

Abstract
Lecithin:cholesterol acyltransferase (LCAT) plays a key role in reverse cholesterol transport by transferring an acyl group from phosphatidylcholine to cholesterol, promoting the maturation of high-density lipoproteins (HDL) from discoidal to spherical particles. LCAT is activated through an unknown mechanism by apolipoprotein A-I (apoA-I) and other mimetic peptides that form a belt around HDL. Here, we report the crystal structure of LCAT with an extended lid that blocks access to the active site, consistent with an inactive conformation. Residues Thr-123 and Phe-382 in the catalytic domain form a latch-like interaction with hydrophobic residues in the lid. Because these residues are mutated in genetic disease, lid displacement was hypothesized to be an important feature of apoA-I activation. Functional studies of site-directed mutants revealed that loss of latch interactions or the entire lid enhanced activity against soluble ester substrates, and hydrogen–deuterium exchange (HDX) mass spectrometry revealed that the LCAT lid is extremely dynamic in solution. Upon addition of a covalent inhibitor that mimics one of the reaction intermediates, there is an overall decrease in HDX in the lid and adjacent regions of the protein, consistent with ordering. These data suggest a model wherein the active site of LCAT is shielded from soluble substrates by a dynamic lid until it interacts with HDL to allow transesterification to proceed.
Keywords: acyltransferase, apolipoprotein, cholesterol, conformational change, crystallography, high-density lipoprotein (HDL), inhibitor, structural biology, HDX MS, lecithin
Introduction
Lecithin:cholesterol acyltransferase (LCAT)3 (EC 2.3.1.43) catalyzes the first step of reverse cholesterol transport, a process wherein cholesterol is moved from macrophages in arterial plaques via high-density lipoprotein (HDL) to the liver for excretion (1). LCAT transfers the sn-2 acyl group of phosphatidylcholine (PC) to cholesterol to create a more hydrophobic cholesteryl ester that accumulates within the core of HDL particles (referred to as α-LCAT activity), thus driving maturation of discoidal pre-β-HDL to spherical α-HDL and promoting further cholesterol efflux from arterial plaques (2). LCAT also acts on cholesterol within apo-B containing lipoproteins (β-LCAT activity) such as low-density lipoprotein (LDL) (3, 4). To date, over 90 genetic mutations of LCAT have been described, which lead to one of two diseases: familial LCAT deficiency (FLD) or fish eye disease (FED) (5, 6). Both diseases are characterized by low levels of HDL cholesterol and corneal opacities, but FLD is more severe with additional symptoms such as anemia, proteinuria, and ultimately renal failure (7, 8).
LCAT is activated by apoA-I, an apolipoprotein that forms a belt around HDL particles (9, 10) and contains 10 tandem amphipathic α-helices. Previous work suggests that the central helices of apoA-I are responsible for LCAT binding and activation (10, 11). Furthermore, a 22-residue LCAT-activating peptide (ESP24218) derived from an apoA-I consensus peptide is able to form synthetic HDL particles and activate LCAT to levels similar to apoA-I (12, 13). Biochemical studies with HDLs have led to a model where LCAT first binds the HDL lipid bilayer and then interacts with apoA-I to become activated (10). Certain surface-exposed residues, such as Thr-123 and Phe-382, are implicated in apoA-I activation because of their involvement in LCAT genetic diseases and their specific loss of α- but not β-LCAT activity (14–16). However, the mechanism by which LCAT is activated by HDL particles is not understood.
LCAT is 50% identical in sequence to lysosomal phospholipase A2 (LPLA2), an acyltransferase found in lysosomes that prefers N-acetylsphingosine as its acyl acceptor. We have reported a series of crystal structures for LPLA2 and a low-resolution structure of LCAT (17). Both enzymes contain a catalytic triad-containing α/β hydrolase domain, consisting of an eight-stranded β-sheet sandwiched between α-helices (18, 19). LCAT and LPLA2 contain two additional domains, termed the membrane-binding and cap domains (17), and all three domains of the enzyme contribute structural elements to a broad hydrophobic pocket that contains the catalytic triad and substrate-binding sites. We hypothesized that a dynamic loop within the cap domain could act as a lid in a structurally analogous way to those found in bacterial triacylglycerol lipases (20–23), which shield the active site until the enzyme comes into contact with a lipid bilayer. Retraction of the lid could therefore represent an aspect of interfacial activation, a phenomenon wherein the presence of a lipid/solvent interface stimulates enzyme activity on soluble substrates (20, 24, 25). Recently, higher resolution structures of human LCAT in complex with one (26) or two (27) Fab fragments have been reported and confirm the overall domain structure of LCAT, but, unlike LPLA2, they contain largely disordered but conformationally distinct lid regions. The presence of bound Fab and a crystal contact formed in the active site also complicates interpretation of how lid conformation correlates with activity in these structures.
Here, we report the 3.1-Å crystal structure of LCAT, in the absence of antibodies, wherein the lid is ordered and packs over the active site, forming hydrophobic latch-like interactions with FED-associated residues Thr-123 and Phe-382 (16, 28, 29). We hypothesized that apoA-I is able to activate LCAT via displacement of this lid, perhaps by interacting with or perturbing residues that form the interface between the lid and the α/β hydrolase domain. We confirmed through site-directed mutagenesis that perturbation of the lid and the latch residues indeed leads to enhanced hydrolysis of soluble esters. These variants exhibited a loss of HDL-dependent activity, confirming the importance of these sites in acyl transfer. Hydrogen–deuterium exchange (HDX) mass spectrometry (MS) showed that the lid region was highly dynamic, but it has less HDX in the presence of isopropyl dodecylfluorophosphonate (IDFP), a covalent inhibitor that mimics an acyl intermediate. The results of these experiments support a role for the lid in protecting the active site from inappropriate substrates as well as in driving interactions with HDL and lipid substrates.
Results
Structure of ligand-free LCAT reveals a lid that covers the active site
Crystals of full-length, fully glycosylated LCAT were obtained, which diffracted to 3.1 Å spacings (Table 1). Initial phases were estimated via molecular replacement with an LCAT homology model derived from LPLA2 (17). Electron density is observed starting at residue 20 and ending at 399, similar to that observed in previous structural determinations (Fig. 1A) (17, 26, 27). The enzyme crystallized with four copies in the asymmetric unit, with an average root mean square deviation (r.m.s.d.) of 0.12 Å for Cα atoms in the individual subunits (30). The overall domain architecture is conserved with LPLA2 and previously reported LCAT structures, with an α/β hydrolase domain positioning the active site at the center of the enzyme, in a cavity flanked by the cap and membrane-binding domains (Fig. 1A) (17, 26, 27). Surprisingly, residues 226–246 in the cap domain of LCAT (referred to as the “lid”), which includes the dynamic “lid loop” of LPLA2 (residues 226–236 in LCAT), adopt a different conformation from all prior structures of LCAT and LPLA2 and is almost fully ordered (Fig. 1 and supplemental Fig. S1). Here, the lid extends over the active site such that it would hinder substrate and solvent access. The initial segment of the lid (residues 226–234) is better ordered than the remainder (Fig. 1B), and residues Pro-232 and Met-234 form extensive interactions with residues Thr-123 and Phe-382 of the α/β hydrolase domain. The latter portion of the lid (residues 235–246, with the density at 240–241 too weak to model with confidence) corresponds to a sequence that instead forms a prominent α-helix (a4) that packs between the membrane-binding and cap domains in all reported LPLA2 structures (supplemental Fig. S1B). It is not clear whether an analogous conformation for the lid can occur in LPLA2 because its αA–αA′ loop, which is two residues longer than in LCAT, would sterically clash with the path of lid in LCAT (supplemental Fig. S1B). The lid adopts a similar conformation in all four unique subunits of LCAT, suggesting that it is not simply a consequence of crystal lattice contacts, although residues 235–239 in the lid form a similar crystal contact in each protomer via a crystallographic 2-fold interaction. Despite its closed configuration and contacts, the lid still exhibits a high level of flexibility as judged by its relatively high B-factors compared with the rest of the structure (Fig. 1D).
Table 1.
Data collection and refinement statistics
The structure was solved by merging data from two crystals.
| Closed lid LCAT | |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 95.9, 123.5, 114.8 |
| α, β, γ (°) | 90.0, 96.2, 90.0 |
| Resolution (Å) | 48.3–3.1 (3.2–3.1)a |
| Rmerge | 0.176 (1.26) |
| I/σI | 6.6 (1.1) |
| Completeness (%) | 99.6 (98.3) |
| Redundancy | 4.1 (4.2) |
| CC1/2 | 0.99 (0.58) |
| Refinement | |
| Resolution (Å) | 20–3.10 (3.18–3.10) |
| No of reflections | 45,564 (4,489) |
| Rwork/Rfree | 0.250/0.267 (0.373/0.371) |
| No. of atoms | |
| Protein | 12,156 |
| Sugar | 240 |
| Water | 14 |
| B-factors | |
| Protein | 90.11 |
| Sugar | 125.49 |
| Water | 39.18 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.32 |
| Ramachandran statistics (%) | |
| Favored | 92.8 |
| Allowed | 6.9 |
| Outliers | 0.3 |
a Values in parentheses are for the highest-resolution shell.
Figure 1.
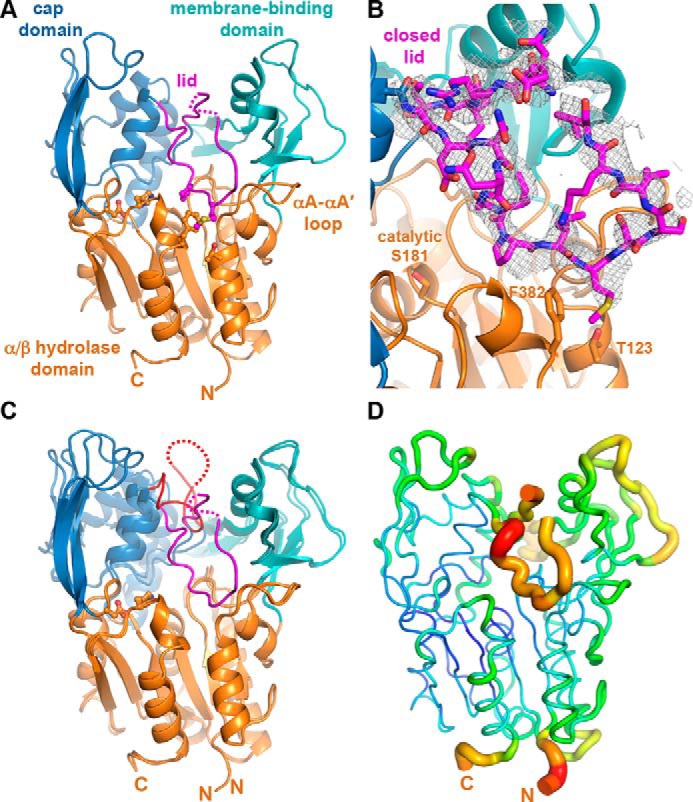
LCAT has a lid that shields the active site. A, 3.1-Å X-ray crystal structure of LCAT in a closed, extended lid conformation. Residues 240–241 (dashed line) were not modeled. Side chains of assayed residues in the lid, lid latch, and catalytic triad are shown as ball and stick models. B, |Fo| − |Fc| omit map contoured at 2 σ for the LCAT lid (residues 226–246). C, closed conformation overlaid with the open-2Fab LCAT structure (PDB entry 5BV7), with red lid and unmodeled residues 236–242 indicated with a dashed line. D, closed lid structure shown using the B-factor putty representation in PyMOL. The blue to green coloring and small tube width indicates lower B-factors, and the yellow to red coloring and wide tube indicates higher B-factors.
Comparison with other LCAT structures reveals active and inactive conformations
Two other moderate-resolution LCAT crystal forms have been reported since our initial low-resolution model (Fig. 1C and supplemental Fig. S1A) (26, 27). In the first, LCAT was crystallized in complex with a Fab fragment that recognizes the membrane-binding domain. This crystal form (Protein Data Bank (PDB) entries 4XX1 and 4XWG, referred to henceforth as closed-Fab) exhibits a conformation similar to our new closed, ligand-free structure, although lid residues 231–237 were not modeled (supplemental Fig. S1A). In the second (PDB entry 5BV7, referred to henceforth as open-2Fab), LCAT is additionally in complex with a second agonistic Fab fragment that binds primarily to the α/β hydrolase domain. In this structure the lid adopts a retracted conformation more similar to prior structures of LPLA2 (Fig. 1C and supplemental Fig. S1C). This “open” lid conformation could be a consequence of either the agonist antibody or an N-terminal pentapeptide (with mutations L4F/N5D) that docks within the active site from a neighboring crystal lattice contact, which would prevent the lid from assuming any of the other previously observed lid conformations.
In light of these new structures, we re-evaluated our original 8.7 Å LCAT structure (PDB entry 4X96) of an N- and C-terminally truncated variant (ΔNΔC, residues 21–397). Because LCAT in this low-resolution structure crystallized as a trimer with Trp-48 from the membrane-binding domain docking in the active site of each symmetry-related subunit, it is also not compatible with a closed lid configuration like those observed in our structures or the closed-Fab crystal structures (supplemental Fig. S2). Therefore, this low-resolution trimeric LCAT structure requires an open lid, although this could not be directly observed given the low resolution. Thus, the lid region of LCAT seems to be very dynamic, with the ability to assume configurations that either hinder (current and closed-Fab structures) or allow full access (low-resolution and open-2Fab structures) to the active site (Fig. 1 and supplemental Fig. S1). The transformation between these two conformations is modeled in supplemental Movie S1, which morphs between the closed lid conformation we report here and the open-2Fab structure.
Examination of LCAT variants for thermal stability
The conformational flexibility of the LCAT lid and its proximity to the active site suggests that it could be important for regulation of activity by apoA-I. For example, the binding of apoA-I either to Thr-123 and Phe-382 could serve to release the lid and allow easier access of substrates to the catalytic site. To test this hypothesis, we created a panel of LCAT variants with site-directed mutations either in the lid or at other interacting or nearby surface-exposed sites previously postulated to be involved in apoA-I binding (e.g. Thr-123 (28), Phe-382 (29), Asn-131 (31), and Asn-391 (32, 33)) (Fig. 2, A and B, and supplemental Fig. S3). Residues were mutated to alanine and/or to known disease-associated mutations as applicable. Residues 227–246 of the lid were also deleted and replaced with a three-residue SGS linker (Δlid), and a chimeric enzyme was created with the lid substituted with the analogous region of LPLA2 (residues 229–246; LPLA2lid). A second chimeric enzyme contained the analogous two-residue longer αA–αA′ loop from LPLA2 (residues 111–119, LPLA2αA), which we hypothesized would prevent lid closure in that enzyme. Finally, we generated control variants with mutations in regions well-established to be important for HDL and/or membrane binding, such as exposed hydrophobic positions in the membrane-binding domain (Trp-48 and Leu-70), and the ΔNΔC LCAT variant, which consists of a core domain similar to full-length LPLA2 but lacks the N and C termini (17). The N terminus is critical for LCAT activity on HDLs, but truncating the C-terminal portion of LCAT has no effect on activity (34–36).
Figure 2.

Disruption of the lid and its interactions with the catalytic core enhances LCAT esterase activity. A and B, close-up view of LCAT, highlighting residues examined in biochemical studies from our closed (A) and the open-2Fab (B) structures. C, change in Tm relative to WT LCAT as measured by DSF. Error bars are the S.D. of at least three independent experiments performed in triplicate (see supplemental Table S1). (*, 0.01 < p < 0.05; **, 0.001 < p < 0.01 via two-tailed t test). D, soluble esterase activity. pNPB hydrolysis is shown for each variant. The dashed line indicates the rate of WT LCAT for ease of comparison. Error bars represent the standard deviation (S.D.) of at least three independent experiments. (*, 0.01 < p < 0.05; **, 0.001 < p < 0.01; ***, p < 0.001; ****, p < 0.0001 via a two-tailed t test).
We first determined the relative melting temperature (Tm) of each variant versus wild type (WT) with differential scanning fluorimetry (DSF). Decreases in Tm may indicate structural defects that may underlie loss of function. All of the variants exhibited a Tm within 3 °C relative to WT (Tm = 54.7 ± 1.8 °C), indicating that none of the mutations disrupted the global fold of the enzyme (Fig. 2C and supplemental Table S1). However, N131D (an FED mutation) and N391A exhibited a 2 °C lower Tm, and N131A and N391S (FED mutation) were destabilized by 0.7 and 1.4 °C, respectively. These variants likely disrupt local protein fold, consistent with loss of hydrogen bonds formed between the side chains of Asn-131 and -391. The I233E mutation decreased the Tm by 2 °C, whereas I233A was not significantly destabilized, indicating that a glutamate at this position is incompatible with local structure. Notably, Ile-233 is pointed toward the hydrophobic active site in the open-2Fab structure (Fig. 2B). The LPLA2lid and LPLA2αA chimeras were destabilized by 2 and 1.3 °C, respectively, suggesting that the swapped residues lost compatibility in the background of LCAT. Some variants exhibited higher Tm values such as W48A (3.1 °C) and T123I (1.2 °C). Surprisingly, the Δlid variant exhibited a 1.3 °C higher Tm, indicating that removal of the entire lid does not destabilize the protein.
Lid inhibits access of a small ester substrate
We next tested whether the LCAT variants could accelerate the hydrolysis of a generic soluble short-chain ester substrate p-nitrophenyl butyrate (pNPB) in the absence of HDL (Fig. 2D and supplemental Table S1). Consistent with the idea of the lid blocking access to the active site, variants predicted to disrupt the latch had an increased ability to hydrolyze pNPB relative to WT, such as F382A (240%), F382E (190%), Δlid (200%), and lid variants G230R (37), I233A, and I233, (160, 190, and 180%, respectively). The combination of F382A and Δlid (F382A/Δlid) exhibited a similar increase in rate versus WT (230%) as either variant on its own, suggesting that the mechanism for increased esterase activity is the same in each case. The FED-associated mutation F382V had a rate similar to WT, suggesting that valine can similarly stabilize a closed lid conformation. Surprisingly, variants created to disrupt HDL/membrane binding also had increased activity, such as ΔNΔC (180%), W48A (170%), and L70S (200%). Because each of these mutations eliminates solvent-exposed hydrophobic residues, this may reflect a lower propensity to aggregate in solution and therefore an enhanced ability to process soluble substrates.
Mutation of the active-site serine (S181A) yielded a 60% decrease in esterase activity, thus indicating that the remaining activity is due to background hydrolysis of substrate. Disease-associated mutations N131D and N391S, which do not interact with the lid, exhibited 50 and 30% decreases in activity, consistent with destabilization as suggested by DSF.
Lid variants exhibit defects in HDL binding
We hypothesized that the residues involved in the interface between the lid and the surface of the α/β hydrolase domain may help dictate affinity for HDL particles. The ability of each variant to bind HDLs was therefore compared using bio-layer interferometry (BLI). Recombinant HDLs were formed with either full-length apoA-I protein or with the ESP24218 LCAT-activating peptide. Biotinylated phosphatidylethanolamine was incorporated into the HDLs to allow for immobilization on streptavidin biosensor tips (supplemental Fig. S4) (14). The magnitude of the interference shift was different for the apoA-I versus the peptide HDLs, but the overall trends were the same for each (Fig. 3A and supplemental Table S2). This result suggests that either that LCAT and its variants are not probing a specific binding site on apoA-I or that the consensus peptide-based HDL provides a very similar LCAT-binding site as apoA-I.
Figure 3.

HDL binding and activity defects in LCAT variants. A, maximal response for LCAT variants binding to HDLs made with apoA-I (solid) or peptide HDLs (dashed) normalized with respect to WT LCAT. B, representative BLI data used for generation of binding analysis in A. A single experiment is depicted with apoA-I HDLs and all LCAT variants at 0.2 μm. C, WT LCAT analyzed with apoA-I HDLs at different concentrations to determine Kd values. D, most LCAT variants have defects in DHE-ester formation. Acyltransferase data for a subset of LCAT variants at 7.5 μg/ml mixed with peptide HDLs containing DHE.
As a preliminary screen to identify variants for more extensive quantitative binding studies, we first compared the total response of each variant binding to the immobilized HDL particles. Variants that perturbed known HDL/membrane-binding elements such as ΔNΔC, W48A, and L70S were severely impacted in their ability to bind to either kind of HDL (Fig. 3A and supplemental Table S2) as expected. However, mutation of residues previously proposed to be important for LCAT activation by apoA-I such as Phe-382 and Thr-123 (14, 16) did not affect the magnitude of binding to either type of HDL (Fig. 3, A and B). Mutation of Asn-131 and Asn-391 had a more pronounced defect, consistent with the deficiencies of these variants observed in the pNPB assay and DSF. Mutations in the lid region (G230R, P232A, I233A, I233E, and M234A) and Δlid, as well as the chimeric LPLA2lid and LPLA2αA variants, also exhibited defects in the total response.
For WT and a subset of the LCAT variants, kon, koff, and Kd values were determined using apoA-I HDLs (Fig. 3C, supplemental Fig. S5, and Table 2). The Kd of 0.72 μm obtained for WT LCAT is consistent with previous studies (14, 38, 39). ΔNΔC was the most defective in this analysis, decreasing the Kd value by an order of magnitude, consistent with a role for the N terminus in HDL binding. W48A was also highly compromised (6-fold), confirming a role for the membrane-binding domain in HDL binding. S181A, T123I, F382A, and M234A all bound similarly to WT (Kd of ∼1 μm). Variants testing the role of the lid in HDL binding, I233A and Δlid, had more pronounced defects. The Kd for I233A was 2.6 μm, with the loss of affinity due to an increase in the koff (as in the case of W48A). Alternatively, Δlid had an ∼30-fold reduced kon and a 3-fold slower koff, although the low signal for this variant makes the Kd determination unreliable (Table 2). This result indicates a role for the lid in binding to HDLs, but this assay cannot differentiate whether there is a defect in binding to lipid bilayers or apoA-I. Overall, these results are inconsistent with our hypothesis that Thr-123 and Phe-382 are involved in binding apoA-I; however, it is possible that apoA-I interactions do not contribute to the overall Kd value here, especially as there is no significant difference between the apoA-I and ESP24218-based HDLs.
Table 2.
Kd determination for LCAT binding to HDLs via BLI
| Variant | kon | koff | Kd | Sig.a | n |
|---|---|---|---|---|---|
| s−1 μm−1 | s−1 | μm | |||
| WT-peptide HDLs | 0.13 ± 0.02b | 0.065 ± 0.01 | 0.50 | NS | 3 |
| WT | 0.079 ± 0.005 | 0.057 ± 0.008 | 0.72 | 3 | |
| ΔNΔC | 0.011 ± 0.02 | 0.38 ± 0.1 | 35 | ** | 4 |
| W48A | 0.067 ± 0.02 | 0.27 ± 0.02 | 4.0 | ** | 3 |
| S181A | 0.064 ± 0.004 | 0.061 ± 0.01 | 0.95 | NS | 3 |
| T123I | 0.077 ± 0.005 | 0.070 ± 0.01 | 0.91 | ** | 3 |
| F382A | 0.088 ± 0.02 | 0.072 ± 0.02 | 0.82 | NS | 3 |
| I233A | 0.084 ± 0.01 | 0.22 ± 0.04 | 2.6 | * | 3 |
| M234A | 0.094 ± 0.005 | 0.091 ± 0.01 | 0.97 | ** | 3 |
| Δlid | 0.0044 ± 0.001 | 0.021 ± 0.004 | 4.8 | NS | 3 |
a Sig. represents significance for the Kd values determined individually for each replicate and then compared with WT using a two-tailed t test. NS indicates not significant. *, 0.01 < p < 0.05; **, 0.001 < p < 0.01.
b Error represents standard deviation.
Role for the lid in acyltransferase activity
The panel of variants was next analyzed for acyltransferase activity using the fluorescent sterol dehydroergosterol (DHE) in place of cholesterol (40). As in the binding studies, HDLs were made with both apoA-I and the ESP24218 peptide, but there were no drastic differences in kinetics between the two (Fig. 3D, Table 3, and supplemental Table S3). Because the signal was better for the ESP24218-based HDLs, we focussed on these results. In this assay, the full mechanism of sterol esterification is examined, because LCAT must be able to bind to the HDL, react with lipid, and subsequently transfer the acyl chain to DHE. Thus, variants with any defect in HDL binding, cholesterol binding, or esterase activity will potentially exhibit a defect in acyl transfer (Fig. 3D and Table 3). Variants targeting known HDL-binding elements, such as ΔNΔC or W48A and L70S, were severely or moderately affected, respectively. For the latter two variants, this defect was measured as an increase in Km, consistent with their observed reduction in HDL binding. All of the disease-causing variants had little to no activity in this assay, as reported previously (14, 16, 28, 41–43). Many of the lid variants displayed only modest defects, with I233A showing a Km defect consistent with its HDL-binding defect. The Δlid, LPLA2lid, and LPLA2αA variants all had low activity. Thus, single-point mutations to the lid may therefore not be enough to disrupt the function of the lid, but larger structural changes diminish its ability to be activated by apoA-I or, potentially, to form specific interactions with acyl transfer substrates.
Table 3.
Acyl transfer on peptide HDL substrates
MBD indicates membrane-binding domain; apoA-I? indicates disease-related surface-exposed residues that could potentially bind apoA-I.
| Variant | Location | Vmax | Km | Vmax/Kd | Sig.a | n |
|---|---|---|---|---|---|---|
| μm/h | μm | |||||
| WT | 20 ± 0.3b | 6.2 ± 0.3 | 3.2 | 21 | ||
| ΔNΔC | NDc | ND | ** | 3 | ||
| S181A | Active site | ND | ND | ** | 3 | |
| W48A | MBD | 55 ± 17 | 173 ± 67 | 0.32 | * | 3 |
| L70S | MBD | 21 ± 6 | 68 ± 30 | 0.31 | * | 3 |
| T123A | ApoA-I? | 4.1 ± 1 | 18 ± 10 | 0.23 | ** | 3 |
| T123Id | ApoA-I? | 2.3 ± 2 | 26 ± 60 | 0.088 | * | 3 |
| F382A | ApoA-I? | 0.72 ± 0.2 | 0.14 ± 1 | 5.1 | ** | 6 |
| F382Vd | ApoA-I? | ND | ND | ** | 3 | |
| F382E | ApoA-I? | 0.95 ± 0.2 | ND | ** | 3 | |
| N131A | ApoA-I? | 36 ± 1 | 10 ± 0.9 | 3.6 | NS | 3 |
| N131Dd | ApoA-I? | 2.6 ± 0.9 | 9.2 ± 9 | 0.28 | ** | 3 |
| N391A | ApoA-I? | 3.4 ± 0.4 | 4.0 ± 2 | 0.85 | ** | 3 |
| N391Sd | ApoA-I? | 6.2 ± 0.5 | 3.6 ± 1 | 1.7 | * | 3 |
| G230Rd | Lid | 1.8 ± 0.3 | 2.3 ± 2 | 0.78 | ** | 3 |
| P232A | Lid | 7.6 ± 0.7 | 6.2 ± 2 | 1.2 | * | 3 |
| I233A | Lid | 12 ± 1 | 29 ± 5 | 0.41 | * | 3 |
| I233E | Lid | 1.1 ± 0.2 | ND | ** | 3 | |
| M234A | Lid | 23 ± 0.9 | 9.4 ± 1 | 2.4 | NS | 3 |
| S235A | Lid | 7.80 ± 0.5 | 9.3 ± 2 | 0.84 | * | 3 |
| S236A | Lid | 23 ± 0.9 | 11 ± 1 | 2.1 | NS | 3 |
| Δlid | Lid | 0.78 ± 0.4 | 10 ± 20 | 0.078 | ** | 9 |
| Δlid/F382A | Lid/ApoA-I? | 0.28 ± 0.2 | ND | ** | 3 | |
| LPLA2lid | Lid | 0.48 ± 0.2 | ND | ** | 3 | |
| LPLA2αA | αA–αA′ loop | 0.57 ± 0.1 | ND | ** | 3 |
a Sig. represents significance compared with WT using a two-tailed t-test. NS indicates not significant. *, 0.01 < p < 0.05; **, 0.001 < p < 0.01.
b Error represents standard deviation.
c ND = not determined; signal was too low for an accurate fit.
d Entry indicates disease-related mutants.
HDX MS supports a dynamic lid in LCAT
Next, HDX MS was used to investigate the dynamics of LCAT and its response to binding a covalent substrate analog. Sequence coverage of 95% was achieved from pepsin digestions of LCAT treated with and without IDFP, with a 5.55 redundancy score (supplemental Fig. S6). In the absence of IDFP, the membrane-binding and cap domains showed relatively fast exchange compared with the α/β hydrolase domain, wherein most of helices and sheets are well protected from exchange (Fig. 4). The initial portion of the lid region (residues 223–236), along with moieties of the membrane-binding and cap domains that bracket the lid region (residues 63–75 and 238–277, respectively) showed the fastest increase in HDX from 28 to 62% over the time course. This result confirms the dynamic nature of the lid as indicated by B-factors in crystal structures.
Figure 4.

LCAT dynamics using HDX MS. A, relative deuterium uptake is shown using a color gradient for each of the five different time points. The most dynamic regions are highlighted using dashed boxes. Most of the α/β hydrolase domain, including catalytic triad residues (Ser-181, Asp-345, and His-377, red dots), has a relatively rigid conformation. B, relative percent deuterium uptake is mapped on the closed structure at 10 s, 10 min, and 4 h with the same coloring as in A. The N and C termini are not observed in the crystal structures but are indicated with dashed lines colored to show their relative deuterium uptake.
The most N-terminal peptide (residues 1–5) and the C-terminal region (residues 404–412), which are not ordered in any reported crystal structure (except for in the open-2Fab structure, where residues 4–8 are involved in a crystal contact), were immediately deuterated by 10 s at a high percentage (40–57 and 40–72%, respectively), indicating that their backbone amides are highly solvent-accessible. This is notable for the N-terminal region because it is known to be critical for LCAT activity, possibly by mediating contacts with apoA-I and/or lipids in the HDL particle (34). Indeed, HDX comparing full-length LCAT and a variant truncated after the first five N-terminal residues (Δ5) revealed no detectable difference in structural dynamics (supplemental Fig. S7A), whereas Δ5 lacks activity on HDL.
Covalent inhibitor alters the dynamics of LCAT
Insights into changes in dynamics that occur upon formation of a covalent intermediate were obtained by comparing native LCAT HDX data with that of LCAT treated with IDFP (Fig. 5 and supplemental Fig. S8). IDFP is composed of a fluorophosphonate group attached to a 12-carbon acyl chain (supplemental Fig. S9A). When IDFP reacts with Ser-181, the result is a complete loss of LCAT activity (supplemental Fig. S9, B and C) (17). We hypothesized that once IDFP binds to LCAT, the lid will retract into a more open, stable conformation such as that observed in the open-2Fab structure, which would result in a decrease in HDX and other associated regions of the protein. Such a stabilization is consistent with the 7 °C increase in LCAT-melting temperature following IDFP derivatization (supplemental Fig. S9D) (17) and the idea that the lid and adjacent structural elements coalesce to form a binding site for the acyl chain of IDFP. Indeed, HDX profiles revealed that multiple regions in LCAT are stabilized by IDFP, with IDFP-bound LCAT showing less HDX overall (Fig. 5 and supplemental Fig. S7B). The largest decrease in HDX was observed in the lid region, followed by residues 243–254 in the cap domain in agreement with their predicted role in directly interacting with IDFP. Peptides within the membrane-binding domain (residues 67–72) topographically adjacent to the lid also exhibited strong protection from HDX upon IDFP binding. Another region on the “front” surface of the protein (Fig. 5B) that becomes protected upon ligand binding is the αA–αA′ loop (residues 113–118), potentially due to a combination of direct contact with IDFP and/or a consequence of the retracted lid conformation. However, all of these regions were still relatively dynamic with respect to most of the rest of the protein, consistent with IDFP altering the equilibrium of conformation states rather than trapping unique conformational states.
Figure 5.

HDX MS differential heat map due to IDFP binding mapped on the primary and tertiary structures of LCAT. A, differences in deuterium uptake between IDFP-bound and unbound LCAT by sequence. The blue color in the sequence heat map represents decreased deuterium uptake upon IDFP binding. B, differences mapped on the open-2Fab structure (PBD entry 5BV7). The location of peptides with increased protection are indicated in magenta for the back of LCAT, and in blue and black for the front. Residues 243–254 (black) exhibited the highest degree of protection. The position of Ser-181 is indicated by a yellow sphere. C, examples of individual peptide deuterium uptake plots.
As expected, elements that directly contribute to the active site of LCAT also became protected from HDX upon IDFP binding. Residues 29–36 exhibited low solvent accessibility in native LCAT (Fig. 4) and even lower when reacted with IDFP (Fig. 5). This is likely explained by direct protection of residues 29–36 by IDFP, which covalently attaches to Ser-181 and occupies the oxyanion hole (17). Peptides containing Ser-181 (177–187 and 179–188) showed no difference in protection, but the solvent accessibility of this region was already extremely low in the IDFP-free state.
Interestingly, we observed IDFP-induced HDX protection on the “back” side of the protein (Fig. 5B), remote from the active site, including residues 146–156, 192–198, and 283–297. Most of these are situated at interfaces between the catalytic core, the cap domain, and the membrane-binding domain, and thus they are not expected to make direct contacts with IDFP. We hypothesize that as IDFP binds to LCAT, these regions tighten up and interact more strongly with each other primarily due to retraction of the lid.
Finally, to compare the HDX to the crystallographic data, we aligned the α/β hydrolase domains of our closed and the open-2Fab structures and then measured the distance between corresponding Cα atoms (Fig. 6, A and B, and supplemental Movie S1). Notably, the lid and cap domains show the largest changes between the two structures, which correlates well with the high HDX observed in these regions, as well as the protection conferred upon addition of IDFP. This is best illustrated in the region surrounding the lid, where the largest Cα–Cα distance is 27 Å (Ser-235), and a region of the cap domain (residues 274–295) moves 2.0–3.4 Å. The second helix in this span (residues 288–295) moves to accommodate the lid opening nearby, and it also exhibits a large degree of HDX protection in the IDFP-bound state. As flexible elements situated around the active site, these regions are all strongly implicated in interfacial activation and substrate binding.
Figure 6.

Changes associated with lid-opening and IDFP binding. A, changes in Cα–Cα distance are graphed for residues between our closed LCAT structure and the open-2Fab structure, aligned on their α/β hydrolase domains. There is a gap in the plot due to unmodeled residues in the two compared crystal structures (residues 236–240). Residues with Cα–Cα differences above 4 Å are colored in red, distances between 2 and 4 Å in orange, and distances 1.5 and 2 Å in yellow. B, Cα–Cα distances from A mapped onto the structures with the same color scheme. The filled arrowhead points to the closed lid, and the open arrowhead points to the open lid. C and D, IDFP-bound LPLA2 (PDB code 4X91) was aligned with the open-2Fab structure using PyMOL. The two tracks observed for IDFP, as observed in the LPLA2 structure, are shown with track A in C and track B in D. IDFP is modeled as purple spheres bound to the active-site serine.
Discussion
Active (open) and inactive (closed) conformations are well known characteristics of hydrolases. In the case of lipases, transitions between these states are hypothesized to be key aspects of interfacial activation, where full activity is achieved only when the enzyme interacts with the appropriate membrane surface (24, 25). Herein, we report the structure of LCAT with a nearly fully ordered lid that covers the active-site cleft of the enzyme, which we propose represents a soluble, lower activity conformation of the enzyme. HDX MS showed that regions with the most exchange (e.g. the lid) correspond to regions that exhibit the highest B-factors in this structure (Fig. 1D) and the most variable conformation among the available crystal structures (Fig. 6, A and B). Upon covalent reaction with an inhibitor (IDFP) that mimics a covalent intermediate, the lid was significantly stabilized. Based on these results, we hypothesize that a dynamic, extended LCAT lid helps to shield the hydrophobic active site until it comes in contact with HDL and then retracts into a more ordered conformation that can accommodate and form specific interactions with substrates (see model, Fig. 7).
Figure 7.
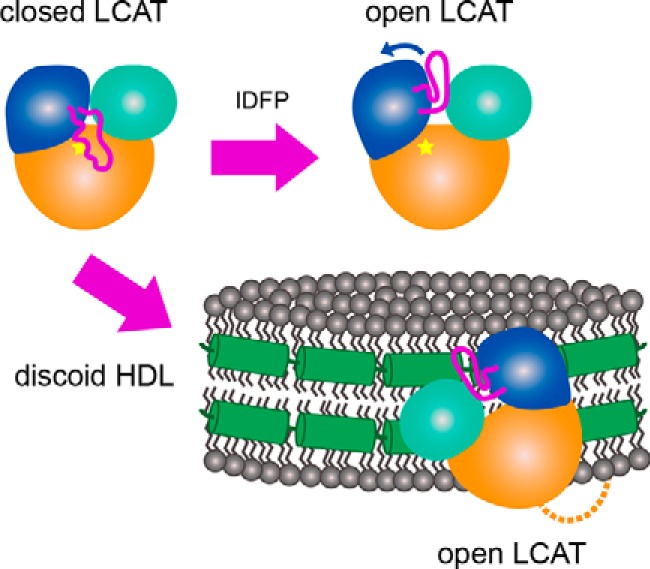
A model describing the proposed conformational change induced by IDFP and HDL. In solution, LCAT is in a closed state (top left) where the dynamic lid is extended over the active site (indicated by the star). As IDFP or other large hydrophobic molecules bind, we propose that the lid retracts into an open conformation (top right). The bottom arrow indicates HDL binding, which based on these data would involve the N-terminal region (dashed line), lid (magenta), and membrane-binding domain (teal). LCAT would then be positioned such that the hydrophobic active site is exposed to HDL to extract substrate for acyl transfer. The specific region of LCAT that contacts apoA-I (green helices) on HDL is unknown.
The trajectory of lid in the new Fab-free LCAT structure reported here is most similar to that of the closed-Fab structures, which contain lids that are disordered between residues 230 and 238 (supplemental Fig. S1A) (26). A prominent feature of our new LCAT structure is the interaction between hydrophobic residues in the lid and the side chains of residues Thr-123 and Phe-382, which are mutated in human disease. Mutation of residues that form the lid interface as well as deletion of the entire lid both led to significant increases in esterase activity against a soluble substrate, consistent with the lid shielding the active site against soluble substrates. Also consistent with our findings, the T123A and T123I mutations were previously shown to exhibit an increase in activity with a soluble, monomeric bis-pyrene PC esterase substrate (16, 44).
Mutations of Thr-123 and Phe-382 were previously reported to specifically affect acyltransferase and phospholipase reactions on HDL, but not acyltransferase activity on LDL (14, 16, 28, 42, 43). Because apoA-I is more abundant in HDL, this suggested a role for these residues in apoA-I binding or at least in a downstream allosteric pathway. However, our results show that although mutation of Thr-123 and Phe-382 causes a severe defect in acyltransferase activity on HDL, their mutation has no significant defect in HDL binding as measured by the maximal response in BLI or by changes in Kd (Fig. 3, supplemental Fig. S5, and Table 2). One explanation for this result is that there is little to no contribution of apoA-I to the overall Kd value, which is instead dominated by phospholipid bilayer interactions, as suggested by previous studies (14, 38, 45). If so, Thr-123 and Phe-382 are more critical for a subsequent step, such as activation by apoA-I, which may include a conformational change (46), or in binding specific substrates. Mutations within the lid itself did, however, show striking decreases in HDL binding. Given the proximity of the lid to residues in the membrane-binding domain already known to play a role in membrane interactions (e.g. Trp-48), our data support a model where interactions with the acyl phase of the membrane promotes retraction of the lid. This may also be accompanied by allosteric changes in the membrane-binding domain, facilitating access of lipid substrates into the catalytic cleft.
Somewhat surprisingly, the entire LCAT lid was dispensable for activity with the small soluble substrate pNPB, nor did its removal adversely affect thermal stability. The lid therefore is not critical for the structural integrity of the enzyme or in the binding and hydrolysis of short acyl chain esters. However, our BLI and acyl transfer data showed that this region is critical for both binding to HDLs and transesterification. With the exception of I233A and I233E, most point mutations within the lid did not cause a dramatic decrease in HDL binding or enzymatic activity, which may indicate that individual substitutions are not strong enough to disrupt lid function on their own. As the lid retracts, Ile-233 appears poised to contribute to the substrate-binding pocket (Fig. 2B), whereas other positions in the lid may directly or indirectly contribute to membrane binding or interactions with adjacent membrane-binding domains.
The functional importance of the lid is further supported by LCAT mutations that cause genetic disease such as N228K (14, 47), G230R (37), and R244G/R244H/R244C (48, 49). All three of these positions are likely involved in stabilizing the open state in WT LCAT, because Asn-228 forms a hydrogen bond with Asp-335, and Arg-244 forms hydrogen bonds with the backbone carbonyls of Leu-223 and Leu-285 in the open-2Fab structure. Asn-228 and Gly-230 may also be involved in acyl acceptor recognition, as proposed previously (17); however, they do not appear to be in position to help form a cholesterol-binding site in the open-2Fab structure.
Throughout this paper, we have been careful to not describe LCAT as having a single open and single closed conformation. Instead, the HDX MS data suggest that an equilibrium within each state is likely. After all, in the three reported high-resolution crystal forms of LCAT, the lid region adopts distinct conformations. The dynamic nature of the lid is also likely required for facilitating the overall reaction catalyzed by LCAT, as structurally diverse ligands (PC, acylated intermediate, and cholesterol) must be selectively accommodated in the active site.
We used an IDFP-bound LPLA2 crystal structure to model possible paths for acyl chains in the open-2Fab structure and to understand how protection from HDX might occur (Fig. 6, C and D) (17, 27). Certain regions on the front side of LCAT are more likely protected directly by IDFP, as they form the active site where IDFP binds. These regions include the oxyanion hole (residues 28–36), the lid region (residues 223–236), a segment in the cap domain (residues 247–254), and the αA–αA′ loop (residues 113–119). Regions that are more distal from the active site are most likely protected from HDX due to allosteric mechanisms. In the open conformation, the lid packs between the membrane-binding and cap domains, which explains the observed protection in regions that contact the retracted lid, as well as regions on the back of the protein (residues 146–156, 192–198, and 287–297), most of which are found at interfaces between these domains.
In conclusion, our data provide strong evidence that HDL binding by LCAT involves conformational changes that favor membrane binding concomitant with the retraction and stabilization of a dynamic lid that shields the active site while the enzyme is in its soluble state (Fig. 7). The lid, either directly or indirectly, is also important for interactions with HDL. Future studies will focus on probing how LCAT and HDL particles interact and the molecular role of apoA-I in activating LCAT.
Experimental procedures
Protein production and purification
The codon-optimized human LCAT gene with a C-terminal His6 tag in pcDNA4 was transiently transfected in HEK293F (Invitrogen) cells as described previously (17). The cells were grown in suspension, and conditioned media were harvested 5 days later. The secreted recombinant protein was purified via nickel-nitrilotriacetic acid and dialyzed against reaction buffer (20 mm HEPES, pH 7.5, 150 mm NaCl). For LCAT used in HDX MS, the cDNA sequence of human LCAT was inserted into an expression vector along with a glutamine-synthetase selectable marker for stable expression under l-methionine sulfoximine selection in a glutamine-free medium. The production cell line was constructed by introduction of the linearized expression plasmid DNA into a CHO-S host cell line. The resulting cells were cloned through a single round of FACS single-cell deposition cloning. The clone selection was performed using MedImmune's proprietary fed-batch process, and the top four clones were selected based on the final product titer. The final clone was selected based on productivity and specific activity of unpurified LCAT. Recombinant human LCAT was then produced in a bioreactor using a proprietary perfusion process with chemically defined media optimized for productivity and specific activity of unpurified LCAT.
Crystallization and structure determination
Crystals of full-length LCAT (10 mg/ml in 20 mm HEPES, pH 7.5, 150 mm NaCl) were obtained via sitting drop vapor diffusion using the JCSG+ Suite (Qiagen) in a condition containing 0.2 m ammonium formate and 20% PEG 3350. Crystals formed at 4 °C in a 1-μl drop with a protein to mother liquor ratio of 1:1. The crystals were cryoprotected by addition of dry glucose to the mother liquor to a final concentration of 30% (w/v). Two data sets were collected at the Advanced Photon Source (APS) at Argonne National Laboratories on the LS-CAT (λ = 0.97919) and GM/CA (λ = 0.98400) beam lines from crystals frozen in nylon cryoloops (Hampton). The data were processed and scaled with XDS (50), and Aimless (51) in the CCP4 (52) suite was used to merge the data sets. An LCAT homology model based on LPLA2 (17) generated by MODELLER (53) was used as a search model in molecular replacement using PHASER (54) to generate initial phases. Non-crystallographic symmetry restraints were applied to the four copies of LCAT per asymmetric unit during refinement in REFMAC5 (55). Manual model building was performed with Coot (56). The final model was validated for stereochemical correctness with MolProbity (57).
Differential scanning fluorimetry
Tm values were determined using a Thermofluor Analyzer (Johnson & Johnson), as described previously (17), with three replicates performed at least in triplicate. LCAT at 0.1 mg/ml was diluted into reaction buffer containing 100 μm 1-anilinonaphthalene-8-sulfonic acid (Sigma) in a final volume of 9 μl in black 384-well PCR plates (Thermo Fisher Scientific, AB-1384/K). 2 μl of silicone oil was overlaid on the reactions to prevent evaporation. The reactions were run from 25 to 80 °C in an up/down mode (30 s per hold) with an image taken at every degree. As the baseline Tm was not consistent (range of 50.7–56.3 °C for WT LCAT) but the ΔTm was consistent, WT LCAT was included on each plate, and the change in Tm with respect to WT LCAT was calculated for each experiment. A one-sample two-tailed t test was used to compare the ΔTm means to a hypothetical value of zero using GraphPad Prism, because the WT LCAT sample had no error values as it was used to establish a baseline of 0.
Soluble esterase assay with pNPB
The esterase assay was performed as described previously (17) at least in triplicate (see Table S1 for number of replicates). pNPB (Sigma) was diluted to 10 mm into reaction buffer containing 10% dimethyl sulfoxide. The reaction was started by addition of 40 μl of 1 μm LCAT to 10 μl of pNPB, and the increase in absorbance at 400 nm was monitored on a Spectramax plate reader. An unpaired two-tailed t test was used to compare each variant to WT LCAT using GraphPad Prism.
HDL preparation
HDLs were prepared with both the ESP24218 peptide (GenScript) with the sequence PVLDLFRELLNELLEALKQKLK and apoA-I purified from human serum (58). 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 16:0 biotinyl Cap PE (biotin PE) were purchased from Avanti Polar lipids, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) from NOF America, and dehydroergosterol (DHE) from Sigma.
For the peptide HDLs, DPPC (8.3 mg, 11 μmol), POPC (8.6 mg, 11 μmol), and either biotin PE (for peptide-biotin HDLs, 0.24 mg, 0.23 μmol) or DHE (for peptide-DHE HDLs, 0.5 mg, 1.3 μmol) were dissolved in chloroform. Peptide (8.2 mg) was dissolved in methanol/water 1:1 (v/v). The lipid solution and peptide solution were mixed and vortexed in a glass vial and dried under nitrogen flow at room temperature for 4 h before being placed in a vacuum oven overnight at room temperature to remove residual solvent. The lipid film was rehydrated with 2.5 ml of 20 mm phosphate buffer containing 1 mm EDTA, pH 7.4, followed by water bath sonication (5 min, room temperature) and probe sonication (2 min × 50 watts, room temperature) to obtain peptide-biotin HDL, with a final biotin PE concentration of 0.09 mm, or peptide-DHE HDL, with a final DHE concentration of 0.5 mm.
For the apoA-I HDLs, DPPC (8.3 mg, 11 μmol), POPC (8.6 mg, 11 μmol), and either biotin PE (for apoA-I-biotin HDLs, 0.24 mg, 0.23 μmol) or DHE (for apoA-I-DHE HDLs, 0.5 mg, 1.3 μmol) were dissolved in chloroform. The lipid solution was dried under nitrogen flow at room temperature for 4 h before being placed in a vacuum oven overnight (room temperature) to remove residual chloroform. 1.38 ml of Tris/HCl buffer (20 mm Tris/HCl, 1 mm EDTA, and 0.02% NaN3, pH 8) was added to the lipid film, preheated to 55 °C, and vortexed for 10 s, followed by water bath sonication (20 min, room temperature). The suspension was then mixed with 0.44 ml of 30 mg/ml sodium cholate in Tris/HCl buffer (20 mm Tris/HCl, 1 mm EDTA, and 0.02% NaN3, pH 8) and heated in a 50 °C water bath for 1 min, followed by water bath sonication (10 min, room temperature) to obtain a micelle solution. The micelle solution (1.82 ml, preheated to 50 °C) was mixed with 0.7 ml of apoA-I (9 mg/ml, preheated to 50 °C) with gentle shaking, followed by three rounds of thermal cycling between 48 °C (10 min) and at room temperature (10 min). Finally, the sodium cholate was removed via two rounds of incubation with Biobeads SM-2 (Bio-Rad), during which they were rotated for 2 h at room temperature (20 rpm/min).
Bio-layer interferometry
A FortéBio Octet RED system was used to measure the binding of LCAT to the peptide-biotin or apoA-I-biotin HDLs. All steps were performed at 25 °C, which reduces the ability of LCAT to react with the HDL substrate and thereby minimizes consumption of the HDL during measurement (38). Streptavidin-coated tips were soaked for at least 10 min to rehydrate the streptavidin prior to HDL loading. Peptide-biotin HDLs were diluted 1:40, whereas apoA-I-biotin HDLs were diluted 1:20 in assay buffer (1× PBS, pH 7.4, 1 mm EDTA, 60 μm fatty acid-free bovine serum albumin (BSA, Sigma)), and then immobilized on the streptavidin tips for 600 s with shaking at 1000 rpm, followed by a wash in assay buffer for 600 s at 1000 rpm to remove unbound HDLs. A baseline was next established for 30 s (1000 rpm). The HDL-bound tips were then dipped into LCAT protein at 0.2 μm in assay buffer or buffer alone as a control and allowed to associate for 300 s (1000 rpm), and then dissociated in assay buffer for 600 s (1000 rpm). These experiments were performed at least in duplicate for each type of HDL (see supplemental Table S2). When determining the Kd values for LCAT variants with apoA-I-biotin-HDLs, LCAT was titrated from 0.2 to 2.4 μm and from 0.6 to 6 μm for ΔNΔC LCAT. The association time was increased to 600 s, and the dissociation time was increased to 1000 s to ensure full association and dissociation. All experiments were performed at least in triplicate (see Table 3).
Changes in refractive index were analyzed with FortéBio's Data Analysis 7.0, and reference sensor measurements without LCAT were used for baseline subtraction. The association and dissociation curves were individually fit using GraphPad Prism using a two-phase model, with a slow phase determined to be LCAT binding non-specifically to the streptavidin tips, as LCAT binds to the tips in the absence of HDL with the same rate. Initially, the plateau value for the fit was used as a general indication of binding in comparison with WT LCAT for each protein variant at 0.2 μm. For these experiments, WT LCAT was included as a positive control to allow for direct comparison. To determine Kd values, the data were fit to a two-phase model as before, and the kobs (from association) and koff (from dissociation) rate constants were determined at each concentration for the fast phase. The kobs were plotted against LCAT concentration, and the slope of the line was evaluated as kon, using the equation kobs = kon [LCAT] + koff. The experimental koff values were averaged together for each variant, and the final Kd value was calculated from koff/kon. For statistical analysis, the Kd for each replicate was determined individually, and the results were compared with WT binding to apoA-I HDLs using an unpaired two-tailed t test in GraphPad Prism.
DHE acyltransferase assay
The LCAT sterol esterification assay was modified from a published protocol (40) to minimize the amount of LCAT and HDL required. The assay was performed in 384-well low-volume black microplates (Corning 4514) with a total assay volume of 16 μl and a final volume of 20 μl after the addition of 4 μl of stop solution (1× PBS with 1 mm EDTA, 5 units/ml cholesterol oxidase (COx), and 7% Triton X-100). In each reaction, LCAT was diluted in assay buffer to 15 μg/ml (see above), whereas the DHE HDLs were diluted in 1× PBS with 1 mm EDTA and 5 mm β-mercaptoethanol. 8 μl of the HDLs were added across the plate, and the reactions were initiated with 8 μl of LCAT, so that LCAT was assayed at 7.5 μg/ml with a range of DHE concentrations from 0 to 50 μm. The peptide–DHE HDLs were stopped after 25 min at 37 °C, whereas the apoA-I–DHE HDLs required a 60-min assay due to the lower signal achieved, likely due to the heating step in HDL preparation. Following the addition of stop solution, the plates were incubated for another 30–60 min at 37 °C to allow for the COx to react. After the plates were re-equilibrated at room temperature, fluorescence was determined on a SpectraMax M5 plate reader with excitation at 325 nm and emission at 425 nm, with a 420-nm cutoff. WT LCAT was included as a positive control on each plate, whereas reactions without LCAT were used for background subtraction, and reactions without LCAT and stop solution lacking COx were used to generate a standard curve for DHE. Reactions were performed in triplicate with three independent experiments per LCAT variant. Data were processed via background subtraction to remove excess fluorescence that results from the higher concentrations of DHE. These values were divided by the slope of the line from the standard curve, which yields the amount of DHE-ester that resulted in each well, and then by the assay time to determine the rate. An unpaired two-tailed t test was used to compare each variant to WT LCAT using GraphPad Prism.
LCAT derivatization with IDFP
IDFP (17 mm) in methyl acetate was first diluted to 10 mm with DMSO and then to 200 mm with HDX buffer consisting of 10 mm sodium phosphate, pH 7.2, 300 mm sucrose. LCAT was then added from a stock of 19 mg/ml to a final concentration of 0.5 mg/ml in a 3-ml total reaction volume. The mixture was reacted for 110 min at room temperature and then concentrated to 8.4 mg/ml using a 30,000 MWCO filter. The soluble esterase assay with pNPB was used with HDX buffer to confirm that LCAT had completely reacted with IDFP and had no residual activity. Data from at least three independent experiments were analyzed using a paired t test. The derivatization with IDFP was further supported via DSF, again in HDX buffer (17, 26). Three independent experiments were performed in triplicate and analyzed with a paired t test.
Hydrogen–deuterium exchange mass spectrometry
The undeuterated protein sample was buffer-exchanged with HDX buffer. The peptide coverage map for both LCAT alone and IDFP bound was obtained from the undeuterated controls as follows: 2.5 μl of sample was diluted with 47.5 μl of buffer solution at room temperature followed by addition of 100 μl of ice-cold quench (4 m guanidine-HCl, 250 mm tris(2-carboxyethyl)phosphine, pH 2.5). Quenched samples were immediately injected into Waters nanoACQUITY UPLC system (59). The on-line pepsin digestion was performed using a 2.0 × 30 mm Ezymate BEH pepsin column (Waters) for 4 min in 0.15% formic acid in H2O at a flow rate of 100 μl/min at 25 °C, and the peptic peptides were trapped and desalted on line using an ACQUITY UPLC BEH C18 1.7-μm VanGuard Pre-column (Waters) at 0 °C. The peptides were eluted into a 1.0 × 100-mm ACQUITY UPLC BEH C18 column (Waters) held at 0 °C and were separated at a flow rate of 40 μl/min in a step gradient: 8–15% acetonitrile gradient in the first 1-min followed by 7 min of linear acetonitrile gradient (15–32%) and held at 40% acetonitrile for 1 min. The eluent was directed into a Waters Xevo Q-Tof G2 XS mass spectrometer with electrospray ionization and lock mass correction. Peptic peptides were acquired in MSE mode and identified using Waters ProteinLynx Global Server 3.0 (60–62), and the PLGS outputs were processed in Waters DynamX 3.0 software. Supplemental Fig. S10 shows the workflow for peptides 240–253 as an example.
For deuterated samples, the dilution and quench steps were the same as for undeuterated controls. The labeling solution (10 mm sodium phosphate, pD 7.2 (= pH 6.8), 300 mm sucrose in D2O) was introduced to the protein stock solution in 20-fold excess, incubated at room temperature for 10 s, 1 and 10 min, and 1 and 4 h, and the labeled samples were immediately quenched at a 1:1 ratio. The deuterium uptake in Da and percentage were determined from the mass shift between deuterated and undeuterated peptide ions pertaining to the intensity weighted center mass using DynamX software (supplemental Fig. S10). For each peptide, the relative deuterium uptake was plotted without back-exchange correction. The average back-exchange was observed to be 24% using a highly deuterated 9-peptide standard, similar to previous data (63–65). Entire labeled samples were duplicated, and undeuterated controls were acquired in triplicate. A comparability profile for the HDX difference between LCAT treated with and without IDFP was plotted in supplemental Fig. S9B (66).
Structural analysis
Crystal structures were visualized using PyMOL (Schrödinger) and in-house developed PyMOL plug-in (HDX toolbox) to display the HDX data on the crystal structures. The open-2Fab and closed structures were aligned on their α/β hydrolase domains using LSQKAB in the CCP4 suite to measure Cα–Cα distances (30, 52). Supplemental Movie S1 was created by using Chimera (67) to create a trajectory between the common residues in the closed structure described here and open-2Fab. The movie was rendered using PyMOL.
Quantification and statistical analysis
Statistical analysis was performed using an unpaired two-tailed t test by comparing the data for each variant to WT LCAT, except for the DSF assay which compared the ΔTm for each variant to a hypothetical value of zero. A paired t test was used to compare the HDX samples pre- and post-IDFP derivatization. GraphPad Prism was used to calculate p values, which are indicated in the figure legends for each experiment. The statistical parameters for each experiment are indicated in the table or supplemental table that corresponds to each experiment, the figure legends, and methods.
Author contributions
K. A. M., A. G., and L. C. performed protein expression and purification. A. G. performed crystallization, data collection, and structure determination. K. A. M., A. G., and J. J. G. T. performed structure refinement. K. A. M. and T. D. M. performed cloning, esterase, and DSF assays. W. Y. made the HDLs for binding and acyltransferase assays. K. A. M. designed and performed HDL binding and acyltransferase assays, IDFP derivatization, and supervised T. D. M. J. A. collected and analyzed the HDX MS data. K. A. M., J. A., and J. J. G. T. wrote the manuscript, with contributions from all authors. C. L. supported the project. J. J. G. T., M. J. A., A. S., and J. A. S. provided project supervision. All authors approved the final paper.
Supplementary Material
Acknowledgments
We thank the organizers of the CCP4/APS Summer School 2014 and the staff at both the GM/CA and LS-CAT beamlines. We also thank Yiming Li, Michaela Wendeler, and Jeong Lee for LCAT purification and expression and Rick Remmele for helpful input.
This work was supported by National Institutes of Health Grants HL071818 and HL122416 (to J. J. G. T.) and AR056991 (to J. A. S.), F32 Ruth L. Kirschstein NRSA F32HL131288 (to K. A. M.), American Heart Association Postdoctoral Fellowships 15POST24870001 (to K. A. M.) and 16POST27760002 (to W. Y.), American Heart Association Scientist Development Grant 13SDG17230049 (to A. S.), University of Michigan Chemical Biology Postdoctoral Fellowship (to A. G.), Veterans Affairs Merit Review Award 1I01BX002021 (to J. A. S.), and MedImmune. J. A., C. L., and M. J. A. declare that they are employees of MedImmune, the global biologics R&D arm of AstraZeneca, and own stock/stock options in AstraZeneca. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S10, Tables S1–S3 and Movie S1.
The atomic coordinates and structure factors (code 5TXF) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LCAT
- lecithin:cholesterol acyltransferase
- apoA-I
- apolipoprotein A-I
- PC
- phosphatidylcholine
- FLD
- familial LCAT deficiency
- FED
- fish eye disease
- LPLA2
- lysosomal phospholipase A2
- HDX MS
- hydrogen–deuterium exchange mass spectrometry
- IDFP
- isopropyl dodecylfluorophosphonate
- r.m.s.d.
- root mean square deviation
- PDB
- Protein Data Bank
- DSF
- differential scanning fluorimetry
- pNPB
- p-nitrophenyl butyrate
- BLI
- bio-layer interferometry
- DHE
- dehydroergosterol
- IDFP
- isopropyl dodecylfluorophosphonate
- DPPC
- 1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- COx
- cholesterol oxidase
- PE
- phosphatidylethanolamine.
References
- 1. Calabresi L., Simonelli S., Gomaraschi M., and Franceschini G. (2012) Genetic lecithin:cholesterol acyltransferase deficiency and cardiovascular disease. Atherosclerosis 222, 299–306 [DOI] [PubMed] [Google Scholar]
- 2. Glomset J. A. (1968) The plasma lecithins:cholesterol acyltransferase reaction. J. Lipid Res. 9, 155–167 [PubMed] [Google Scholar]
- 3. Chen C. H., and Albers J. J. (1982) Distribution of lecithin-cholesterol acyltransferase (LCAT) in human plasma lipoprotein fractions. Evidence for the association of active LCAT with low-density lipoproteins. Biochem. Biophys. Res. Commun. 107, 1091–1096 [DOI] [PubMed] [Google Scholar]
- 4. Zhao Y., Thorngate F. E., Weisgraber K. H., Williams D. L., and Parks J. S. (2005) Apolipoprotein E is the major physiological activator of lecithin-cholesterol acyltransferase (LCAT) on apolipoprotein B lipoproteins. Biochemistry 44, 1013–1025 [DOI] [PubMed] [Google Scholar]
- 5. Kuivenhoven J. A., Pritchard H., Hill J., Frohlich J., Assmann G., and Kastelein J. (1997) The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J. Lipid Res. 38, 191–205 [PubMed] [Google Scholar]
- 6. Rousset X., Vaisman B., Amar M., Sethi A. A., and Remaley A. T. (2009) Lecithin:cholesterol acyltransferase–from biochemistry to role in cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 16, 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rousset X., Shamburek R., Vaisman B., Amar M., and Remaley A. T. (2011) Lecithin cholesterol acyltransferase: an anti- or pro-atherogenic factor? Curr. Atheroscler. Rep. 13, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahsan L., Ossoli A. F., Freeman L., Vaisman B., Amar M. J., Shamburek R. D., and Remaley A. T. (2014) in The HDL Handbook: Biological Functions and Clinical Implications (Komoda T., ed) pp. 159–194, Elsevier, New York [Google Scholar]
- 9. Fielding C. J., Shore V. G., and Fielding P. E. (1972) A protein cofactor of lecithin:cholesterol acyltransferase. Biochem. Biophys. Res. Commun. 46, 1493–1498 [DOI] [PubMed] [Google Scholar]
- 10. Jonas A. (2000) Lecithin cholesterol acyltransferase. Biochim. Biophys. Acta 1529, 245–256 [DOI] [PubMed] [Google Scholar]
- 11. Sorci-Thomas M. G., Bhat S., and Thomas M. J. (2009) Activation of lecithin:cholesterol acyltransferase by HDL apoA-I central helices. Clin. Lipidol. 4, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dassuex J.-L., Sekul R., Buttner K., Cornut I., Metz G., and Dufourcq J. (December 21, 1999) Apolipoprotein A-I agonists and their use to treat dyslipidemic disorders. U. S. Patent 6,004,925 [Google Scholar]
- 13. Li D., Gordon S., Schwendeman A., and Remaley A. T. (2015) in Apolipoprotein mimetics in management of human disease (Anantharamaiah G.M, and Goldberg D., eds), pp. 29–42, Springer International Publishing, New York [Google Scholar]
- 14. Adimoolam S., Jin L., Grabbe E., Shieh J. J., and Jonas A. (1998) Structural and functional properties of two mutants of lecithin-cholesterol acyltransferase (T123I and N228K). J. Biol. Chem. 273, 32561–32567 [DOI] [PubMed] [Google Scholar]
- 15. Peelman F., Verschelde J. L., Vanloo B., Ampe C., Labeur C., Tavernier J., Vandekerckhove J., and Rosseneu M. (1999) Effects of natural mutations in lecithin:cholesterol acyltransferase on the enzyme structure and activity. J. Lipid Res. 40, 59–69 [PubMed] [Google Scholar]
- 16. Vanloo B., Peelman F., Deschuymere K., Taveirne J., Verhee A., Gouyette C., Labeur C., Vandekerckhove J., Tavernier J., and Rosseneu M. (2000) Relationship between structure and biochemical phenotype of lecithin:cholesterol acyltransferase (LCAT) mutants causing fish-eye disease. J. Lipid Res. 41, 752–761 [PubMed] [Google Scholar]
- 17. Glukhova A., Hinkovska-Galcheva V., Kelly R., Abe A., Shayman J. A., and Tesmer J. J. (2015) Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase. Nat. Commun. 6, 6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hiraoka M., Abe A., and Shayman J. A. (2005) Structure and function of lysosomal phospholipase A2: identification of the catalytic triad and the role of cysteine residues. J. Lipid Res. 46, 2441–2447 [DOI] [PubMed] [Google Scholar]
- 19. Peelman F., Vinaimont N., Verhee A., Vanloo B., Verschelde J. L., Labeur C., Seguret-Mace S., Duverger N., Hutchinson G., Vandekerckhove J., Tavernier J., and Rosseneu M. (1998) A proposed architecture for lecithin cholesterol acyl transferase (LCAT): identification of the catalytic triad and molecular modeling. Protein Sci. 7, 587–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schrag J. D., Li Y., Cygler M., Lang D., Burgdorf T., Hecht H. J., Schmid R., Schomburg D., Rydel T. J., Oliver J. D., Strickland L. C., Dunaway C. M., Larson S. B., Day J., and McPherson A. (1997) The open conformation of a Pseudomonas lipase. Structure 5, 187–202 [DOI] [PubMed] [Google Scholar]
- 21. Kim K. K., Song H. K., Shin D. H., Hwang K. Y., and Suh S. W. (1997) The crystal structure of a triacylglycerol lipase from Pseudomonas cepacia reveals a highly open conformation in the absence of a bound inhibitor. Structure 5, 173–185 [DOI] [PubMed] [Google Scholar]
- 22. Lang D. A., Mannesse M. L., de Haas G. H., Verheij H. M., and Dijkstra B. W. (1998) Structural basis of the chiral selectivity of Pseudomonas cepacia lipase. Eur. J. Biochem. 254, 333–340 [DOI] [PubMed] [Google Scholar]
- 23. Nardini M., Lang D. A., Liebeton K., Jaeger K. E., and Dijkstra B. W. (2000) Crystal structure of Pseudomonas aeruginosa lipase in the open conformation. The prototype for family I.1 of bacterial lipases. J. Biol. Chem. 275, 31219–31225 [DOI] [PubMed] [Google Scholar]
- 24. Brzozowski A. M., Derewenda U., Derewenda Z. S., Dodson G. G., Lawson D. M., Turkenburg J. P., Bjorkling F., Huge-Jensen B., Patkar S. A., and Thim L. (1991) A model for interfacial activation in lipases from the structure of a fungal lipase-inhibitor complex. Nature 351, 491–494 [DOI] [PubMed] [Google Scholar]
- 25. van Tilbeurgh H., Egloff M. P., Martinez C., Rugani N., Verger R., and Cambillau C. (1993) Interfacial activation of the lipase-procolipase complex by mixed micelles revealed by X-ray crystallography. Nature 362, 814–820 [DOI] [PubMed] [Google Scholar]
- 26. Piper D. E., Romanow W. G., Gunawardane R. N., Fordstrom P., Masterman S., Pan O., Thibault S. T., Zhang R., Meininger D., Schwarz M., Wang Z., King C., Zhou M., and Walker N. P. (2015) The high-resolution crystal structure of human LCAT. J. Lipid Res. 56, 1711–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gunawardane R. N., Fordstrom P., Piper D. E., Masterman S., Siu S., Liu D., Brown M., Lu M., Tang J., Zhang R., Cheng J., Gates A., Meininger D., Chan J., Carlson T., et al. (2016) Agonistic human antibodies binding to lecithin-cholesterol acyltransferase modulate high-density lipoprotein metabolism. J. Biol. Chem. 291, 2799–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Funke H., von Eckardstein A., Pritchard P. H., Albers J. J., Kastelein J. J., Droste C., and Assmann G. (1991) A molecular defect causing fish eye disease: an amino acid exchange in lecithin-cholesterol acyltransferase (LCAT) leads to the selective loss of α-LCAT activity. Proc. Natl. Acad. Sci. U.S.A. 88, 4855–4859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nanjee M. N., Stocks J., Cooke C. J., Molhuizen H. O., Marcovina S., Crook D., Kastelein J. P., and Miller N. E. (2003) A novel LCAT mutation (Phe382→Val) in a kindred with familial LCAT deficiency and defective apolipoprotein B-100. Atherosclerosis 170, 105–113 [DOI] [PubMed] [Google Scholar]
- 30. Kabsch W. (1976) A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A 32, 922–923 [Google Scholar]
- 31. Kuivenhoven J. A., van Voorst tot Voorst E. J., Wiebusch H., Marcovina S. M., Funke H., Assmann G., Pritchard P. H., and Kastelein J. J. (1995) A unique genetic and biochemical presentation of fish-eye disease. J. Clin. Invest. 96, 2783–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frohlich J., Hoag G., McLeod R., Hayden M., Godin D. V., Wadsworth L. D., Critchley J. D., and Pritchard P. H. (1987) Hypoalphalipoproteinemia resembling fish eye disease. Acta Med. Scand. 221, 291–298 [DOI] [PubMed] [Google Scholar]
- 33. Hill J. S. (1994) The Molecular Pathology of Lecithin:Cholesterol Acyltransferase Deficiency. Doctoral dissertation, University of British Columbia [Google Scholar]
- 34. Vickaryous N. K., Teh E. M., Stewart B., and Dolphin P. J. (2003) Deletion of N-terminal amino acids from human lecithin: cholesterol acyltransferase differentially affects enzyme activity toward α-and β-substrate lipoproteins. Biochim. Biophys. Acta 1646, 164–172 [DOI] [PubMed] [Google Scholar]
- 35. Lee Y. P., Adimoolam S., Liu M., Subbaiah P. V., Glenn K., and Jonas A. (1997) Analysis of human lecithin-cholesterol acyltransferase activity by carboxyl-terminal truncation. Biochim. Biophys. Acta 1344, 250–261 [DOI] [PubMed] [Google Scholar]
- 36. Francone O. L., Evangelista L., and Fielding C. J. (1996) Effects of carboxy-terminal truncation on human lecithin:cholesterol acyltransferase activity. J. Lipid Res. 37, 1609–1615 [PubMed] [Google Scholar]
- 37. Miettinen H. E., Gylling H., Tenhunen J., Virtamo J., Jauhiainen M., Huttunen J. K., Kantola I., Miettinen T. A., and Kontula K. (1998) Molecular genetic study of Finns with hypoalphalipoproteinemia and hyperalphalipoproteinemia: a novel Gly230Arg mutation (LCATFin) of lecithin:cholesterol acyltransferase (LCAT) accounts for 5% of cases with very low serum HDL cholesterol levels. Arterioscler. Thromb. Vasc. Biol. 18, 591–598 [DOI] [PubMed] [Google Scholar]
- 38. Jin L., Shieh J.-J., Grabbe E., Adimoolam S., Durbin D., and Jonas A. (1999) Surface plasmon resonance biosensor studies of human wild-type and mutant lecithin cholesterol acyltransferase interactions with lipoproteins. Biochemistry 38, 15659–15665 [DOI] [PubMed] [Google Scholar]
- 39. Gu X., Wu Z., Huang Y., Wagner M. A., Baleanu-Gogonea C., Mehl R. A., Buffa J. A., DiDonato A. J., Hazen L. B., Fox P. L., Gogonea V., Parks J. S., DiDonato J. A., and Hazen S. L. (2016) A systematic investigation of structure/function requirements for the apolipoprotein A-I/lecithin cholesterol acyltransferase interaction loop of high-density lipoprotein. J. Biol. Chem. 291, 6386–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Homan R., Esmaeil N., Mendelsohn L., and Kato G. J. (2013) A fluorescence method to detect and quantitate sterol esterification by lecithin:cholesterol acyltransferase. Anal. Biochem. 441, 80–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klein H. G., Duverger N., Albers J. J., Marcovina S., Brewer H. B. Jr., and Santamarina-Fojo S. (1995) In vitro expression of structural defects in the lecithin-cholesterol acyltransferase gene. J. Biol. Chem. 270, 9443–9447 [DOI] [PubMed] [Google Scholar]
- 42. O K, Hill J. S., Wang X., and Pritchard P. H. (1993) Recombinant lecithin:cholesterol acyltransferase containing a Thr123→Ile mutation esterifies cholesterol in low-density lipoprotein but not in high-density lipoprotein. J. Lipid Res. 34, 81–88 [PubMed] [Google Scholar]
- 43. Qu S. J., Fan H. Z., Blanco-Vaca F., and Pownall H. J. (1995) In vitro expression of natural mutants of human lecithin:cholesterol acyltransferase. J. Lipid Res. 36, 967–974 [PubMed] [Google Scholar]
- 44. Christiaens B., Vanloo B., Gouyette C., Van Vynckt I., Caster H., Taveirne J., Verhee A., Labeur C., Peelman F., Vandekerckhove J., Tavernier J., and Rosseneu M. (2000) Headgroup specificity of lecithin cholesterol acyltransferase for monomeric and vesicular phospholipids. Biochim. Biophys. Acta 1486, 321–327 [DOI] [PubMed] [Google Scholar]
- 45. Bolin D. J., and Jonas A. (1994) Binding of lecithin: cholesterol acyltransferase to reconstituted high-density lipoproteins is affected by their lipid but not apolipoprotein composition. J. Biol. Chem. 269, 7429–7434 [PubMed] [Google Scholar]
- 46. Reshetnyak Y., Tchedre K. T., Nair M. P., Pritchard P. H., and Lacko A. G. (2006) Structural differences between wild-type and fish eye disease mutant of lecithin:cholesterol acyltransferase. J. Biomol. Struct. Dyn. 24, 75–82 [DOI] [PubMed] [Google Scholar]
- 47. Gotoda T., Yamada N., Murase T., Sakuma M., Murayama N., Shimano H., Kozaki K., Albers J. J., Yazaki Y., and Akanuma Y. (1991) Differential phenotypic expression by three mutant alleles in familial lecithin:cholesterol acyltransferase deficiency. Lancet 338, 778–781 [DOI] [PubMed] [Google Scholar]
- 48. Charlton-Menys V., Pisciotta L., Durrington P. N., Neary R., Short C. D., Calabresi L., Calandra S., and Bertolini S. (2007) Molecular characterization of two patients with severe LCAT deficiency. Nephrol. Dial. Transplant. 22, 2379–2382 [DOI] [PubMed] [Google Scholar]
- 49. Pisciotta L., Calabresi L., Lupattelli G., Siepi D., Mannarino M. R., Moleri E., Bellocchio A., Cantafora A., Tarugi P., Calandra S., and Bertolini S. (2005) Combined monogenic hypercholesterolemia and hypoalphalipoproteinemia caused by mutations in LDL-R and LCAT genes. Atherosclerosis 182, 153–159 [DOI] [PubMed] [Google Scholar]
- 50. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 52. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 54. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lerch P. G., Förtsch V., Hodler G., and Bolli R. (1996) Production and characterization of a reconstituted high-density lipoprotein for therapeutic applications. Vox Sang. 71, 155–164 [DOI] [PubMed] [Google Scholar]
- 59. Wales T. E., Fadgen K. E., Gerhardt G. C., and Engen J. R. (2008) High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal. Chem. 80, 6815–6820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li G. Z., Vissers J. P., Silva J. C., Golick D., Gorenstein M. V., and Geromanos S. J. (2009) Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 9, 1696–1719 [DOI] [PubMed] [Google Scholar]
- 61. Geromanos S. J., Vissers J. P., Silva J. C., Dorschel C. A., Li G. Z., Gorenstein M. V., Bateman R. H., and Langridge J. I. (2009) The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data depandant LC-MS/MS. Proteomics 9, 1683–1695 [DOI] [PubMed] [Google Scholar]
- 62. Silva J. C., Denny R., Dorschel C., Gorenstein M. V., Li G. Z., Richardson K., Wall D., and Geromanos S. J. (2006) Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: a sweet tale. Mol. Cell. Proteomics 5, 589–607 [DOI] [PubMed] [Google Scholar]
- 63. Iacob R. E., Chen G., Ahn J., Houel S., Wei H., Mo J., Tao L., Cohen D., Xie D., Lin Z., Morin P. E., Doyle M. L., Tymiak A. A., and Engen J. R. (2014) The influence of adnectin binding on the extracellular domain of epidermal growth factor receptor. J. Am. Soc. Mass Spectrom. 25, 2093–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Iacob R. E., Bou-Assaf G. M., Makowski L., Engen J. R., Berkowitz S. A., and Houde D. (2013) Investigating monoclonal antibody aggregation using a combination of H/DX-MS and other biophysical measurements. J. Pharm. Sci. 102, 4315–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ahn J., Jung M. C., Wyndham K., Yu Y. Q., and Engen J. R. (2012) Pepsin immobilized on high-strength hybrid particles for continuous flow online digestion at 10,000 psi. Anal. Chem. 84, 7256–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Houde D., Berkowitz S. A., and Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.