramatic chemical shift differences in Ca2+–CaM alone compared with a CaM–Cav3.2 or CaM–LCav3 CaMB peptide complex is illustrated in Fig. 6C.
Intracellular dialysis of gating brake peptides, but not mutated gating brake peptides, causes a hyperpolarizing shift in voltage dependence and faster gating kinetics in Cav3.2 channels
We tested whether the observed nanomolar affinity of CaM for the gating brake governs the unique gating properties of Cav3 T-type channels such as its low voltage of activation. We challenged cells expressing Cav3.2 T-type channels with 5 μm Cav3.2 CaMB peptides pre-loaded in the intracellular pipette, and we evaluated the change of gating behavior in whole-cell patch-clamp recording of transfected Cav3.2 T-type channels in HEK-293T cells, before and after equilibration of the intracellular dialysis of gating brake peptide (after 15–20 min) (Fig. 7). We conducted parallel control experiments with the intracellular dialysis of mutant gating brake peptide (Cav3.2 CaMBmut) (mutant sequence in Figs. 2B and 4B). The Cav3 CaMB peptide challenge to Cav3.2 channels causes a phenotype change, which resembles the 33- or 111-amino acid deletion mutant phenotype of Cav3 channels lacking the gating brake, Cav3.2(D453–491) and Cav3.2(D429–539) (23), respectively (Fig. 7). The gating brake is so named because it is considered a unique regulator of Cav3 T-type channels, maintaining them in the closed state at resting membrane potentials (23–26), where disruptions of the gating brake cause the voltage threshold for activation and inactivation gating to be shifted 20–30 mV in the hyperpolarizing direction (e.g. see Figs. 8 and 9B) (23, 25, 26, 45), with dramatically accelerated activation and inactivation kinetics (23–26). Activation curves are shifted to negative potentials by an average of 8 mV (a 50% activation voltage change from −55.28 ± 1.33 to −63.10 ± 1.29 mV) after dialysis of Cav3.2 CaMB peptide, whereas no corresponding voltage shifts are observed with mutated CaMB peptides (Cav3.2 CaMBmut) (Fig. 7A). Steady-state inactivation curves of HEK-293T cell-transfected Cav3.2 channels are also shifted toward negative potentials by an average of 16 mV (a 50% inactivation voltage change from −83.19 ± 2.35 mV to −99.62 ± 1.37 mV) after 20 min of dialysis with Cav3.2 CaMB peptide but not after dialysis with mutated peptide (Cav3.2 CaMBmut)-treated cells (Figs. 7B and 8B). Intracellular dialysis of Cav3.2 CaMB peptide also confers a significant speeding up of channel activation and inactivation kinetics, compared with Cav3.2 channels dialyzed with mutated peptide (Cav3.2 CaMBmut). This is quantified as significant reduction in the τ for exponential fits of the kinetics of activation (Fig. 7A) and inactivation (Fig. 7B) in the presence of Cav3.2 CaMB peptide.
Figure 7.

Intracellular dialysis of 5 μm Cav3 CaMB peptide but not mutated (CaMBmut) peptide causes a hyperpolarization shift and faster channel kinetics in Cav3.2 channels. Transfected Cav3.2 channels in HEK-293T cells were evaluated by whole-cell patch-clamp electrophysiology for activation (A) and inactivation (B) at start of patch recording (time 0) and after 20 min of intracellular dialysis of 26-mer Cav3 CaM-binding (CaMB) peptide or mutated CaM-binding (CaMBmut) peptide (peptide sequences in Figs. 2B and 4B). Representative current traces after 20-min dialyses are shown. Calcium currents were measured for their current-voltage relationships and τ mono-exponential fits for the kinetics of activation (A) and inactivation (B). Activation and steady-state inactivation curves were created with peak currents generated from a step depolarization from −110 to −80 to −10 mV and to −30 mV from holding potentials ranging from −140 to −40 mV, respectively. Statistical significance (p < 0.05) using a non-parametric Wilcoxon matched-pairs signed rank test for the kinetic data is shown by * measured before and after equilibration of the intracellular dialysis of Cav3 CaMB peptide or mutated CaM binding (CaMBmut) peptide. n values for current-voltage relationship Cav3 CaMB peptide (n = 7) and CaMBmut (n = 6). n values for inactivation Cav3 CaMB peptide (n = 5) and CaMBmut (n = 6).
Figure 8.

Intracellular dialysis of 5 μm Cav3 CaMB peptides cause a hyperpolarizing shift in voltage dependence but only with Cav3.2 channels containing an intact gating brake. Scatterplot (left) with mean ± S.E. (right) values of the voltages of 50% activation (A) and steady-state inactivation (B) taken from Boltzmann fits of activation and steady-state inactivation curves for wild-type Cav3.2 channels and Cav3.2 channels with a deleted gating brake (deleted sequence from 429 to 539, see Fig. 2A). Representative current traces at time 0 min and after 20-min dialysis of Cav3.2 CaMB peptide are shown. Dialysis of CaMB peptide (after 20 min) generates a hyperpolarizing shift in voltage dependence of wild-type Cav3.2 channels, but it does not promote additional hyperpolarizing shifts in voltage dependence on the Cav3.2 channels that are highly hyperpolarizing shifted after gating brake deletion. The statistical significance after dialyses of CaMB peptide in Cav3.2 channels containing an intact gating brake is p = 0.0156 (*) and p = 0.0313 (*) for V50% activation and V50% inactivation, respectively, in a Wilcoxon matched-pairs signed rank test.
Figure 9.

Overexpression of apo-CaM/CaM1234 (Ca2+-binding deficient CaM) causes a hyperpolarizing shift in voltage sensitivities and faster channel kinetics in Cav3.2 channels only in the presence of an intact gating brake in Cav3.2. A and B, Cav3.2 channels with and without co-expressed CaM1234 were measured for their current-voltage relationships and τ for mono-exponential fits of the kinetics of activation (A) and inactivation (B). Representative current traces are shown. Results are presented as mean ± S.E., and n = the number of cells. Statistical analyses were first performed with the Student's t test or with one-way ANOVA combined with a Tukey post-test for multiple comparisons (*, p < 0.05; **, p < 0.01; ***, p < 0.001). C, scatter plot (left) with mean ± S.E. (right) values illustrate the 50% activation and inactivation values taken from Boltzmann fits of activation and steady-state inactivation curves of Cav3.2 channels alone (−), and Cav3.2 channels with co-expressed apo-calmodulin (CaM1234) or calmodulin (CaM). Voltage responses to co-expressed apo-calmodulin (CaM1234) are also illustrated for Cav3.2 deletion mutants spanning sequences 429–539 or 453–491, lacking the 111 or 39 amino acids, respectively, flanking the gating brake in the I–II linker. The region of deletion mutants spanning the gating brake of Cav3.2 channels are illustrated in Fig. 2B. The statistical significance of CaM1234 overexpression compared with wild-type conditions was p = 0.0028 (***) and p = 0.0308 (*) for V50% activation and V50% inactivation, respectively, using a Kruskal-Wallis test followed by a Dunn's multiple comparisons test. The effect of CaM1234 overexpression compared with wild-type conditions for Cav3.2 deletion mutants (D429–539 or D453–491) were non-significant using a Mann-Whitney test.
Dialyzed gating brake peptides specifically target the gating brake of Cav3.2 channels
We observed that the intracellular dialysis of Cav3.2GB peptide does not confer additional hyperpolarizing shifts in voltage sensitivity of activation (Fig. 8A) or steady-state inactivation (Fig. 8B) in Cav3.2 channels possessing the gating brake deletion Cav3.2(D429–539). These observations are consistent with the specific targeting of Cav3.2 CaMB peptide to helix-2 of the gating brake region in the I–II linker, a region that possesses a nanomolar affinity for CaM to promote the observed hyperpolarizing shifts in voltage sensitivities of Cav3 T-type channels.
Results of overexpression experiments with mutated CaM (CaM1234) strongly suggest that CaM regulates Cav3.2 channels at the gating brake
ITC measurements suggest that the gating brake associates with both N- and C-terminal Ca2+-binding EF hand pairs. CaM binds to the gating brake in the absence of viable calcium-binding sites (CaM1234). This CaM association for Cav3.1 and LCav3 CaMB peptide in the absence of calcium is at an ∼12- or ∼349-fold reduced affinity, respectively, compared with wild-type CaM (Fig. 6A). We tested whether calcium occupancy in CaM lobes is required to prevent the gating brake phenotype. We overexpressed a plasmid construct coding for CaM1234 to compete with native CaM (46). The CaM1234 overexpression construct included the bicistronic marker mRFP (red color fluorescent protein) (46) to be used as a visual estimate of the expected level of CaM1234 overexpression, after mercury lamp excitation with the epifluorescence-inverted microscope used for patch-clamp recording. The CaM1234 overexpression construct also contained two shRNA sequences to knock down CaM (CALM1,2,3 genes) (46–48). The shRNA knockdown sequences is reported to be 70% complete in disabling native CaM expression (46–48). However, knockdown was not effective under our experimental conditions, as we observed no significant physiological consequence after transfection of the shRNA CaM knockdown alone. The combined construct of shRNA knockdown of wild-type CaM and overexpression of CaM1234 had a dominant effect, compared with the overexpression of a similar construct with wild-type CaM (Fig. 9). CaM1234 causes a hyperpolarizing shift in voltage dependence for activation and inactivation and more rapid gating kinetics (Fig. 9), resembling the phenotype after saturating CaM-binding sites with dialyzed gating brake peptides (Figs. 7 and 8) and resembling the runaway gating phenotype of T-type channels after deletion or mutation of their gating brake sequences (Fig. 8, A and B) (23–26). The hyperpolarizing voltage shift conferred by CaM1234 appear to require association with an intact gating brake, because additional hyper-polarizing shifts in voltage dependence are lacking in Cav3 channels containing deletion of 39 and 111 amino acids spanning the gating brake, Cav3.2(D453–491) and Cav3.2(D429–539), respectively (Fig. 9B) (23).
Discussion
Ca2+–CaM readily complexes with full-length Cav3 T-type channels in vitro
We show here that the two most common isoforms of Cav3 T-type channels expressed in both the mammalian brain and heart (Cav3.1 and Cav3.2) (49), in their full-length configurations, complex with Ca2+–CaM in vitro. We illustrate using electron microscopy that purified full-length Cav3.1 binds calcium–CaM, data supported by co-immunoprecipitation experiments. A recent study by Lee et al. reported that a rat Cav3.3 splice variant co-immunoprecipitates with CaM (50), adding support for a universality in CaM associating with all Cav3 T-type channels, in the same manner as CaM serves as a ubiquitous resident calcium sensor at the C-terminal IQ tails of Cav1 and Cav2 calcium channels (1–5) and Nav1 sodium channels (6). Cav3 T-type channels (like all eukaryotic calcium and sodium channels), are large proteins (∼250 kDa) that do not readily express well enough for the purposes of extensive in vitro biochemical analyses. We chose candidate peptide fragments based on in silico prediction for resolving finer details of the CaM regulation of Cav3 T-type channels. Examination of candidate peptides is the standard approach used for other CaM targets (51).
All known Cav3 T-type channels have a predicted high-affinity CaM-binding site in helix2 of the helix–loop–helix of the gating brake in the I–II cytoplasmic linker
Cav3 channels lack the canonical C-terminal IQ motif (FILV)QXXX(RK)GXXX(RK)XX(FILVWY) of Cav1 L-type Ca2+ channels or (FILV)QXXX(RK)XXXXXXXX Nav1 sodium channels for CaM association (51). One of the criteria for a potential CaM-binding motif is its omnipresence from microbial eukaryotes to humans such as the IQ motif within different Cav1 and Nav channels (9). This criterion was satisfied with the predicted CaM association in the second helix of the helix–loop–helix motif forming the Cav3 gating brake in the I–II linker of almost all known Cav3 channels. All 26-mer gating brake sequences are similar (e.g. 71–76% between pond snail and human Cav3 isoforms) but are also completely lacking the string of identical and canonical amino acids conserved in the IQ motif in different Cav1 and Nav channels. Interestingly, the most basal of multicellular organisms, the placozoan species T. adherens, possess a Cav3 T-type channel (40) but lack an identifiable gating brake motif (only 10% similarity to pond snail and human Cav3 isoforms). In its place, the Trichoplax channel sequence is similar to the IQ motifs of L-type calcium channels (48% to the human Cav1.2). It is highly suggestive that the earliest eukaryotic Cav3 T-type channels had a CaM-binding IQ motif shared with other Cav and Nav channels, before its divergence and establishment of its unique identity in Cav3 T-type channels, as the gating brake.
Conformational changes in both Ca2+–CaM and CaMB peptides are observed upon their high nanomolar affinity association
Nanomolar and specific CaM binding to Cav3 CaMB peptides are corroborated in the ability of Ca2+–CaM to increasingly associate with Cav3 CaMB peptides corresponding to increasing concentrations of the Cav3 CaMB peptides illustrated by gel-mobility-shift assays. Cav3 CaMB peptides appear to undergo dramatic conformational changes (i.e. increasing α-felicity) on association with Ca2+–CaM illustrated by circular dichroism. Although ellipticity changes in the circular dichroism likely involve the induction of α-helices in the peptide upon binding CaM, some of the changes may be induced by tertiary structural changes in the proteins themselves (43). The association of the Cav3 CaMB peptides with CaM generates dramatic conformational changes in Ca2+–CaM, illustrated by amide chemical shifts of CaM's amino acids in 1H-15N HSQC NMR spectra spanning both N- and C-terminal lobes of CaM. Thermodynamic properties measured within the isothermal cell of the Ca2+–CaM complexing with Cav3 CaMB peptides illustrate that Ca2+–CaM and Cav3 CaMB peptides associate in a 1:1 stoichiometry (N value) at a high nanomolar association constant (Ka).
Highest affinity association of Ca2+–CaM CaMB peptides requires both lobes of CaM and their Ca2+-binding sites
Further evidence of the specificity of the Cav3 CaMB peptides to Ca2+–CaM is in the requirement of intact Ca2+–CaM for high nanomolar affinity binding. We show that the N-lobes and C-lobes of CaM alone will associate with Cav3 CaMB peptides, but at a lower measurable affinity compared with both CaM lobes. The high-affinity of CaM for Cav3 CaMB peptides requires intact Ca2+-binding sites for CaM, as evidenced by the greatly weakened affinity of CaM in the CaM1234 configuration for the Cav3 CaMB peptides.
Different sequences for CaMB peptides from sample invertebrate LCav3 and mammalian Cav3 channels possess a high nanomolar affinity and similar binding properties in complexing with Ca2+–CaM
Differing sequences of CaMB peptides for snail LCav3 and the three human Cav3 gene isoforms are 71–76% similar to one another and possess similar binding properties with Ca2+–CaM. Snail LCav3 and the different human Cav3 isoforms of the CaMB peptides associate with a nanomolar affinity and in a 1:1 stoichiometry with Ca2+–CaM. All CaMB peptides assume a greater α-helicity upon association of Ca2+–CaM, except the alanine-rich Cav3.1 isoform, and both snail LCav3 and mammalian Cav3.2 CaMB peptides generate similar and large conformational changes to both N- and C-lobes of CaM as measured by the amide chemical shifts of CaM's amino acids in 1H-15N HSQC NMR spectra. Both snail LCav3 and mammalian Cav3.2 CaMB peptides also associate with Ca2+–CaM at a much weakened affinity when Ca2+-binding sites are mutated in CaM.
Differing sequences of CaMB peptides differ in their relative affinities for Ca2+–CaM
The sequence differences between the Cav3 CaMB peptides contributed to significant differences in binding affinities, which varied from rank order of highest to lowest binding affinities of LCav3 > Cav3.1 > Cav3.2 > Cav3.3 of 12, 43, 187, and 383 nm, respectively. The rank order of binding affinities measured by the thermodynamic changes resulting from the 1:1 complexing of Cav3 CaMB peptides and Ca2+–CaM measured in the isothermal cell is also the same observed rank order of the fold concentration of Cav3 CaMB peptides required for saturation of Ca2+–CaM with Cav3 CaMB peptides for a complete gel-mobility shift.
Nanomolar affinities of Cav3 CaMB peptides are approximately equivalent to the measured high affinities of the IQ motif of Cav1.2 and Cav2.1 channels and greater than the affinity of Ca2+–CaM for Nav1.5 channels
Our measured binding affinities of Cav3 CaMB peptides for N- and C-lobes of Ca2+–CaM (in nm) of 31 ± 1.9 and 690 ± 42, respectively, is in the relative range of nanomolar affinities for CaM as the C-terminal IQ domain of Cav1.2 of 57.6 ± 35.5 and 2.63 ± 0.07 (52) and the C-terminal IQ domain of Cav2.1 of 51 ± 20 and 4.32 ± 0.39 (53). The higher nanomolar binding affinity of the Cav3.2 channel CaMB peptides for N- and C-lobes of Ca2+–CaM is greater than NSCaTE, a secondary CaM-binding site of Cav1.2 channels (in nm): 650 ± 60 and 1330 ± 300 (16), respectively. Cav3.2 channel CaMB peptides are also much higher affinity than the micromolar binding affinity range for the N- and C-lobes of Ca2+–CaM of Nav1.5 sodium channel fragments (in nm), such as its C-terminal IQ domain of 2170 ± 400 and 6130 ± 900 (6) and the III–IV linker of 19,270 ± 940 and >500,000, respectively (54).
Phenotype after dialysis of Cav3 CaMB peptides corresponds to immobilizing the gating brake in the I–II linker
Our biochemical studies reflect that Ca2+–CaM associates with helix-2 of the gating brake in both invertebrate and all mammalian isoforms of Cav3 channels, in a high nanomolar affinity with binding characteristics that is reminiscent of the canonical binding of Ca2+-CaM to the proximal C-terminal IQ motif of Cav1 and Cav2 channels. We then tracked the consequences of introducing the high-affinity CaMB peptide in expressed Cav3.2 channels in real time, by comparing the biophysical changes to the Cav3.2 channels from the start of a stabilized patch-clamp recording (0 min) and after complete dialysis of 5 μm CaMB peptides from our patch pipette (15–20 min). Biochemically, we observed that equivalent mutated CaMB peptide did not associate with Ca2+–CaM, so it served as a control to compare the effects of the CaMB peptide. We observed ∼8- and ∼16-mV hyperpolarization shifts in activation and inactivation curves, respectively, as a result of CaMB peptide dialysis. Faster channel kinetics were also observed, which are a likely a consequence of the hyperpolarizing shift, because corresponding kinetic changes are usually associated with shifts in voltage sensitivities within Cav3 T-type channels (37). The target for the CaMB peptide effect was the gating brake, as we observed no additional hyperpolarizing shift in voltage sensitivities after CaMB peptide dialysis in Cav3.2 channel mutants lacking the gating brake. We carried out the complete set of electrophysiology experiments using mammalian Cav3.2 because of the availability of gating brake mutants. We will carry out future analyses to confirm the hyperpolarizing shifts in voltage sensitivities in response to CaMB peptide dialyses are shared among other Cav3 T-type channels, including invertebrate LCav3, all of which have a common nanomolar affinity for CaM binding to the gating brake.
Overexpression of Ca2+-binding deficient CaM (i.e. CaM1234) reveals that CaMB peptides likely compete for CaM binding at the gating brake in the I–II linker of Cav3 channels
Involvement of CaM regulation of ion channels has been supported by introducing a Ca2+-insensitive mutant form of CaM (CaM1234), which possess point mutations within both pairs of EF hands in the N- and C-lobes of CaM to eliminate Ca2+ binding (44). It is believed that CaM pre-associates onto its target in the apo-CaM form (lacking Ca2+ occupied in its EF hands) (55–57). CaMB peptides have a measurable affinity with CaM1234, albeit at levels that are ∼12- and ∼349-fold lower compared with wild-type CaM, for Cav3.1 and LCav3 CaMB peptides. Apo-CaM bound to its channel target is considered a necessary antecedent to the subsequent regulation of CaM by Ca2+. Overexpression of the mutant apo-CaM, which is incapable of binding Ca2+, is thus considered a competitive inhibitor to silence the effects of Ca2+–CaM. For example, the dominant-negative effects of CaM1234 overexpression have been used to illustrate the importance of Ca2+–CaM in the gating of small-conductance Ca2+-activated K+ channels (44), and the necessity of Ca2+–CaM in the Ca2+-dependent inactivation of Cav1 L-type channels (58, 59) as well as the calcium-dependent facilitation of Cav2 channels (60). We observe that CaM1234 causes a modest (6.7 and 7.2 mV) hyperpolarizing shift in activation and inactivation curves of Cav3.2 channels, respectively, compared with wild-type CaM overexpression. The likely target for the CaM1234 effect is the gating brake, as we observed no additional CaM1234 causing hyperpolarizing shifts on Cav3.2 channels lacking the gating brake.
Combined biochemistry and electrophysiology data highly suggest that Ca2+–CaM is a high-affinity resident sensor at the gating brake of Cav3 channels
To summarize our data, we illustrate that CaM1234, considered as a substitute for apo-CaM (CaM in the absence of calcium), associates with helix-2 of the gating brake at a low micromolar affinity. Upon binding Ca2+, CaMB peptides and CaM both undergo dramatic conformational changes, leading to CaM's tighter association at the gating brake at a higher nanomolar affinity. Our data suggest that Ca2+–CaM is behaving as a similar high-affinity and resident sensor at the gating brake in the I–II linker of Cav3 T-type channels, in both sample invertebrate and the three human gene isoforms, in a universal manner similar to the calcium–CaM regulation on the IQ motif containing C-terminal tails of Cav1 and Cav2 calcium channels (1–5) and Nav1 sodium channels (6). Introducing CaMB peptides or CaM1234 competitively inhibits Ca2+–CaM at the gating brake of Cav3.2 channels, generating a runaway gating phenotype, with hyperpolarized shifts in voltage sensitivities that are even greater than observed after deletion of the brake from Cav3 channels. Because the introduced CaMB peptides or CaM1234 have no effect on Cav3.2 channels with a missing gating brake, it suggests that the primary target for Ca2+–CaM is the gating brake of Cav3 channels. We did not observe the complexing of full-length Cav3.1 and Cav3.2 channels with CaM in vitro in the absence of calcium ions, but we suspect that it may relate to the structural requirements for calcium ions in recapitulating the native behavior of full-sized, Cav3 channels in vitro, the sensitivities of our assays, and the technical challenges in working with whole ion channel proteins of such large sizes (∼259 kDa) in vitro.
Cav3.3 exhibits a similar calcium-dependent CaM regulation as other calcium and sodium channels
Consistent with our findings here, Lee et al. (50) report a hyperpolarizing shift in activation and inactivation curves of Cav3.3 channels and faster inactivation kinetics, under conditions where intracellular free calcium is at a higher (1 μm) but not lower (27 nm) calculated concentration. Lee et al. (50) also observe a faster inactivation kinetics in Cav3.3 channels associated with external calcium compared with barium as the charge carrier. The much faster inactivating mammalian T-type channels, Cav3.1 and Cav3.2, do not appear to exhibit the same calcium-dependent change in biophysical properties as Cav3.3 channels but possess a more pronounced voltage-dependent inactivation (61). Lee et al. (50) illustrate that rat Cav3.3 channels possess a unique C-terminal sequence that associates with Ca2+–CaM, conferring the unique CaM regulation observed for Cav3.3 channels.
Observed variability of the Ca2+-dependent regulation within Cav3 channels is consistent with the variability in Ca2+–CaM binding within the Cav1 L-type channels
A suggested hypothesis is that all Cav3 channels possess a central, high-affinity Ca2+–CaM binding at the gating brake in the I–II linker, with additional modulatory, intracellular Ca2+–CaM-binding sites elsewhere, which can confer novel attributes, such as the Ca2+–CaM regulation at the C terminus for Cav3.3 channels (50). A highly customizable form of Ca2+–CaM regulation is observed within the “IQ motif centric” calcium (Cav1 and Cav2) and sodium (Nav1) channels. The preoccupancy of CaM at the primary IQ motif in the absence of Ca2+ ions is one form of regulation. The specialized Cav1.4 homolog localized in vertebrate photoreceptors possesses a highly conserved IQ motif but exhibits almost no calcium-dependent inactivation compared with closely-related Cav1.2 channels. Cav1.4 possess a distal C-terminal auto-inhibitory domain, which effectively competes for apo-CaM for binding, thus preventing Ca2+–CaM regulation at Cav1.4's conserved proximal C-terminal IQ motif (62). Cav1.3 possess a similar auto-inhibitory domain in the distal C terminus generated through alternative splicing, which competes with apo-CaM (63, 64).
Another source of variability is in the semi-autonomy and cooperativity among CaM's pairs of Ca2+-binding EF hands in the N- and C-lobes, each of which is capable of conferring unique attributes. For example, the Ca2+–CaM-binding site NSCaTE shared within the N terminus of invertebrate Cav1 channels (9) and mammalian Cav1.2 and Cav1.3 channels (16, 17) interacts specifically with CaM's N-lobes, conferring a capacity for more global Ca2+-sensing and buffer-resistant calcium-dependent inactivation in classical L-type channels. The C-lobe of CaM is occupied with the IQ motif, imparting a local, more rapid, and spatially restricted calcium-sensing of Ca2+ ions (16, 17).
Observed variability of the Ca2+-dependent regulation within Cav3 channels is consistent with the variability in Ca2+–CaM binding within the Cav2 synaptic Ca2+ channels and Na+ channels
The bipartite nature of CaM's N- and C-lobes is also evident in the synaptic Cav2 channel family, where Ca2+ regulation manifests as a rapid, calcium-dependent facilitation of the calcium current triggered at CaM's C-lobe and a slower-developing calcium-dependent inactivation imparted by CaM's N-lobe (60, 65).
One confounding issue is a potential interplay between calcium- and voltage-dependent properties, such as the consequences of a Ca2+–CaM-binding site identified in the III–IV linker of Nav1.5 sodium channels, which is also the region responsible for the fast voltage-dependent inactivation within sodium channels (6, 54, 66). The contributions of both calcium- and voltage-dependent properties can obscure the typical U-shaped calcium-dependent inactivation (39).
Technical challenges in measuring the Ca2+ occupancy of CaM in vivo
Observations in vitro often do not correlate to those gathered within different cell types too, where different factors influence CaM's effects within the cellular environment. For example, when CaM has many high-affinity targets within a cell, it has been reported that CaM availability is likely to be limiting in many cell types (67, 68). In such cases, CaM regulation may be unavailable because individual CaMs may be preoccupied at different high-affinity cellular targets. There is also likely competition among differing calcium-sensing proteins. A good example is calcium-binding protein expressed in the vertebrate nervous system, whose expression can antagonize CaM-dependent regulation of Cav1 L-type channels (69).
The last confounding issue with studies involving CaM is in the technical challenge in modeling calcium availability for CaM regulation. It is challenging to precisely measure native calcium-buffering conditions because they are variable within a particular cell or among different cell types. Ion channel studies usually involve intracellular dialysis in the whole-cell patch-clamp configuration of intracellular calcium chelators (e.g. EGTA and BAPTA), which impose artificial calcium-buffering conditions, whose calcium affinity and on-off rates are not known in comparison with the native buffering conditions in vivo.
Universal “calmodulation” among sodium and Ca2+ channels also involves Cav3 channels
Cav3 channels are the least well studied of the calcium or sodium channels, which means that Cav3 channels are interpreted through the lens of what is understood for other calcium and sodium channels (37). A universal IQ centric calmodulation of sodium and calcium channels has more variations than commonality by the many interactions that fine-tune the Ca2+–CaM regulation outside of the primary C-terminal IQ motif. So it should not be surprising that a functional Ca2+–CaM regulation is only evident within one of three mammalian Cav3 channels in preliminary analyses, and lacking evidence so far within Cav3.1 and Cav3.2 channels (50). Despite possessing proximal C-terminal Ca2+–CaM binding in the IQ motif and the III–IV linker, cardiac muscle Nav1.5 channels possess no obvious Ca2+–CaM regulation, whereas skeletal muscle Nav1.4 has a robust calcium-dependent inactivation (2, 70, 71). A deeper molecular analysis uncovered, for example, a Ca2+–CaM regulation of classical L-type channels within Cav1.4 channels, after deletion of its C-terminal auto-inhibitory domain (62).
Potential implications for Cav3 channels possessing Ca2+–CaM regulation at the gating brake
Cav3 channels are equally distant structurally to both calcium and sodium channels (37), and it should come as no surprise that its Ca2+–CaM regulation is also strikingly different, where the equivalence to the C-terminally positioned IQ motif is transposed onto a different cytoplasmic region. The gating brake is a unique element within all Cav3 T-type channels, which tunes the unique low voltage sensitivity of this channel class (72). The gating brake is a position that overlaps with the Alpha1 Interaction Site of Cav1 and Cav2 calcium channels where accessory Cavβ subunits regulate their expression levels and biophysical properties (25). Cav3 T-type channels lack equivalency of the high-nanomolar affinity Cavβ subunit-binding site within Cav1 and Cav2 calcium channels (73). It is possible to confer Cavβ subunit regulation onto Cav3 T-type channels after transplanting the I–II linker from Cav2 channels (45). But in their native state, Cav3 T-type channels possess remarkably similar properties as their in vivo counterparts without requirement of co-expressed accessory subunits in invertebrates (74) and mammals (45, 75). Cavβ subunits will boost the level of expression of invertebrate (76) and mammalian (75) Cav3 T-type channels, but this is considered to be the result of indirect and generalized consequences of overexpressing a multifunctional, scaffolding, and regulatory protein, such as the Cavβ subunit in vitro (77).
With a lack of equivalent accessory subunit interactions within Cav3 channels, opportunities are created for a Ca2+–CaM regulation within the gating brake of the I–II linker. The gating brake is placed in the I–II loop, just downstream of a stiff α-helical linker extending from the segment 6 of domain II, which contributes to pore opening (72). Affixing Ca2+–CaM to the I–II loop may have structural implications because the I–II loop is immobilized at both ends compared with the C-terminal tails containing the IQ motif of sodium and calcium channels, which is linked only a single end. A more proximate location of the gating brake to the intracellular pore mouth may also have implications, contributing to a unique Ca2+–CaM regulation within Cav3 channels.
Potential importance of Ca2+–CaM regulation of Cav3 channels for human physiology and disease
The unique high-affinity binding and regulation of CaM at the gating brake of Cav3 T-type channels has dramatic potential consequences, because of the gating brake's role as a primary regulator of the voltage sensitivity of Cav3 T-type channels. Cav3 T-type channels are unique among sodium and calcium channels by the low voltage range of activation that is close to typical resting membrane potentials. CaM at the gating brake is perfectly positioned in Cav3 channels for fine-tuning the channels' responsiveness to voltage, providing a means to govern the capacity of Cav3 T-type channels to facilitate pacemaker rhythms where T-type channels have established roles, such as the thalamo-cortical circuits of the brain (where Cav3 T-type channel expression is highest) and the conducting system in the heart (49). Human mutations in the gating brake are associated with arrhythmic vulnerabilities such as epilepsy (27, 28) or autism spectrum disorder (78), and it is the platform where Ca2+ and CaM can modulate the low-voltage gating behavior in T-type channels, in a manner that is unique from the regulation of CaM universally at the IQ motif in the C-terminal tails of Cav1 and Cav2 calcium channels (1–5) and Nav1 sodium channels (6).
Outside of Ca2+–CaM regulation, Cav3 channels are very different from other calcium and sodium channels
Cav3 channels uniquely different Ca2+–CaM regulation goes alongside its many observed differences from other sodium and calcium channels. This includes their ion selectivity, which can range from mostly calcium-conducting (Cav3.1 and Cav3.2), equally sodium- and calcium-conducting (Cav3.3) and as sodium-conducting as Nav1 sodium channels (protostome invertebrate Cav3 channel isoforms).
We have shown that most non-vertebrate Cav3 T-type channels have alternative splicing of the L5 extracellular turret in domain II, which converts the mostly calcium-conducting T-type channel into a highly sodium-conducting isoform, whose expression in the invertebrate heart serves as the primary sodium current (74). The plasticity of invertebrate Cav3 channels in assuming calcium- or sodium-conducting pore configurations is to expand their niche in the absence of Nav1 channel genes expressed outside of the nervous systems of many invertebrates, and in the many species of non-vertebrates completely lacking any Nav1 channel in their genomes (nematodes, echinoderms, hemichordates, and parasitic flatworms) (74). It is an interesting question to address how Ca2+–CaM regulation varies when Na+ is the primary conducting ion rather than Ca2+ through differing invertebrate Cav3 channel isoforms.
Potential role for calcium sensing at the gating brake is in influencing calcium ion selectivity within Cav3 channels
We have shown that the Cav3 channels are endowed with ∼2-fold increase in the number of cysteines in L5 and L6 extracellular turrets, contributing to uniquely folded extracellular scaffolds (79). In particular, alternative turrets with unique cysteine configurations in both DII L5 and DIV L6 regulate the degree of sodium and calcium conductances through invertebrate Cav3 channels.4 Deletion of these unique L5 cysteines in domain II, alters drug affinity, divalent cation block with Ni2+ and Zn2+, and also increasing the apparent pore size, allowing for the accommodation of a simultaneous flow of both monovalent and divalent cations.5 The simultaneous flow of differing divalent and monovalent ions may only be possible within Cav3 T-type channels, given its likely wider, more accommodating EEDD selectivity filter compared with other Cav1 and Cav2 calcium channels with an EEEE selectivity filter (49). One of the possible implications of a high-affinity calcium sensor, such as CaM near the internal mouth of the pore, is in its potential in influencing calcium ion conductance. We observe an apparent simultaneous flow of monovalent and divalent ions through Cav3 channels containing mutated cysteines in the domain II, L5 extracellular loop, which is only possible if the divalent cation alongside the monovalent (e.g. Na+) is either Sr2+ or Ba2+ but not Ca2+.5 A high-affinity calcium sensor, like CaM at the pore mouth, may contribute to the explanation for the observations that Ca2+ appears as the dominant and preferred divalent ion carrier through the Cav3 channel pore, even when the channel is highly sodium-conducting.5
Experimental procedures
Expression of Cav3 constructs
The cDNAs encoding for Cav3.2 were previously characterized (61) as well as hemagglutinin (HA) epitope-tagged Cav3 subunits expressed using pcDNA 3.1 vector (23, 75). We illustrate how to maintain HEK-293T cell lines and introduce calcium channel expressible plasmids for transient transfection, expression, and whole-cell patch-clamp recording in an on-line video (JoVE) format (82). HEK-293T cells were cultured as described previously (23, 75). For optimal transfection, cells were plated at 50–70% confluence. Cells were transfected using the JetPEI transfection reagent (QBiogene) according to the manufacturer's protocol. In all conditions, corresponding empty plasmids were used to adjust the quantity of the transfected material (1 μg of each plasmid/35-mm dish). Gating brake deletion mutants for Cav3.2, D429–539, and D453–491 were used previously (23).
Expression of CaM, CaM mutant, and CaM knockdown constructs
We have used human CaM and associated CaM mutants that we have described before (9), CaM(1–44) and partial CaMs: N-CaM(1–74), C-CaM(75–148), and CaMs that simulate CaM binding in the absence of calcium, by substitution of glutamate residues in the two calcium-binding sites in the N-terminal lobe (CaM12) or C-terminal lobe (CaM34), or mutation of all four calcium-binding sites (CaM1234) of CaM. CaM–GFP used for Western blotting is a construct consisting of GFP fused to the C terminus of CaM expressed in pcDNA3.1 (Cell Signaling Technology) (83). EGFP expression vector was obtained by subcloning EGFP from pEGFP-C1 (BD Biosciences) into the multiple cloning site of pcDNA3.1. The CaM1234 overexpression construct contained bicistronic marker mRFP (to generate a red color) (46) to distinguish from the channel expression with bicistronic EGFP (to generate a green color). The CaM1234 overexpression construct also containing CaM shRNA knockdown includes two short hairpin sequences described above to knock down CaM (Calm1,2,3 genes) (46–48). The shRNA knockdown sequences is reported to be 70% complete in disabling of native CaM expression (46–48), but this knockdown was insufficient for our purposes, as we observed no significant physiological consequence after transfection of the shRNA CaM knockdown alone.
Synthetic peptide experiments
We purified bacterially expressed His-tagged CaM (CaM) and measured in vitro association with 26-mer synthetic peptides (BioBasic) spanning the gating brake of human Cav3.x, Cav3.2GBmut, and snail LCav3 in gel-shift mobility assays, differential circular dichroism, and isothermal titration calorimetry. We followed protocols that we have described previously in an analysis of CaM binding with snail or human L-type calcium channel peptides in the presence of 0.1 or 0.5 mm CaCl2-containing solution (9).
CaM protein expression for biochemistry experiments
Rat CaM was purified using phenyl-Sepharose 6 Fast-Flow (High Sub) (GE Healthcare), packed to ∼10 ml volume, by gravity flow at 4 °C. Codon-optimized CaM was cloned in pET9d, a T7 Escherichia coli expression vector. CaM was transformed into BL21 DE3 E. coli and grown at 37 °C and 200 rpm to OD of 0.4–0.6 (for Luria Broth, LB) or 0.5–0.9 (for SuperBroth, SB), at which point samples were induced by 1 mm added isopropyl β-d-1-thiogalactopyranoside and grown for another 4 (for SB) to 6 (for LB) h. Robust expression of soluble CaM was confirmed by SDS-PAGE. Cells were collected by centrifugation at ∼4000 × g, then lysed in high-salt buffer (0.1 m Tris-Cl, pH 7.5, 0.5 m NaCl, 0.5 m ammonium sulfate, 1 mm DTT) using an Avestin Emulsiflex homogenizer at 15,000–20,000 p.s.i. with jacketed water cooling, through which the cells were passed ∼3× on average. The cell lysate was clarified by centrifugation at 20,000 rpm in an SS-34 rotor (∼50,000 × g) at 4 °C for 30 min. To the cleared lysate, CaCl2 was added to a final concentration of 10 mm. The lysate was then applied to the gravity phenyl-Sepharose column equilibrated with the same high salt buffer (and 1 mm CaCl2). The column was washed with high-salt buffer. CaM was then eluted using 10 mm Tris-Cl and EDTA solution, pH 7.5. The highest fractions were pooled, concentrated, and applied to the preparation grade Superdex 75 16/60 column (GE Healthcare) on an AKTA FPLC system. Typical injection volume was 1 ml, and the gel filtration buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1 mm CaCl2) was set to flow at 1 ml/min. Wild-type CaM reproducibly elutes at ∼68 ml on this column. Elution peak width is dependent on the amount of CaM present; routinely up to 10 1-ml fractions were obtained of concentrations 200–400 μm; these were pooled and concentrated using the spin concentrators from Sartorius or Amicon (10,000 molecular weight cutoff). Extinction coefficient of 3006 m−1 cm−1 at A276 was used (66), and the final protein stock was aliquoted to 500 μl and stored at −80 °C.
Purification of Ca2+-deficient CaM proteins
The interaction of CaM with hydrophobic interaction chromatography media depends on its exposure of the hydrophobic residues upon Ca2+ binding. The phenyl-Sepharose method is not nearly as efficient for the purification of CaM12, CaM34, or CaM1234. So for Ca2+-deficient CaM proteins, we used a combination of ion-exchange chromatography and subsequent gel filtration for purification. A Q-Sepharose (quaternary ammonium) 6-ml column (GE Healthcare) was used for ion exchange. Lysis and low-salt buffer consisted of 5 mm BisTris, pH 6.0, with no Ca2+ or EDTA. Lysis buffer also had DTT added to 1 mm final concentration to maintain CaM's methionines in the reduced state. CaM12, CaM34, and CaM1234 (in pET28a vector) were expressed and harvested in same manner as wild-type CaM. Cells were lysed using the Avestin Emulsiflex homogenizer (>15,000 p.s.i.), clarified by centrifugation at ∼ 50,000 × g, and applied to the column (ion exchange was done using the AKTA FPLC system, same as for gel-filtration chromatography). The column was then briefly washed with low-salt buffer (5–10 column volumes), and a shallow salt gradient was applied (100 mm NaCl over 10 column volumes). Subsequently, the column proteins were eluted with a steep gradient (1.6 m NaCl over 2 column volumes, held for 2 more column volumes), after which the column was re-equilibrated or cleaned. The resulting fractions were analyzed by SDS-PAGE and read for absorbance levels at 278 nm. Best candidates were pooled and concentrated using YM-10 spin columns (Millipore) and further purified by gel-filtration chromatography. Purity of the CaM proteins was confirmed by SDS-PAGE and ESI-MS (electrospray ionization-mass spectrometry), and their concentrations were determined using the modified Bradford assay (84) (with wild-type CaM as the standard), aliquoted, and stored in gel filtration buffer at −80 °C.
Bioinformatic analyses of peptide sequences
Protein parameters (pI and molecular weight) were determined using the prediction tool in ExPaSy (http://web.expasy.org/protparam/),6 and the aggregation propensity was determined using the on-line software at Centre for Genomic Regulation (http://tango.crg.es/).6 All phylogenetic alignments were done using variations of the BLAST tool on the GenBankTM web site. The CaM-binding site prediction database (http://calcium.uhnres.utoronto.ca/ctdb/contacts/index.htm)6 was used to search for other possible “hits” in the channel ORFs (39) and to design optimal peptide sequences for synthesis.
Native Trp fluorescence
Steady-state measurements were obtained using a Photon Technologies International Quantamaster Fluorimeter (London, Ontario, Canada) in a 50-μl 1-mm fluorescence cuvette (Hellma) at room temperature. The same buffer was used as for gel filtration and ITC experiments (50 mm Tris-Cl, pH 7.5, 150 mm NaCl) with either 1 mm CaCl2 or 10 mm EDTA; buffer was used as a baseline for subtraction. To the cuvette containing the starting peptide solution (25 μm), increasing amounts of CaM were added (in increments corresponding to ∼2.5 μm). Stepwise mission scans from 300 to 400 nm using a 280-nm excitation wavelength and 1-nm slit width (averaging over 1s each) were performed.
Dansyl-CaM fluorescence
Dansyl (5-(dimethylamino) naphthalene-1-sulfonyl chloride) is an amine-reactive fluorescent dye. Dansyl-CaM was prepared as described previously (85). CaM (1 mg/ml) was transferred into 10 mm NaHCO3, 1 mm EDTA, pH 10.0, at 4 °C. 30 μl of 6 mm dansyl-chloride (1.5 mol/mol of CaM) in DMSO was added to 2 ml of CaM, with stirring. After incubation for 12 h at 4 °C, the mixture was first dialyzed against 500 volumes of 150 mm NaCl, 1 mm EDTA, 20 mm Tris-HCl, pH 7.5, at 4 °C, and then dialyzed against 500 volumes of water. Labeling yields were determined from absorbance spectra using the ϵ320 of 3,400 m−1 cm−1 and were compared with protein concentrations determined using the Bradford method with wild-type CaM used as the protein standard (86) ESI-MS was used to confirm successful dansyl-labeling of CaM protein. The concentration of dansyl-CaM in all experiments was 2 μm. Steady-state fluorescence was performed in a similar manner as the Trp experiments, except that a newer Fluorolog 3-22 (Horiba Scientific, Ltd.) fluorimeter was used, and the buffer used was 10 mm HEPES, pH 7.0, with supplemented 0.1 mm CaCl2.
Differential circular dichroism
CD measurements were performed using a Jasco-715 spectropolarimeter (Jasco Instruments, Easton, MD), using the following parameters: 250–190-nm range, 50 nm/min speed, with 1-s response time, 1-nm bandwidth, and 100-millidegree sensitivity at room temperature. 16–25 accumulations were gathered depending on signal quality. Recordings were made in a 1-mm cuvette, in 10 mm sodium phosphate buffer with 0.1 mm added CaCl2, and used as baseline, which was subtracted from each recording. For CaM and CaMB peptide spectra, the same solution of 10 μm CaM was scanned before stock (1 mm) peptide was added in incremental amounts before being scanned again. The resulting spectra were subtracted (e.g. CaM and CaMB peptide-CaM alone) to extract the spectrum corresponding to the change associated with adding the peptide. Every recording was an average of at least 16 accumulations and subsequently corrected for protein concentration (converted to units of mean residue ellipticity). These were smoothed and exported into ASCII format for analysis with Excel and either SOMCD or Dichroweb K2d algorithm (http://dichroweb.cryst.bbk.ac.uk)6 (92, 93). TFE recordings were performed using pure 2,2,2-trifluoroethanol (Sigma) at 10, 25, and 50% after thorough pre-mixing and incubation with peptides in the phosphate buffer, baseline-corrected against the same TFE/buffer solution without peptide. LCav3 and Cav3.2 CaMB peptides were contained in PBS buffer (10 mm sodium phosphate, pH 7.3, 137 mm NaCl, 2 mm KH2PO4, and 2.7 mm KCl). Cav3.1, Cav3.3, CaMB peptides were characterized in low ionic strength buffers due to severe aggregation issues in PBS. Low ionic strength buffer for Cav3.1 and Cav3.3 CaMB peptides was composed of 10 mm HEPES, pH 7. A 0.1-cm quartz cuvette (Hellma) was used for CD measurements. CaM-binding experiments were performed by adding increments of stock (1 mm) CaMB peptide to the same cuvette containing 10 μm CaM (300 μl). Subsequent scans would then have the CaM-only trace subtracted and corrected for total peptide concentration, smoothed, and converted to mean residue ellipticity units.
Gel mobility-shift assays
Gel assays were run in native conditions with 4 m urea, containing 15% acrylamide separating and 4% stacking gels. Running buffer contained 192 mm glycine, 50 mm Tris-Cl, 0.1 mm CaCl2, pH 8.3. Separating gel buffer (2×) contained 0.7 m Tris-Cl and 1 mm CaCl2, pH 8.8. Stacking buffer (2×) contained 0.5 m Tris-Cl and 1 mm CaCl2, pH 6.7. Peptides were incubated with CaM for ≥1 h at 4 °C in gel filtration buffer (150 mm NaCl, 50 mm Tris-Cl, pH 7.5, 1 mm CaCl2) prior to loading. 50% glycerol was used in the gel filtration buffer, and <0.1% bromphenol blue (as tracker dye) was used at 1:2 sample volume just prior to loading for each sample. Gels were run at 100 V in 4 °C jacketed/ice-bath conditions for 6–8 h as needed. All gel-mobility-shift assays were confirmed in three replicate experiments.
Isothermal titration calorimetry
Heat gain or heat requirements in the sample cell (for exothermic binding or endothermic binding, respectively) correspond to the direction of the injection peaks measured in the isothermal titration calorimeter (MicroCal iTC200, Malvern Instruments, UK). The peaks were integrated and plotted as a sigmoidal plot of μcal (concentration of the ligand in the cell) versus μmol of injectant (protein concentration in the syringe). Origin 2017 software (OriginLab) was used to fit the resulting data through iterative least-squares-type regression. Due to the tendency of CaMB peptides to aggregate in any salt, we reconstituted the CaMB peptides for ITC experiments in a low ionic strength (10 mm HEPES, pH 7.0, 0.1 mm CaCl2) buffer for all CaMB peptides. All experiments were performed at 25 °C, with 1000 rpm stirring, 5 μcal/s reference power, and high-feedback gain. Injection conditions were typically 1.2–1.8 μl each (22–28 injections), and cell/syringe concentrations were optimized for individual experiments (c value between 10 and 1000) (87–89) but typically between 10 and 50 μm for the cell and 100 to 500 μm for the syringe. Periodically, water-water and buffer-buffer controls were conducted to ensure the syringe and cell were clean and in good working order. Most of the endothermic titrations had to be baseline-subtracted against the final injection heats (end-weighted auto-baseline or manual baseline for Cav3.3 correct for the heat of dilution) due to fitting artifacts from the isotherm passing through the x axis. Baseline-corrected curves were all fitted to a one-set-of-sites model in Origin 2017 (OriginLab) software with satisfactory results.
Electron microscopy
Full-length mammalian Cav3.1 was expressed in Sf9 insect cells and purified as described previously (38). Recombinant human Cav3.1 was incubated (1:1 molar ratio) with biotin–CaM (Calbiochem) in 1.5 mm CaCl2 after which a streptavidin–gold conjugate (5 nm) was added (38). Samples were examined using standard negative staining techniques with 2% (w/v) uranyl acetate. Images of the Cav3.1–CaM complexes were recorded on a Tecnai 12 transmission electron microscope operated at 100 kV.
NMR
Complex formation of gating brake peptide with CaM in a 1:1 ratio was monitored by acquiring a 1H-15N heteronuclear single-quantum coherence (HSQC) spectrum in a manner previously published with CaM and peptides (90). NMR spectra were recorded at 25 °C on Bruker 600 and 700 MHz DRX spectrometers equipped with XYZ-gradient triple-resonance probes (Bruker, Billerica, MA), and analyzed with CARA (computer-aided resonance assignment). CaM for NMR experiments was expressed in M9 minimal media (11.03 g/liter Na2HPO4·7H2O, 3.0 g/liter KH2PO4, 0.5 g/liter NaCl, 2 mm MgSO4, 0.1 mm CaCl2, 5 mg/ml thiamine, 100 μg/ml kanamycin) containing 2 g/liter [13C]glucose and 1 g/liter 15NH4Cl; 13C-15N CaM was purified. The samples were prepared for NMR experiments via a buffer exchange into NMR solution (100 mm KCl, 10 mm CaCl2, 0.2 mm NaN3, 90% H2O, 10% 2H2O) at pH 6.0 using a YM10 centrifugal filter device (Millipore, Billerica). All NMR samples contained 300 μm CaM in a total volume of 500 μl. The samples were transferred into 5-mm NMR sample tubes and stored at 4 °C until required for NMR experiments. NMR experiments on the complexes were conducted on CaM samples titrated with each peptide to saturation in a 1:1 CaM/peptide ratio. Complex formation was monitored after each addition by acquisition of a 1H-15N HSQC pulse sequence. NMR spectra were recorded at 25 °C on Bruker 600 and 700 MHz DRX spectrometers equipped with XYZ-gradients triple-resonance probes (Bruker, Billerica, MA.). Spectra were analyzed using the program CARA.
Electrophysiological recordings
Macroscopic currents were recorded at room temperature using an Axopatch 200B amplifier (Molecular Devices). Borosilicate glass pipettes had a resistance of 1.5–2.5 megaohms when filled with an internal solution containing (in mm): 140 CsCl, 10 EGTA, 10 HEPES, 3 Mg-ATP, 0.6 GTP-Na (pH adjusted to 7.25 with CsOH, ∼315 mosm). The extracellular solution contained (in mm): 145 tetraethylammonium-Cl, 10 HEPES, and 2 CaCl2 (pH adjusted to 7.25 with tetraethylammonium-OH, ∼330 mosm). Recordings were filtered at 2 kHz. Data were analyzed using pCLAMP9 (Molecular Devices), GraphPad Prism (GraphPad), and Origin 2017 (OriginLab) software. Current-voltage relationships were generated in 5- or 10-mV voltage steps from a holding potential of −100 mV. Steady-state inactivation curves were constructed by plotting normalized peak currents (peak test pulse current/peak pre-pulse current) as a function of inactivating potentials. Kinetics of activation and inactivation were determined by fitting mono-exponential functions over the growing or decaying phases of each current trace.
Statistical analyses
Results are presented as mean ± S.E., and n = the number of cells. Statistical analysis was first performed with the Student's t test or with one-way ANOVA combined with a Tukey post-test for multiple comparisons (*, p < 0.05; **, p < 0.01; ***, p < 0.001). For the electrophysiology data sets as paired data of treatment values compared with control values at time 0 and after 20 min of CaMB peptide dialyses in Figs. 7 and 8, we also used a non-parametric Wilcoxon matched-pairs signed rank test. In Fig. 9, there were three sets of data to compare between wild type, CaM overexpression, and CaM1234 overexpression, so we used a Kruskal-Wallis test here followed by a Dunn's multiple comparison test or a Mann-Whitney test. The statistical analyses using parametric and non-parametric analyses produced similar results.
Co-immunoprecipitation experiments
Forty eight hours after transfection, cells cultured in 35-mm dishes were lysed on ice for 20 min with Nonidet P-40 buffer containing 50 mm Tris-HCl, pH 7.4, 125 mm NaCl, 25 mm MgCl2, 33.3 μm CaCl2, 5% glycerol, 1% Nonidet P-40. Cell lysates were centrifuged at 10,000 × g for 30 min at 4 °C. Clarified cell lysates were then incubated with anti-GFP-conjugated magnetic beads (Thermo Fisher Scientific) for 4 h at 4 °C. The beads were washed four times with lysis buffer and resuspended in 2× loading buffer. The immunoprecipitates were then analyzed by SDS-PAGE and Western blotting.
Western blotting
Samples were loaded on SDS-PAGE. Proteins were then transferred onto nitrocellulose membranes and blocked with 5% powdered nonfat milk in PBS-T (Tween 0.1%). Primary antibody was incubated overnight at 4 °C in 5% powdered nonfat milk in PBS-T (Tween 0.1%). After three washes in 5% powdered nonfat milk in PBS-T, the secondary HRP-coupled antibodies were incubated for 1 h at room temperature in the same buffer. The signal was detected using the Super Signal West Pico chemiluminescent system (Thermo Fisher Scientific).
Antibodies and reagents
The antibodies used in the study were as follows: rat anti-HA (3F10, Roche Applied Science; 1:1000) and secondary goat anti-rat antibody coupled to horseradish peroxidase (112-036-072 Jackson ImmunoResearch; 1:5000); rabbit anti-GFP (TP401, Torrey Pines Biolabs; 1:5000); and secondary donkey anti-rabbit coupled to horseradish peroxidase (NA934 GE Healthcare; 1:10,000).
Author contributions
The CaV3.1-baculovirus construct was made and Sf9 cells harvested in the laboratory of A. C. D., which were used for the single particular cryo-EM carried out in the laboratory of A. K. The co-immunoprecipitation experiments were carried out by A. M. in his laboratory. The gel-mobility-shift assays, circular-dichroism spectroscopy, and isothermal-titration calorimetry experiments were carried out by V. T. in the laboratory of J. G. G. Most electrophysiology experiments of mammalian Cav3.2 channels were conducted by J. C. in his laboratory, based on preliminary discoveries by the electrophysiology work of R. F. S. and W. G. in the laboratory of J. D. S. Cav3.2 channel mutants were provided by E. P. R., who was especially helpful in the design of electrophysiology experiments involving the gating brake. The NMR experiments were conducted by M. P. in the laboratory of T. D. Vectors for CaM1234 overexpression and shRNA knockdown of Calm1,2,3 genes were provided by Z. P. P. All authors contributed to the writing of the manuscript. Team management and the bulk of the writing of the manuscript was conducted by J. D. S.
Acknowledgments
We thank Conor P. Walsh from Dr. Kitmitto's laboratory for contributing to the electron microscopy work and to Philippe Lory, the research group leader of J. C. and A. M., for his continuous support.
This work was supported by a Heart and Stroke Foundation of Canada grant-in-aid (to J. D. S.), Natural Sciences and Engineering Research Council of Canada Discovery grants (to J. D. S., T. D., and J. G. G.), British Heart Foundation (to A. K.), LabEx “Ion Channel Science and Therapeutics” and ANR-10-BLAN-1601 (to J. C. and A. M.), National Institutes of Health Grant NS067456 (to E. P.-R.), an NSERC Canada Graduate Scholarship (to V. T.), and research incentive funds from University of Waterloo. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
R. F. Stephens, W. Guan, O. Mourad, A. Mehta, and J. D. Spafford, manuscript in revision.
W. Guan, R. F. Stephens, and J. D. Spafford, manuscript in revision.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- CaM
- calmodulin
- aa
- amino acid
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ITC
- isothermal titration calorimetry
- CaMB
- CaM binding
- ANOVA
- analysis of variance
- TFE
- trifluoroethanol
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- NSCaTE
- N-terminal spatial Ca2+-transforming element
- RFP
- red fluorescent protein
- HSQC
- heteronuclear single-quantum coherence
- dansyl
- 5-(dimethylamino)naphthalene-1-sulfonyl chloride
- CARA
- computer-aided resonance assignment
- ESI-MS
- electrospray ionization-mass spectrometry
- EGFP
- enhanced GFP.
References
- 1. Ben-Johny M., Dick I. E., Sang L., Limpitikul W. B., Kang P. W., Niu J., Banerjee R., Yang W., Babich J. S., Issa J. B., Lee S. R., Namkung H., Li J., Zhang M., Yang P. S., et al. (2015) Towards a unified theory of calmodulin regulation (calmodulation) of voltage-gated calcium and sodium channels. Curr. Mol. Pharmacol. 8, 188–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ben-Johny M., Yang P. S., Niu J., Yang W., Joshi-Mukherjee R., and Yue D. T. (2014) Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell 157, 1657–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Budde T., Meuth S., and Pape H. C. (2002) Calcium-dependent inactivation of neuronal calcium channels. Nat. Rev. Neurosci. 3, 873–883 [DOI] [PubMed] [Google Scholar]
- 4. Halling D. B., Aracena-Parks P., and Hamilton S. L. (2006) Regulation of voltage-gated Ca2+ channels by calmodulin. Sci. STKE 2006, er1. [DOI] [PubMed] [Google Scholar]
- 5. Minor D. L. Jr., and Findeisen F. (2010) Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels 4, 459–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarhan M. F., Tung C. C., Van Petegem F., and Ahern C. A. (2012) Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. U.S.A. 109, 3558–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Leon M., Wang Y., Jones L., Perez-Reyes E., Wei X., Soong T. W., Snutch T. P., and Yue D. T. (1995) Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science 270, 1502–1506 [DOI] [PubMed] [Google Scholar]
- 8. Lee A., Scheuer T., and Catterall W. A. (2000) Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J. Neurosci. 20, 6830–6838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taiakina V., Boone A. N., Fux J., Senatore A., Weber-Adrian D., Guillemette J. G., and Spafford J. D. (2013) The calmodulin-binding, short linear motif, NSCaTE is conserved in L-type channel ancestors of vertebrate Cav1.2 and Cav1.3 channels. PLoS One 8, e61765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kink J. A., Maley M. E., Preston R. R., Ling K. Y., Wallen-Friedman M. A., Saimi Y., and Kung C. (1990) Mutations in paramecium calmodulin indicate functional differences between the C-terminal and N-terminal lobes in vivo. Cell 62, 165–174 [DOI] [PubMed] [Google Scholar]
- 11. Brehm P., and Eckert R. (1978) Calcium entry leads to inactivation of calcium channel in paramecium. Science 202, 1203–1206 [DOI] [PubMed] [Google Scholar]
- 12. Brehm P., Dunlap K., and Eckert R. (1978) Calcium-dependent repolarization in paramecium. J. Physiol. 274, 639–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brehm P., and Eckert R. (1978) An electrophysiological study of the regulation of ciliary beating frequency in paramecium. J. Physiol. 283, 557–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jeziorski M. C., Greenberg R. M., Clark K. S., and Anderson P. A. (1998) Cloning and functional expression of a voltage-gated calcium channel α1 subunit from jellyfish. J. Biol. Chem. 273, 22792–22799 [DOI] [PubMed] [Google Scholar]
- 15. Ben-Johny M., and Yue D. T. (2014) Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol. 143, 679–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dick I. E., Tadross M. R., Liang H., Tay L. H., Yang W., and Yue D. T. (2008) A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Z., and Vogel H. J. (2012) Structural basis for the regulation of L-type voltage-gated calcium channels: interactions between the N-terminal cytoplasmic domain and Ca2+-calmodulin. Front. Mol. Neurosci. 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liebeskind B. J., Hillis D. M., and Zakon H. H. (2011) Evolution of sodium channels predates the origin of nervous systems in animals. Proc. Natl. Acad. Sci. U.S.A. 108, 9154–9159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson P. A., Holman M. A., and Greenberg R. M. (1993) Deduced amino acid sequence of a putative sodium channel from the scyphozoan jellyfish Cyanea capillata. Proc. Natl. Acad. Sci. U.S.A. 90, 7419–7423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spafford J. D., Spencer A. N., and Gallin W. J. (1998) A putative voltage-gated sodium channel α subunit (PpSCN1) from the hydrozoan jellyfish, Polyorchis penicillatus: structural comparisons and evolutionary considerations. Biochem. Biophys. Res. Commun. 244, 772–780 [DOI] [PubMed] [Google Scholar]
- 21. Liang H., DeMaria C. D., Erickson M. G., Mori M. X., Alseikhan B. A., and Yue D. T. (2003) Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron 39, 951–960 [DOI] [PubMed] [Google Scholar]
- 22. Perez-Reyes E. (2010) Characterization of the gating brake in the I–II loop of CaV3 T-type calcium channels. Channels 4, 453–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vitko I., Bidaud I., Arias J. M., Mezghrani A., Lory P., and Perez-Reyes E. (2007) The I–II loop controls plasma membrane expression and gating of Ca(v) 3.2 T-type Ca2+ channels: a paradigm for childhood absence epilepsy mutations. J. Neurosci. 27, 322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vitko I., Chen Y., Arias J. M., Shen Y., Wu X. R., and Perez-Reyes E. (2005) Functional characterization and neuronal modeling of the effects of childhood absence epilepsy variants of CACNA1H, a T-type calcium channel. J. Neurosci. 25, 4844–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arias-Olguín I. I., Vitko I., Fortuna M., Baumgart J. P., Sokolova S., Shumilin I. A., Van Deusen A., Soriano-García M., Gomora J. C., and Perez-Reyes E. (2008) Characterization of the gating brake in the I–II loop of Ca(v) 3.2 T-type Ca2+ channels. J. Biol. Chem. 283, 8136–8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baumgart J. P., Vitko I., Bidaud I., Kondratskyi A., Lory P., and Perez-Reyes E. (2008) I–II loop structural determinants in the gating and surface expression of low voltage-activated calcium channels. PLoS One 3, e2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Y., Lu J., Pan H., Zhang Y., Wu H., Xu K., Liu X., Jiang Y., Bao X., Yao Z., Ding K., Lo W. H., Qiang B., Chan P., Shen Y., and Wu X. (2003) Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann. Neurol. 54, 239–243 [DOI] [PubMed] [Google Scholar]
- 28. Liang J., Zhang Y., Chen Y., Wang J., Pan H., Wu H., Xu K., Liu X., Jiang Y., Shen Y., and Wu X. (2007) Common polymorphisms in the CACNA1H gene associated with childhood absence epilepsy in Chinese Han population. Ann. Hum. Genet. 71, 325–335 [DOI] [PubMed] [Google Scholar]
- 29. Mesirca P., Torrente A. G., and Mangoni M. E. (2014) T-type channels in the sino-atrial and atrioventricular pacemaker mechanism. Pflugers Arch. 466, 791–799 [DOI] [PubMed] [Google Scholar]
- 30. Kim D., Song I., Keum S., Lee T., Jeong M. J., Kim S. S., McEnery M. W., and Shin H. S. (2001) Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking α(1G) T-type Ca2+ channels. Neuron 31, 35–45 [DOI] [PubMed] [Google Scholar]
- 31. Cheong E., and Shin H. S. (2013) T-type Ca2+ channels in normal and abnormal brain functions. Physiol. Rev. 93, 961–992 [DOI] [PubMed] [Google Scholar]
- 32. Carbone E., Calorio C., and Vandael D. H. (2014) T-type channel-mediated neurotransmitter release. Pflugers Arch. 466, 677–687 [DOI] [PubMed] [Google Scholar]
- 33. Kuo I. Y., Howitt L., Sandow S. L., McFarlane A., Hansen P. B., and Hill C. E. (2014) Role of T-type channels in vasomotor function: team player or chameleon? Pflugers Arch. 466, 767–779 [DOI] [PubMed] [Google Scholar]
- 34. Dreyfus F. M., Tscherter A., Errington A. C., Renger J. J., Shin H. S., Uebele V. N., Crunelli V., Lambert R. C., and Leresche N. (2010) Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J. Neurosci. 30, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gray L. S., Schiff D., and Macdonald T. L. (2013) A model for the regulation of T-type Ca2+ channels in proliferation: roles in stem cells and cancer. Expert Rev. Anticancer Ther. 13, 589–595 [DOI] [PubMed] [Google Scholar]
- 36. Cribbs L. (2010) T-type calcium channel expression and function in the diseased heart. Channels 4, 447–452 [DOI] [PubMed] [Google Scholar]
- 37. Senatore A., Guan W., and Spafford J. D. (2014) Cav3 T-type channels: regulators for gating, membrane expression, and cation selectivity. Pflugers Arch. 466, 645–660 [DOI] [PubMed] [Google Scholar]
- 38. Walsh C. P., Davies A., Butcher A. J., Dolphin A. C., and Kitmitto A. (2009) Three-dimensional structure of CaV3.1: comparison with the cardiac L-type voltage-gated calcium channel monomer architecture. J. Biol. Chem. 284, 22310–22321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yap K. L., Kim J., Truong K., Sherman M., Yuan T., and Ikura M. (2000) Calmodulin target database. J. Struct. Funct. Genomics 1, 8–14 [DOI] [PubMed] [Google Scholar]
- 40. Smith C. L., Abdallah S., Wong Y. Y., Le P., Harracksingh A. N., Artinian L., Tamvacakis A. N., Rehder V., Reese T. S., and Senatore A. (2017) Evolutionary insights into T-type Ca2+ channel structure, function, and ion selectivity from the Trichoplax adhaerens homologue. J. Gen. Physiol. 149, 483–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jasanoff A., and Fersht A. R. (1994) Quantitative determination of helical propensities from trifluoroethanol titration curves. Biochemistry 33, 2129–2135 [DOI] [PubMed] [Google Scholar]
- 42. Sönnichsen F. D., Van Eyk J. E., Hodges R. S., and Sykes B. D. (1992) Effect of trifluoroethanol on protein secondary structure: an NMR and CD study using a synthetic actin peptide. Biochemistry 31, 8790–8798 [DOI] [PubMed] [Google Scholar]
- 43. Gagné S. M., Tsuda S., Li M. X., Chandra M., Smillie L. B., and Sykes B. D. (1994) Quantification of the calcium-induced secondary structural changes in the regulatory domain of troponin-C. Protein Sci. 3, 1961–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xia X. M., Fakler B., Rivard A., Wayman G., Johnson-Pais T., Keen J. E., Ishii T., Hirschberg B., Bond C. T., Lutsenko S., Maylie J., and Adelman J. P. (1998) Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395, 503–507 [DOI] [PubMed] [Google Scholar]
- 45. Arias J. M., Murbartián J., Vitko I., Lee J. H., and Perez-Reyes E. (2005) Transfer of β subunit regulation from high to low voltage-gated Ca2+ channels. FEBS Lett. 579, 3907–3912 [DOI] [PubMed] [Google Scholar]
- 46. Pang Z. P., Xu W., Cao P., and Südhof T. C. (2010) Calmodulin suppresses synaptotagmin-2 transcription in cortical neurons. J. Biol. Chem. 285, 33930–33939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pang Z. P., Cao P., Xu W., and Südhof T. C. (2010) Calmodulin controls synaptic strength via presynaptic activation of calmodulin kinase II. J. Neurosci. 30, 4132–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sun T., Wu X. S., Xu J., McNeil B. D., Pang Z. P., Yang W., Bai L., Qadri S., Molkentin J. D., Yue D. T., and Wu L. G. (2010) The role of calcium/calmodulin-activated calcineurin in rapid and slow endocytosis at central synapses. J. Neurosci. 30, 11838–11847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Perez-Reyes E. (2003) Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 83, 117–161 [DOI] [PubMed] [Google Scholar]
- 50. Lee N., Jeong S., Kim K. C., Kim J. A., Park J. Y., Kang H. W., Perez-Reyes E., and Lee J. H. (2017) Ca2+ regulation of Cav3.3 T-type Ca2+ channel is mediated by calmodulin. Mol. Pharmacol. 92, 347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kovalevskaya N. V., van de Waterbeemd M., Bokhovchuk F. M., Bate N., Bindels R. J., Hoenderop J. G., and Vuister G. W. (2013) Structural analysis of calmodulin binding to ion channels demonstrates the role of its plasticity in regulation. Pflugers Arch. 465, 1507–1519 [DOI] [PubMed] [Google Scholar]
- 52. Van Petegem F., Chatelain F. C., and Minor D. L. Jr. (2005) Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat. Struct. Mol. Biol. 12, 1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim E. Y., Rumpf C. H., Fujiwara Y., Cooley E. S., Van Petegem F., and Minor D. L. Jr. (2008) Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure 16, 1455–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sarhan M. F., Van Petegem F., and Ahern C. A. (2009) A double tyrosine motif in the cardiac sodium channel domain III–IV linker couples calcium-dependent calmodulin binding to inactivation gating. J. Biol. Chem. 284, 33265–33274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Erickson M. G., Alseikhan B. A., Peterson B. Z., and Yue D. T. (2001) Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron 31, 973–985 [DOI] [PubMed] [Google Scholar]
- 56. Erickson M. G., Liang H., Mori M. X., and Yue D. T. (2003) FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron 39, 97–107 [DOI] [PubMed] [Google Scholar]
- 57. Pitt G. S., Zühlke R. D., Hudmon A., Schulman H., Reuter H., and Tsien R. W. (2001) Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J. Biol. Chem. 276, 30794–30802 [DOI] [PubMed] [Google Scholar]
- 58. Peterson B. Z., DeMaria C. D., Adelman J. P., and Yue D. T. (1999) Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22, 549–558 [DOI] [PubMed] [Google Scholar]
- 59. Zühlke R. D., Pitt G. S., Deisseroth K., Tsien R. W., and Reuter H. (1999) Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399, 159–162 [DOI] [PubMed] [Google Scholar]
- 60. DeMaria C. D., Soong T. W., Alseikhan B. A., Alvania R. S., and Yue D. T. (2001) Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411, 484–489 [DOI] [PubMed] [Google Scholar]
- 61. Chemin J., Monteil A., Perez-Reyes E., Bourinet E., Nargeot J., and Lory P. (2002) Specific contribution of human T-type calcium channel isotypes (α(1G), α(1H) and α(1I)) to neuronal excitability. J. Physiol. 540, 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tan G. M., Yu D., Wang J., and Soong T. W. (2012) Alternative splicing at C terminus of Ca(V)1.4 calcium channel modulates calcium-dependent inactivation, activation potential, and current density. J. Biol. Chem. 287, 832–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tan B. Z., Jiang F., Tan M. Y., Yu D., Huang H., Shen Y., and Soong T. W. (2011) Functional characterization of alternative splicing in the C terminus of L-type CaV1.3 channels. J. Biol. Chem. 286, 42725–42735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Singh A., Gebhart M., Fritsch R., Sinnegger-Brauns M. J., Poggiani C., Hoda J. C., Engel J., Romanin C., Striessnig J., and Koschak A. (2008) Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 283, 20733–20744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee A., Zhou H., Scheuer T., and Catterall W. A. (2003) Molecular determinants of Ca2+/calmodulin-dependent regulation of Ca(v) 2.1 channels. Proc. Natl. Acad. Sci. U.S.A. 100, 16059–16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Potet F., Chagot B., Anghelescu M., Viswanathan P. C., Stepanovic S. Z., Kupershmidt S., Chazin W. J., and Balser J. R. (2009) Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J. Biol. Chem. 284, 8846–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Romoser V. A., Hinkle P. M., and Persechini A. (1997) Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J. Biol. Chem. 272, 13270–13274 [DOI] [PubMed] [Google Scholar]
- 68. Persechini A., and Cronk B. (1999) The relationship between the free concentrations of Ca2+ and Ca2+-calmodulin in intact cells. J. Biol. Chem. 274, 6827–6830 [DOI] [PubMed] [Google Scholar]
- 69. Yang P. S., Johny M. B., and Yue D. T. (2014) Allostery in Ca2+ channel modulation by calcium-binding proteins. Nat. Chem. Biol. 10, 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang C., Chung B. C., Yan H., Lee S. Y., and Pitt G. S. (2012) Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure 20, 1167–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang C., Chung B. C., Yan H., Wang H. G., Lee S. Y., and Pitt G. S. (2014) Structural analyses of Ca2+/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat. Commun. 5, 4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Perez-Reyes E., and Lee J. H. (2014) Ins and outs of T-channel structure function. Pflugers Arch. 466, 627–633 [DOI] [PubMed] [Google Scholar]
- 73. Bae J., Suh E. J., and Lee C. (2010) Interaction of T-type calcium channel Ca(V)3.3 with the β-subunit. Mol. Cells 30, 185–191 [DOI] [PubMed] [Google Scholar]
- 74. Senatore A., Guan W., Boone A. N., and Spafford J. D. (2014) T-type channels become highly permeable to sodium ions using an alternative extracellular turret region (S5-P) outside the selectivity filter. J. Biol. Chem. 289, 11952–11969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dubel S. J., Altier C., Chaumont S., Lory P., Bourinet E., and Nargeot J. (2004) Plasma membrane expression of T-type calcium channel α(1) subunits is modulated by high voltage-activated auxiliary subunits. J. Biol. Chem. 279, 29263–29269 [DOI] [PubMed] [Google Scholar]
- 76. Dawson T. F., Boone A. N., Senatore A., Piticaru J., Thiyagalingam S., Jackson D., Davison A., and Spafford J. D. (2014) Gene splicing of an invertebrate αβ subunit (LCavβ) in the N-terminal and HOOK domains and its regulation of LCav1 and LCav2 calcium channels. PloS one 9, e92941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Buraei Z., and Yang J. (2010) The αβ subunit of voltage-gated Ca2+ channels. Physiol. Rev. 90, 1461–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Splawski I., Yoo D. S., Stotz S. C., Cherry A., Clapham D. E., and Keating M. T. (2006) CACNA1H mutations in autism spectrum disorders. J. Biol. Chem. 281, 22085–22091 [DOI] [PubMed] [Google Scholar]
- 79. Stephens R. F., Guan W., Zhorov B. S., and Spafford J. D. (2015) Selectivity filters and cysteine-rich extracellular loops in voltage-gated sodium, calcium, and NALCN channels. Front. Physiol. 6, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Deleted in proof.
- 81. Deleted in proof.
- 82. Senatore A., Boone A. N., and Spafford J. D. (2011) Optimized transfection strategy for expression and electrophysiological recording of recombinant voltage-gated ion channels in HEK-293T cells. J. Vis. Exp. 47, 2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schauer-Vukasinovic V., Deo S. K., and Daunert S. (2002) Purification method for recombinant proteins based on a fusion between the target protein and the C terminus of calmodulin. Anal. Bioanal. Chem. 373, 501–507 [DOI] [PubMed] [Google Scholar]
- 84. Ernst O., and Zor T. (2010) Linearization of the Bradford protein assay. J. Vis. Exp. 38, 1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kincaid R. L., Vaughan M., Osborne J. C. Jr, and Tkachuk V. A. (1982) Ca2+-dependent interaction of 5-dimethylaminonaphthalene-1-sulfonyl-calmodulin with cyclic nucleotide phosphodiesterase, calcineurin, and troponin I. J. Biol. Chem. 257, 10638–10643 [PubMed] [Google Scholar]
- 86. Chen R. F. (1968) Dansyl labeled proteins: determination of extinction coefficiency and number of bound residues with radioactive dansyl chloride. Anal. Biochem. 25, 412–416 [DOI] [PubMed] [Google Scholar]
- 87. Turnbull W. B., and Daranas A. H. (2003) On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J. Am. Chem. Soc. 125, 14859–14866 [DOI] [PubMed] [Google Scholar]
- 88. Freiburger L. A., Auclair K., and Mittermaier A. K. (2009) Elucidating protein binding mechanisms by variable-c ITC. Chembiochem 10, 2871–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Broecker J., Vargas C., and Keller S. (2011) Revisiting the optimal c value for isothermal titration calorimetry. Anal. Biochem. 418, 307–309 [DOI] [PubMed] [Google Scholar]
- 90. Piazza M., Taiakina V., Guillemette S. R., Guillemette J. G., and Dieckmann T. (2014) Solution structure of calmodulin bound to the target peptide of endothelial nitric oxide synthase phosphorylated at Thr495. Biochemistry 53, 1241–1249 [DOI] [PubMed] [Google Scholar]
- 91. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 92. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 393–400 [DOI] [PubMed] [Google Scholar]
- 93. Whitmore L., and Wallace B. A. (2004) The Peptaibol Database: a database for sequences and structures of naturally occurring peptaibols. Nucleic Acids Res. 89, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]