

Abstract
In photosynthetic water oxidation, two water molecules are converted into one oxygen molecule and four protons at the Mn4CaO5 cluster in photosystem II (PSII) via the S-state cycle. Efficient proton exit from the catalytic site to the lumen is essential for this process. However, the exit pathways of individual protons through the PSII proteins remain to be identified. In this study, we examined the involvement of a hydrogen-bond network near the redox-active tyrosine YZ in proton transfer during the S-state cycle. We focused on spectroscopic analyses of a site-directed variant of D1-Asn-298, a residue involved in a hydrogen-bond network near YZ. We found that the D1-N298A mutant of Synechocystis sp. PCC 6803 exhibits an O2 evolution activity of ∼10% of the wild-type. D1-N298A and the wild-type D1 had very similar features of thermoluminescence glow curves and of an FTIR difference spectrum upon YZ oxidation, suggesting that the hydrogen-bonded structure of YZ and electron transfer from the Mn4CaO5 cluster to YZ were little affected by substitution. In the D1-N298A mutant, however, the flash-number dependence of delayed luminescence showed a monotonic increase without oscillation, and FTIR difference spectra of the S-state cycle indicated partial and significant inhibition of the S2 → S3 and S3 → S0 transitions, respectively. These results suggest that the D1-N298A substitution inhibits the proton transfer processes in the S2 → S3 and S3 → S0 transitions. This in turn indicates that the hydrogen-bond network near YZ can be functional as a proton transfer pathway during photosynthetic water oxidation.
Keywords: cyanobacteria, Fourier transform IR (FTIR), photosynthesis, photosystem II, site-directed mutagenesis, D1-Asn298, YZ, proton trasfer, thermoluminescence, water oxidation
Introduction
Plants and cyanobacteria utilize water as an ultimate electron donor to reduce carbon dioxide in the synthesis of sugars (1). This reaction of water oxidation is performed in photosystem II (PSII)3 protein complexes, which are embedded in thylakoid membranes. Water oxidation produces protons and molecular oxygen (2–9); protons are released into the thylakoid lumen, making a proton gradient across the membrane, and are used to synthesize ATP, whereas molecular oxygen is liberated to the air to be the major oxygen source of the atmosphere.
In PSII, photochemical reactions start with light-induced charge separation between the primary donor chlorophyll (Chl), P680, and the pheophytin electron acceptor, Pheo (10, 11). An electron is transferred from Pheo to the primary quinone electron acceptor, QA, and then the secondary quinone electron acceptor, QB (12). On the electron donor side, the P680 cation abstracts an electron from the redox-active tyrosine, YZ (D1-Tyr-161), which immediately becomes a neutral radical (YZ•) by shifting a proton to the neighboring D1-His-190 through a strong hydrogen bond, forming a protonated His cation (13–16). YZ• then oxidizes the water-oxidizing center (WOC), the catalytic site of water oxidation, which consists of a Mn4CaO5 cluster as an inorganic core, its amino acid (D1-Asp-170, D1-Glu-189, D1-His-332, D1-Glu-333, D1-Asp-342, D1-Ala-344, and CP43-Glu-354) and water (W1–W4) ligands, two Cl ions (Cl-1 and Cl-2), a hydrogen-bonded network formed by nearby water molecules and amino acid residues (Fig. 1A) (17–21). The water-oxidation reaction proceeds by a light-driven cycle of five intermediates, called Si-state (i = 0–4; with a larger i value representing a higher oxidation state), in which the one-electron-oxidized S1-state is the most stable in the dark (Fig. 1B) (22–23). Each S-state advances to the next state upon one-electron oxidation, except that the S4-state, which is the most oxidized but unstable intermediate, immediately relaxes to the S0-state, by releasing an O2 molecule (2–9). During this S-state cycle, four protons are released to the lumen with a stoichiometry of 1:0:1:2 for the S0 → S1:S1 → S2:S2 → S3:S3 → S0 transitions (24–26). Because no proton is released in the S1→ S2 transition, an excessive positive charge is accumulated on the Mn4CaO5 cluster in the S2- and S3-states.
Figure 1.

A, hydrogen-bond network around the Mn4CaO5 cluster and putative proton transfer pathways. The X-ray free electron laser structure of Thermosynechococcus vulcanus at 1.95-Å resolution (PDB code 4UB6 (18)) was used to draw the picture. Mn, purple; Ca, orange; Cl, green; O, red; N, blue. Oxygen atoms of water molecules are colored cyan. The numbering of the Mn and oxygen atoms in the Mn4CaO5 cluster and its water ligands follows the numbering in Umena et al. (17). D1-Asn-298, which was replaced with Ala in this work, is circled with a red dotted line. Magenta arrows represent putative proton pathways, which were designated Cl path, O4 path, YZ-O1 path, and YZ-Asn-298 path in this work. Hydrogen bonds are shown with green dotted lines. Amino acid residues with labels are on the D1 subunit except for Lys-317 and Glu-312 on the D2 subunit, and Glu-354 and Ala-411 on the CP43 subunit. B, the S-state cycle of water oxidation.
Efficient proton exit from the catalytic site is essential in the mechanism of water oxidation. However, it remains unclear how the four protons are transferred from WOC to the lumen through the PSII proteins during the S-state cycle. The X-ray structures of PSII complexes (17, 18, 27, 28) and theoretical calculations based on them have shown that there are several channels suitable for proton transfer around the Mn4CaO5 cluster leading to the lumen (Fig. 1A) (29–39). These channels involve a number of water molecules forming hydrogen-bond networks, as revealed by the recent high-resolution X-ray structures that resolved water oxygen atoms (17–21). Near the entrance of each channel, either D1-Asp-61 or YZ is located, and a water cluster is formed between these two residues including water ligands to Mn4 (W1 and W2) and Ca2+ (W3 and W4).
The channel from D1-Asp-61 to PsbO through the Cl-1 site and the D1-Glu-65/D2-Glu-312/D1-Arg-334 triad has been most extensively studied and proposed to function as a pathway for protons or substrate water (designated “Cl path” in the present report; Fig. 1A) (29–37, 39). Theoretical calculations suggested that D1-Asp-61 plays a crucial role in proton transfer (40, 41), and that the Cl− ion has a function to regulate proton transfer through this pathway (34). D1-Asp-61 is also located near the entrance of a water chain starting at O4 and leading to PsbU (designated “O4 path”; Fig. 1A), which has been proposed to function as a proton exit pathway especially in the S0 → S1 transition (38, 39). The recent X-ray free electron laser study for the S3-state showed the displacement of a water molecule near O4, suggesting the involvement of water molecules near O4 in proton transfer (21). The hydrogen-bond network near YZ has also been suggested to be a proton pathway (16, 17, 42, 43). YZ form a hydrogen-bonded triad with D1-His-190 and D1-Asn-298, which is connected with a hydrogen-bond network leading to PsbV through D1-Asn-322 (designated “YZ-Asn-298 path”; Fig. 1A). Molecular dynamics (MD) simulations (36, 39) suggested a tight hydrogen-bond network of this pathway, which is advantageous to proton transfer. A large water cluster interacting the YZ/D1-His-190/D1-Asn-298 triad also forms another hydrogen-bond network through O1 and D1-Glu-329 leading to PsbV (designated “YZ-O1 path”). MD simulation showed that this channel contains a number of mobile water molecules (33, 39). It is noted that the above key amino acid residues in the hydrogen-bond networks in cyanobacterial PSII (Fig. 1A) are all conserved in higher plants whose PSII-LHCII structures were recently resolved by cryo-electron microscopy (44, 45).
The finding of several channels around the Mn4CaO5 cluster raises a question, which proton pathways are used for individual protons released in the S-state transitions? The most direct experimental method to answer this question is examining the effects of site-directed mutations, by which amino acid residues on putative proton channels are altered to different amino acids, on the reactions of individual S-state transitions. Many mutants of amino acid residues in putative channels have been investigated so far (46–60). Most of them are located on the Cl path; mutations of D1-Asp-61 to Ala and Asn significantly retarded the kinetic rate of O2 release in the S3 → S0 transition (47, 53, 58) and changed the property of a nearby water network (57). Mutations of D2-Lys-317 and D1-Asn-181, which are ligands to Cl-1, also retarded the O2 kinetics or decreased the efficiency of the S3 → S0 transition (54, 55, 59). In addition, mutants of any of the D1-Glu-65/D2-Glu-312/D1-Arg-334 triad substantially inhibited the S3 → S0 transition (49, 52). These data suggest that the Cl path is involved in proton or water transfer at least in the S3 → S0 transition, although a caution is necessary in the interpretation of results for charged residues, whose mutations could affect the redox potential of the Mn4CaO5 cluster and hence the electron transfer reaction. In contrast, fewer mutagenesis studies have been performed for the channels near the YZ site. Mutation of D1-Gln-165, which is hydrogen-bonded with W4 near YZ, to Glu decreased the efficiency of the S3 → S0 transition (56), whereas mutation of D1-Glu-329 on the YZ-O1 path to Gln little affected the S-state transitions (52). Fourier transform infrared (FTIR) analysis of the S-state cycle using various mutants by Debus and co-workers (52, 54, 56, 57) further showed the presence of an extensive hydrogen-bond network around the Mn4CaO5 cluster.
D1-Asn-298 is located at a crucial position to keep a hydrogen-bond network involving water molecules in the YZ-Asn-298 and YZ-O1 pathways (Fig. 1A). This has also been proposed to be a key residue to determine the hydrogen-bonded structure of YZ and the rate of its proton-coupled electron transfer through the interaction with D1-His-190 (15, 16). Thus, mutation of D1-Asn-298 could affect the proton transfer from the catalytic site as well as electron transfer from the Mn4CaO5 cluster to YZ• during S-state transitions. Yamasato et al. (50) previously found that the D1-N298I mutant obtained by random mutagenesis inhibited electron flow from water to the electron acceptor side. More recently, Kuroda et al. (60) generated 19 site-directed mutants of D1-Asn-298 using Chlamydomonas reinhardtii, and showed that only seven mutants (D1-N298G, -A, -C, -M, -S, -Q, and -H) have O2 evolution activity but with significantly low rates. However, detailed properties of the mutants of D1-Asn-298, such as specific inhibition of either electron or proton transfer in individual S-state transitions, have not yet been investigated, and hence no clear answer was obtained about the involvement of pathways near the YZ site in the water oxidation mechanism.
In this work, we investigated whether the hydrogen-bond network near YZ is functional or not in proton transfer during water oxidation, by constructing a D1-N298A mutant using a cyanobacterium Synechocystis sp. PCC 6803, and analyzing this mutant by means of light-induced FTIR difference spectroscopy and detection of thermoluminescence (TL) and delayed luminescence (DL). Because Asn and Ala are both non-charged amino acids and the replacement with a small Ala side chain can avoid the undesirable alterations of main chains, only the perturbation of the hydrogen-bond network is expected to occur without changes in the electrostatic interaction and protein conformations. FTIR difference spectroscopy is a powerful method for detecting the structural perturbations of amino acid residues, water molecules, and the hydrogen-bond network around the Mn4CaO5 cluster, as well as monitoring the inhibition of the S-state transitions (61–70). The results provided solid evidence for the involvement of D1-Asn-298, and hence the hydrogen-bond network near YZ, in proton transfer in the S2 → S3 and S3 → S0 transitions of water oxidation.
Results
In vivo analyses of the D1-N298A mutant
The strains of Synechocystis sp. PCC 6803 used in the present study lacked the psbA1 and psbA3 genes, and hence PsbA2 was expressed as a D1 protein (71). In addition, a histidine tag was attached to the C terminus of the CP47 protein. Thus, the strain having a PsbA2 protein with a native sequence is designated “WT*” hereafter. In contrast, the Asn-298 of PsbA2 was replaced by Ala in the D1-N298A mutant. The D1-N298A mutant grew photoautotrophically with a rate slightly slower than the WT* (Fig. 2A). The O2 evolution activities of WT* and D1-N298A were 600–670 and 60–70 μmol of O2 (mg of Chl)−1 h−1, respectively, in cells, and 2800–3300 and 190–300 μmol of O2 (mg of Chl)−1 h−1, respectively, in isolated PSII complexes. Thus, the O2 evolution activity of the D1-N298A mutant is about 10% of WT* in both cells and PSII core complexes. This reduced activity of the D1-N298A mutant is in good agreement with that of a corresponding mutant of a green alga C. reinhardtii, which showed a 11.5% activity of WT in cells (60).
Figure 2.

In vivo characterization of WT* (black) and D1-N298A (red) cells. A, growth curves of cells. B, TL glow curves in the (a) presence and (b) absence of DCMU representing charge recombination of the S2QA− and S2QB− charged pairs, respectively. C, flush-number dependence of DL induced by 10 successive flashes (1 Hz). DL was measured at 25 °C and the amplitude at 0.8 s after each flash was plotted.
TL glow curves of PSII in the presence and absence of DCMU originate from charge recombination of S2QA− (Q band) and S2QB− (B band), respectively (72). WT* cells showed Q and B bands at about 14 and 34 °C, respectively, and they were virtually unaffected by the D1-N298A mutation (Fig. 2B). In contrast, the flash-number dependence of DL, which was monitored at 0.8 s after each flash, showed significantly different patterns between WT* and D1-N298A cells (Fig. 2C). Whereas WT* cells showed a typical period-four oscillation of DL signals, D1-N298A cells did not show any oscillation and the DL amplitude monotonically increased with a flash number.
FTIR difference spectrum upon YZ oxidation
Light-induced FTIR difference spectra upon YZ oxidation (YZ•/YZ difference spectra) were obtained using manganese-depleted PSII core complexes of WT* and D1-N298A (Fig. 3). Characteristic bands of the YZ•/YZ spectra were virtually identical between the two species. The prominent positive band at 1512 cm−1 has been assigned to the CO stretching vibration of the oxidized YZ• radical, whereas a negative band at 1258–1257 cm−1 has been assigned to the coupled CO stretching/COH bending vibration of neutral YZ (16, 73). A differential signal at 1707/1700 cm−1 and a negative peak at 1113–1110 cm−1 have been attributed to the keto C = O vibration of PD1 in P680 affected by YZ oxidation (71) and the imidazole CN stretch of D1-His-190 hydrogen bonded with YZ (73), respectively. Bands in the 1680–1630 cm−1 region, and a band at 1541 cm−1 can be assigned to amide I and II vibrations, respectively, representing the perturbations of protein backbones. Except for changes in other minor bands and some intensity changes in the amide I and II bands, a notable change by the mutation was observed only in the appearance of a differential signal at 1244/1234 cm−1 in the spectrum of D1-N298A, although the origin of this signal is unknown at the present stage. The absence of significant changes in the characteristic bands upon the D1-N298A mutation indicates that the hydrogen-bonded interaction of YZ was virtually unaffected by this mutation.
Figure 3.
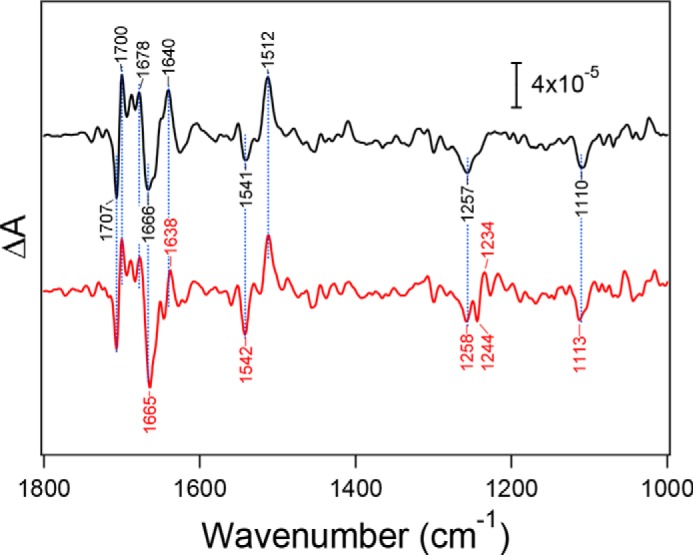
Flash-induced FTIR difference spectra upon YZ oxidation of manganese-depleted PSII core complexes from WT* (black line) and D1-N298A (red line). Spectra were measured at 250 K. The spectrum of D1-N298A was scaled to adjust the intensity of the ferricyanide CN peak at 2116 cm−1 (not shown) to that in the WT* spectrum.
Flash-induced FTIR difference spectra of S-state transitions
Light-induced FTIR difference spectra of the S-state cycle of WOC were measured by applying four flashes on the O2-evolving PSII core complexes from WT* and D1-N298A. The first-, second-, third-, and fourth-flash spectra of the WT* PSII complexes in a moderately hydrated film (Fig. 4A, black lines) represent the structural changes upon the S1 → S2, S2 → S3, S3 → S0, and S0 → S1 transitions, respectively. The spectral features were very similar to those of previously reported spectra (52, 54–57, 74–81). Bands at 1700–1600 cm−1 mainly arise from the amide I vibrations (CO stretching vibrations of backbone amides), representing the perturbations of polypeptide chains near the Mn4CaO5 cluster (78, 82). Characteristic bands at 1450–1350 cm−1 are due to the symmetric COO− stretching vibrations (78, 80–85), whereas the coupled asymmetric COO− stretching bands appear at 1600–1500 cm−1, overlapping the amide II bands (NH bending and CN stretching vibrations of backbone amides) (78, 82). The changes of these COO− bands during the S-state cycle are attributed to the structural changes of carboxylate residues around the Mn4CaO5 cluster (80, 81, 83–85). The obtained spectra showed characteristic features depending on the flash number, especially in the symmetric COO− region around 1400 cm−1, reflecting high efficiencies of the S-state transitions.
Figure 4.
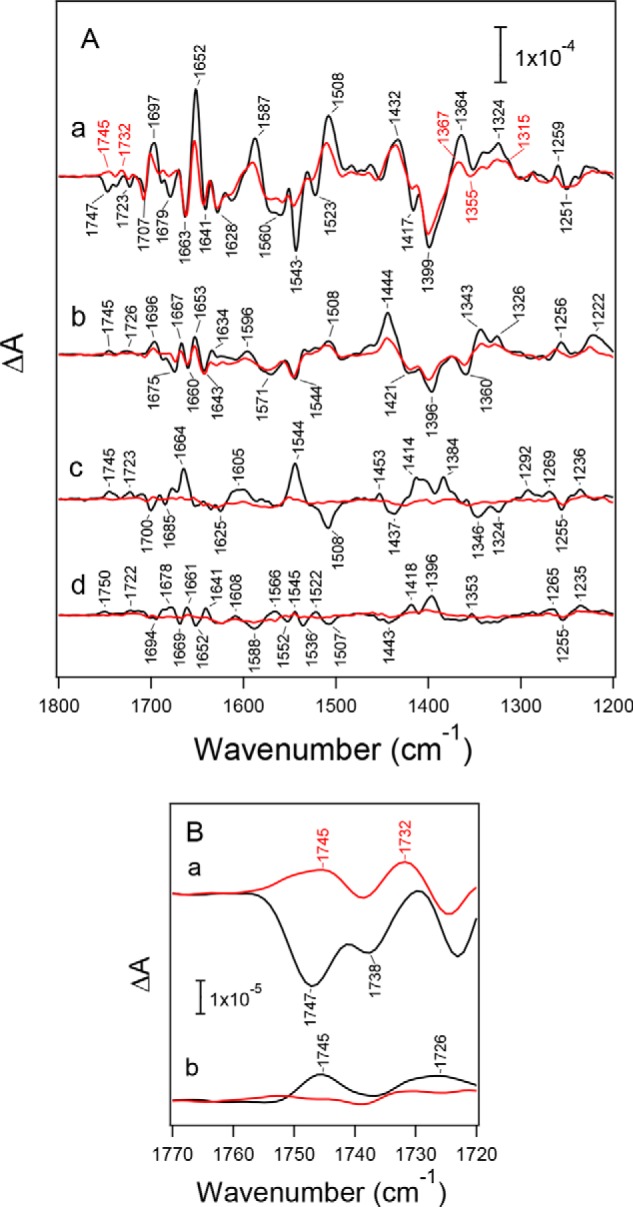
Flash-induced FTIR difference spectra of the S-state cycle of PSII core complexes from WT* (black lines) and D1-N298A (red lines) in moderately hydrated films. A, mid-infrared region (1800–1200 cm−1) of protein vibrations. Difference spectra were measured upon the (a) first, (b) second, (c) third, and (d) fourth flashes. B, expanded view of the C = O stretching region of COOH groups (1770–1720 cm−1) at the (a) first and (b) second flashes. The spectra of D1-N298A were scaled with a factor to adjust the intensity of the ferricyanide CN peak at 2116 cm−1 (not shown) in the first-flash spectrum to that of WT*.
Spectra of the S-state cycle of the D1-N298A mutant at the first to fourth flashes are also shown in Fig. 4A (red lines) superimposing those of WT* (black lines). These spectra of D1-N298A were scaled using a factor to adjust the intensity of the CN band of ferricyanide at 2116 cm−1 at the first-flash (not shown) to that of WT*, representing the same extent of an electron flow in PSII in the S1 → S2 transition. Although the first-flash spectrum due to the S1 → S2 transition showed overall features similar to those of WT*, some differences were observed in several bands (Fig. 4A, a). In the symmetric COO− region, band intensities at 1417(−) and 1364(+) cm−1 were significantly reduced. In addition, bands at 1652(+), 1587(+), 1543(−), 1523(−), and 1508(+) cm−1 in the amide I and the asymmetric COO−/amide II regions decreased their intensities. Furthermore, a negative band at 1747 cm−1, which has been assigned to the C = O stretching vibration of an unidentified COOH group involved in a hydrogen-bond network around the Mn4CaO5 cluster (52, 56), diminished and is replaced by a positive band at 1745 cm−1 (Fig. 4B, a). In contrast, spectral features of the second-flash spectra of D1-N298A were very similar to those of WT* (Fig. 4Ab). However, an overall intensity in the COO− region (1450–1300 cm−1) was ∼60% of WT*. A small positive peak at 1745 cm−1 due to a COOH group, which has a different origin from the negative band at 1747 cm−1 at the first flash (56), also diminished (Fig. 4B, b).
The most striking change in the spectra of the D1-N298A mutant is that the third- and fourth-flash spectra showed very small intensities (Fig. 4A, c and d), indicating that the S3 → S0 transition was significantly inhibited in this mutant. Assuming that the FTIR spectrum of the S3 → S0 transition in the D1-N298A mutant has an identical feature with that of WT*, spectral fitting in the COO− region of the third-flash spectrum of the mutant using the second- and third-flash spectra of WT* (76) provided the ∼20% contribution of the S3 → S0 transition at the third flash. With the ∼60% contribution of the S2 → S3 transition at the second flash, it follows that the efficiency of the S3 → S0 transition in the D1-N298A mutant is ∼30% of WT*. Previously, the D2-K317R mutant also showed similar intensity decreases in the third- and fourth-flash FTIR spectra in a moderately hydrated film (55), but the intensities recovered in a solution sample. It was suggested that partial dehydration reduced the efficiency of the S3 → S0 transition in the K317R mutant (55). To examine the similar dehydration effect, FTIR spectra of the S-state cycle were measured in solution samples of WT* and D1-N298A (Fig. 5). All the spectral changes by the D1-N298A mutation mentioned above were also observed in the solution samples, indicating that these changes are not ascribed to partial dehydration but are attributed to direct effects of the D1-N298A mutation irrespective of sample conditions.
Figure 5.
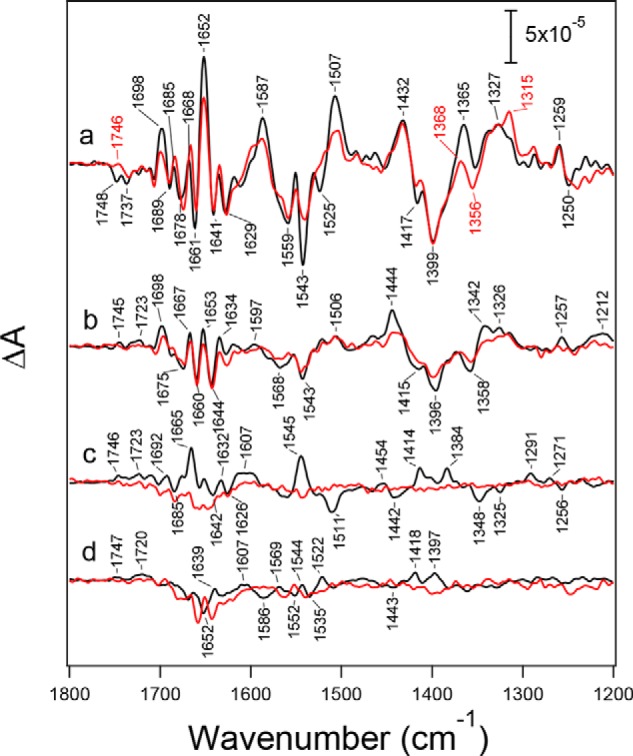
Flash-induced FTIR difference spectra (1800–1200 cm−1) of the S-state cycle of PSII core complexes from WT* (black lines) and D1-N298A (red lines) in solutions measured upon the (a) first, (b) second, (c) third, and (d) fourth flashes. The spectra of D1-N298A were scaled with a factor to adjust the intensity of the ferricyanide CN peak at 2116 cm−1 (not shown) in the first-flash spectrum to that of WT*.
The mutation effects were also observed in water vibrations obtained in hydrated PSII films. In the high frequency region of 3700–3500 cm−1 (Fig. 6A), weakly hydrogen-bonded OH vibrations of water molecules (77, 86, 87) showed a differential signal at 3607/3588 cm−1 and a negative band at 3662 cm−1 in the first-flash spectrum of WT*, analogously to the previous reports (57, 77, 80, 86, 87). In the D1-N298A mutant, the negative peak at 3662 cm−1 diminished, whereas another negative peak at 3588 cm−1 seems unchanged (Fig. 6A, a). At the second flash, a broad negative band at ∼3620 cm−1 in WT* (57, 77) seemed slightly downshifted upon the D1-N298A mutation (Fig. 6A, b).
Figure 6.
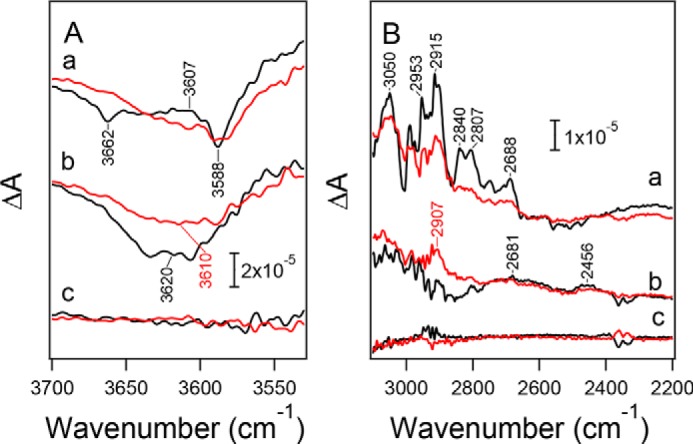
Flash-induced FTIR difference spectra of PSII core complexes from WT* (black lines) and D1-N298A (red lines) in moderately hydrated films in the regions of (A) weakly hydrogen-bonded OH vibrations of water (3700–3530 cm−1) and (B) strongly hydrogen-bonded OH/NH vibrations (3100–2200 cm−1). a, first flash; b, second flash; c, dark-minus-dark difference spectra representing noise levels. The intensities of the spectra of D1-N298A were scaled with the same factor as the corresponding spectra in Fig. 4. Signals around 2350 cm−1 in panel B are artifacts by CO2 absorption.
In the region of vibrations of polarizable protons in strong hydrogen bonds (3100–2200 cm−1) (77, 87), a broad positive feature with several sharp peaks was observed in the first-flash spectrum of WT* (Fig. 6B, a). This band feature was attributed to the OH stretching vibrations of water molecules around the Mn4CaO5 cluster (87), overlapping the NH stretching vibration with Fermi resonance peaks of a His side chain (D1-His-337) hydrogen-bonded with the O3 oxo bridge of the Mn4CaO5 cluster (88). The intensities of these bands were weakened upon the D1-N298A mutation. In the second-flash spectra (Fig. 6B, b), WT* exhibited broad positive features at 2681 and 2456 cm−1 (57, 77), whereas a new positive band appeared at 2907 cm−1 upon the D1-N298A mutation leaving the broad features.
Discussion
D1-Asn-298 is a crucial amino acid residue in retaining a hydrogen-bond network near YZ. It supports a water chain that leads from W4 to PsbV through D1-Asn-191 and D1-Asn-322 (YZ-Asn-298 path), and a water cluster that interacts with O1 of the Mn4CaO5 cluster and D1-Glu-329 leading to PsbV (YZ-O1 path) (Fig. 1A), both of which have been candidates of proton/water pathways (31–33, 35–37, 39). Thus, mutation of D1-Asn-298 to non-hydrogen bonding Ala is expected to significantly perturb the structures of these hydrogen-bond networks by reorganizing nearby water molecules. In addition, D1-Asn-298 is a key amino acid residue in determining the hydrogen-bonded structure of YZ. The amide C = O of the Asn-298 side chain accepts an hydrogen bond from the Nπ-H of D1-His-190, whereas the Nτ of His-190 functions as a hydrogen-bond acceptor of the OH of YZ (15–18). This hydrogen-bonded structure of the YZ/D1-His-190/D1-Asn-298 triad realizes rapid proton-coupled electron transfer of YZ, in which upon oxidation by P680+, the proton of YZ is immediately shifted to D1-His-190 forming a HisH+ cation (15, 16). The D1-N298A mutation hence could also perturb the hydrogen-bonded structure of YZ and its reaction. In contrast, mutation from Asn to Ala, both of which are non-charged amino acids, should have little electrostatic effect on YZ and the Mn4CaO5 cluster to change their redox potentials. Furthermore, a relatively remote location (>10 Å) of D1-Asn-298 from the Mn4CaO5 cluster without any direct interactions even with the first shell ligands makes less likely the possibility that its mutation has a direct effect on the catalytic reaction of water oxidation.
We examined the effect of the D1-N298A mutation on the hydrogen-bonded structure of YZ by comparing the YZ•/YZ FTIR difference spectrum of the manganese-depleted PSII core complex from the D1-N298A mutant with that from WT*. The results showed that they had very similar features (Fig. 3). In particular, the vibrational frequencies of the CO stretching band of YZ• (1512 cm−1) and the CO stretching/COH bending band of neutral YZ (1257 cm−1), which are sensitive to changes in hydrogen-bonding interactions, were identical within 1 cm−1, indicating that the hydrogen-bonded interaction of YZ was little affected by this mutation. It is presumed that the replacement of Asn with a smaller Ala produced enough space for a new water molecule to bind to the Nπ of D1-His-190 instead of the Asn C = O. Thus, the hydrogen-bonding pattern of the YZ/D1-His-190/D1-Asn-298 triad was probably unchanged in the YZ/D1-His-190/H2O triad in the mutant, although the possibility remains that in intact PSII with the Mn4CaO5 cluster, the mutation has some effect on the YZ interaction through the hydrogen-bond network around the Mn4CaO5 cluster.
The reaction process of the S1 → S2 transition is thought to be simple electron transfer from the Mn4CaO5 cluster to YZ•. Hence, if electron transfer from the Mn4CaO5 cluster to YZ• is impaired by the D1-N298A mutation, the efficiency of the S1 → S2 transition should also be affected. The S2/S1 FTIR difference spectra of WT* and the D1-N298A mutant at the first flash (Figs. 4A, a, and 5a) showed similar overall features but with intensity changes and slight frequency shifts in some bands. At the second flash, the spectrum of D1-N298A showed very similar features to the spectrum of WT* representing the S2 → S3 transition (Figs. 4A, b, and 5b), although the mutant spectrum had a smaller overall intensity (∼60% of WT*). The absence of the features of the S2/S1 spectrum in the second-flash spectrum of the mutant (e.g. 1432 cm−1 peak characteristic of the first-flash spectrum was not involved in the second-flash spectrum) indicates that the S1 → S2 efficiency in D1-N298A is as high as that in WT*. In addition, the peak temperatures of the TL glow curves at 14 and 34 °C arising from charge recombination of S2QA− (Q band) and S2QB− (B band), respectively, little changed upon the D1-N298A mutation (Fig. 2B). This indicates that the redox potential of the S1 → S2 transition of the Mn4CaO5 cluster, which should regulate the electron transfer reaction, is virtually unchanged by this mutation. These observations suggest that the D1-N298A mutation little affected the electron transfer from the Mn4CaO5 cluster to YZ• at least during the S1 → S2 transition.
In contrast to unaffected electron transfer, the hydrogen-bond network around the Mn4CaO5 cluster was clearly affected by the D1-N298A mutation, as revealed by the following observations. (i) The symmetric COO− region (1450–1300 cm−1) in the S2/S1 FTIR difference spectra showed characteristic changes, such as intensity decreases of the 1417 and 1364 cm−1 bands by the mutation (Figs. 4A, a, and 5a). These bands were recently assigned to the symmetric COO− vibrations of D1-Asp-342/D1-Asp-61 and D1-Asp-170/D1-Glu-333/D1-Asp-342/D1-Glu-189, respectively, by quantum mechanics/molecular mechanics (QM/MM) calculations (85), suggesting slight perturbations of these carboxylate groups by the mutation. (ii) A negative band at 1747 cm−1 in the S2/S1 spectrum and a positive band at 1745 cm−1 in the S3/S2 spectrum (Fig. 4B), which have been assigned to the CO stretching vibrations of unidentified COOH groups (52, 56), diminished in the spectra of the D1-N298A mutant, indicating the pKa changes of these COOH groups were located on a common hydrogen-bond network involving D1-Asn-298. (iii) The intensities of the amide II bands in the 1590–1500 cm−1 region together with an amide I band at 1652 cm−1 in the S2/S1 spectrum significantly decreased (Figs. 4A, a, and 5a), suggesting the perturbation of the backbone interactions near D1-Asn-298. (iv) A negative band at 3662 cm−1 in the weakly hydrogen-bonded OH region of water (57, 77, 87) in the S2/S1 spectrum diminished and a broader negative band at 3620 cm−1 in the S3/S2 spectrum (57, 77) seemed slightly downshifted (Fig. 6A). (v) The intensity of the feature in the 3000–2600 cm−1 region, which have been assigned to strongly hydrogen-bonded OH vibrations of water molecules (77, 87) overlapping the NH vibration of protonated D1-His-337 (88), in the S2/S1 difference spectrum decreased by the D1-N298A mutation (Fig. 6B, a). In addition, in the similar region in the S3/S2 spectrum, a positive band newly appeared at 2907 cm−1 (Fig. 6B, b). These spectral changes in (iv) and (v) represent some perturbations in the hydrogen-bonded structure of a water network near the Mn4CaO5 cluster by the D1-N298A mutation.
Similar changes in the symmetric COO−, COOH, and amide II regions in FTIR difference spectra have been previously observed in site-directed mutants of D1-Asp-61, D1-Glu-65, D2-Lys-317, D2-Glu-312, and D1-Arg-334 on the Cl path, and those of D1-Gln-165 near YZ and D1-Glu-329 on the YZ-O1 path (Fig. 1A) (52, 54–57). In addition, the similar decrease in the weakly hydrogen-bonded OH band at ∼3662 cm−1 and the broad feature in the 3000–2400 cm−1 region in the S2/S1 difference spectrum was observed in the D1-D61A mutant (57). Indeed, the contribution of W1, which directly interacts with D1-Asp-61, to the bands at ∼3662 and 3000–2400 cm−1 was suggested by the QM/MM calculation (87). Recently, diminishing of the ∼3662 cm−1 band in the S2/S1 spectrum was also observed in PSII in which Ca2+ is substituted with Sr2+ (89). All of the above observations, therefore, indicate that a hydrogen-bond network structurally coupled with the Mn4CaO5 cluster is widely extended across the channels near Cl and YZ involving D1-Asn-298.
The S3/S2 FTIR difference spectrum at the second flash decreased its overall intensity to ∼60% of WT* by the D1-N298A mutation (Figs. 4A, b, and 5b). Furthermore, the FTIR spectra at the third and fourth flashes showed very small intensities (Figs. 4A and 5). These observations were identical between samples in hydrated films (Fig. 4) and solutions (Fig. 5). These FTIR data of isolated PSII core complexes are consistent with the in vivo data of DL measurements using D1-N298A cells, in which the DL intensity monotonically increased with the flash number and reached near maximum by 4 or 5 flashes, in contrast to WT* cells that showed a clear period-four oscillation (Fig. 2C). In addition, the 10-fold decrease in the O2 evolution activity by the D1-N298A mutation was identical between cells and PSII core complexes. These results indicate that the intensity changes in the FTIR spectra by mutation are ascribed to neither the core preparations nor the measurement conditions, but represent the real changes in efficiencies in the S-state transitions. It is thus concluded that the efficiency of the S2 → S3 transition is lowered to ∼60% and the S3 → S0 transition is significantly inhibited upon the D1-N298A mutation.
It is generally accepted that one and two protons are released in the S2 → S3 and S3 → S0 transitions, respectively (24–26). With the absence of the prominent effect on the electron transfer from the Mn4CaO5 cluster to YZ• in the S1 → S2 transition, together with the unlikeliness of direct involvement of D1-Asn-298 in the catalytic reaction at the Mn4CaO5 cluster because of its remote distance, the observed efficiency decreases in the S2 → S3 and S3 → S0 transitions suggest that the proton transfer from the catalytic site are partially and significantly inhibited in the S2 → S3 and S3 → S0 transitions, respectively, in the D1-N298A mutant. Alternative possibilities, however, cannot be excluded at the present stage that electron transfer reactions are affected in the S-state transitions other than S1 → S2 and the mutation provides some indirect effect on the catalytic reaction at the Mn4CaO5 cluster through a hydrogen-bond network. So far, various mutants at residues near the putative proton/water channels, such as D1-D61A/N/E (46–48, 53, 57, 58), D2-K317R/A/E/Q (54, 55), D1-N181A/S (59), D1-D59N (48), D1-E65A (52), D2-E312A (52), D1-R334A/V/E (49, 56), D1-V185N (58), CP43-R357K (51), D1-Q165E (56), and D1-E329Q (52), have been examined to investigate the effects of mutations on the S-state transitions. However, none of these mutants apparently blocked the S2 → S3 transition. Although some changes in the symmetric COO− stretching region of the second-flash FTIR spectra in mutants at the D1-Glu-65/D2-Glu-312/D1-Arg-334 triad were interpreted to be due to a decreased S2 → S3 efficiency (52, 56), electrostatic effects by mutations of charged amino acid residues have not been identified. In addition, although the D1-D61N mutation slowed the S2 → S3 transition by a factor of 2–4, the S1 → S2 transition was also slowed by a similar extent, and hence this retardation was suggested to be caused by the increase in the redox potential of the Mn4CaO5 cluster (47, 53). The partial inhibition of the S2 → S3 efficiency in the D1-N298A mutant observed in our study suggests that when the main proton pathway was blocked by this mutation, another pathway was used for the proton exit in the S2 → S3 transition or this transition originally uses multiple proton pathways. Such flexible proton transfer through different pathways may be realized by a widely extended hydrogen-bond network involving several different pathways around the Mn4CaO5 cluster mentioned above.
Because significant inhibition by the D1-N298A mutation was observed in the S3 → S0 transition, at least one of two protons released in this transition most likely uses the pathway near YZ perturbed by this mutation. The other proton may use the Cl path because mutations at D1-Asp-61, D2-Lys-317, D1-Asn-181, and the D1-Glu-65/D2-Glu-312/D1-Arg-334 triad substantially lowered the efficiency of this transition or significantly retarded the O2 evolution kinetics.
A tight hydrogen-bond network is advantageous to rapid proton transfer using the Grotthuss mechanism rather than a network of mobile water molecules. Previous MD simulation by Ogata et al. (36) showed that water molecules in the YZ-Asn-298 path (“path 1” in Ref. 36) showed only small fluctuations. In addition, Sakashita et al. (39) recently showed by MD simulation of water accessibility that water molecules near D1-Asn-298 are less exchangeable in contrast to exchangeable water molecules in the YZ-O1 path and the Cl path (“O1-water chain” and “Glu-65/Glu-312 channel,” respectively, in Ref. 39), and suggested the involvement of the hydrogen-bond network near D1-Asn-298 in proton transfer. In the D1-N298A mutant, the tight hydrogen-bond network near D1-Asn-298 should be disrupted and may be replaced by a loose water network, which would retard the proton transfer from the catalytic site. Although this mutation may also loosen the structure of a water cluster near O1 in the YZ-O1 path, the effect would be minor because water molecules in this channel are originally mobile as shown in previous MD simulations (33, 35, 36, 39). In contrast to proton transfer, channels involving mobile water molecules are advantageous to water transfer. Although insertion of substrate water to the catalytic site has been suggested to occur in the S2 → S3 and S3 → S0 transitions (40, 42, 43, 66, 76, 90–93), the loosened water network possibly produced by the D1-N298A mutation would not inhibit water insertion. Thus, the N298A-induced inhibition of the S2 → S3 and S3 → S0 transitions observed in the present study is more likely caused by the impairment of proton transfer rather than water transfer. One possible mechanism of proton transfer through the YZ site has been proposed from FTIR and QM/MM analyses (16, 70). QM/MM calculations showed that upon YZ oxidation, the network of nearby water molecules is rearranged and one water molecule moves toward D1-His-190. The proton that was shifted from YZ to the Nτ of D1-His-190 has a large polarizability as revealed by a broad feature at ∼2800 cm−1 in a YZ•/YZ FTIR spectrum (16). It was thus proposed that this proton between YZ• and HisH+ may have a chance to hop to the nearby water, probably using NH bending vibration, followed by immediate proton transfer through a water chain supported by D1-Asn-298. An alternative proposal was proton transfer from W4, a ligand to Ca2+, or a nearby water to the water chain near D1-Asn-298. The transferred proton from D1-His-190 or W4 will be immediately replenished by a proton from a substrate water through a water network between YZ and Mn4 (Fig. 1A) using coupled OH stretching vibrations extended to several water molecules (87). Note that this proton transfer mechanism is different from the previous so-called hydrogen abstraction model (94), in which the proton of YZ is first released to the bulk upon its oxidization. The driving force of the above proton transfer process is charge repulsion between the protonated cation of D1-His-190 and the excess positive charge on the Mn4CaO5 cluster in the S2 and S3 states. Also, proton release needs to take place before electron transfer in the S2 → S3 and S3 → S0 transitions to decrease the redox potential of the Mn4CaO5 cluster (42). Thus, this proton transfer mechanism through the YZ site can be functional in the S2 → S3 transition and for the first proton in the S3 → S0 transition. Indeed, proton transfer as a lag phase (∼30 μs) before electron transfer (42) or as a rate-limiting step (∼350 μs) coupled to electron transfer (43) have been proposed in the S2 → S3 transition, whereas a lag phase (∼200 μs) before the electron transfer/O2 evolution phase in the S3 → S0 transfer has been attributed to a proton transfer process (58, 95–97). These putative proton transfer processes during the S2 → S3 and S3 → S0 transitions take place in the similar time regime of tens or hundreds of microseconds (42, 43, 58, 95–97), suggesting that they are performed by a similar proton transfer mechanism. However, the extent of inhibition by the D1-Asn-298 mutation was different between these transitions: the S2 → S3 transition is only partially inhibited, whereas the S3 → S0 transition is more significantly blocked. This difference could be related to different hydrogen-bonded structures around the Mn4CaO5 cluster between the S2- and S3-states and their changes upon mutation, which are revealed by a broad positive feature in the strongly hydrogen-bonded OH region at 2800–2400 cm−1 in the S3/S2 FTIR difference spectrum (57, 77) and a change in the mutant such as the appearance of a positive feature at 2907 cm−1 (Fig. 6B, b).
In conclusion, spectroscopic analyses of the D1-N298A mutant suggest that the hydrogen-bond network near YZ is functional in proton transfer during water oxidation, especially in the S2 → S3 and S3 → S0 transitions. D1-Asn-298 plays a pivotal role in forming a tight hydrogen-bond network near YZ, which is crucial for efficient proton transfer. At the present stage, however, possibilities other than proton transfer as an inhibited process by the mutation cannot be fully excluded. Further studies using this mutant as well as other site-directed mutants of residues on putative proton pathways, in combination with spectroscopic analyses such as time-resolved infrared spectroscopy, are necessary for identifying proton release pathways for four individual protons in the S-state cycle. Such studies of proton transfer are crucial for full understanding of the mechanism of photosynthetic water oxidation.
Experimental procedures
Construction of site-directed mutant
The wild-type control strain of the D1 subunit (WT*) and the D1-N298A strain were constructed in Synechocystis sp. PCC 6803 with an analogous method of Nagao et al. (71). Plasmid pRN123, which involved the coding region of psbA2, was used as a parental vector for site-directed mutagenesis. A host Synechocystis strain, which was transformed with pRN123 to obtain the WT* strain, lacked all of the three psbA genes (ΔpsbA1/ΔpsbA2/ΔpsbA3) and contains a His6 tag attached to the C terminus of CP47 (71). Mutation of D1-Asn-298 to Ala was introduced into pRN123 by replacing an AAC codon at a target site with a GCC codon, and the resultant plasmid was introduced into the host ΔpsbA1/ΔpsbA2/ΔpsbA3 strain.
The WT* and D1-N298A strains were maintained on BG-11 (98) agar plates containing 5 μg ml−1 of kanamycin (Km), 5 μg ml−1 of chloramphenicol (Cm), 5 μg ml−1 of erythromycin (Em), and 5 μg ml−1 of spectinomycin (Sm) in the presence of 5 mm glucose and 10 μm 3-(3,4)-dichlorophenyl-1,1-dimethylurea (DCMU) under a continuous low-light condition. The genotype of the D1-N298A mutant was confirmed by PCR analysis and DNA sequencing in both cases of in vivo analyses and preparation of PSII core complexes for FTIR measurements. No trace of the wild-type psbA2 gene was detected in any cultures of the D1-N298A strain.
Cell growth
WT* and D1-N298A cells were grown photoautotrophically in 40 ml of a BG-11 medium (98) supplemented with 4 mm Hepes-NaOH (pH 7.5) and 5 μg ml−1 Km/Cm/Em/Sm by bubbling with air containing 1% (v/v) CO2 at 30 °C under continuous illumination (20 μmol of photons m−2 s−1) by white fluorescence lamps. The cell density was monitored as an optical density at 730 nm (A730) using a spectrophotometer (Shimadzu UV-3100PC). For in vivo analyses, cells grown to the concentration of A730 = 0.5–1.0 were inoculated into 40 ml of a fresh BG-11 medium without antibiotics (A730 = ∼0.1), and were cultured under the same condition. Log-phase cells were collected by centrifugation (1000 × g for 5 min at 25 °C) and suspended in a BG-11 medium. For PSII preparation, cells were grown in an 8-liter culture bottle without antibiotics under the photoautotrophic growth condition. Cells cultured in six bottles (total volume of 48 liters) were used for preparation of PSII core complexes from each strain.
Preparation of PSII core complexes
O2-evolving PSII core complexes were purified using the method by Nagao et al. (71) with a minor modification. Thylakoid membranes suspended in a buffer (pH 6.0) containing 50 mm Mes-NaOH, 5 mm CaCl2, 10 mm MgCl2, and 25% (w/v) glycerol (buffer A) were solubilized with 1% (w/v) n-dodecyl β-d-maltoside (DM) at a Chl concentration of 1.0 mg ml−1 by stirring for 10 min on ice. After centrifugation at 27,000 × g for 15 min, the resultant supernatant was applied to a Ni2+ affinity column equilibrated with buffer A containing 0.04% DM (buffer B). The column was washed with 1 volume of buffer B containing 5 mm l-histidine followed by further washing with buffer B. PSII complexes were eluted with buffer B containing 50 mm l-histidine and then concentrated by ultrafiltration (Vivaspin 20, 100 kDa MWCO, Sartorius Stedim). The isolated PSII complexes were stored in liquid nitrogen.
Measurement of O2 evolution activity
The O2 evolution activity was measured using a Clark-type oxygen electrode at 30 °C under a saturating light condition. The activities of cell samples (10 μg of Chl) were measured in BG-11 medium in the presence of 1 mm DCBQ and 1 mm potassium ferricyanide as electron acceptors, whereas those of PSII complexes (3.5–5 μg of Chl) were measured in a buffer (pH 6.0) containing 1 m sucrose, 5 mm CaCl2, 10 mm NaCl, and 50 mm Mes-NaOH in the presence of 0.1 mm DCBQ and 4 mm potassium ferricyanide as electron acceptors.
TL and DL measurements
TL and DL were measured using a laboratory-built apparatus, as described previously (99). Before measurements, cells suspended in BG-11 medium (250 μg of Chl ml−1) were exposed to white continuous light (200 μmol of photons m−2 s−1; ∼16 milliwatt cm−2 at the sample point) for 30 s at 30 °C, followed by incubation at this temperature for 5 min in the dark. A cell suspension (70 μl) was then loaded onto a piece of filter paper. For detection of a TL B band (S2QB− recombination), the sample was illuminated by a single saturating flash from a Xe lamp (SL-230S, Sugawara) at 5 °C, whereas for a TL Q band (S2QA− recombination), the sample in the presence of 50 μm DCMU was illuminated with continuous white light (∼55 milliwatt cm−2 at the sample point) from a halogen lamp (MEJIRO PRECISION PHL-150) for 10 s at −20 °C. The illuminated sample was quickly cooled down and then warmed at a rate of 40 °C min−1 to record a TL glow curve.
For DL measurement, the sample was illuminated by a series of saturating Xe flashes (1 Hz) at 25 °C, and DL emission upon each flash was recorded. The DL amplitude at 0.8 s after each flash was plotted against a flush number. The measurement was performed once for each sample, and the data obtained using three different samples were averaged.
FTIR measurements
Light-induced FTIR difference spectra were recorded using a Bruker IFS-66/S spectrophotometer equipped with an MCT detector (InfraRed D313-L) at 4 cm−1 resolution. Flash illumination was performed by a Q-switched Nd:YAG laser (Quanta-Ray GCR-130; 532 nm, ∼7 ns full width at half-maximum) with a power of ∼7 mJ pulse−1 cm−2 at a sample point.
FTIR measurements of the S-state transitions in PSII core complexes were performed following the previous methods (26, 74, 76, 77). PSII complexes were washed with a buffer (pH 6.0) containing 10 mm Mes-NaOH, 5 mm NaCl, 5 mm CaCl2, 40 mm sucrose, and 0.06% DM (buffer C) and concentrated to ∼2.5 mg of Chl ml−1 by ultrafiltration (Apollo 7-ml High-Performance Centrifugal Concentrators, 150 kDa MWCO, Orbital Biosciences). Two sample types, solutions and moderately hydrated films, were used for measurements. For solution samples, 10 μl of the PSII solution (∼2.5 mg of Chl ml−1) in buffer C mixed with 1 μl of 100 mm potassium ferricyanide (total volume: 11 μl) was lightly dried on a CaF2 plate (25 mm in diameter) under N2 gas flow. The resultant sample was then mixed with 1 μl of Milli-Q water, and sandwiched with another CaF2 plate with a circular groove (14-mm inner diameter; 1-mm width). The sample cell was sealed with silicone grease in the outer part of the groove, where a tiny piece of aluminum foil was placed as a spacer. For preparation of a hydrated film, 4.5 μl of PSII solution (∼2.5 mg of Chl ml−1) in buffer C mixed with additional 5.5 μl of buffer C and 1 μl of 100 mm potassium ferricyanide (total volume: 11 μl) was dried on a CaF2 plate (25 × 25 mm) in a circle shape (8 mm in diameter) under N2 gas flow. The resultant sample was sealed using another CaF2 plate and a silicone spacer (0.5 mm in thickness), enclosing 2 μl of 40% (v/v) glycerol solution (95% relative humidity) without touching the sample to moderately hydrate the film (76). For both types of samples, the sample temperature was kept at 10 °C by circulating cold water through a copper holder.
In the measurement of a moderately hydrated film, two preflashes with a 1-s interval were first applied to the sample followed by dark adaptation for 15 min to synchronize all centers to the S1 state. Four flashes were then applied with intervals of 10 s; a single-beam spectrum with 20 scans (10-s scan) was measured twice before the first flash and once after individual flashes. The whole measurement scheme was repeated 12 times, and the spectra were averaged using five different samples (total 1200 scans). The measurement of a solution sample was performed using the same scheme, but with the dark interval of 10 min. The measurement was repeated 24 times using one sample, and the spectra were averaged (total 480 scans). To obtain better signal-to-noise ratios in the 3700–2200 cm−1 region of the spectra in the S1 → S2 and S2 → S3 transitions, measurements by two flashes were performed using the hydrated films. In this case, preflashes except for the first ones were omitted because the S2- and S3-states returned back to the S1-state during dark adaptation for 15 min. Other conditions were the same as the four-flash measurements of the hydrated films. The measurement was repeated 120 times, and the spectra using four samples were averaged (total 8400 scans). These average spectra were used to calculate flash-induced spectra of the S-state transitions together with a dark-minus-dark spectrum representing a noise level.
YZ•/YZ FTIR difference spectra were measured following the method described previously (16) with a slight modification. Mn depletion was performed by 10 mm NH2OH treatment for 1 h on ice, followed by washing four times with a buffer (pH 6.5) containing 20 mm Mes-NaOH, 5 mm NaCl, and 0.06% DM using ultrafiltration (Vivaspin 500, 100 kDa MWCO, Sartorius Stedim), which finally concentrated the sample to 2.5 mg of Chl ml−1. An aliquot (8 μl) of the sample solution, which was mixed with 1 μl of 100 mm potassium ferricyanide, was dried on a BaF2 plate (13 mm in diameter) under a N2 gas flow, and then sandwiched with another BaF2 plate together with 0.85 μl of Milli-Q water. The sample temperature was adjusted to 250 K in a cryostat (Oxford DN1704). Single-beam spectra with 50 scans (25-s scan) were recorded twice before and once after a single flash. This measurement was repeated 90 and 160 times for WT* and D1-N298A, respectively, with a dark interval of 225 s, and the average spectra were used to calculate a YZ•/YZ spectrum together with a dark-minus-dark spectrum representing a noise level.
Author contributions
R. N. and T. N. designed the study and wrote the manuscript. R. N. and H. U.-N. performed the experiments. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by the Grants-in-Aid for Scientific Research from Japan Society for the Promotion of Science 26840091 and 17K07442 (to R. N.) and 17H03662 and 17H06435 (to T. N.). The authors declare that they have no conflicts of interest with the contents of this article.
- PSII
- photosystem II
- Chl
- chlorophyll
- DCBQ
- 2,6-dichloro-p-benzoquinone
- DL
- delayed luminescence
- DM
- n-dodecyl β-d-maltoside
- MD
- molecular dynamics
- QM/MM
- quantum mechanics/molecular mechanics
- TL
- thermoluminescence
- DCMU
- 3-(3,4)-dichlorophenyl-1,1-dimethylurea
- Km
- kanamycin
- Cm
- chloramphenicol
- Em
- erythromycin
- Sm
- spectinomycin.
References
- 1. Blankenship R. E. (2002) Molecular Mechanisms of Photosynthesis, Blackwell Science, Oxford, UK [Google Scholar]
- 2. Debus R. J. (1992) The manganese and calcium ions of photosynthetic oxygen evolution. Biochim. Biophys. Acta 1102, 269–352 [DOI] [PubMed] [Google Scholar]
- 3. McEvoy J. P., and Brudvig G. W. (2006) Water-splitting chemistry of photosystem II. Chem. Rev. 106, 4455–4483 [DOI] [PubMed] [Google Scholar]
- 4. Dau H., and Haumann M. (2008) The manganese complex of photosystem II in its reaction cycle: basic framework and possible realization at the atomic level. Coord. Chem. Rev. 252, 273–295 [Google Scholar]
- 5. Renger G. (2012) Mechanism of light induced water splitting in Photosystem II of oxygen evolving photosynthetic organisms. Biochim. Biophys. Acta 1817, 1164–1176 [DOI] [PubMed] [Google Scholar]
- 6. Messinger J., Noguchi T., and Yano J. (2012) in Molecular Solar Fuels (Wydrzynski T. J., and Hillier W., eds) pp. 163–207, Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 7. Cox N., and Messinger J. (2013) Reflections on substrate water and dioxygen formation. Biochim. Biophys. Acta 1827, 1020–1030 [DOI] [PubMed] [Google Scholar]
- 8. Yano J., and Yachandra V. (2014) Mn4Ca cluster in photosynthesis: where and how water is oxidized to dioxygen. Chem. Rev. 114, 4175–4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen J.-R. (2015) The structure of photosystem II and the mechanism of water oxidation in photosynthesis. Annu. Rev. Plant Biol. 66, 23–48 [DOI] [PubMed] [Google Scholar]
- 10. Diner B. A., and Rappaport F. (2002) Structure, dynamics, and energetics of the primary photochemistry of photosystem II of oxygenic photosynthesis. Annu. Rev. Plant Biol. 53, 551–580 [DOI] [PubMed] [Google Scholar]
- 11. Renger G., and Holzwarth A. R. (2005) in Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase (Wydrzynski T. J., and Satoh K., eds) pp. 139–175, Springer, Dordrecht, The Netherlands [Google Scholar]
- 12. Petrouleas V., and Crofts A. R. (2005) in Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase (Wydrzynski T. J., and Satoh K., eds) pp. 177–206, Springer, Dordrecht, The Netherlands [Google Scholar]
- 13. Diner B. A., and Britt R. D. (2005) in Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase (Wydrzynski T. J., and Satoh K., eds) pp. 207–233, Springer, Dordrecht, The Netherlands [Google Scholar]
- 14. Styring S., Sjöholm J., and Mamedov F. (2012) Two tyrosines that changed the world: Interfacing the oxidizing power of photochemistry to water splitting in photosystem II. Biochim. Biophys. Acta 1817, 76–87 [DOI] [PubMed] [Google Scholar]
- 15. Saito K., Shen J.-R., Ishida T., and Ishikita H. (2011) Short hydrogen bond between redox-active tyrosine YZ and D1-His190 in the photosystem II crystal structure. Biochemistry 50, 9836–9844 [DOI] [PubMed] [Google Scholar]
- 16. Nakamura S., Nagao R., Takahashi R., and Noguchi T. (2014) Fourier transform infrared detection of a polarizable proton trapped between photooxidized tyrosine YZ and a coupled histidine in photosystem II: relevance to the proton transfer mechanism of water oxidation. Biochemistry 53, 3131–3144 [DOI] [PubMed] [Google Scholar]
- 17. Umena Y., Kawakami K., Shen J.-R., and Kamiya N. (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60 [DOI] [PubMed] [Google Scholar]
- 18. Suga M., Akita F., Hirata K., Ueno G., Murakami H., Nakajima Y., Shimizu T., Yamashita K., Yamamoto M., Ago H., and Shen J.-R. (2015) Native structure of photosystem II at 1.95-Å resolution viewed by femtosecond X-ray pulses. Nature 517, 99–103 [DOI] [PubMed] [Google Scholar]
- 19. Young I. D., Ibrahim M., Chatterjee R., Gul S., Fuller F. D., Koroidov S., Brewster A. S., Tran R., Alonso-Mori R., Kroll T., Michels-Clark T., Laksmono H., Sierra R. G., Stan C. A., Hussein R., et al. (2016) Structure of photosystem II and substrate binding at room temperature. Nature 540, 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanaka A., Fukushima Y., and Kamiya N. (2017) Two different structures of the oxygen-evolving complex in the same polypeptide frameworks of photosystem II. J. Am. Chem. Soc. 139, 1718–1721 [DOI] [PubMed] [Google Scholar]
- 21. Suga M., Akita F., Sugahara M., Kubo M., Nakajima Y., Nakane T., Yamashita K., Umena Y., Nakabayashi M., Yamane T., Nakano T., Suzuki M., Masuda T., Inoue S., Kimura T., et al. (2017) Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 543, 131–135 [DOI] [PubMed] [Google Scholar]
- 22. Joliot P., Barbieri G., and Chabaud R. (1969) A new model of the photochemical centers in system II. Photochem. Photobiol. 10, 309–329 [Google Scholar]
- 23. Kok B., Forbush B., and McGloin M. (1970) Cooperation of charges in photosynthetic O2 evolution: I. a linear four step mechanism. Photochem. Photobiol. 11, 457–475 [DOI] [PubMed] [Google Scholar]
- 24. Förster V., and Junge W. (1985) Stoichiometry and kinetics of proton release upon photosynthetic water oxidation. Photochem. Photobiol. 41, 183–190 [Google Scholar]
- 25. Schlodder E., and Witt H. T. (1999) Stoichiometry of proton release from the catalytic center in photosynthetic water oxidation: reexamination by a glass electrode study at pH 5.5–7.2. J. Biol. Chem. 274, 30387–30392 [DOI] [PubMed] [Google Scholar]
- 26. Suzuki H., Sugiura M., and Noguchi T. (2009) Monitoring proton release during photosynthetic water oxidation in photosystem II by means of isotope-edited infrared spectroscopy. J. Am. Chem. Soc. 131, 7849–7857 [DOI] [PubMed] [Google Scholar]
- 27. Ferreira K. N., Iverson T. M., Maghlaoui K., Barber J., and Iwata S. (2004) Architecture of the photosynthetic oxygen-evolving center. Science 303, 1831–1838 [DOI] [PubMed] [Google Scholar]
- 28. Guskov A., Kern J., Gabdulkhakov A., Broser M., Zouni A., and Saenger W. (2009) Cyanobacterial photosystem II at 2.9-Å resolution and the role of quinones, lipids, channels and chloride. Nat. Struct. Mol. Biol. 16, 334–342 [DOI] [PubMed] [Google Scholar]
- 29. Ishikita H., Saenger W., Loll B., Biesiadka J., and Knapp E.-W. (2006) Energetics of a possible proton exit pathway for water oxidation in photosystem II. Biochemistry 45, 2063–2071 [DOI] [PubMed] [Google Scholar]
- 30. Murray J. W., and Barber J. (2007) Structural characteristics of channels and pathways in photosystem II including the identification of an oxygen channel. J. Struct. Biol. 159, 228–237 [DOI] [PubMed] [Google Scholar]
- 31. Ho F. M., and Styring S. (2008) Access channels and methanol binding site to the CaMn4 cluster in Photosystem II based on solvent accessibility simulations, with implications for substrate water access. Biochim. Biophys. Acta 1777, 140–153 [DOI] [PubMed] [Google Scholar]
- 32. Gabdulkhakov A., Guskov A., Broser M., Kern J., Müh F., Saenger W., and Zouni A. (2009) Probing the accessibility of the Mn4Ca cluster in photosystem II: channels calculation, noble gas derivatization, and cocrystallization with DMSO. Structure 17, 1223–1234 [DOI] [PubMed] [Google Scholar]
- 33. Vassiliev S., Comte P., Mahboob A., and Bruce D. (2010) Tracking the flow of water through photosystem II using molecular dynamics and streamline tracing. Biochemistry 49, 1873–1881 [DOI] [PubMed] [Google Scholar]
- 34. Rivalta I., Amin M., Luber S., Vassiliev S., Pokhrel R., Umena Y., Kawakami K., Shen J.-R., Kamiya N., Bruce D., Brudvig G. W., Gunner M. R., and Batista V. S. (2011) Structural-functional role of chloride in photosystem II. Biochemistry 50, 6312–6315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vassiliev S., Zaraiskaya T., and Bruce D. (2012) Exploring the energetics of water permeation in photosystem II by multiple steered molecular dynamics simulations. Biochim. Biophys. Acta 1817, 1671–1678 [DOI] [PubMed] [Google Scholar]
- 36. Ogata K., Yuki T., Hatakeyama M., Uchida W., and Nakamura S. (2013) All-atom molecular dynamics simulation of photosystem II embedded in thylakoid membrane. J. Am. Chem. Soc. 135, 15670–15673 [DOI] [PubMed] [Google Scholar]
- 37. Linke K., and Ho F. M. (2014) Water in Photosystem II: Structural, functional and mechanistic considerations. Biochim. Biophys. Acta 1837, 14–32 [DOI] [PubMed] [Google Scholar]
- 38. Saito K., Rutherford A. W., and Ishikita H. (2015) Energetics of proton release on the first oxidation step in the water-oxidizing enzyme. Nat. Commun. 6, 8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sakashita N., Watanabe H. C., Ikeda T., Saito K., and Ishikita H. (2017) Origins of water molecules in the photosystem II crystal structure. Biochemistry 56, 3049–3057 [DOI] [PubMed] [Google Scholar]
- 40. Siegbahn P. E. (2012) Mechanisms for proton release during water oxidation in the S2 to S3 and S3 to S4 transitions in photosystem II. Phys. Chem. Chem. Phys. 14, 4849–4856 [DOI] [PubMed] [Google Scholar]
- 41. Amin M., Vogt L., Szejgis W., Vassiliev S., Brudvig G. W., Bruce D., and Gunner M. R. (2015) Proton-coupled electron transfer during the S-state transitions of the oxygen-evolving complex of photosystem II. J. Phys. Chem. B 119, 7366–7377 [DOI] [PubMed] [Google Scholar]
- 42. Klauss A., Haumann M., and Dau H. (2012) Alternating electron and proton transfer steps in photosynthetic water oxidation. Proc. Natl. Acad. Sci. U.S.A. 109, 16035–16040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sakamoto H., Shimizu T., Nagao R., and Noguchi T. (2017) Monitoring the reaction process during the S2 → S3 transition in photosynthetic water oxidation using time-resolve infrared spectroscopy. J. Am. Chem. Soc. 139, 2022–2029 [DOI] [PubMed] [Google Scholar]
- 44. Wei X., Su X., Cao P., Liu X., Chang W., Li M., Zhang X., and Liu Z. (2016) Structure of spinach photosystem II-LHCII supercomplex at 3.2-Å resolution. Nature 534, 69–74 [DOI] [PubMed] [Google Scholar]
- 45. Su X., Ma J., Wei X., Cao P., Zhu D., Chang W., Liu Z., Zhang X., and Li M. (2017) Structure and assembly mechanism of plant C2S2M2-type PSII-LHCII supercomplex. Science 357, 815–820 [DOI] [PubMed] [Google Scholar]
- 46. Chu H.-A., Nguyen A. P., and Debus R. J. (1995) Amino acid residues that influence the binding of manganese or calcium to photosystem II: 1. the lumenal interhelical domains of the D1 polypeptide. Biochemistry 34, 5839–5858 [DOI] [PubMed] [Google Scholar]
- 47. Hundelt M., Hays A.-M., Debus R. J., and Junge W. (1998) Oxygenic photosystem II: the mutation D1-D61N in Synechocystis sp. PCC 6803 retards S-state transitions without affecting electron transfer from YZ to P680+. Biochemistry 37, 14450–14456 [DOI] [PubMed] [Google Scholar]
- 48. Qian M., Dao L., Debus R. J., and Burnap R. L. (1999) Impact of mutations within the putative Ca2+-binding lumenal interhelical a-b loop of the photosystem II D1 protein on the kinetics of photoactivation and H2O-oxidation in Synechocystis sp PCC6803. Biochemistry 38, 6070–6081 [DOI] [PubMed] [Google Scholar]
- 49. Li Z., and Burnap R. L. (2002) Mutations of basic arginine residue 334 in the D1 protein of Photosystem II lead to unusual S2 state properties in Synechocystis sp. PCC 6803. Photosynth. Res. 72, 191–201 [DOI] [PubMed] [Google Scholar]
- 50. Yamasato A., Kamada T., and Satoh K. (2002) Random mutagenesis targeted to the psbAII gene of Synechocystis sp. PCC 6803 to identify functionally important residues in the D1 protein of the photosystem II reaction center. Plant Cell Physiol. 43, 540–548 [DOI] [PubMed] [Google Scholar]
- 51. Hwang H. J., Dilbeck P., Debus R. J., and Burnap R. L. (2007) Mutation of arginine 357 of the CP43 protein of photosystem II severely impairs the catalytic S-state cycle of the H2O oxidation complex. Biochemistry 46, 11987–11997 [DOI] [PubMed] [Google Scholar]
- 52. Service R. J., Hillier W., and Debus R. J. (2010) Evidence from FTIR difference spectroscopy of an extensive network of hydrogen bonds near the oxygen-evolving Mn4Ca cluster of photosystem II involving D1-Glu65, D2-Glu312, and D1-Glu329. Biochemistry 49, 6655–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dilbeck P. L., Hwang H. J., Zaharieva I., Gerencser L., Dau H., and Burnap R. L. (2012) The D1-D61N mutation in Synechocystis sp. PCC 6803 allows the observation of pH-sensitive intermediates in the formation and release of O2 from photosystem II. Biochemistry 51, 1079–1091 [DOI] [PubMed] [Google Scholar]
- 54. Pokhrel R., Service R. J., Debus R. J., and Brudvig G. W. (2013) Mutation of lysine 317 in the D2 subunit of photosystem II alters chloride binding and proton transport. Biochemistry 52, 4758–4773 [DOI] [PubMed] [Google Scholar]
- 55. Suzuki H., Yu J., Kobayashi T., Nakanishi H., Nixon P. J., and Noguchi T. (2013) Functional roles of D2-Lys317 and the interacting chloride ion in the water oxidation reaction of photosystem II as revealed by Fourier transform infrared analysis. Biochemistry 52, 4748–4757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Service R. J., Hillier W., and Debus R. J. (2014) Network of hydrogen bonds near the oxygen-evolving Mn4CaO5 cluster of photosystem II probed with FTIR difference spectroscopy. Biochemistry 53, 1001–1017 [DOI] [PubMed] [Google Scholar]
- 57. Debus R. J. (2014) Evidence from FTIR difference spectroscopy that D1-Asp61 influences the water reactions of the oxygen-evolving Mn4CaO5 cluster of photosystem II. Biochemistry 53, 2941–2955 [DOI] [PubMed] [Google Scholar]
- 58. Bao H., and Burnap R. L. (2015) Structural rearrangements preceding dioxygen formation by the water oxidation complex of photosystem II. Proc. Natl. Acad. Sci. U.S.A. 112, E6139–E6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pokhrel R., Debus R. J., and Brudvig G. W. (2015) Probing the effect of mutations of asparagine 181 in the D1 subunit of photosystem II. Biochemistry 54, 1663–1672 [DOI] [PubMed] [Google Scholar]
- 60. Kuroda H., Kodama N., Sun X.-Y., Ozawa S., and Takahashi Y. (2014) Requirement for Asn298 on D1 protein for oxygen evolution: Analyses by exhaustive amino acid substitution in the green alga Chlamydomonas reinhardtii. Plant Cell Physiol. 55, 1266–1275 [DOI] [PubMed] [Google Scholar]
- 61. Chu H.-A., Hillier W., Law N. A., and Babcock G. T. (2001) Vibrational spectroscopy of the oxygen-evolving complex and of manganese model compounds. Biochim. Biophys. Acta 1503, 69–82 [DOI] [PubMed] [Google Scholar]
- 62. Noguchi T., and Berthomieu C. (2005) in Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase (Wydrzynski T. J., and Satoh K., eds) pp. 367–387, Springer, Dordrecht, The Netherlands [Google Scholar]
- 63. Noguchi T. (2007) Light-induced FTIR difference spectroscopy as a powerful tool toward understanding the molecular mechanism of photosynthetic oxygen evolution. Photosynth. Res. 91, 59–69 [DOI] [PubMed] [Google Scholar]
- 64. Debus R. J. (2008) Protein ligation of the photosynthetic oxygen-evolving center. Coord. Chem. Rev. 252, 244–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Noguchi T. (2008) Fourier transform infrared analysis of the photosynthetic oxygen-evolving center. Coord. Chem. Rev. 252, 336–346 [Google Scholar]
- 66. Noguchi T. (2008) FTIR detection of water reactions in the oxygen-evolving center of photosystem II. Phil. Trans. R. Soc. B 363, 1189–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chu H.-A. (2013) Fourier transform infrared difference spectroscopy for studying the molecular mechanism of photosynthetic water oxidation. Front. Plant Sci. 4, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Noguchi T. (2013) Monitoring the reactions of photosynthetic water oxidation using infrared spectroscopy. Biomed. Spectrosc. Imaging 2, 115–128 [Google Scholar]
- 69. Debus R. J. (2015) FTIR studies of metal ligands, networks of hydrogen bonds, and water molecules near the active site Mn4CaO5 cluster in Photosystem II. Biochim. Biophys. Acta 1847, 19–34 [DOI] [PubMed] [Google Scholar]
- 70. Noguchi T. (2015) Fourier transform infrared difference and time-resolved infrared detection of the electron and proton transfer dynamics in photosynthetic water oxidation. Biochim. Biophys. Acta 1847, 35–45 [DOI] [PubMed] [Google Scholar]
- 71. Nagao R., Yamaguchi M., Nakamura S., Ueoka-Nakanishi H., and Noguchi T. (2017) Genetically introduced hydrogen bond interactions reveal an asymmetric charge distribution on the radical cation of the special-pair chlorophyll P680. J. Biol. Chem. 292, 7474–7486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vass I., and Govindjee (1996) Thermoluminescence from the photosynthetic apparatus. Photosynth. Res. 48, 117–126 [DOI] [PubMed] [Google Scholar]
- 73. Berthomieu C., Hienerwadel R., Boussac A., Breton J., and Diner B. A. (1998) Hydrogen bonding of redox-active tyrosine Z of photosystem II probed by FTIR difference spectroscopy. Biochemistry 37, 10547–10554 [DOI] [PubMed] [Google Scholar]
- 74. Noguchi T., and Sugiura M. (2001) Flash-induced Fourier transform infrared detection of the structural changes during the S-state cycle of the oxygen-evolving complex in photosystem II. Biochemistry 40, 1497–1502 [DOI] [PubMed] [Google Scholar]
- 75. Hillier W., and Babcock G. T. (2001) S-state dependent Fourier transform infrared difference spectra for the photosystem II oxygen evolving complex. Biochemistry 40, 1503–1509 [DOI] [PubMed] [Google Scholar]
- 76. Noguchi T., and Sugiura M. (2002) Flash-induced FTIR difference spectra of the water oxidizing complex in moderately hydrated photosystem II core films: effect of hydration extent on S-state transitions. Biochemistry 41, 2322–2330 [DOI] [PubMed] [Google Scholar]
- 77. Noguchi T., and Sugiura M. (2002) FTIR detection of water reactions during the flash-induced S-state cycle of the photosynthetic water-oxidizing complex. Biochemistry 41, 15706–15712 [DOI] [PubMed] [Google Scholar]
- 78. Noguchi T., and Sugiura M. (2003) Analysis of flash-induced FTIR difference spectra of the S-state cycle in the photosynthetic water-oxidizing complex by uniform 15N and 13C isotope labeling. Biochemistry 42, 6035–6042 [DOI] [PubMed] [Google Scholar]
- 79. Kimura Y., Mizusawa N., Ishii A., and Ono T. (2005) FTIR detection of structural changes in a histidine ligand during S-state cycling of photosynthetic oxygen-evolving complex. Biochemistry 44, 16072–16078 [DOI] [PubMed] [Google Scholar]
- 80. Shimada Y., Suzuki H., Tsuchiya T., Tomo T., Noguchi T., and Mimuro M. (2009) Effect of a single-amino acid substitution of the 43 kDa chlorophyll protein on the oxygen-evolving reaction of the cyanobacterium Synechocystis sp. PCC 6803: analysis of the Glu354Gln mutation. Biochemistry 48, 6095–6103 [DOI] [PubMed] [Google Scholar]
- 81. Service R. J., Yano J., McConnell I., Hwang H. J., Niks D., Hille R., Wydrzynski T., Burnap R. L., Hillier W., and Debus R. J. (2011) Participation of glutamate-354 of the CP43 polypeptide in the ligation of manganese and the binding of substrate water in photosystem II. Biochemistry 50, 63–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kimura Y., Mizusawa N., Ishii A., Yamanari T., and Ono T. (2003) Changes of low-frequency vibrational modes induced by universal 15N- and 13C-isotope labeling in S2/S1 FTIR difference spectrum of oxygen-evolving complex. Biochemistry 42, 13170–13177 [DOI] [PubMed] [Google Scholar]
- 83. Noguchi T., Ono T., and Inoue Y. (1995) Direct detection of a carboxylate bridge between Mn and Ca2+ in the photosynthetic oxygen-evolving center by means of Fourier transform infrared spectroscopy. Biochim. Biophys. Acta 1228, 189–200 [Google Scholar]
- 84. Chu H.-A., Hillier W., and Debus R. J. (2004) Evidence that the C-terminus of the D1 polypeptide of photosystem II is ligated to the manganese ion that undergoes oxidation during the S1 to S2 transition: an isotope-edited FTIR study. Biochemistry 43, 3152–3166 [DOI] [PubMed] [Google Scholar]
- 85. Nakamura S., and Noguchi T. (2016) Quantum mechanics/molecular mechanics simulation of the ligand vibrations of the water-oxidizing Mn4CaO5 cluster in photosystem II. Proc. Natl. Acad. Sci. U.S.A. 113, 12727–12732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Noguchi T., and Sugiura M. (2000) Structure of an active water molecule in the water-oxidizing complex of photosystem II as studied by FTIR spectroscopy. Biochemistry 39, 10943–10949 [DOI] [PubMed] [Google Scholar]
- 87. Nakamura S., Ota K., Shibuya Y., and Noguchi T. (2016) Role of a water network around the Mn4CaO5 cluster in photosynthetic water oxidation: a Fourier transform infrared spectroscopy and quantum mechanics/molecular mechanics calculation study. Biochemistry 55, 597–607 [DOI] [PubMed] [Google Scholar]
- 88. Nakamura S., and Noguchi T. (2017) Infrared determination of the protonation state of a key histidine residue in the photosynthetic water oxidizing center. J. Am. Chem. Soc. 139, 9364–9375 [DOI] [PubMed] [Google Scholar]
- 89. Kim C. J., and Debus R. J. (2017) Evidence from FTIR difference spectroscopy that a substrate H2O molecule for O2 formation in photosystem II is provided by the Ca ion of the catalytic Mn4CaO5 cluster. Biochemistry 56, 2558–2570 [DOI] [PubMed] [Google Scholar]
- 90. Suzuki H., Sugiura M., and Noguchi T. (2008) Monitoring water reactions during the S-state cycle of the photosynthetic water-oxidizing center: Detection of the DOD bending vibrations by means of Fourier transform infrared spectroscopy. Biochemistry 47, 11024–11030 [DOI] [PubMed] [Google Scholar]
- 91. Isobe H., Shoji M., Shen J.-R., and Yamaguchi K. (2015) Strong coupling between the hydrogen bonding environment and redox chemistry during the S2 to S3 transition in the oxygen-evolving complex of photosystem II. J. Phys. Chem. B 119, 13922–13933 [DOI] [PubMed] [Google Scholar]
- 92. Cox N., Retegan M., Neese F., Pantazis D. A., Boussac A., and Lubitz W. (2014) Electronic structure of the oxygen-evolving complex in photosystem II prior to O-O bond formation. Science 345, 804–808 [DOI] [PubMed] [Google Scholar]
- 93. Capone M., Narzi D., Bovi D., and Guidoni L. (2016) Mechanism of water delivery to the active site of photosystem II along the S2 to S3 transition. J. Phys. Chem. Lett. 7, 592–596 [DOI] [PubMed] [Google Scholar]
- 94. Hoganson C. W., and Babcock G. T. (1997) A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science 277, 1953–1956 [DOI] [PubMed] [Google Scholar]
- 95. Rappaport F., Blanchard-Desce M., and Lavergne J. (1994) Kinetics of electron transfer and electrochromic change during the redox transitions of the photosynthetic oxygen-evolving complex. Biochim. Biophys. Acta 1184, 178–192 [Google Scholar]
- 96. Haumann M., Liebisch P., Müller C., Barra M., Grabolle M., and Dau H. (2005) Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science 310, 1019–1021 [DOI] [PubMed] [Google Scholar]
- 97. Noguchi T., Suzuki H., Tsuno M., Sugiura M., and Kato C. (2012) Time-resolved infrared detection of the proton and protein dynamics during photosynthetic oxygen evolution. Biochemistry 51, 3205–3214 [DOI] [PubMed] [Google Scholar]
- 98. Stanier R. Y., Kunisawa R., Mandel M., and Cohen-Bazire G. (1971) Purification and properties of unicellular blue-green algae (order Chroococcales). Bacteriol. Rev. 35, 171–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Noguchi T., Katoh M., and Inoue Y. (2002) A new system for detection of thermoluminescence and delayed luminescence from photosynthetic apparatus with precise temperature control. Spectroscopy 16, 89–94 [Google Scholar]