

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most commonly prescribed drugs worldwide—more than 111 million prescriptions were written in the United States in 2014. NSAIDs allosterically inhibit cytosolic sulfotransferases (SULTs) with high specificity and therapeutically relevant affinities. This study focuses on the interactions of SULT1A1 and mefenamic acid (MEF)—a potent, highly specific NSAID inhibitor of 1A1. Here, the first structure of an NSAID allosteric site—the MEF-binding site of SULT1A1—is determined using spin-label triangulation NMR. The structure is confirmed by site-directed mutagenesis and provides a molecular framework for understanding NSAID binding and isoform specificity. The mechanism of NSAID inhibition is explored using molecular dynamics and equilibrium and pre-steady-state ligand-binding studies. MEF inhibits SULT1A1 turnover through an indirect (helix-mediated) stabilization of the closed form of the active-site cap of the enzyme, which traps the nucleotide and slows its release. Using the NSAID-binding site structure of SULT1A1 as a comparative model, it appears that 11 of the 13 human SULT isoforms harbor an NSAID-binding site. We hypothesize that these sites evolved to enable SULT isoforms to respond to metabolites that lie within their metabolic domains. Finally, the NSAID-binding site structure offers a template for developing isozyme-specific allosteric inhibitors that can be used to regulate specific areas of sulfuryl-transfer metabolism.
Keywords: allosteric regulation, kinetics, nuclear magnetic resonance (NMR), structural model, sulfotransferase, NSAID, acetylsalicylic, mefenamic, spin label
Introduction
Tens of billions of NSAID2 tablets are consumed annually in the United States by the more than 65 million adults that routinely consume them (1). Given these statistics, it is perhaps not surprising that NSAIDs are the most prevalent pharmaceutical contaminants in United States aquifers (2). The populace of the United States is under the persistent metabolic influence of these drugs. For more than 3 millennia, humans have consumed NSAIDs to reduce inflammation, pain, and fever (3). These therapeutic properties are attributed to the NSAID inhibition of COX1 and COX2 (4), which are essential for prostaglandin biosynthesis. In addition to traditional therapeutic applications, the antiplatelet activity of NSAIDs has led to their widespread use in the prevention of myocardial infarction, blood clot formation, and cardiovascular disease (5–7), and their anticancer effects (8, 9) (for which there is no known mechanism (10, 11)) recommend them as “anti-cancer drugs for the masses (7).”
Human cytosolic sulfotransferases (SULTs) are allosterically inhibited by NSAIDs (12). SULTs regulate the activities and terminal half-lives of thousands of metabolites, including many signaling small molecules, via transfer of the sulfuryl-moiety (–SO3) from the nucleotide donor (3′-phosphoadenosine 5′-phosphosulfate (PAPS)) to the hydroxyls and amines of acceptors (13). In addition to their homeostatic and detoxifying functions, SULTs catalyze in situ synthesis of carcinogens by sulfonating compounds (typically low-pKa acceptors) that disproportionate into sulfate and highly reactive carbocations that covalently modify protein and DNA (14, 15). It is notable that the risk of prostate cancer is elevated 5–8-fold in individuals that express SULTs at high levels (16), and it is reduced significantly by regular NSAID use (17, 18).
Thirteen SULT isoforms are encoded in the human genome. The substrate specificities of these enzymes are broad, somewhat overlapping, and center on different areas of metabolism. To date, NSAID inhibition of four isoforms has been confirmed: 1A1 (19); 1E1 (20); 1A3 (21); and 2A1 (22). SULT1A1, the focus of this work, is the broadest specificity SULT. 1A1 is found at its highest levels in liver, where it is present in near-gram quantities (23), and in mature enterocytes that line the small intestine (24), where the enzyme screens and detoxifies xenobiotics en route to plasma. The specificities of 1A3, 2A1, and 1E1 are centered, respectively, on catecholamine neurotransmitters (25), hydroxysteroids (26), and estrogens (27). The variable sensitivity of SULTs toward NSAID inhibition (19–21) reveals the uniqueness of the sites and is consistent with the concept that they evolved to bind metabolites that lie within the metabolic purview of the isoforms in which they reside—such is the case with catechin SULT-allosteric sites (28, 29). This study defines, for the first time, the structure of an NSAID allosteric site—the mefenamic acid (MEF)-binding site of SULT1A1—and the molecular mechanism of NSAID inhibition. The structure provides a template for identifying endogenous metabolite allosteres and as a basis for the rational design of isoform-specific SULT allosteric inhibitors that might allow independent control of the numerous and disease-relevant metabolic areas in which SULTs operate.
Results and discussion
Structure of the SULT1A1 MEF-binding site
Overview
We have developed a method that allows the structure of a protein's ligand-binding site to be determined from the 1D solution–NMR spectrum of the ligand (28, 29). The technique involves determining distances from ligand nuclei (typically protons) to the unpaired electrons of three covalently-attached spin labels that are positioned such that each proton can be mapped (± ∼2.5 Å) to the protein surface by triangulation. Proton/spin-label interactions are detectable over distances of ≤ ∼25 Å; thus, each unpaired electron can be considered the center of an “interaction sphere” of diameter ∼50 Å—a dimension comparable with that of many protein subunits. In favorable cases, spin labels can be positioned to “coat” the entire protein surface in a detectable paramagnetic field without altering the protein's catalytic functions. The final structure of the binding site is refined using NMR distance-constrained molecular dynamics (MD) docking.
Spin-label insertion sites
In previous work with SULT1A1 (29), it proved possible to remove reactive cysteines from the wild-type scaffold and to install and spin-label cysteine at six well-separated, solvent-exposed positions without altering the initial-rate parameters of the enzyme. Taken together, the paramagnetic fields of these six constructs provide essentially complete coverage of the enzyme's surface. The line-broadening effects of these constructs on the 1D solution spectrum of MEF were used to approximate the position of the MEF-binding site and to identify the insertion sites to be used in triangulation studies. Two of the selected sites, residues 29 and 234, were previously characterized (29). A third previous site (site 151) (29) was positioned close enough to bound MEF that its solution line widths were broadened beyond the point of accurate measurement. The 1A1 scaffold was studied to identify a possible alternative Cys-insertion point—a geometrically well-positioned, solvent-exposed residue with a stable backbone (i.e. low MD-predicted RMSF). The analysis suggested an E198C conversion would prove a suitable third, spin-label attachment site; consequently, the E198C mutation was inserted into a 1A1. To assess whether Cys insertion and spin-label attachment at positions 29, 198, or 234 alters the catalytic behavior of the enzyme, the initial-rate and MEF-inhibition parameters of the spin-labeled constructs were determined and compared with the wild-type enzyme. The parameters of the labeled-mutant and wild-type enzymes are, within error, indistinguishable (see Table 1).
Table 1.
Initial-rate parameters for WT and spin-labeled SULT1A1 mutants
| Enzyme | kcata | Km, 1-HPa | Ki, MEFa |
|---|---|---|---|
| min−1 | nm | nm | |
| WT | 120 (17)b | 22 (3.1) | 28 (2.5) |
| 29c | 110 (15) | 25 (3.5) | 33 (1.9) |
| 198 | 108 (11) | 28 (2.6) | 25 (1.8) |
| 234 | 109 (15) | 24 (3.2) | 27 (2.6) |
a kcat and Km values were determined at saturating PAPS (500 μm, 17 × Km).
b Values in parentheses indicate 1 S.D.
c Cys residue at which spin label is attached.
Distance measurements
Distances between protein-bound ligand protons and covalently attached spin labels are calculated (using the Solomon Bloembergen equation (30)) from the line broadening effects of the unpaired electron on the proton resonance. When the frequency of ligand exchange between the protein surface and solution is comparable to or greater than the Larmor frequency difference between the bound and free proton, line broadening is observed in the ligand's solution spectrum. In such cases, the observed transverse relaxation (R2 obs), which is calculated from the line width, depends linearly on the fraction of bound ligand (FB) according to Equation 1 (30, 31),
| (Eq. 1) |
where R2B and R3F are the bound- and free-proton relaxation rates, respectively, and R2 ex is the chemical exchange contribution to the relaxation.
Relaxation of a protein-bound ligand proton is enhanced primarily by Brownian rotation of the protein, which causes the proton to experience magnetic field fluctuations at or near its Larmor frequency. For a spin-labeled enzyme, such fluctuations arise from the motion of the unpaired electron and/or nuclear spins. Distance calculations require the contribution of the unpaired electron to the proton relaxation. To isolate the electron's contribution, one constructs observed relaxation (R2 obs) versus fraction ligand-bound (FB) plots for a paramagnetic construct and a diamagnetic homologue, in which the spin-label PROXYL moiety is replaced with a cyclohexyl group (29, 32), and subtracts the slope of the diamagnetic construct plot from that of the paramagnetic construct (33, 34).
The five MEF protons used in the triangulation study are highlighted in red in Fig. 1A, which presents the structure and solution 1H NMR spectrum of MEF (35). These protons span the body of MEF and thus allow the molecule to be oriented relative to the spin labels. The line-broadening effects of the Cys-234 spin label on the H7 peak of MEF are shown as a function of the percent of enzyme-bound MEF in Fig. 1B. To complete the H7 triangulation, similar experiments were performed using the Cys-198 and Cys-29 spin labels and a diamagnetic construct. Line widths were determined by fitting using NMRPipe (36), and the results were used to construct the R2 obs versus FB plots (Fig. 1C). Similar experiments were performed for each of the remaining highlighted protons (Fig. 1A), and the associated R2 obs versus FB plots are presented in Fig. S1. Each titration used in constructing R2 obs versus FB plots was performed in duplicate. The distances used to triangulate the protons, and thus position and orient MEF on the enzyme, were calculated using the Solomon-Bloembergen equation (30) and are compiled in Table 2.
Figure 1.
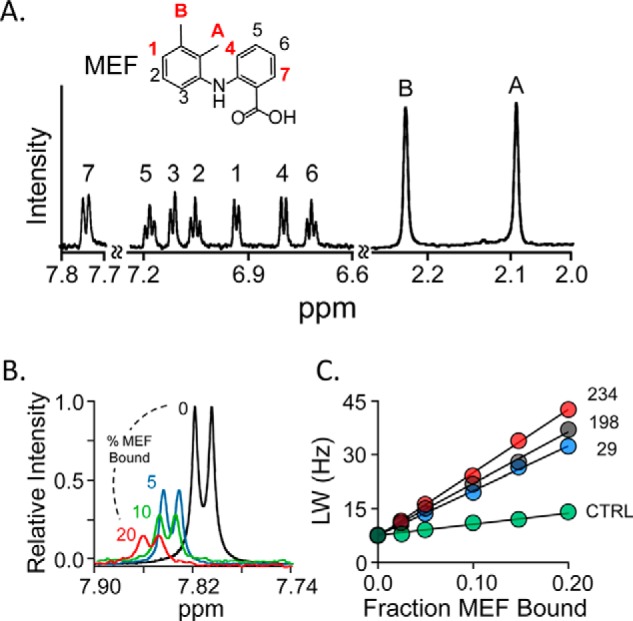
NMR measurements. A, the structure and 600-MHz 1H NMR spectrum of MEF. MEF protons are labeled in the spectrum and structure. Red labels identify the proton positions used in NMR distance measurements. Conditions: MEF (100 μm), KPO4 (50 mm), pD 7.4, D2O (>98%), 25 ± 1 °C. B, spin-label effects on the H7-proton peak. The solution 1H NMR spectrum (600 MHz) of the MEF H7 peak is shown as a function of percent MEF bound to spin-labeled Cys-234-SULT1A1. Conditions: MEF (100 μm), spin-labeled Cys-234-SULT1A1 (0 μm (black), 20 μm (red), 10 μm (green), and 5 μm (blue)), PAP (500 μm, 17 × Kd), KPO4 (50 mm), pD 7.4, 25 ± 1 °C. The enzyme is saturated at all MEF concentrations (Kd MEF = 20 nm). Peak amplitudes are normalized to MEF concentration. C, line-width versus fraction-MEF-bound plot. The effects of paramagnetic spin labels (4-maleimido-PROXYL attached at Cys-234 (red), Cys-198 (black), or Cys-29 (blue) and diamagnetic control (N-cyclohexylmaleimide attached at Cys-234 (green)) on the line width of the H7-proton peak are plotted as a function of the fraction of enzyme-bound MEF. Each dot represents the average of two independent measurements. The straight line through the data represent the least-squares best-fit to the full (not averaged) dataset. Conditions are given in B of this legend.
Table 2.
Proton to spin label distances (Å)
| Proton | Spin-label attachment residue |
||
|---|---|---|---|
| 29 | 198 | 234 | |
| HA | 21 (19–23)a | 17 (15–19) | 21 (19–23) |
| HB | 20 (18–22) | 16 (14–18) | 16 (14–19) |
| H1 | 13 (11–15) | 19 (17–22) | 22 (20–24) |
| H4 | 22 (20–24) | 17 (15–19) | 20 (18–23) |
| H7 | 25 (22–27) | 12 (10–14) | 17 (15–19) |
a Values in parentheses indicate 1 S.D.
Refining the structure
NMR distance–constrained MD docking was used to obtain the final structure of the MEF-binding site. The NMR-determined distances represent vectors between the time-averaged position of the spin-label oxygen atoms (calculated using GROMACS) and a given proton. Each proton represents the point of intersection of the three vectors that position it. The standard errors (±1 σ) associated with the distance measurements (see Table 2) define the magnitudes of the principal axes of an error ellipsoid that centers on the proton. MD docking is constrained, using distance_restraints (GROMACS (37, 38)), by applying a 50 kJ mol−1 Å−1 restoring force that drives the proton toward the ellipsoid center if any part of its van der Waals surface lies outside the ellipsoid. Distance_restraints was parameterized to utilize time-averaged (1/r6)-weighted restraints, as is appropriate for NMR spin–spin interaction measurements (30, 31, 39). The five triangulated MEF protons were constrained simultaneously during docking, which was repeated 10 times. Nearly identical structures were achieved in each case (the r.m.s.d. for the 10 MEF structures was 1.3 Å), and the resulting structure did not change once the distance constraints were removed.
Structure
The MD-refined structure of the SULT1A1·MEF·PAPS·pNP complex is presented in Fig. 2, A and B (model Archive (40, 41) accession number 9lofhczzif5mpb7u1). The surface layer of the MEF-binding pocket is highlighted by a semi-transparent surface. The MEF-binding pocket is formed primarily by four planar R-groups that directly contact MEF. Three of the residues (Tyr-140, His-141, and His-144) interact with benzyl-moieties of MEF and are located in helix 6 (H6 (42)); the fourth, Trp-155, forms a contact between its face and the MEF C6-methyl group. Finally, His-141 and His-144 are positioned such that they may hydrogen bond with the MEF carboxyl. It should be noted that H6, which is adjacent to the active-site cap of SULT1A1 (orange), appears to couple MEF binding to the inhibition of the enzyme (see under “Inhibition mechanism”).
Figure 2.
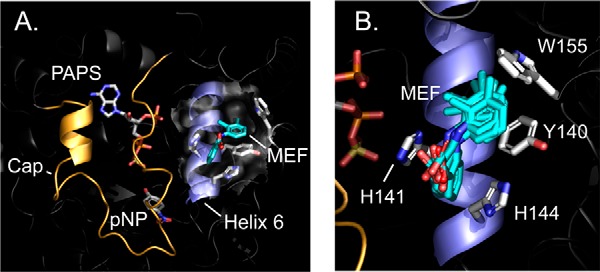
SULT1A1·MEF·PAPS·pNP structure. A, the complex. MEF (teal) is shown docked into helix 6 (blue). The active-site cap (gold) is shown in its closed position over the nucleotide (PAPS) and acceptor (para-nitrophenol (pNP)). B, putative near-interactions. An ensemble of MD-predicted MEF structures is shown interacting with the four direct-contact residues. A and B structures are the result of proton/spin-label triangulation measurements followed by NMR distance–constrained MD-docking experiments (see “Results and discussion”).
Structure validation
To validate the MD-predicted structure, each residue in direct contact with MEF (Tyr-140, His-14, His-144, and Trp-155) was mutated either to a conservative replacement (Y140L and H141S) or a residue found at the identical position in a homologous SULT (H144R and W155R). The mutations significantly (1.6–5.5-fold) affected Ki values of MEF and the fraction turnover at saturating inhibitor (kcat inh/kcat), but did not alter kcat (see Table 3, Fig. S2, and under “Experimental procedures”). The effects were small enough to be explained on the basis of indirect interactions and thus did not convincingly support the binding site structure. It seemed either the mutations were remote from the MEF-binding site or altering only one of the four contact residues left enough of the site intact to allow MEF to bind and inhibit, albeit more weakly.
Table 3.
NSAID inhibition parameters for wild-type and mutant SULT1A1
| Inhibitor | SULT1A1 | Ki | kcat | kcat inh/kcata |
|---|---|---|---|---|
| nm | min−1 | |||
| MEFb | Wild type | 27 (1)c | 120 (8) | 0.15 (0.02) |
| Y140L | 44 (2) | 122 (6) | 0.44 (0.04) | |
| H141S | 148 (10) | 118 (8) | 0.52 (0.02) | |
| H144R | 93 (4) | 120 (9) | 0.48 (0.02) | |
| W155R | 140 (6) | 118 (8) | 0.38 (0.01) | |
| ASAd | Wild type | 260 (17) | 119 (4) | 0.18 (0.02) |
| Y140L | 450 (23) | 119 (5) | 0.22 (0.03) | |
| H141S | 5560 (110) | 116 (6) | 0.22 (0.01) | |
| H144R | 7510 (160) | 122 (9) | 0.20 (0.01) | |
| W155R | 440 (36) | 117 (6) | 0.21 (0.01) |
a kcat inh indicates turnover at [inhibitor] = 20 × Ki; kcat is turnover at [inhibitor] = 0. Values were determined at PAPS (500 μm, 17 × Km) and 1-HP (2.0 μm, 100 × Km).
b MEF is mefenamic acid.
c Values in parentheses indicate standard error.
d ASA is acetylsalicylic acid.
To resolve the remote-versus-direct interaction issue, an inhibition study using the same mutants was performed using an NSAID, acetylsalicylic acid (ASA), that contains only the benzyl ring of MEF (Fig. 3A). The residues that directly contact MEF separate into two pairs (141/144 and 140/155), each of which interacts with a different MEF ring. We reasoned that a single-ring NSAID might interact preferentially with only one of the pairs—perhaps, as in the case of ASA, the same pair that binds the MEF benzyl-moiety (i.e. 141/144). If so, a mutation in the 141/144 pair should largely abolish the ASA-binding site, whereas mutations in the “unused” pair will have little effect. The results of ASA-inhibition studies (Table 3 and Fig. S2) strongly support this theory—mutating either residue of the 141/144 pair substantially decreases Ki, ASA (21–28-fold), and altering the 140/155 pair has only slight (<2-fold) effects. To ensure that ASA and MEF bind at the same site, the ligands were tested for competitive binding. At saturating MEF, turnover of the Y140L mutant is 44% of that seen at zero inhibitor, whereas ASA saturation reduces turnover to 22%. If the ligands compete, titrating MEF into a solution of ASA-saturated Y140L will cause the velocity to increase to a maximum of 2-fold (i.e. 22–44%) as ASA is replaced by MEF. As is seen in Fig. 3B, this is precisely what is observed. In conclusion, the mutagenesis findings support the MEF-binding site structure seen in Fig. 2 and suggest that ASA, the most commonly consumed NSAID, binds at the His-141/144 subsection of the NSAID-binding site.
Figure 3.
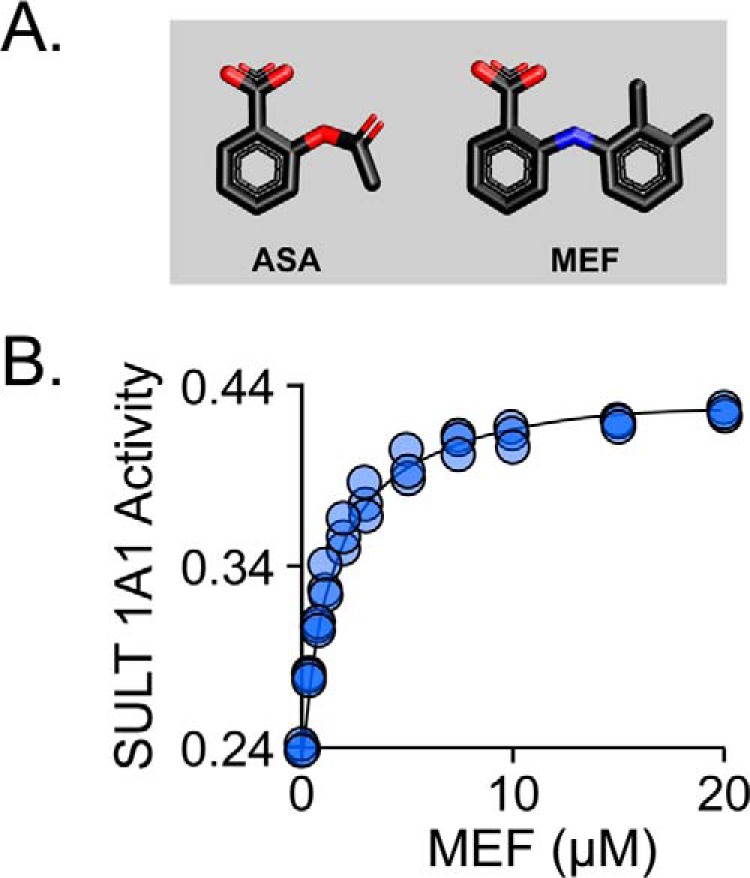
Mutagenic confirmation of structure and the aspirin (ASA)-binding site. A, the structures of ASA and MEF. B, MEF and ASA bind competitively. Reaction progress was monitored via sulfonation-induced changes in 1-HP fluorescence (λex = 325 nm and λem = 370 nm). Rates were normalized to the rate in the absence of all inhibitors. Reaction conditions: SULT1A1 Y140L (25 nm, dimer), ASA (15.0 μm, 33 × Ki), MEF (concentration indicated), PAPS (0.50 mm, 17 × Km), 1-HP (2.0 μm, 100 × Km), MgCl2 (5.0 mm), KPO4 (50 mm), pH 7.5, 25 ± 2 °C. Less than 5% of the 1-HP converted at the reaction end point was consumed during the rate measurements. The line through the data points represents the behavior predicted by the constants compiled in Table 3.
Conservation of the NSAID-binding site
The SULT family is highly structurally conserved (43). The SULT1A1 residues that directly contact MEF are listed in Table 4 and are compared with the analogous residues in the 12 remaining catalytically active members of the SULT family. The SULTs listed in bold italic font in Table 4 have been shown experimentally to be inhibited by NSAIDs (1A1 (19), 1A3 (21), 1E1 (20), 2A1 (22)). As can be seen, the direct contact residues of the remaining nine SULTs are conserved with respect to at least one of the experimentally verified isoforms. Thus, it appears that most if not all SULTs harbor the NSAID-binding site. In vivo studies reveal that NSAIDs significantly alter metabolite sulfonation in humans (44–47). These findings raise important questions regarding the nature and extent of NSAID influences on human sulfuryl-transfer metabolism and on the many sulfonated Food and Drug Administration-approved drugs (48).
Table 4.
Alignment of SULT NSAID-contact residues
| SULT | Residuea |
|||
|---|---|---|---|---|
| 140 | 141 | 144 | 155 | |
| 1A1b | Tyr | His | His | Trp |
| 1A2 | Tyr | His | His | Trp |
| 1A3b | Tyr | His | Arg | Trp |
| 1A4 | Tyr | His | Arg | Trp |
| 1B1 | Tyr | His | Leu | Trp |
| 1C1 | Tyr | His | Arg | Trp |
| 1C2 | Tyr | His | Arg | Trp |
| 1C3 | Tyr | His | Arg | Trp |
| 1C4 | Tyr | His | Arg | Trp |
| 1E1b | Tyr | Tyr | Leu | Phe |
| 2A1b | Tyr | Phe | Lys | Trp |
| 2B1a | Tyr | His | Lys | Pro |
| 2B1b | Tyr | His | Lys | Pro |
a Numbering corresponds to SULT1A1 sequence.
b NSAID inhibition was experimentally verified.
Inhibition mechanism
Helix 6 harbors the majority of the NSAID-binding sites and is adjacent to the SULT1A1 active-site cap (Fig. 2, A and B). The cap is conserved across the SULT family, and its structure and dynamics are intimately linked to substrate selectivity and turnover (49–53). When the cap is closed, it encapsulates nucleotide and must therefore open for nucleotide to escape (53, 54). SULT1A1 turnover is rate-limited by PAP release (49, 55, 56), which, in turn, is determined by the fraction of the time the cap spends in the open position(s). Given that H6 interacts with both MEF and the cap, we reasoned that MEF might slow turnover via H6-mediated stabilization of the closed form of the cap.
Previous binding studies reveal that MEF and PAP do not significantly influence one another's affinity—their interaction energy is ∼0 (12). Hence, MEF does not slow turnover by enhancing PAP affinity. If, instead, MEF independently stabilized the closed confirmation of the cap, nucleotide binding and release would be slowed to the same extent (because nucleotide can neither enter nor escape once the cap is closed), and turnover would thus decrease without altering PAP affinity. To explore this hypothesis, cap behavior was simulated with and without ligand (MEF or PAP) using all-atom MD. As is seen in Fig. 4A, the cap is predicted to remain open in the absence of ligand and to close and form a similar pore in response to the binding of either MEF or nucleotide. To test the prediction that MEF independently fosters cap closure, nucleotide on- and off-rate constants were determined in the presence and absence of saturating MEF using stopped-flow fluorescence (49, 53, 56). The results confirm the prediction—nucleotide on- and off-rate constants are decreased by the same factor (∼7.5) in the presence of MEF (see Fig. 4B and Table 5). Given that nucleotide release is largely rate-limiting (12), the 7.5-fold decrease in off-rate constant predicts that turnover will decrease to 13% at saturating MEF, which is in excellent agreement with the observed value of 15% (see Table 3). These findings strongly support that MEF slows turnover via an indirect, H6-mediated stabilization of the cap-closed form of SULT1A1.
Figure 4.
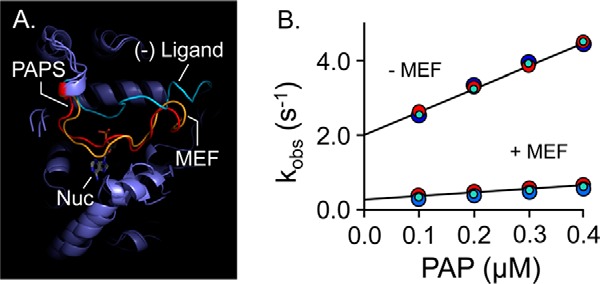
Mechanism of MEF inhibition. A, simulated MEF-induced SULT1A1 cap closure. All-atom MD was performed with SULT1A1, SULT1A1·PAPS, and SULT1A1·MEF. The predicted structures are superposed, and the active-site caps of the structures are highlighted as follows: SULT1A1 (cyan); SULT1A1·PAPS (red); SULT1A1·MEF (gold). B, MEF affects nucleotide-binding rate constants. Binding-reaction progress curves were monitored using a stopped-flow fluorimeter (λex = 290 nm and λem ≥ 330 nm (cutoff filter)). Reactions were initiated by mixing (1:1 v/v) a solution containing SULT1A1 (50 nm, dimer), MEF (0, or 1.0 μm (37 × Kd)), MgCl2 (5.0 mm), KPO4 (50 mm), pH 7.5, 25 ± 2 °C, with a solution that was identical except that it lacked SULT1A1 and contained PAP at twice the indicated concentrations. kobs values were obtained by fitting five averaged progress curves using Pro-K analysis software. Each kobs value is the average of three independent determinations. kon and koff were obtained from the slopes and intercepts predicted by linear least-squares analysis and are compiled in Table 5.
Table 5.
Rate constants governing nucleotide binding to SULT1A1
| Nucleotide | (−) MEF |
(+) MEF |
||||
|---|---|---|---|---|---|---|
| kon | koff | Kd | kon | koff | Kd | |
| μm−1s−1 | s−1 | μm | μm−1s−1 | s−1 | μm | |
| PAPS | 6.8 (0.2)a | 2.1 (0.2) | 0.31 (0.03) | 1.0 (0.02) | 0.28 (0.01) | 0.28 (0.01) |
| PAP | 5.9 (0.4) | 2.1 (0.2) | 0.36 (0.04) | 0.9 (0.06) | 0.27 (0.02) | 0.30 (0.03) |
a Values in parentheses indicate standard error.
Conclusions
The structure of the first SULT NSAID-binding site has been determined—that of SULT1A1. Only four residues at the binding site directly contact MEF (Tyr-140, His-141, His-144, and Trp-155). These four planar residues separate into two pairs (140/141 and 140/155), each of which binds a different MEF ring moiety. Mutagenesis and inhibition studies strongly suggest that the 141/144 pair forms the docking site for the single ring of aspirin (ASA). Mechanism work reveals that MEF binding inhibits turnover by stabilizing the closed form of the active-site cap of the enzyme indirectly through an intervening helix (H6). Finally, sequence comparisons indicate that most if not all of the 13 SULT isoforms harbor the NSAID-binding site, which raises the question of the extent to which NSAIDs influence human sulfuryl-transfer metabolism and biology.
Experimental procedures
Materials
The materials and sources used in this study are as follows: 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), DTT, EDTA, l-glutathione (reduced), 1-HP, imidazole, IPTG, LB media, lysozyme, 4-maleimido-PROXYL, N-cyclohexylmaleimide, pepstatin A, and potassium phosphate were the highest grade available from Sigma. Acetylsalicylic acid, ampicillin, HEPES, KCl, KOH, MgCl2, and phenylmethylsulfonyl fluoride (PMSF) were purchased from Thermo Fisher Scientific. Glutathione- and nickel-chelating resins were obtained from GE Healthcare. Competent Escherichia coli (BL21(DE3)) was purchased from Novagen. Mefenamic acid was obtained from Santa Cruz Biotechnology. PAPS and PAP were synthesized as described previously (57) and were ≥99% pure as assessed by anion-exchange HPLC.
Computer and software
Molecular dynamics simulations were performed on a Parallel Quantum Solutions QS32-2670C-XS8 computer. PQS Molecular Builder was purchased from Parallel Quantum Solutions (58). A Genetically Optimized Ligand Docking (GOLD) (59–61) license was obtained from the Cambridge Crystallographic Data Center. The source code for GROningen MAchine for Chemical Simulation (GROMACS) 4.5 (37, 38, 62) was downloaded from http://www.GROMACS.org3 under the GROMCAS General Public License (GPL). Pro-K software was obtained from Applied Photophysics Ltd.
Methods
SULT1A1 constructs
The E. coli codon-optimized SULT1A1 coding region was inserted into a triple-tag pGEX-6P expression vector containing an (N-terminal)-His/GST/MBP tag. To create the Cys-insertion mutants used for regiospecific attachment of maleimide-based labels, a Cys-light 1A1 plasmid was first constructed by replacing reactive Cys-287 and Cys-70 (both of which react with DTNB (29)) with serines; three single-Cys mutants were then created by inserting Cys into the Cys-light 1A1 scaffold at residues Gly-29, Glu-198, and Lys-234, respectively. The mutants for testing the hypothetical interaction of mefenamic acid with SULT1A1 were constructed by mutating the wild-type SULT1A1-coding region as follows: Y140L, H141S, H144R, and W155R. All mutagenesis projects used standard PCR mutagenesis protocols (51).
Protein purification
SULT expression and purification were performed as described previously (12, 52, 54). Briefly, E. coli cells (BL21(DE3)) harboring a SULT1A1 expression plasmid were grown at 37 °C in LB medium, induced with IPTG (0.30 mm) at A600 ∼0.6, and cultured at 17 °C for 18 h following induction. The cells were pelleted, resuspended in lysis buffer (PMSF (290 μm), pepstatin A (1.5 μm), lysozyme (0.10 mg/ml), EDTA (2.0 mm), KCl (400 mm), KPO4 (50 mm), pH 7.5), sonicated, and centrifuged (10,000 × g, 1.0 h) at 4 °C. MgCl2 (5.0 mm) was then added to chelate EDTA, and the supernatant was passed through a chelating Sepharose Fast Flow column charged with Ni2+. The column was washed (imidazole (10 mm), KCl (400 mm), and KPO4 (50 mm), pH 7.5); enzyme was eluted (imidazole (250 mm), KCl (400 mm), and KPO4 (50 mm), pH 7.5) and loaded directly onto a glutathione-Sepharose column, which was washed (DTT (2.0 mm), KCl (400 mm), and KPO4 (50 mm), pH 7.5) before eluting the tagged enzyme (reduced glutathione (10 mm), DTT (2.0 mm), KCl (400 mm), and Tris (100 mm), pH 8.0). The fusion protein was digested using PreScission protease overnight at 4 °C, and run through a GST column to remove the tag. The protein was >95% pure as judged by SDS-PAGE. The protein was then concentrated, and its concentration was determined by UV absorbance (ϵ280 = 53.9 mm−1 cm−1) (51). The final protein was flash-frozen and stored at −80 °C.
Covalent tagging
3-Maleimido-PROXYL or N-cyclohexylmaleimide (diamagnetic control label) was added to a solution containing enzyme in a 20:1 ratio over reactive Cys (29, 32). PAP was added to the reaction solution to enhance enzyme stability during labeling. Reaction conditions were as follows: SUTL1A1 (50 μm, monomer), 3-maleimido-PROXYL or N-cyclohexylmaleimide (1.0 mm), PAP (0.50 mm), KPO4 (50 mm), pH 7.5, 4 ± 2 °C. Reactions were monitored using DTNB to detect unreacted cysteine and were considered complete when >98% of the cysteine in the protein had been labeled. Following reaction completion, the reaction mixtures were dialyzed against PAP (0.50 mm), KPO4 (50 mm), pD 7.4, D2O (>95%), 4 ± 2 °C.
Initial rate studies
Initial rate parameters were determined using a previously described 1-HP assay (12). Briefly, reactions were initiated by addition of PAPS (0.50 mm, 17 × Km) to a solution containing enzyme (10 nm, active site), 1-HP (2.0 μm, ∼100 × Km), and KPO4 (50 mm), pH 7.5, 25 ± 2 °C. Reaction progress was monitored via the sulfonation-dependent change in 1-HP fluorescence (λex = 325 nm and λem = 370 nm). Km and Vmax values were determined using progress curve analysis (48). In the MEF and ASA inhibition studies, the inhibitor concentrations ranged from 0.2 to 20 × Ki values, and Ki value was obtained from weighted least-squares fitting (64) using the following partial non-competitive inhibition equation (64): v/vo = ([S]·(Ki + (α·[I])))/(([S] + Km)·(Ki + [I]))), where vo is the initial rate at [I] = 0, and α is the ratio of initial rates at saturating substrate with/without saturating inhibitor.
NMR measurements
MEF 1D-proton spectra were acquired at 298 K using a Bruker 600 MHz spectrometer equipped with a TCI H/F-cryogenic probe. Spectra were acquired at the following conditions: spin-labeled SULT1A1 (20, 15, 10, 5.0, and 2.5 μm active sites), MEF (100 μm), PAP (500 μm, 17 × Kd), KPO4 (50 mm), pD 7.4, 25 ± 1 °C. Peak widths were obtained by fitting using NMRdraw configured to use a Lorentzian peak shape (36).
Molecular dynamics modeling
SWISS-MODEL (65) was used to create a ligand-free model of SULT1A1 from the SULT1A1·PAP (Protein Data Bank code 4GRA (52)) structure. The model was protonated (pH 7.4) and energy-minimized using GROMACS (37, 38, 62), as described previously (28, 29). GROMAS57 energy-parameter files were created using Automated Topology Builder (63) for MEF, PAPS, and a spin-labeled cysteine analogue in which the nitroxyl moiety was replaced by a hydroxyl group. The spin-labeled cysteine analogue was added to the GROMAS57 energy field as a non-canonical amino acid and inserted into SULT1A1 by replacing residues Gly-29, Glu-198, and Lys-234. PAPS was positioned in the active site using GOLD (59–61). The system was equilibrated (298 K, NaCl (50 mm), pH 7.4) in 100-ps increments using GROMACS. Once equilibrated, MEF was randomly positioned in a simulated box of water (52 × 52 × 52 Å) containing the spin-labeled SULT1A1·PAPS construct and docked using GROMACS. Docking was constrained using NMR-determined spin-label/MEF-proton distances (see under “Results and discussion”). The simulation exercise was repeated 10 times, and identical MEF-bound structures were obtained in each case.
Ligand- binding molecular dynamics modeling
The model of native SULT1A1 (i.e. without attached spin labels) was constructed as described above. Apo-SULT1A1 was equilibrated over 1.2 ns in 100-ps increments at NaCl (50 mm), 298 K, pH 7.4, using GROMACS (37, 38, 62). The r.m.s.d. of the system stabilized between 0.2 and 0.3 ns. The behavior of the ligand complexes (SULT·PAPS and SULT·MEF) was simulated by positioning ligands at their binding sites in the equilibrated apo-structure and allowing the complexes to equilibrate for an additional 1.0 ns.
Effect of PAP(S) binding by MEF at pre-steady state
Pre-steady-state binding of nucleotide to SULT1A1 was monitored via ligand-induced enzyme fluorescence change using an Applied Photophysics SX20 stopped-flow spectrofluorimeter (56). SULT1A1 fluorescence was excited at 290 nm and detected above 330 nm using a cutoff filter. kon and koff values of PAP(S) binding to SULT1A1 were obtained by rapidly mixing (1:1, v/v) a solution containing SULT1A1 (50 nm, dimer), MEF (0 or 1.0 μm, 37 × Kd), KPO4 (50 mm), pH 7.5, T = 25 ± 2 °C with a solution that was identical except that it contained PAP(S) and MEF (0 or 1.0 μm, 37 × Kd) and was without enzyme. Three independently determined progress curves (each of which is an average of five binding reactions) were collected at four separate nucleotide concentrations. The observed rate constant (kobs) at a given [nucleotide] was obtained by fitting the average of the three curves to Pro-K analysis software. kon and koff values were obtained from the slopes and intercepts predicted by linear least-squares analysis of four-point kobs versus [nucleotide] plots.
Author contributions
T. S. L. conceived the project. T. W., I. C., and T. S. L. designed the experiments. T. W. and I. C. executed the experiments. T. W., I. C., and T. S. L. interpreted the results. T. W. and I. C. helped T. S. L. write the manuscript. All authors revised and agreed to the final version of the manuscript.
Supplementary Material
Acknowledgments
The Bruker 600 NMR used in these studies is located in the Einstein NMR Facility and was acquired using the National Institutes of Health Shared Instrumentation Grant 1S10OD016305.
This work was supported by National Institutes of Health Grants GM121849 and GM106158. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- NSAID
- non-steroidal anti-inflammatory drug
- MEF
- mefenamic acid
- ASA
- acetylsalicylic acid
- 1-HP
- 1-hydroxypyrene
- SULT
- sulfotransferase
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- IPTG
- isopropyl thio-β-d-galactopyranoside
- r.m.s.d.
- root mean square deviation
- MD
- molecular dynamics
- DTNB
- 5,5′-dithiobis-2-nitrobenzoic acid
- PAP
- 3′-phosphoadenosine 5′-phosphate.
References
- 1. Campbell C. L., Smyth S., Montalescot G., and Steinhubl S. R. (2007) Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 297, 2018–2024 [DOI] [PubMed] [Google Scholar]
- 2. Strauch K. A. (2011) Invisible pollution: the impact of pharmaceuticals in the water supply. AAOHN J. 59, 525–532 [DOI] [PubMed] [Google Scholar]
- 3. Fuster V., and Sweeny J. M. (2011) Aspirin: a historical and contemporary therapeutic overview. Circulation 123, 768–778 [DOI] [PubMed] [Google Scholar]
- 4. Mitchell J. A., and Warner T. D. (2006) COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat. Rev. Drug Discov. 5, 75–86 [DOI] [PubMed] [Google Scholar]
- 5. Mora S., and Manson J. E. (2016) Aspirin for primary prevention of atherosclerotic cardiovascular disease: advances in diagnosis and treatment. JAMA Intern. Med. 176, 1195–1204 [DOI] [PubMed] [Google Scholar]
- 6. Richman I. B., and Owens D. K. (2017) Aspirin for primary prevention. Med. Clin. North Am. 101, 713–724 [DOI] [PubMed] [Google Scholar]
- 7. Zhou Y., Boudreau D. M., and Freedman A. N. (2014) Trends in the use of aspirin and nonsteroidal anti-inflammatory drugs in the general U.S. population. Pharmacoepidemiol. Drug Saf. 23, 43–50 [DOI] [PubMed] [Google Scholar]
- 8. Patrignani P., and Patrono C. (2016) Aspirin and cancer. J. Am. Coll. Cardiol. 68, 967–976 [DOI] [PubMed] [Google Scholar]
- 9. Walczak K., Marciniak S., and Rajtar G. (2017) Cancer chemoprevention–selected molecular mechanisms. Postepy Hig. Med. Dosw. (Online) 71, 149–161 [DOI] [PubMed] [Google Scholar]
- 10. Baron J. A., and Sandler R. S. (2000) Nonsteroidal anti-inflammatory drugs and cancer prevention. Annu. Rev. Med. 51, 511–523 [DOI] [PubMed] [Google Scholar]
- 11. Gurpinar E., Grizzle W. E., and Piazza G. A. (2014) NSAIDs inhibit tumorigenesis, but how? Clin. Cancer Res. 20, 1104–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cook I., Wang T., Falany C. N., and Leyh T. S. (2015) The allosteric binding sites of sulfotransferase 1A1. Drug Metab. Dispos. 43, 418–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duffel M. W., and Jakoby W. B. (1981) On the mechanism of aryl sulfotransferase. J. Biol. Chem. 256, 11123–11127 [PubMed] [Google Scholar]
- 14. Glatt H. (1997) Sulfation and sulfotransferases 4: bioactivation of mutagens via sulfation. FASEB J. 11, 314–321 [DOI] [PubMed] [Google Scholar]
- 15. Glatt H. (2000) Sulfotransferases in the bioactivation of xenobiotics. Chem. Biol. Interact. 129, 141–170 [DOI] [PubMed] [Google Scholar]
- 16. Nowell S., Ratnasinghe D. L., Ambrosone C. B., Williams S., Teague-Ross T., Trimble L., Runnels G., Carrol A., Green B., Stone A., Johnson D., Greene G., Kadlubar F. F., and Lang N. P. (2004) Association of SULT1A1 phenotype and genotype with prostate cancer risk in African-Americans and Caucasians. Cancer Epidemiol. Biomarkers Prev. 13, 270–276 [DOI] [PubMed] [Google Scholar]
- 17. Huang W. T., Erickson S. R., Hansen R. A., and Wu C. H. (2016) The association between regular use of aspirin and the prevalence of prostate cancer: results from the National Health Interview Survey. Medicine 95, e3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith C. J., Dorsey T. H., Tang W., Jordan S. V., Loffredo C. A., and Ambs S. (2017) Aspirin use reduces the risk of aggressive prostate cancer and disease recurrence in African-American men. Cancer Epidemiol. Biomarkers Prev. 26, 845–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vietri M., De Santi C., Pietrabissa A., Mosca F., and Pacifici G. M. (2000) Inhibition of human liver phenol sulfotransferase by nonsteroidal anti-inflammatory drugs. Eur. J. Clin. Pharmacol. 56, 81–87 [DOI] [PubMed] [Google Scholar]
- 20. King R. S., Ghosh A. A., and Wu J. (2006) Inhibition of human phenol and estrogen sulfotransferase by certain non-steroidal anti-inflammatory agents. Curr. Drug Metab. 7, 745–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vietri M., Vaglini F., Pietrabissa A., Spisni R., Mosca F., and Pacifici G. M. (2002) Sulfation of R(−)-apomorphine in the human liver and duodenum, and its inhibition by mefenamic acid, salicylic acid and quercetin. Xenobiotica 32, 587–594 [DOI] [PubMed] [Google Scholar]
- 22. Wang L. Q., and James M. O. (2005) Sulfotransferase 2A1 forms estradiol-17-sulfate and celecoxib switches the dominant product from estradiol-3-sulfate to estradiol-17-sulfate. J. Steroid Biochem. Mol. Biol. 96, 367–374 [DOI] [PubMed] [Google Scholar]
- 23. Riches Z., Stanley E. L., Bloomer J. C., and Coughtrie M. W. (2009) Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie”. Drug Metab. Dispos. 37, 2255–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Teubner W., Meinl W., Florian S., Kretzschmar M., and Glatt H. (2007) Identification and localization of soluble sulfotransferases in the human gastrointestinal tract. Biochem. J. 404, 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eisenhofer G., Coughtrie M. W., and Goldstein D. S. (1999) Dopamine sulphate: an enigma resolved. Clin. Exp. Pharmacol. Physiol. Suppl. 26, S41–S53 [PubMed] [Google Scholar]
- 26. Falany C. N., Vazquez M. E., and Kalb J. M. (1989) Purification and characterization of human liver dehydroepiandrosterone sulphotransferase. Biochem. J. 260, 641–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aksoy I. A., Wood T. C., and Weinshilboum R. (1994) Human liver estrogen sulfotransferase: identification by cDNA cloning and expression. Biochem. Biophys. Res. Commun. 200, 1621–1629 [DOI] [PubMed] [Google Scholar]
- 28. Cook I., Wang T., and Leyh T. S. (2017) Tetrahydrobiopterin regulates monoamine neurotransmitter sulfonation. Proc. Natl. Acad. Sci. U.S.A. 114, E5317–E5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cook I., Wang T., Girvin M., and Leyh T. S. (2016) The structure of the catechin-binding site of human sulfotransferase 1A1. Proc. Natl. Acad. Sci. U.S.A. 113, 14312–14317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Solomon I. (1955) Relaxation processes in a system of two spins. Phys. Rev. 99, 559–566 [Google Scholar]
- 31. Battiste J. L., and Wagner G. (2000) Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear Overhauser effect data. Biochemistry 39, 5355–5365 [DOI] [PubMed] [Google Scholar]
- 32. Girvin M. E., and Fillingame R. H. (1995) Determination of local protein structure by spin label difference 2D NMR: the region neighboring Asp61 of subunit c of the F1F0 ATP synthase. Biochemistry 34, 1635–1645 [DOI] [PubMed] [Google Scholar]
- 33. Guan J. Y., Keizers P. H., Liu W. M., Löhr F., Skinner S. P., Heeneman E. A., Schwalbe H., Ubbink M., and Siegal G. (2013) Small-molecule binding sites on proteins established by paramagnetic NMR spectroscopy. J. Am. Chem. Soc. 135, 5859–5868 [DOI] [PubMed] [Google Scholar]
- 34. John M., Pintacuda G., Park A. Y., Dixon N. E., and Otting G. (2006) Structure determination of protein-ligand complexes by transferred paramagnetic shifts. J. Am. Chem. Soc. 128, 12910–12916 [DOI] [PubMed] [Google Scholar]
- 35. Venkataraman H., den Braver M. W., Vermeulen N. P., and Commandeur J. N. (2014) Cytochrome P450-mediated bioactivation of mefenamic acid to quinoneimine intermediates and inactivation by human glutathione S-transferases. Chem. Res. Toxicol. 27, 2071–2081 [DOI] [PubMed] [Google Scholar]
- 36. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 37. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 38. Berendsen H. J., Vanderspoel D., and Vandrunen R. (1995) Gromacs–a message-passing parallel molecular-dynamics implementation. Comput. Phys. Commun. 91, 43–56 [Google Scholar]
- 39. Schmitz U., Ulyanov N. B., Kumar A., and James T. L. (1993) Molecular dynamics with weighted time-averaged restraints for a DNA octamer. Dynamic interpretation of nuclear magnetic resonance data. J. Mol. Biol. 234, 373–389 [DOI] [PubMed] [Google Scholar]
- 40. Schwede T., Sali A., Honig B., Levitt M., Berman H. M., Jones D., Brenner S. E., Burley S. K., Das R., Dokholyan N. V., Dunbrack R. L. Jr., Fidelis K., Fiser A., Godzik A., Huang Y. J., et al. (2009) Outcome of a workshop on applications of protein models in biomedical research. Structure 17, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haas J., Roth S., Arnold K., Kiefer F., Schmidt T., Bordoli L., and Schwede T. (2013) The Protein Model Portal–a comprehensive resource for protein structure and model information. Database 2013, bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gamage N. U., Tsvetanov S., Duggleby R. G., McManus M. E., and Martin J. L. (2005) The structure of human SULT1A1 crystallized with estradiol. An insight into active site plasticity and substrate inhibition with multi-ring substrates. J. Biol. Chem. 280, 41482–41486 [DOI] [PubMed] [Google Scholar]
- 43. Allali-Hassani A., Pan P. W., Dombrovski L., Najmanovich R., Tempel W., Dong A., Loppnau P., Martin F., Thornton J., Thonton J., Edwards A. M., Bochkarev A., Plotnikov A. N., Vedadi M., and Arrowsmith C. H. (2007) Structural and chemical profiling of the human cytosolic sulfotransferases. PLos Biol. 5, e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whitehouse L. W., Paul C. J., and Thomas B. H. (1975) Effect of aspirin on fate of 14C-acetaminophen in guinea pigs. J. Pharm. Sci. 64, 819–821 [DOI] [PubMed] [Google Scholar]
- 45. Whitehouse L. W., Paul C. J., Wong L. T., and Thomas B. H. (1977) Effect of aspirin on a subtoxic dose of 14C-acetaminophen in mice. J. Pharm. Sci. 66, 1399–1403 [DOI] [PubMed] [Google Scholar]
- 46. Thomas B. H., Coldwell B. B., Zeitz W., and Solomonraj G. (1972) Effect of aspirin, caffeine, and codeine on the metabolism of phenacetin and acetaminophen. Clin. Pharmacol. Ther. 13, 906–910 [DOI] [PubMed] [Google Scholar]
- 47. Levy G., and Yamada H. (1971) Drug biotransformation interactions in man. 3. Acetaminophen and salicylamide. J. Pharm. Sci. 60, 215–221 [DOI] [PubMed] [Google Scholar]
- 48. Cook I., Wang T., Falany C. N., and Leyh T. S. (2013) High accuracy in silico sulfotransferase models. J. Biol. Chem. 288, 34494–34501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang T., Cook I., and Leyh T. S. (2016) Isozyme specific allosteric regulation of human sulfotransferase 1A1. Biochemistry 55, 4036–4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leyh T. S., Cook I., and Wang T. (2013) Structure, dynamics and selectivity in the sulfotransferase family. Drug Metab. Rev. 45, 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cook I., Wang T., Almo S. C., Kim J., Falany C. N., and Leyh T. S. (2013) Testing the sulfotransferase molecular pore hypothesis. J. Biol. Chem. 288, 8619–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cook I., Wang T., Almo S. C., Kim J., Falany C. N., and Leyh T. S. (2013) The gate that governs sulfotransferase selectivity. Biochemistry 52, 415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cook I., Wang T., Falany C. N., and Leyh T. S. (2012) A nucleotide-gated molecular pore selects sulfotransferase substrates. Biochemistry 51, 5674–5683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cook I., Wang T., Wang W., Kopp F., Wu P., and Leyh T. S. (2016) Controlling sulfuryl-transfer biology. Cell Chem. Biol. 23, 579–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang T., Cook I., and Leyh T. (2016) The design and interpretation of human SULT1A1 assays. Drug Metab. Dispos. 44, 481–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang T., Cook I., and Leyh T. S. (2014) 3′-Phosphoadenosine 5′-phosphosulfate allosterically regulates sulfotransferase turnover. Biochemistry 53, 6893–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun M., and Leyh T. S. (2010) The human estrogen sulfotransferase: a half-site reactive enzyme. Biochemistry 49, 4779–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baker J., Wolinski K., Malagoli M., Kinghorn D., Wolinski P., Magyarfalvi G., Saebo S., Janowski T., and Pulay P. (2009) Quantum chemistry in parallel with PQS. J. Comput. Chem. 30, 317–335 [DOI] [PubMed] [Google Scholar]
- 59. Verdonk M. L., Chessari G., Cole J. C., Hartshorn M. J., Murray C. W., Nissink J. W., Taylor R. D., and Taylor R. (2005) Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 48, 6504–6515 [DOI] [PubMed] [Google Scholar]
- 60. Verdonk M. L., Berdini V., Hartshorn M. J., Mooij W. T., Murray C. W., Taylor R. D., and Watson P. (2004) Virtual screening using protein-ligand docking: avoiding artificial enrichment. J. Chem. Inf. Comput. Sci. 44, 793–806 [DOI] [PubMed] [Google Scholar]
- 61. Verdonk M. L., Cole J. C., Hartshorn M. J., Murray C. W., and Taylor R. D. (2003) Improved protein-ligand docking using GOLD. Proteins 52, 609–623 [DOI] [PubMed] [Google Scholar]
- 62. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Malde A. K., Zuo L., Breeze M., Stroet M., Poger D., Nair P. C., Oostenbrink C., and Mark A. E. (2011) An automated force field topology builder (ATB) and repository, Version 1.0. J. Chem. Theory Comput. 7, 4026–4037 [DOI] [PubMed] [Google Scholar]
- 64. Whiteley C. G. (1999) Enzyme kinetics: partial and complete non-competitive inhibition. Biochemical Education 27, 15–18 [DOI] [PubMed] [Google Scholar]
- 65. Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.