

Abstract
Pathologies including diabetes and conditions such as exercise place an unusual demand on liver energy metabolism, and this demand induces a state of energy discharge. Hepatic AMP-activated protein kinase (AMPK) has been proposed to inhibit anabolic processes such as gluconeogenesis in response to cellular energy stress. However, both AMPK activation and glucose release from the liver are increased during exercise. Here, we sought to test the role of hepatic AMPK in the regulation of in vivo glucose-producing and citric acid cycle–related fluxes during an acute bout of muscular work. We used 2H/13C metabolic flux analysis to quantify intermediary metabolism fluxes in both sedentary and treadmill-running mice. Additionally, liver-specific AMPK α1 and α2 subunit KO and WT mice were utilized. Exercise caused an increase in endogenous glucose production, glycogenolysis, and gluconeogenesis from phosphoenolpyruvate. Citric acid cycle fluxes, pyruvate cycling, anaplerosis, and cataplerosis were also elevated during this exercise. Sedentary nutrient fluxes in the postabsorptive state were comparable for the WT and KO mice. However, the increment in the endogenous rate of glucose appearance during exercise was blunted in the KO mice because of a diminished glycogenolytic flux. This lower rate of glycogenolysis was associated with lower hepatic glycogen content before the onset of exercise and prompted a reduction in arterial glucose during exercise. These results indicate that liver AMPKα1α2 is required for maintaining glucose homeostasis during an acute bout of exercise.
Keywords: energy metabolism, fibroblast growth factor (FGF), intermediary metabolism, isotopic tracer, liver metabolism, metabolic flux analysis
Introduction
The dysregulation of glucose homeostasis is associated with adverse consequences and is of great concern in pathologies such as diabetes (1). In addition to disease states, physiological conditions such as exercise challenge the ability of the glucoregulatory system to maintain blood glucose within a narrow range (2). Systemic glucose disposal is elevated during short-term, moderate exercise without appreciably changing arterial glucose (3–6). This is due to a concurrent increase in glucose release from the liver (5, 7, 8).
Hepatic glucose production during exercise is influenced by multiple factors, including endocrine action (9). In particular, glucagon increases hepatic glycogenolysis and gluconeogenesis (7). It also stimulates gluconeogenic precursor extraction by the liver (7, 8). Of note, glucose synthesis from three-carbon sources is energetically demanding, making ATP of great importance for this anabolic process (10). Consequently, exercise promotes liver energetic stress, as indicated by an increase in the AMP/ATP ratio (11).
Within the cell, AMP-activated protein kinase (AMPK)2 responds to changes in the AMP/ATP ratio (12). This heterotrimeric serine/threonine kinase is composed of a catalytic α subunit and regulatory β and γ subunits. Two catalytic subunit isoforms are present in the liver: α1 and α2 (13–15). Kinase activity of the α subunits is enhanced by energetic stress and post-translational modification. AMP binds the AMPK γ subunit (16). This promotes a conformational change in the enzyme that positions itself for phosphorylation on threonine residue 172 of the α subunit (αThr-172) and protects against αThr-172 dephosphorylation (17–19). Additionally, ATP competitively impedes AMPK activation by AMP (20). In response to energy discharge, AMPK promotes oxidative metabolism to maintain energy status (13, 21). For example, loss of hepatic AMPK attenuates oxygen consumption in vitro (22, 23), suggesting that AMPK activity is critical for appropriate mitochondrial oxidative phosphorylation and ATP provision.
Experimental and physiological conditions with elevated glucagon action, including exercise, promote AMPK activation (11, 24, 25). This is consistent with the need for adequate ATP supply to support gluconeogenesis during muscular work. It is important to note that many hypotheses regarding the physiological influence of hepatic AMPK on endogenous glucose production are factious (26). Many studies have concluded that AMPK action inhibits rather than promotes glucose release from the liver (26). As such, the current study employed in vivo 2H/13C metabolic flux analysis (MFA) to characterize the dynamic alterations in nutrient fluxes contributing to endogenous glucose production in C57Bl/6J mice in response to an acute treadmill running bout. Subsequently, liver-specific AMPKα1α2 KO and WT littermates were used to test the hypothesis that AMPK is required for the increment in hepatic glucose producing and citric acid-cycle (CAC)–related fluxes during exercise. Mice lacking hepatic AMPK catalytic subunits exhibited an impaired ability to maintain glucose homeostasis during a 30-min treadmill run. This was associated with lower liver glycogen and glycogenolytic response to exercise. Gluconeogenesis-related fluxes during sedentary and exercise conditions were not altered by deletion of hepatic AMPKα1α2.
Results
Exercise promotes changes in circulating hormones and liver energy metabolism
Initial studies characterized the exercise response in C57Bl/6J mice during an acute treadmill running bout at 45% of maximal running speed. Body weights of sedentary (SED) and treadmill-running (exercising; EX) mice were comparable (Table 1). Maximal running speed was similar between the SED and EX groups at 36.5 ± 1.3 and 38.5 ± 1.2 m min−1, respectively (Table 1). During stable isotope infusions, SED mice remained on a stationary treadmill, whereas the exercising mice ran on a treadmill at 17.3 ± 1.3 m min−1 for 30 min (Table 1). Characteristic hormonal responses to exercise were observed in a separate cohort of mice undergoing the study conditions. Arterial insulin declined following the onset of exercise (Table 1). Plasma glucagon was elevated at 30 min of treadmill running (Table 1). Liver glycogen was reduced in the EX group at the conclusion of the treadmill run (Table 1). The exercise bout promoted changes in hepatic energy status. Liver ATP was lower and AMP was higher in EX mice (Table 1). The AMP/ATP ratio was increased and energy charge was decreased in EX mice (Table 1). It is important to note that liver ATP relative to ADP and AMP are lower in SED mice than that reported in perfused livers (27, 28). Livers in this study were harvested and freeze-clamped in under 20 s following euthanization. It is possible that compromised oxygen availability during that time frame may have induced energy discharge.
Table 1.
Biometric characteristics of sedentary and treadmill-running C57Bl/6J mice
Shown are body weight (g) before food and water withdrawal at −510 min in SED and EX mice as well as maximal running speed (m min−1) and experimental running speed (45% of maximal running speed for EX group; m min−1). Liver glycogen (mg g−1), liver adenine nucleotides (TAN = ATP + ADP + AMP; μmol g−1), and AMP/ATP and energy charge ([ATP + 0.5 ADP]/[TAN]) were determined in SED and EX mice. Plasma insulin and glucagon at 10 min before commencing the acute exercise bout and 30 min of treadmill running at 45% of maximal running speed were obtained from a separate cohort of age-matched, male C57Bl/6J mice. Data are mean ± S.E. n = 5–9 mice/group.
| Biometric and running characteristics | SED | EX |
|---|---|---|
| Body weight (g) | 27.7 ± 0.4 | 27.6 ± 0.3 |
| Maximal running speed (m min−1) | 35.3 ± 1.6 | 38.5 ± 1.2 |
| Experimental running speed (m min−1) | 0.0 ± 0.0 | 17.3 ± 0.5a |
| Liver metabolites | ||
| Glycogen (mg g−1) | 10.4 ± 2.1 | 1.8 ± 0.5a |
| ATP (μmol g−1) | 1.6 ± 0.1 | 1.1 ± 0.1a |
| ADP (μmol g−1) | 2.0 ± 0.1 | 2.1 ± 0.1 |
| AMP (μmol g−1) | 1.3 ± 0.1 | 1.8 ± 0.1a |
| TAN (μmol g−1) | 4.9 ± 0.1 | 5.0 ± 0.1 |
| AMP/ATP | 0.8 ± 0.1 | 1.7 ± 0.2a |
| Energy charge | 0.53 ± 0.01 | 0.43 ± 0.02a |
| Arterial hormones | ||
| Plasma insulin (ng ml−1) | 0.42 ± 0.09 | 0.17 ± 0.03a |
| Plasma glucagon (pg ml−1) | 11 ± 4 | 38 ± 10a |
a p < 0.05 versus SED.
Treadmill running stimulates hepatic nutrient fluxes in vivo
2H/13C MFA allowed glucose-producing and associated nutrient fluxes to be determined in the SED and EX mice. The studies achieved isotopic steady state in sedentary mice and a new isotopic dynamic steady state during exercise conditions. The time course of the mass isotopomer distribution (MID) for the fragment ion range m/z 301–305 is provided in Fig. 1. The remaining five fragment ions of glucose derivatives used for 2H/13C MFA also show isotopic steady state (data not shown). Pronounced disparities between the SED and EX mice in the glucose-producing fluxes estimated in vivo (Fig. 2A) were identified. Blood glucose was higher at 10 min and lower at 30 min in EX mice (Fig. 2B). Treadmill running enhanced endogenous glucose production (VEndoRa) at the 10-, 20-, and 30-min time points (Fig. 2C). Fluxes from glycogen (VPYGL; Fig. 2D) and gluconeogenic sources (VAldo; Fig. 2E) were elevated with exercise. Interestingly, the increased gluconeogenic flux was due to substrate provision from phosphoenolpyruvate (PEP; VEnol) but not glycerol (VGK) (Fig. 2, F and G).
Figure 1.
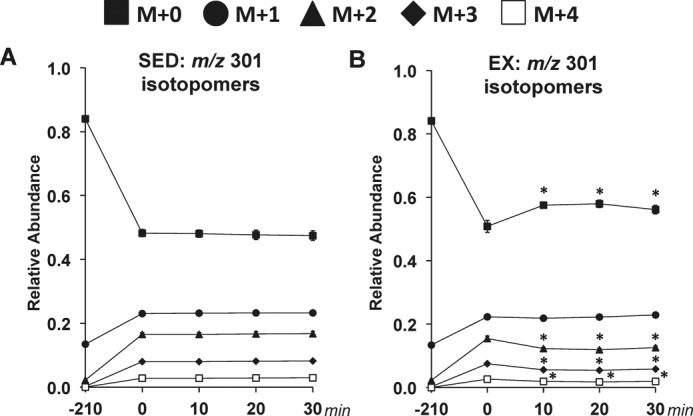
Time course of MID of glucose fragment ions in sedentary and treadmill-running C57Bl/6J mice. A time course of the average, uncorrected relative abundance of m/z 301 isotopomers used in 2H/13C metabolic flux analysis is shown. A, m/z 301 isotopomers for SED mice. B, m/z 301 isotopomers for EX mice. M+n indicates the mass shift from the unlabeled state for a given fragment ion with M+0 to M+4 shown. Data are mean ± S.E. (error bars); n = 8–9 mice/group. *, p < 0.05 versus 0-min time point.
Figure 2.

Glucose-producing and CAC-related fluxes in sedentary and treadmill-running C57Bl/6J mice. A, schematic representation of select glucose producing and CAC-related fluxes used for 2H/13C metabolic flux analysis. B, a time course of arterial blood glucose concentrations in SED and EX (45% of maximal running speed) mice. Shown are model-estimated, absolute nutrient fluxes (μmol kg−1 min−1) in SED and EX mice: VEndoRa (C), VPYGL (D), VAldo (E), VGK (F), VEnol (G), VPK+ME (H), VLDH (I), VPC (J), VPCK (K), VCS (L), VPCC (M), and VSDH (N). Shown is a time course for VPYGL/VEndoRa (O), VAldo/VEndoRa (P), and plasma NEFAs (Q) in SED and EX mice. Data are mean ± S.E. n = 6–9 mice/group. *, p < 0.05 versus SED at a specified time point. †, p < 0.05 versus 0-min time point within group.
In addition to glucose synthesis, CAC-related reactions were estimated at rest and during treadmill running. The return of PEP to pyruvate increased during exercise (VPK+ME; Fig. 2H). Flux from unlabeled substrates to pyruvate (VLDH) was ∼1.7-fold higher throughout the running bout (Fig. 2I), which contributed to the increase in anaplerotic flux from pyruvate to oxaloacetate (VPC; Fig. 2J). Cataplerosis (VPCK) exhibited an increment at each time point during exercise (Fig. 2K). Similarly, flux from oxaloacetate and acetyl-CoA to citrate (VCS), flux from propionyl-CoA to succinyl-CoA (VPCC), and flux from succinyl-CoA to oxaloacetate (VSDH) were greater in the EX group compared with the SED group (Fig. 2, L–N). Of note, VPCC was higher at the 30-min time point compared with the 0-min time point in the SED mice (Fig. 2M).
Assessment of the relative contributions of glucose sources to VEndoRa showed that the increase in VPYGL throughout the 30-min treadmill run was associated with a higher VPYGL/VEndoRa ratio (Fig. 2O). The greater contribution of glycogen to VEndoRa mediated a decline in the contribution of gluconeogenesis to VEndoRa, as indicated by the VAldo/VEndoRa ratio at the 10-, 20-, and 30-min time points in the EX mice (Fig. 2P). It should also be noted that plasma non-esterified fatty acids (NEFA) were similar between groups (Fig. 2Q).
Loss of AMPK does not influence running capacity
Previous work has shown that treadmill running increases liver AMPK phosphorylation (11). Initial work aimed to assess the role of hepatic AMPKα on functional parameters. WT mice and littermates with a liver-specific deletion of AMPK α1 and α2 subunits (KO) were used. Body weight (27.0 ± 0.5 versus 26.0 ± 0.4 g), maximal running speed (36.0 ± 1.7 versus 38.4 ± 1.0 m min−1), and experimental running speed (16.2 ± 0.8 versus 17.3 ± 0.5 m min−1) were similar between WT and KO mice (data are mean ± S.E. for n = 6–9 mice/group, WT versus KO).
AMPK is required for glucose homeostasis during exercise
Whereas mice of both genotypes performed exercise at the same intensity, the absence of hepatic AMPK caused marked alterations in estimated glucose-related fluxes (Fig. 3A). Plasma NEFAs increased similarly while running at 45% of maximal running speed in WT and KO mice (Fig. 3B). Under sedentary conditions, blood glucose levels were comparable between groups; however, KO mice exhibited a reduced arterial glucose during exercise (Fig. 3C). The increment in VEndoRa was blunted in the KO mice (Fig. 3D). The lower endogenous glucose production during exercise in the KO group was accompanied by reduced VPYGL (Fig. 3E). Fluxes of reactions involved in gluconeogenesis, VAldo, VGK, and VEnol, were similar between groups at rest and during the treadmill run (Fig. 3, F–H). CAC-related fluxes were comparable between sedentary and exercising WT and KO mice. Pyruvate cycling (VPK+ME) was elevated during the treadmill running (Fig. 3I) in both groups. Anaplerosis and related fluxes (VLDH, VPC, and VPCC) were raised with exercise in WT and KO mice (Fig. 3, J, K, and N). The generation of PEP (VPCK) was similar between genotypes before exercise (Fig. 3L). During treadmill running, the increase in VPCK was comparable in both groups (Fig. 3L). CAC fluxes, VCS and VSDH, showed similar increases throughout the treadmill running in WT and KO mice (Fig. 3, M and O).
Figure 3.

Glucose-producing and CAC-related fluxes in sedentary and treadmill-running mice lacking liver AMPKα. A, schematic representation of select glucose-producing and CAC-related fluxes for 2H/13C metabolic flux analysis. Shown is a time course of plasma NEFAs (B) and arterial blood glucose concentrations in WT mice and mice exhibiting a liver-specific deletion of AMPK α1 and α2 subunits (KO) (C). Shown are model-estimated, absolute nutrient fluxes (μmol kg−1 min−1) in WT and KO mice at rest and during 30 min of treadmill running at 45% of maximal running speed: VEndoRa (D), VPYGL (E), VAldo (F), VGK (G), VEnol (H), VPK+ME (I), VLDH (J), VPC (K), VPCK (L), VCS (M), VPCC (N), and VSDH (O). Data are mean ± S.E. (error bars); n = 6–9 mice/group. *, p < 0.05 versus WT at a specified time point. †, p < 0.05 versus 0-min time point within group.
AMPK influences liver nutrient partitioning and energy status under sedentary, short-term fasted conditions
Changes in hepatic molecular mediators of metabolism and metabolites were observed in the absence of differences in liver nutrient fluxes between genotypes following 8.5 h of food and water withdrawal. Immunoblotting was performed to confirm deletion of AMPKα subunits (Fig. 4A). Glycogen synthase phosphorylation was similar in both groups (Fig. 4B); however, UDP-glucose pyrophosphorylase 2 (UGP2) was reduced (Fig. 4C). This was associated with a decline in liver glycogen levels in KO mice (Fig. 4D). Mice with a deletion of AMPK α1 and α2 subunits displayed a reduction in liver ATP (Fig. 4E), ADP (Fig. 4F), and total adenine nucleotide (TAN) pool (Fig. 4H). Liver AMP was higher in KO mice (Fig. 4G). The alterations in adenine nucleotides resulted in an increase in the AMP/ATP ratio (Fig. 4I) and a lower energy charge (Fig. 4J) in KO livers. KO mice showed a lower phosphorylated acetyl-CoA carboxylase (pACCS79)/ACC ratio (Fig. 4K). KO mice exhibited changes in liver lipid characteristics (Fig. 4, L–O). Specifically, triglycerides were higher, and phospholipids were reduced with the loss of AMPK α1 and α2 (Fig. 4, L and N).
Figure 4.

Metabolites and molecular mediators of metabolism in sedentary mice lacking hepatic AMPKα. Livers from WT mice and mice exhibiting a liver-specific deletion of AMPK α1 and α2 subunits (KO) were obtained from non-catheterized mice on a stationary treadmill at the 0-min time point. A, liver AMPKα as determined by immunoblotting and representative immunoblot of hepatic AMPKα and β-actin. B, liver phosphorylated glycogen synthase/total glycogen synthase ratio (pGSS641/GS), as determined by immunoblotting and representative immunoblot of hepatic pGSS641, total GS, and β-actin. C, liver UGP2, as determined by immunoblotting and representative immunoblot of hepatic UGP2 and β-actin. D, liver glycogen content (mg g−1) in WT and KO mice. Shown are liver adenine nucleotides ATP (E), ADP (F), AMP (G), and total adenine nucleotide pool (TAN = ATP + ADP + AMP; μmol g−1) (H). I, liver AMP/ATP; J, energy charge ([ATP + 0.5 ADP]/[TAN]). Markers of lipid metabolism were evaluated in WT and KO mice. K, liver phosphorylated acetyl-CoA carboxylase/total acetyl-CoA carboxylase ratio (pACCS79/ACC) as determined by immunoblotting and representative immunoblot of hepatic pACCS79, total ACC, and β-actin. Shown are liver lipids (μg mg−1), including triglycerides (L), diglycerides (M), phospholipids (N), and cholesterol esters (O). Data are mean ± S.E. (error bars); n = 6–10 mice/group. *, p < 0.05 versus WT.
Altered metabolites are retained following exercise in mice lacking liver AMPK
Deletion of hepatic AMPKα subunits was confirmed by immunoblotting (Fig. 5A). Liver glycogen was reduced in the KO mice following 30 min of exercise (Fig. 5B). Energy charge were similar between groups (Fig. 5C). Liver ATP was comparable between genotypes (Fig. 5D). ADP (Fig. 5E), AMP (Fig. 5F), and TAN (Fig. 5G) were lower in livers lacking AMPKα1α2 at the completion of the treadmill run. Triglycerides (Fig. 5H), diglycerides (Fig. 5I), and cholesterol esters (Fig. 5K) were comparable in WT and KO mice. Phospholipids remained lower in the KO mice after exercise (Fig. 5J).
Figure 5.

Metabolites in mice lacking hepatic AMPKα following exercise. Livers from WT mice and mice exhibiting a liver-specific deletion of AMPK α1 and α2 subunits (KO) were obtained from catheterized mice at the 30-min time point following completion of the stable isotope infusions and treadmill running bout (45% of maximal running speed). A, liver AMPKα as determined by immunoblotting and representative immunoblot of hepatic AMPKα and β-actin. B, liver glycogen content (mg g−1). C, energy charge ([ATP + 0.5 ADP]/[TAN]). Shown are hepatic adenine nucleotides ATP (D), ADP (E), AMP (F), and total adenine nucleotide pool (TAN = ATP + ADP + AMP; μmol g−1) (G). Shown are liver lipids (μg mg−1), including triglycerides (H), diglycerides (I), phospholipids (J), and cholesterol esters (K). Data are mean ± S.E. (error bars); n = 6–9 mice/group. *, p < 0.05 versus WT.
AMPK affects liver nutrient partitioning and energy status in long-term fasted and refed mice
The lower glycogen before the exercise bout in KO mice prompted fasting-refeeding experiments to provide insight into the ability of mice lacking liver AMPKα1α2 to synthesize liver glycogen. Deletion of hepatic AMPKα subunits is represented by immunoblotting (Fig. 6A). Liver glycogen was depleted and comparable between genotypes following 24 h of food withdrawal (Fig. 6B). Liver glycogen increased in both groups following 6 h of refeeding compared with fasted levels (Fig. 6B). However, the increase in glycogen stores were attenuated in the KO mice compared with the WT mice in the refed condition (Fig. 6B). Energy charge was comparable between groups (Fig. 6C). No genotype or refeeding effect was observed in ATP (Fig. 6D), ADP (Fig. 6E), and AMP (Fig. 6F). Within the KO mice, TAN was modestly depressed after 6 h of refeeding compared with the 24 h of fasting (Fig. 6G). Triglycerides were lower in KO mice following the 24-h fast compared with WT mice (Fig. 6H). Within the WT mice, the refed conditions exhibited lower liver triglycerides (Fig. 6H), diglycerides (Fig. 6I), phospholipids (Fig. 6J), and total cholesterol esters (Fig. 6K). Cholesterol esters were higher in livers lacking AMPKα compared with control livers in the 24-h fasted conditions (Fig. 6K). 6 h of food availability following the long-term fast lowered cholesterol esters in the KO mice (Fig. 6K).
Figure 6.

Metabolites in fasted and refed mice lacking hepatic AMPKα. Livers from WT mice and mice exhibiting a liver-specific deletion of AMPK α1 and α2 subunits (KO) were obtained from non-catheterized, sedentary mice following 24-h fasted and 24-h-fasted–6-h-refed conditions. A, liver AMPKα as determined by immunoblotting and representative immunoblot of hepatic AMPKα and β-actin. B, liver glycogen content (mg g−1). C, energy charge ([ATP + 0.5 ADP]/[TAN]). Shown are hepatic adenine nucleotides ATP (D), ADP (E), AMP (F), and total adenine nucleotide pool (TAN = ATP + ADP + AMP; μmol g−1) (G). Shown are liver lipids (μg mg−1), including triglycerides (H), diglycerides (I), phospholipids (J), and cholesterol esters (K). Data are mean ± S.E. (error bars); n = 6–7 mice/group. *, p < 0.05 versus WT within fasting conditions. †, p < 0.05 versus 24-h fast within genotype.
Mice lacking hepatic AMPKα1α2 subunits showed changes in fibroblast growth factor 21 (FGF21) physiology
Previous studies in short-term fasted mice lacking liver AMPKα showed metabolic characteristics of long-term food withdrawal (29). FGF21 is secreted by the liver in response to starvation and exercise (30, 31). Furthermore, genetically engineered mice indicate that FGF21 promotes gluconeogenesis (32). Interestingly, livers obtained from sedentary mice in the treadmill at the 0-min time point displayed lower hepatic FGF21 protein as determined by immunoblotting in KO mice compared with the WT mice (Fig. 7A). Hepatic FGF21 was reduced in KO mice compared with WT mice following treadmill running as well (Fig. 7B). To exclude the possibility that the treadmill belt and enclosure may cause a unique FGF21 response, a separate cohort of sedentary, 9-h fasted mice were studied outside the treadmill in a 1-liter plastic container with bedding. This cohort of KO mice displayed elevated liver FGF21 mRNA (Fig. 7C), reduced liver FGF21 protein (Fig. 7D), and increased plasma FGF21 (Fig. 7E) in relation to WT mice. Deletion of hepatic AMPKα subunits was confirmed by immunoblotting (Fig. 7F). The increased plasma FGF21 in KO mice was paralleled by a paradoxical decline in potential mediators of FGF21 action. Hepatic phosphorylation of MEK1/2 (Fig. 7G) and its downstream target, ERK1/2 (Fig. 7H), was reduced in KO mice. Phosphorylation of ERK substrate, the p90 ribosomal S6 kinases (p90RSK), was not statistically different between groups (Fig. 7I). Plasma glucose (Fig. 7J) and insulin (Fig. 7K) were similar in WT and KO mice.
Figure 7.

Altered FGF21 characteristics and dysregulated hepatic FGF21 signaling in sedentary mice lacking hepatic AMPKα. A, liver FGF21 as determined by immunoblotting and representative immunoblot of hepatic FGF21 and β-actin. Livers taken at time = 0 min from non-catheterized, WT mice and mice exhibiting a liver-specific deletion of AMPK α1 and α2 subunits (KO) placed on an enclosed, stationary treadmill. B, liver FGF21 as determined by immunoblotting and representative immunoblot of hepatic FGF21 and β-actin. Livers were taken following stable isotope infusions and 30 min of treadmill running at 45% of maximal running speed from catheterized WT and KO mice. A separate cohort of sedentary, catheterized WT and KO mice fasted for 9 h, not in an enclosed treadmill, was studied to evaluate FGF21 characteristics. C, liver FGF21 mRNA. D, liver FGF21 as determined by immunoblotting and representative immunoblot of hepatic FGF21 and β-actin. E, plasma FGF21. F, liver AMPKα as determined by immunoblotting and representative immunoblot of hepatic AMPKα and β-actin. G, liver phosphorylated MEK1/2-to-total MEK1/2 ratio (pMEKS217/221/MEK1/2) as determined by immunoblotting in and representative immunoblot of hepatic pMEKS217/221, total MEK1/2, and β-actin. H, liver phosphorylated ERK1/2-to-total ERK1/2 ratio (pERKT202/Y204/ERK1/2) as determined by immunoblotting in and representative immunoblot of hepatic pERKT202/Y204, total ERK1/2, and β-actin. I, liver phosphorylated p90RSK/total p90RSK ratio (pp90RSKS380/p90RSK) as determined by immunoblotting and representative immunoblot of hepatic pp90RSKS380, p90RSK, and β-actin. J, plasma glucose (mm). K, plasma insulin (ng ml−1). Data are mean ± S.E. (error bars); n = 6–11 mice/group. *, p < 0.05 versus WT.
Discussion
The predominant hypothesis concerning AMPK's role in the control of endogenous glucose production is that it impedes glucose release from the liver. Deletion of the primary AMPK kinase, liver kinase B1, in the liver of mice results in hyperglycemia (33). Moreover, LKB/AMPK-associated signaling pathways have been reported to negatively regulate the expression of genes involved in glucose production, such as glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) (33–36). Support for the aforementioned hypothesis also arises from the experimental use of biguanides and 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR). These pharmacological agents concurrently activate AMPK and lower glucose release from hepatocytes or blood glucose in vivo (22, 37, 38). Although more recently a growing body of research indicates that the ability of biguanides and AICAR to inhibit hepatic glucose production occurs in an AMPK-independent manner (22, 39, 40).
Similar to these pharmaceutical agents, exercise promotes energy discharge and an increase in AMPK phosphorylation (11). However, this physiological stressor is associated with an increase in glucose release from the liver. The current study aimed to test the requirement for hepatic AMPK to maintain glucose homeostasis through the promotion of endogenous glucose production and associated nutrient fluxes during exercise. A recently developed in vivo 2H/13C MFA technique (41) was used to characterize intermediary metabolism fluxes in the conscious C57Bl/6J mouse during short-term treadmill running at 45% of their maximal running speed. Further experiments utilized mice with a liver-specific deletion of AMPKα1α2 subunits. The results indicate that deletion of liver AMPK α subunits in mice impairs glycogen deposition. This compromises the ability of these mice to increase glycogenolysis (VPYGL) as needed to maintain euglycemia. Loss of hepatic AMPK catalytic subunits results in a decline in ATP and increase in AMP/ATP ratio; however, gluconeogenic and CAC-related responses to the 30-min treadmill running bout are not influenced by genotype.
Under sedentary conditions, arterial glucose concentration and endogenous glucose production were comparable between WT and KO mice. This is consistent with previous studies in which AMPKα and AMPKβ subunits were deleted (22, 39, 42, 43). During an acute exercise bout, endogenous glucose production (VEndoRa) and blood glucose were lower in KO mice. The diminished increment in VPYGL in response to treadmill running in mice lacking hepatic AMPKα1α2 indicates that lower glycogenolysis contributed to reduced endogenous glucose production and impaired glucose homeostasis during muscular work.
In vitro work has shown that AMPK phosphorylates protein targeting to glycogen (R5/PTG), promoting the degradation of this PP1-interacting protein in a laforin-malin–dependent manner (44). Also, AMPK catalytic activity positively regulates the formation of the laforin-malin complex (45). These AMPK actions could facilitate greater glycogen mobilization and contribute to the enhanced glycogenolysis during exercise in mice with hepatic AMPK. However, the reduction in glycogen flux to glucose 6-phosphate precipitated during exercise in KO mice was associated with diminished hepatic glycogen before commencing exercise (8.5-h fast) and upon completion of the 30-min treadmill run. As such, the lower glycogenolytic response to exercise is more likely the result of limited glycogen as a substrate.
Glycogen content is a product of formation and degradation processes. The metabolic cycle of UDP-glucose insertion into glycogen granules and generation of glucose 6-phosphate from glycogen is centered around glycogen synthase and glycogen phosphorylase function, respectively (46). Hepatic glycogen was comparable between genotypes following a 24-h fast. However, a reduction in hepatic glycogen content was observed in AMPKα1α2-deficient livers from 24-h fasted, 6-h refed mice. This suggests that altered hepatic glycogen deposition occurs in the absence of AMPKα1α2. Phosphorylation of the (Ser-641) phosphorylation site, which inhibits enzymatic activity, was not different between genotypes. It is conceivable that another phosphorylation site with inhibitory action is increased (47).
In addition to covalent modification of glycogen synthase, alterations in energy status resulting from loss of AMPKα1α2 hold the potential to influence glycogen synthase activity via allosteric regulation. Glycogen synthase activity diminishes with declines in adenylate energy charge (48). KO mice had lower hepatic energy charge before the onset of exercise. Previous work showed that the energy discharge exhibited by hepatic AMPK deletion was associated with lower glycogen accumulation in response to AICAR infusion (22). In contrast, fasted–refed mice showed that absolute concentrations of hepatic adenine nucleotides and relative energy state ratios were comparable between genotypes despite the persistent reduction in liver glycogen content in AMPK-null livers. These results suggest that the lower glycogen is not mediated by changes in energy state.
Previous studies have identified glucokinase protein as being reduced in hepatocytes lacking AMPKα1α2 (49). This may influence substrate for glycogen deposition. In the current study, a reduction in the provision of glycogen precursors could impact glycogen storage. Liver UGP2 protein was reduced in KO mice. UGP is an enzyme that catalyzes the generation of UDP-glucose, the immediate precursor for glycogen formation (50). Mice overexpressing and lacking AMPKγ3 exhibit an increase and decrease in UGP transcript levels, respectively (51). Muscle glycogen is positively correlated to UGP and AMPK in swine with mutated activation of AMPKγ subunits (52–54). The roles of covalent modification, allosteric control, and substrate availability in mediating the alterations in liver glycogen homeostasis in the absence of AMPK remain to be fully clarified.
As indicated previously, many in vivo and in vitro studies employing genetic tools have implicated AMPK as a negative regulator of gluconeogenesis. This hypothesis is supported by findings in mice with short-term overexpression of constitutively active AMPKα2 in the liver (34). PEPCK and G6Pase mRNA were reduced 48 h following adenovirus-mediated gene transfer of constitutively active AMPKα2 in the liver of mice (34). These molecular changes were associated with lower blood glucose (34). In agreement, 12-week-old mice with hepatic deletion of AMPKα2 displayed elevated endogenous glucose production following 6 h of fasting (55). Interestingly, PEPCK and G6Pase mRNA were not altered by loss of AMPKα2; however, the enzyme activities were elevated (55). In contrast, recent work has added to the growing body of literature refuting such a regulatory role for liver AMPK in glucose homeostasis. We recently reported that 15-week-old mice lacking liver AMPKα1α2 subunits had no effect on endogenous glucose production or gluconeogenic flux following 7 h of fasting (29).
The present study further evaluated the influence of AMPK on gluconeogenesis under acute physiological conditions (8.5–9 h of fasting and acute exercise) in 15–16-week-old mice. WT and KO mice exhibited comparable contributions of PEP and glycerol to glucose production, suggesting that AMPK does not exert control over gluconeogenesis. Means of gluconeogenic regulation include the efficiency of intrahepatic conversion of gluconeogenic precursors to glucose as well as substrate supply and hepatic gluconeogenic precursor extraction (9). The absence of an increase in gluconeogenesis in sedentary, postabsorptive mice lacking hepatic AMPKα1α2 may be related to the altered energy state attenuating the efficacy of gluconeogenic reactions. Our previous work identified integrative mitochondrial oxidative phosphorylation as being impaired in livers lacking AMPK (22). The current study showed a reduction in ATP, TAN, and energy charge in KO mice. The impaired capacity for ATP provision may have prevented uncontrolled CAC-related and gluconeogenic reactions during sedentary conditions. During exercise, hepatic extraction of gluconeogenic precursors may create an abundance of gluconeogenic precursor availability, which may have masked the molecular control AMPK has over CAC-related and gluconeogenic fluxes.
Our previous work using [13C3]propionate at the same dose as in the present study shows no effect on cataplerosis (VPCK) or gluconeogenic flux from PEP (VEnol) (41). Propionate is a precursor of succinyl-CoA. The 2H/13C MFA is sufficiently robust to detect an increase in CAC fluxes (VCS and VSDH) (41). It is important to note that using this methodology, a higher VCS in liver-specific AMPK KO mice compared with WT mice following a 19-h fast was shown (29). Thus, the method is sensitive to experimental and physiological differences in CAC and gluconeogenic fluxes between WT and KO mice. We could use a lower-dose [13C3]propionate if it were necessary, but there would be lower confidence in the flux estimates (41).
KO livers were shown to have reduced glycogen and increased triglycerides at 8.5 h but not 24 h of fasting. The short-term fasting phenotype resembles that of a longer fast (29), suggesting that the KO mice transition to the long-term fasted state quicker than the WT mice following food withdrawal. Extended fasting promotes the expression and secretion of FGF21 from the liver (56). Moreover, FGF21 stimulates gluconeogenesis in the fasted state when insulin is low (32, 57). We hypothesized that hepatic FGF21 action may compensate for the absence of AMPK and preserve appropriate gluconeogenic response in KO mice. Indeed, liver FGF21 mRNA and plasma FGF21 were higher in KO mice. Interestingly, however, FGF21 action seems to be attenuated in KO livers, as indicated by a reduction in MEK1/2 and ERK1/2 phosphorylation. Of note, constitutive activation of hepatic MEK1 and deletion of hepatic ERK1/2 increases and decreases blood glucose in mice, respectively (58). In light of these results, loss of AMPK may impede hepatic MEK/ERK phosphorylation by FGF21 and, subsequently, prevent increased gluconeogenesis. FGF21 administration has been shown to induce AMPK phosphorylation and regulate similar metabolic pathways (59, 60). Alternatively, AMPK and FGF21 action may converge on MEK/ERK signaling. Previous studies have reported that under conditions of energetic stress, such as that induced by AICAR, AMPK is upstream and required for full phosphorylation and activation of the MEK/ERK signaling pathway (61, 62). The apparent FGF21 resistance in livers with attenuated AMPK activation bears a striking resemblance to phenotypes observed in obesity and diabetes models (13, 63).
The use of stable isotopes to probe metabolic systems has strengths and limitations (64, 65). We have performed extensive characterization of 2H/13C MFA using different doses of [13C3]propionate (41). We showed that a range of [13C3]propionate doses have no effects on plasma glucose and hepatic glucose production. This is in agreement with previous studies (66). With this foundation, we used [13C3]propionate in the present study to assess the requirement of AMPK for glucose production and associated nutrient fluxes during exercise. Indeed, propionate can increase pyruvate carboxylase activity (67). Perry et al. (68) recently reported that combining intra-arterial infusion of propionate with an intra-arterial infusion of mass quantities of a precursor for this enzyme ([3-13C]lactate) increases CAC intermediates, endogenous glucose production, and circulating glucose, in the presence of a 6.5-fold increase in plasma insulin. Such effects do not occur with propionate infusion alone. This is consistent with the absence of reported plasma glucose or insulin when they gave propionate alone (68). Clearly combining intra-arterial propionate with an exogenous intra-arterial lactate load causes profound insulin resistance when the metabolic demand of the organism does not match the increment in anaplerotic flux. A confounding aspect of the experimental design of Perry et al. (68) is that propionate and lactate were infused into a carotid artery catheter, where it has a natural first pass to the contralateral carotid artery and other arteries that perfuse the brain. It is possible that the solutes or volume load of multiple infusates can evoke a response that contributes to the effects of combining propionate with a lactate load.
Energy status is an important regulator of glucose metabolism (69, 70). This has prompted substantial scientific investigation into the energy sensor, AMPK, to understand its role in metabolic control (71). The studies presented here aimed to evaluate the AMPK-dependent actions on endogenous glucose production during the physiological energy stress of acute exercise. Initial studies employed 2H/13C MFA in C57Bl/6J mice to determine glucose-producing and -associated nutrient fluxes throughout a 30-min treadmill run. Endogenous glucose production (VEndoRa), glycogenolysis (VPYGL), and gluconeogenesis from PEP (VEnol) were increased at 10, 20, and 30 min of exercise. Concurrent increases in cataplerosis (VPCK), CAC fluxes (VCS and VSDH), and anaplerosis-related fluxes (VPC, VPCC, and VLDH) were shown. Subsequent exercise experiments used mice with a liver-specific deletion of AMPKα subunits. AMPK KO mice were unable to maintain euglycemia during treadmill running. VEndoRa and VPYGL were lower in KO mice. However, VEnol and downstream fluxes were comparable between WT and KO mice throughout muscular work. In conclusion, hepatic AMPK is necessary for the glycogenolytic, but not the gluconeogenic, response to exercise.
Experimental procedures
Animal models
Procedures were approved by the Vanderbilt University Animal Care and Use Committee. 13-Week-old male C57Bl/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and underwent surgical procedures at 14 weeks of age. Mice exhibiting a liver-specific knock-out of AMPK α1 and α2 subunits were generated by crossing mice containing floxed AMPK α1 and α2 subunits on a C57Bl/6J background with AlbCre mice (B6.Cg-Tg(Alb-cre)21Mgn/J). Liver-specific deletion of AMPK α1 and α2 subunits was assessed by genotyping and/or immunoblotting. Floxed AMPKα1α2 mice were used as WT mice. Mice underwent surgical procedures at 14 weeks of age if used for stable isotope infusion studies or for experiments requiring the acquisition of arterial samples. All mice received PicoLab® Laboratory Rodent Diet (5L0D, Purina, Richmond, IN) and had free access to water in temperature- and humidity-controlled housing maintained on a 12-h/12-h light/dark cycle.
Exercise stress test
An exercise stress test was performed to determine maximal running speed, as described previously (72). Briefly, mice were acclimatized to running on an enclosed single-lane treadmill by completing a 10-min exercise bout at 10 m min−1 (0% incline) a day before the exercise stress test and 2 days before surgical procedures. To determine maximal running speed, mice were placed in an enclosed single-lane treadmill (Columbus Instruments, Columbus, OH). Following a 10-min sedentary period, mice initiated running at 10 m min−1 (0% incline). The treadmill speed was increased every 3 min by 4 m min−1 until mice reached exhaustion. Exhaustion was defined as the time point when mice remained on the shock grid at the back of the treadmill for >5 s.
Surgical procedures
At 14 weeks of age, mice underwent catheterization procedures, as performed previously (73). Briefly, the jugular vein and carotid artery were catheterized for venous infusions and arterial sampling, respectively. The free end of each catheter was exteriorized at the back of the neck, flushed with 200 units/ml heparinized saline and 5 mg/ml ampicillin, and sealed with stainless steel plugs. Following surgery, the mice were housed individually and allowed 7–8 days of postoperative recovery before an acclimation treadmill running bout of 10 min at 45% of their predetermined maximal running speed (0% incline). Two days later, the stable isotope infusion studies were performed. All mice were within 10% of presurgical weight on the day of stable isotope infusions. An age-matched cohort of WT and KO mice had only arterial catheters inserted for blood glucose, plasma insulin, and plasma FGF21 measurements at 9 h of fasting. These mice did not undergo treadmill experiments.
Stable isotope infusions
See Fig. 8 for a schematic representation of the in vivo infusion procedures and timeline. Within an hour of the start of the light cycle, food and water were withdrawn for the remainder of the study (9 h). Four hours into the fast, mice were placed in an enclosed single-lane treadmill (Columbus Instruments), and the exteriorized catheters were connected to catheter leads and infusion syringes. At 5 h into the fast, an 80-μl arterial blood sample was obtained to determine natural isotopic enrichment of plasma glucose. Immediately following this sample, a quantitative stable isotope delivery to increase isotopic enrichment above natural isotopic labeling was initiated as performed previously (41). Briefly, a 2H2O (99.9%)-saline bolus was infused for 25 min to enrich total body water to 4.5%. [6,6-2H2]Glucose (99%) was dissolved in the 2H2O-saline bolus to provide a prime (440 μmol kg−1). An independent, continuous infusion of [6,6-2H2]glucose (4.4 μmol kg−1 min−1) was started following the 2H2O-saline bolus and [6,6-2H2]glucose prime. A primed (1.1 mmol kg−1), continuous (0.055 mmol kg−1 min−1) intravenous administration of [13C3]propionate (99%, sodium salt) was initiated 2 h after the 2H2O bolus and [6,6-2H2]glucose prime. Four 100-μl arterial blood samples were obtained 90–120 min following the [13C3]propionate bolus (time = 0–30 min) to determine arterial blood glucose and plasma NEFA levels as well as to perform 2H/13C MFA to determine endogenous glucose and associated nutrient fluxes. The sample taken at 90 min following the [13C3]propionate bolus (time = 0 min) was obtained while mice were in a sedentary state on a stationary treadmill. Samples taken 100–120 min following the [13C3]propionate bolus (time = 10–30 min) were obtained while mice were in a sedentary state or performing an acute treadmill running bout at 45% of their predetermined maximal running speed. Plasma samples were stored at −20 °C until analysis. Donor erythrocytes were provided throughout the duration of the study to ensure that hematocrit did not fall >10%. Immediately following the final sample, mice were removed from the treadmill and sacrificed by cervical dislocation. Liver tissues were rapidly excised, freeze-clamped in liquid nitrogen, and stored at −80 °C until analysis. Stable isotopes were purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA). Each infusate was prepared in a 4.5% 2H2O-enriched saline solution unless otherwise specified.
Figure 8.

Schematic representation of experimental procedures and timeline. Stable isotope infusions and an acute treadmill running bout were performed following 9–10 days of recovery from arterial and jugular catheterization surgeries. At 210 min before the treadmill running bout, a 2H2O bolus was administered into the venous circulation to enrich total body water at 4.5%. Simultaneously, a 440 μmol kg−1 min−1 [6,6-2H2]glucose prime was infused followed by a continuous 4.4 μmol kg−1 min−1 [6,6-2H2]glucose infusion. Ninety minutes before the onset of exercise, a 1.1 mmol kg−1 min−1 prime and 0.055 mmol kg−1 min−1 continuous infusion of [13C3]propionate was initiated. Donor erythrocytes were administered to prevent a decline in hematocrit. Arterial samples were obtained before stable isotope infusion as well as during 30-min sedentary and treadmill running (45% of maximal running speed) conditions for 2H/13C metabolic flux analysis of hepatic intermediary metabolism.
Glucose derivatization and GC-MS analysis
40 μl of plasma from the −210-, 0-, 10-, 20-, and 30-min time points was processed to obtain di-O-isopropylidene propionate, aldonitrile pentapropionate, and methyloxime pentapropionate derivatives of glucose as described previously (41, 74). GC-MS analysis was completed as described previously (41) with minor modifications. A custom MATLAB function was used to integrate derivative peaks to obtain MIDs for six fragment ions. The following fragment ion ranges were used for determining uncorrected MIDs: aldonitrile, m/z 173–178, 259–266, 284–291, and 370–379; methyloxime, m/z 145–149; di-O-isopropylidene, m/z 301–314.
2H/13C metabolic flux analysis
A detailed description of the in vivo metabolic flux analysis methodology employed in these studies has been provided (41). Briefly, a reaction network was constructed using the INCA software package (75) (http://mfa.vueinnovations.com/mfa).3 The reaction network defined the carbon and hydrogen transitions for biochemical reactions linking hepatic glucose production and associated intermediary metabolism reactions. Given the model functions based on steady-state isotopomer balances, it does not depend on metabolite pool sizes. Flux through each network reaction was estimated relative to VCS (fixed at 100) by minimizing the sum of squared residuals between simulated and experimentally determined MIDs of the six fragment ions described previously (41, 76). The formulation of the isotopomer balance equations used to simulate the fragment MIDs has been thoroughly detailed by Antoniewicz et al. (77). Flux estimates were repeated 50 times from random initial values. Goodness of fit was assessed by a χ2 test. Confidence intervals of 95% were determined as described previously (41, 76). Fits were accepted according to a χ2 test (p = 0.05) with 34 degrees of freedom. The infusion rate of [6,6-2H2]glucose (VInf) and mouse weights were used to determine absolute values.
Blood, plasma, and tissue metabolite analyses
Arterial blood glucose was measured using an Accu-Chek® glucometer (Roche Diagnostics). Plasma NEFAs were measured via an enzyme-linked spectrophotometric assay (NEFA C kit, Wako Chemicals USA Inc., Richmond, VA). Liver glycogen was determined enzymatically from tissue homogenates as described previously (78). Hepatic adenine nucleotides were extracted and measured as performed previously (22). Energy charge is defined by the equation, energy charge = ([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP]). Liver lipids, including triglycerides, diglycerides, phospholipids, and cholesterol esters, were measured as described previously (29). A cohort of 15–16-week-old, uncatheterized, male WT and KO mice were placed in an enclosed, stationary single-lane treadmill (Columbus Instruments) 4 h into a fast and were sacrificed for liver glycogen, lipids, adenine nucleotides, and immunoblotting in the sedentary state at 8.5 h of fasting (equivalent to the time = 0 min of infusion studies). A cohort of 15–16-week-old, uncatheterized, male WT and KO mice were 24-h fasted and 24-h fasted–6-h refed. These mice were sacrificed to evaluate liver glycogen, lipids, and adenine nucleotides. A separate cohort of age-matched, male C57Bl/6J mice were studied to determine plasma insulin and glucagon concentrations in response to exercise. Plasma insulin was evaluated by a double antibody method as performed previously (79). Plasma glucagon was assessed as reported (80).
Immunoblotting
Liver tissue was homogenized in an extraction buffer containing 20 mm NaCl, 20 mm Tris-HCl, 0.1 mm EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% β-mercaptoethanol (pH 7.4), and a protease and phosphatase inhibitor mixture (Thermo Fisher Scientific). Liver homogenate was centrifuged (15 min at 800 × g and 4 °C), and the supernatant protein concentration was assessed using the Bradford method (Bio-Rad). Liver (20–40 μg) proteins were denatured and reduced at 70 °C, separated on a NuPAGE 4–12% BisTris gel (Invitrogen), and transferred to a PVDF membrane. Membranes were probed with the following antibodies: phospho-acetyl-CoA carboxylase (Ser-79) (Cell Signaling Technology (Danvers, MA), 3661, lot 10; 1:1000 dilution), acetyl-CoA carboxylase (Cell Signaling Technology, 3662, lot 4; 1:1000 dilution), AMPKα (Cell Signaling Technology, 2532, lot 19; 1:1000 dilution), phospho-p44/42 MAPK (ERK1/2) (Thr-202/Tyr-204) (Cell Signaling Technology, 9101, lot 28; 1:1000 dilution), p44/42 MAPK (ERK1/2) (Cell Signaling Technology, 9102, lot 26; 1:1000 dilution), phospho-glycogen synthase (Ser-641) (Cell Signaling Technology, 3891, lot 2; 1:1000 dilution), glycogen synthase (Cell Signaling Technology, 3893, lot 4; 1:1000 dilution), anti-FGF21 antibody (EPR8314(2)) (abcam (Cambridge, MA), ab171941, lot GR207975-17; 1:1000 dilution), phospho-MEK1/2 (Ser-217/221) (41G9) (Cell Signaling Technology, 9154, lot 14; 1:1000 dilution), MEK1/2 (47E6) (Cell Signaling Technology, 9126, lot 3; 1:1000 dilution), phospho-p90RSK (Ser-380) (D3H11) (Cell Signaling Technology, 11989, lot 4; 1:1000 dilution), RSK1/RSK2/RSK3 (D7A2H) (Cell Signaling Technology, 14813, lot 1; 1:1000 dilution), and UDP-glucose pyrophosphorylase 2 (Santa Cruz Biotechnology, Inc. (Dallas, TX), sc-292069, lot CO111; 1:1000 dilution). β-Actin antibody (Cell Signaling, 4967, lot 7; 1:2000 dilution) was utilized as a loading control. Following incubation with an HRP-conjugated secondary antibody, the membranes were exposed to a chemiluminescent HRP-substrate (Millipore, Billerica, MA) and imaged using a ChemiDocTM imaging system and Image LabTM software (Bio-Rad). Densitometry was completed using ImageJ software.
Real-time PCR
Liver RNA extraction and reverse transcription were performed as described (81). Real-time PCR was completed with FGF21 and housekeeping gene, β-actin, TaqMan® gene expression assays (Thermo Fisher Scientific). Data were analyzed using the 2−ΔΔCt method (82).
Statistical analyses
SigmaStat® statistical software was used to perform two-tailed t tests, ANOVA, one-way repeated measures ANOVA, and two-way repeated measures ANOVA as appropriate to detect statistical differences (p < 0.05) followed by Bonferroni post hoc tests. Comparisons of interest were SED versus EX and WT versus KO. All data are reported as means ± S.E.
Author contributions
C. C. H., B. V., M. F., J. D. Y., and D. H. W. contributed to the conception and design of experiments. C. C. H., F. D. J., D. P. B., and E. P. D. contributed to data acquisition. C. C. H. analyzed and interpreted data. C. C. H. drafted the manuscript. All authors contributed to revising the manuscript for critically important intellectual content. All authors approved the manuscript for publication.
Acknowledgments
We thank the Vanderbilt University Mouse Metabolic Phenotyping Center for core services (supported by NIDDK, National Institutes of Health, Grant DK059637). We acknowledge the Vanderbilt Diabetes Research and Training Center (supported by NIDDK, National Institutes of Health, Grant DK020593).
This work was supported by NIDDK, National Institutes of Health Grants DK050277 (to D. H. W.), DK054902 (to D. H. W.), and DK106348 (to J. D. Y.) and a Diabetes Canada Postdoctoral Fellowship (to C. C. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- AMPK
- AMP-activated protein kinase
- ACC
- acetyl-CoA carboxylase
- CAC
- citric acid cycle
- EX
- exercising
- GS
- glycogen synthase
- G6Pase
- glucose-6-phosphatase
- MFA
- metabolic flux analysis
- MID
- mass isotopomer distribution
- NEFA
- non-esterified fatty acid
- PEP
- phosphoenolpyruvate
- PEPCK
- phosphoenolpyruvate carboxykinase
- SED
- sedentary
- VAldo
- flux from dihydroxyacetone phosphate and glyceraldhyde 3-phosphate to fructose 1,6-bisphosphate
- VCS
- flux from oxaloacetate and acetyl-CoA to citrate
- VEndoRa
- endogenous glucose production
- VEnol
- flux from PEP to glyceraldehyde 3-phosphate
- VGK
- flux from glycerol to dihydroxyacetone phosphate
- VLDH
- non-PEP derived, unlabeled sources of anaplerosis to pyruvate
- VPC
- flux from pyruvate to oxaloacetate
- VPCC
- flux from propionyl-CoA to succinyl-CoA
- VPCK
- flux from oxaloacetate to PEP
- VPK+ME
- contribution of pyruvate kinase (PK) and malic enzyme (ME) to pyruvate
- VPYGL
- flux from glycogen to glucose 6-phosphate
- VSDH
- flux from succinyl-CoA to oxaloacetate
- UGP2
- UDP-glucose pyrophosphorylase 2
- TAN
- total adenine nucleotide
- AICAR
- 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance.
References
- 1. Wasserman D. H. (2009) Four grams of glucose. Am. J. Physiol. Endocrinol. Metab. 296, E11–E21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Camacho R. C., Galassetti P., Davis S. N., and Wasserman D. H. (2005) Glucoregulation during and after exercise in health and insulin-dependent diabetes. Exerc. Sport Sci. Rev. 33, 17–23 [PubMed] [Google Scholar]
- 3. Lee-Young R. S., Ayala J. E., Hunley C. F., James F. D., Bracy D. P., Kang L., and Wasserman D. H. (2010) Endothelial nitric oxide synthase is central to skeletal muscle metabolic regulation and enzymatic signaling during exercise in vivo. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1399–R1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kang L., Lustig M. E., Bonner J. S., Lee-Young R. S., Mayes W. H., James F. D., Lin C. T., Perry C. G., Anderson E. J., Neufer P. D., and Wasserman D. H. (2012) Mitochondrial antioxidative capacity regulates muscle glucose uptake in the conscious mouse: effect of exercise and diet. J. Appl. Physiol. 113, 1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wasserman D. H., Lacy D. B., Bracy D., and Williams P. E. (1992) Metabolic regulation in peripheral tissues and transition to increased gluconeogenic mode during prolonged exercise. Am. J. Physiol. 263, E345–E354 [DOI] [PubMed] [Google Scholar]
- 6. Wolfe R. R., Nadel E. R., Shaw J. H., Stephenson L. A., and Wolfe M. H. (1986) Role of changes in insulin and glucagon in glucose homeostasis in exercise. J. Clin. Invest. 77, 900–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wasserman D. H., Spalding J. A., Lacy D. B., Colburn C. A., Goldstein R. E., and Cherrington A. D. (1989) Glucagon is a primary controller of hepatic glycogenolysis and gluconeogenesis during muscular work. Am. J. Physiol. 257, E108–E117 [DOI] [PubMed] [Google Scholar]
- 8. Wasserman D. H., Williams P. E., Lacy D. B., Goldstein R. E., and Cherrington A. D. (1989) Exercise-induced fall in insulin and hepatic carbohydrate metabolism during muscular work. Am. J. Physiol. 256, E500–E509 [DOI] [PubMed] [Google Scholar]
- 9. Wasserman D. H., and Cherrington A. D. (1991) Hepatic fuel metabolism during muscular work: role and regulation. Am. J. Physiol. 260, E811–E824 [DOI] [PubMed] [Google Scholar]
- 10. Hems R., Ross B. D., Berry M. N., and Krebs H. A. (1966) Gluconeogenesis in the perfused rat liver. Biochem. J. 101, 284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Camacho R. C., Donahue E. P., James F. D., Berglund E. D., and Wasserman D. H. (2006) Energy state of the liver during short-term and exhaustive exercise in C57BL/6J mice. Am. J. Physiol. Endocrinol. Metab. 290, E405–E408 [DOI] [PubMed] [Google Scholar]
- 12. Viollet B., Horman S., Leclerc J., Lantier L., Foretz M., Billaud M., Giri S., and Andreelli F. (2010) AMPK inhibition in health and disease. Crit. Rev. Biochem. Mol. Biol. 45, 276–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steinberg G. R., and Kemp B. E. (2009) AMPK in health and disease. Physiol. Rev. 89, 1025–1078 [DOI] [PubMed] [Google Scholar]
- 14. Stapleton D., Mitchelhill K. I., Gao G., Widmer J., Michell B. J., Teh T., House C. M., Fernandez C. S., Cox T., Witters L. A., and Kemp B. E. (1996) Mammalian AMP-activated protein kinase subfamily. J. Biol. Chem. 271, 611–614 [DOI] [PubMed] [Google Scholar]
- 15. Verhoeven A. J., Woods A., Brennan C. H., Hawley S. A., Hardie D. G., Scott J., Beri R. K., and Carling D. (1995) The AMP-activated protein kinase gene is highly expressed in rat skeletal muscle: alternative splicing and tissue distribution of the mRNA. Eur. J. Biochem. 228, 236–243 [PubMed] [Google Scholar]
- 16. Xiao B., Heath R., Saiu P., Leiper F. C., Leone P., Jing C., Walker P. A., Haire L., Eccleston J. F., Davis C. T., Martin S. R., Carling D., and Gamblin S. J. (2007) Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449, 496–500 [DOI] [PubMed] [Google Scholar]
- 17. Riek U., Scholz R., Konarev P., Rufer A., Suter M., Nazabal A., Ringler P., Chami M., Müller S. A., Neumann D., Forstner M., Hennig M., Zenobi R., Engel A., Svergun D., et al. (2008) Structural properties of AMP-activated protein kinase: dimerization, molecular shape, and changes upon ligand binding. J. Biol. Chem. 283, 18331–18343 [DOI] [PubMed] [Google Scholar]
- 18. Sanders M. J., Grondin P. O., Hegarty B. D., Snowden M. A., and Carling D. (2007) Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 403, 139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suter M., Riek U., Tuerk R., Schlattner U., Wallimann T., and Neumann D. (2006) Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 281, 32207–32216 [DOI] [PubMed] [Google Scholar]
- 20. Corton J. M., Gillespie J. G., Hawley S. A., and Hardie D. G. (1995) 5-Aminoimidazole-4-carboxamide ribonucleoside: a specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565 [DOI] [PubMed] [Google Scholar]
- 21. Viollet B., Guigas B., Leclerc J., Hebrard S., Lantier L., Mounier R., Andreelli F., and Foretz M. (2009) AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol. 196, 81–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hasenour C. M., Ridley D. E., Hughey C. C., James F. D., Donahue E. P., Shearer J., Viollet B., Foretz M., and Wasserman D. H. (2014) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) effect on glucose production, but not energy metabolism, is independent of hepatic AMPK in vivo. J. Biol. Chem. 289, 5950–5959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guigas B., Taleux N., Foretz M., Detaille D., Andreelli F., Viollet B., and Hue L. (2007) AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem. J. 404, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carlson C. L., and Winder W. W. (1999) Liver AMP-activated protein kinase and acetyl-CoA carboxylase during and after exercise. J. Appl. Physiol. 86, 669–674 [DOI] [PubMed] [Google Scholar]
- 25. Kimball S. R., Siegfried B. A., and Jefferson L. S. (2004) Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J. Biol. Chem. 279, 54103–54109 [DOI] [PubMed] [Google Scholar]
- 26. Hasenour C. M., Berglund E. D., and Wasserman D. H. (2013) Emerging role of AMP-activated protein kinase in endocrine control of metabolism in the liver. Mol. Cell. Endocrinol. 366, 152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Woods H. F., Eggleston L. V., and Krebs H. A. (1970) The cause of hepatic accumulation of fructose 1-phosphate on fructose loading. Biochem. J. 119, 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Woods H. F., and Krebs H. A. (1973) The effect of glycerol and dihydroxyacetone on hepatic adenine nucleotides. Biochem. J. 132, 55–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hasenour C. M., Ridley D. E., James F. D., Hughey C. C., Donahue E. P., Viollet B., Foretz M., Young J. D., and Wasserman D. H. (2017) Liver AMP-activated protein kinase is unnecessary for gluconeogenesis but protects energy state during nutrient deprivation. PLoS One 12, e0170382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kliewer S. A., and Mangelsdorf D. J. (2010) Fibroblast growth factor 21: from pharmacology to physiology. Am. J. Clin. Nutr. 91, 254S–257S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tanimura Y., Aoi W., Takanami Y., Kawai Y., Mizushima K., Naito Y., and Yoshikawa T. (2016) Acute exercise increases fibroblast growth factor 21 in metabolic organs and circulation. Physiol. Rep. 10.14814/phy2.12828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Potthoff M. J., Inagaki T., Satapati S., Ding X., He T., Goetz R., Mohammadi M., Finck B. N., Mangelsdorf D. J., Kliewer S. A., and Burgess S. C. (2009) FGF21 induces PGC-1α and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. U.S.A. 106, 10853–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., and Cantley L. C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foretz M., Ancellin N., Andreelli F., Saintillan Y., Grondin P., Kahn A., Thorens B., Vaulont S., and Viollet B. (2005) Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 54, 1331–1339 [DOI] [PubMed] [Google Scholar]
- 35. Koo S. H., Flechner L., Qi L., Zhang X., Screaton R. A., Jeffries S., Hedrick S., Xu W., Boussouar F., Brindle P., Takemori H., and Montminy M. (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 36. Mihaylova M. M., Vasquez D. S., Ravnskjaer K., Denechaud P. D., Yu R. T., Alvarez J. G., Downes M., Evans R. M., Montminy M., and Shaw R. J. (2011) Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim Y. D., Park K. G., Lee Y. S., Park Y. Y., Kim D. K., Nedumaran B., Jang W. G., Cho W. J., Ha J., Lee I. K., Lee C. H., and Choi H. S. (2008) Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 57, 306–314 [DOI] [PubMed] [Google Scholar]
- 38. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., and Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Foretz M., Hébrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G., Sakamoto K., Andreelli F., and Viollet B. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miller R. A., Chu Q., Xie J., Foretz M., Viollet B., and Birnbaum M. J. (2013) Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494, 256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hasenour C. M., Wall M. L., Ridley D. E., Hughey C. C., James F. D., Wasserman D. H., and Young J. D. (2015) Mass spectrometry-based microassay of 2H and 13C plasma glucose labeling to quantify liver metabolic fluxes in vivo. Am. J. Physiol. Endocrinol. Metab. 309, E191–E203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dzamko N., van Denderen B. J., Hevener A. L., Jørgensen S. B., Honeyman J., Galic S., Chen Z. P., Watt M. J., Campbell D. J., Steinberg G. R., and Kemp B. E. (2010) AMPK β1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J. Biol. Chem. 285, 115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Steinberg G. R., O'Neill H. M., Dzamko N. L., Galic S., Naim T., Koopman R., Jørgensen S. B., Honeyman J., Hewitt K., Chen Z. P., Schertzer J. D., Scott J. W., Koentgen F., Lynch G. S., Watt M. J., et al. (2010) Whole body deletion of AMP-activated protein kinase β2 reduces muscle AMPK activity and exercise capacity. J. Biol. Chem. 285, 37198–37209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vernia S., Solaz-Fuster M. C., Gimeno-Alcañiz J. V., Rubio T., García-Haro L., Foretz M., de Córdoba S. R., and Sanz P. (2009) AMP-activated protein kinase phosphorylates R5/PTG, the glycogen targeting subunit of the R5/PTG-protein phosphatase 1 holoenzyme, and accelerates its down-regulation by the laforin-malin complex. J. Biol. Chem. 284, 8247–8255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Solaz-Fuster M. C., Gimeno-Alcañiz J. V., Ros S., Fernandez-Sanchez M. E., Garcia-Fojeda B., Criado Garcia O., Vilchez D., Dominguez J., Garcia-Rocha M., Sanchez-Piris M., Aguado C., Knecht E., Serratosa J., Guinovart J. J., Sanz P., and Rodriguez de Córdoba S. (2008) Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 17, 667–678 [DOI] [PubMed] [Google Scholar]
- 46. Shearer J., and Graham T. E. (2004) Novel aspects of skeletal muscle glycogen and its regulation during rest and exercise. Exerc. Sport Sci. Rev. 32, 120–126 [DOI] [PubMed] [Google Scholar]
- 47. Roach P. J. (1990) Control of glycogen synthase by hierarchal protein phosphorylation. FASEB J. 4, 2961–2968 [PubMed] [Google Scholar]
- 48. Roach P. J., and Larner J. (1976) Rabbit skeletal muscle glycogen synthase. II. Enzyme phosphorylation state and effector concentrations as interacting control parameters. J. Biol. Chem. 251, 1920–1925 [PubMed] [Google Scholar]
- 49. Guigas B., Bertrand L., Taleux N., Foretz M., Wiernsperger N., Vertommen D., Andreelli F., Viollet B., and Hue L. (2006) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 55, 865–874 [DOI] [PubMed] [Google Scholar]
- 50. Roach P. J., Depaoli-Roach A. A., Hurley T. D., and Tagliabracci V. S. (2012) Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441, 763–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nilsson E. C., Long Y. C., Martinsson S., Glund S., Garcia-Roves P., Svensson L. T., Andersson L., Zierath J. R., and Mahlapuu M. (2006) Opposite transcriptional regulation in skeletal muscle of AMP-activated protein kinase γ3 R225Q transgenic versus knock-out mice. J. Biol. Chem. 281, 7244–7252 [DOI] [PubMed] [Google Scholar]
- 52. Scheffler T. L., Park S., Roach P. J., and Gerrard D. E. (2016) Gain of function AMP-activated protein kinase γ3 mutation (AMPKγ3R200Q) in pig muscle increases glycogen storage regardless of AMPK activation. Physiol. Rep. 10.14814/phy2.12802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Luptak I., Shen M., He H., Hirshman M. F., Musi N., Goodyear L. J., Yan J., Wakimoto H., Morita H., Arad M., Seidman C. E., Seidman J. G., Ingwall J. S., Balschi J. A., and Tian R. (2007) Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J. Clin. Invest. 117, 1432–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hedegaard J., Horn P., Lametsch R., Søndergaard Møller H., Roepstorff P., Bendixen C., and Bendixen E. (2004) UDP-glucose pyrophosphorylase is upregulated in carriers of the porcine RN− mutation in the AMP-activated protein kinase. Proteomics 4, 2448–2454 [DOI] [PubMed] [Google Scholar]
- 55. Andreelli F., Foretz M., Knauf C., Cani P. D., Perrin C., Iglesias M. A., Pillot B., Bado A., Tronche F., Mithieux G., Vaulont S., Burcelin R., and Viollet B. (2006) Liver adenosine monophosphate-activated kinase-α2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 147, 2432–2441 [DOI] [PubMed] [Google Scholar]
- 56. Badman M. K., Pissios P., Kennedy A. R., Koukos G., Flier J. S., and Maratos-Flier E. (2007) Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 [DOI] [PubMed] [Google Scholar]
- 57. Markan K. R., and Potthoff M. J. (2016) Metabolic fibroblast growth factors (FGFs): mediators of energy homeostasis. Semin. Cell Dev. Biol. 53, 85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiao P., Feng B., Li Y., He Q., and Xu H. (2013) Hepatic ERK activity plays a role in energy metabolism. Mol. Cell. Endocrinol. 375, 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Salminen A., Kauppinen A., and Kaarniranta K. (2017) FGF21 activates AMPK signaling: impact on metabolic regulation and the aging process. J. Mol. Med. 95, 123–131 [DOI] [PubMed] [Google Scholar]
- 60. Zhu S., Ma L., Wu Y., Ye X., Zhang T., Zhang Q., Rasoul L. M., Liu Y., Guo M., Zhou B., Ren G., and Li D. (2014) FGF21 treatment ameliorates alcoholic fatty liver through activation of AMPK-SIRT1 pathway. Acta Biochim. Biophys. Sin. 46, 1041–1048 [DOI] [PubMed] [Google Scholar]
- 61. Wang H. M., Mehta S., Bansode R., Huang W., and Mehta K. D. (2008) AICAR positively regulate glycogen synthase activity and LDL receptor expression through Raf-1/MEK/p42/44MAPK/p90RSK/GSK-3 signaling cascade. Biochem. Pharmacol. 75, 457–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang J., Whiteman M. W., Lian H., Wang G., Singh A., Huang D., and Denmark T. (2009) A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 284, 21412–21424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fisher F. M., Chui P. C., Antonellis P. J., Bina H. A., Kharitonenkov A., Flier J. S., and Maratos-Flier E. (2010) Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes 59, 2781–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Previs S. F., and Kelley D. E. (2015) Tracer-based assessments of hepatic anaplerotic and TCA cycle flux: practicality, stoichiometry, and hidden assumptions. Am. J. Physiol. Endocrinol. Metab. 309, E727–E735 [DOI] [PubMed] [Google Scholar]
- 65. Metallo C. M., Walther J. L., and Stephanopoulos G. (2009) Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J. Biotechnol. 144, 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Burgess S. C., Jeffrey F. M., Storey C., Milde A., Hausler N., Merritt M. E., Mulder H., Holm C., Sherry A. D., and Malloy C. R. (2005) Effect of murine strain on metabolic pathways of glucose production after brief or prolonged fasting. Am. J. Physiol. Endocrinol. Metab. 289, E53–E61 [DOI] [PubMed] [Google Scholar]
- 67. Scrutton M. C. (1974) Pyruvate carboxylase: studies of activator-independent catalysis and of the specificity of activation by acyl derivatives of coenzyme A for the enzyme from rat liver. J. Biol. Chem. 249, 7057–7067 [PubMed] [Google Scholar]
- 68. Perry R. J., Borders C. B., Cline G. W., Zhang X. M., Alves T. C., Petersen K. F., Rothman D. L., Kibbey R. G., and Shulman G. I. (2016) Propionate increases hepatic pyruvate cycling and anaplerosis and alters mitochondrial metabolism. J. Biol. Chem. 291, 12161–12170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Krebs H. (1964) The Croonian Lecture, 1963: gluconeogenesis. Proc. R. Soc. Lond. B Biol. Sci. 159, 545–564 [DOI] [PubMed] [Google Scholar]
- 70. Atkinson D. E. (1969) Regulation of enzyme function. Annu. Rev. Microbiol. 23, 47–68 [DOI] [PubMed] [Google Scholar]
- 71. Hardie D. G., and Hawley S. A. (2001) AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays 23, 1112–1119 [DOI] [PubMed] [Google Scholar]
- 72. Lee-Young R. S., Griffee S. R., Lynes S. E., Bracy D. P., Ayala J. E., McGuinness O. P., and Wasserman D. H. (2009) Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J. Biol. Chem. 284, 23925–23934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ayala J. E., Bracy D. P., Malabanan C., James F. D., Ansari T., Fueger P. T., McGuinness O. P., and Wasserman D. H. (2011) Hyperinsulinemic-euglycemic clamps in conscious, unrestrained mice. J. Vis. Exp. 10.3791/3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Antoniewicz M. R., Kelleher J. K., and Stephanopoulos G. (2011) Measuring deuterium enrichment of glucose hydrogen atoms by gas chromatography/mass spectrometry. Anal. Chem. 83, 3211–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Young J. D. (2014) INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30, 1333–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Antoniewicz M. R., Kelleher J. K., and Stephanopoulos G. (2006) Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab. Eng. 8, 324–337 [DOI] [PubMed] [Google Scholar]
- 77. Antoniewicz M. R., Kelleher J. K., and Stephanopoulos G. (2007) Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab. Eng. 9, 68–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chan T. M., and Exton J. H. (1976) A rapid method for the determination of glycogen content and radioactivity in small quantities of tissue or isolated hepatocytes. Anal. Biochem. 71, 96–105 [DOI] [PubMed] [Google Scholar]
- 79. Morgan C. R., and Lazarow A. (1965) Immunoassay of pancreatic and plasma insulin following alloxan injection of rats. Diabetes 14, 669–671 [DOI] [PubMed] [Google Scholar]
- 80. Otero Y. F., Lundblad T. M., Ford E. A., House L. M., and McGuinness O. P. (2014) Liver but not adipose tissue is responsive to the pattern of enteral feeding. Physiol. Rep. 2, e00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Williams A. S., Trefts E., Lantier L., Grueter C. A., Bracy D. P., James F. D., Pozzi A., Zent R., and Wasserman D. H. (2017) Integrin-linked kinase is necessary for the development of diet-induced hepatic insulin resistance. Diabetes 66, 325–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]