

Abstract
Trypanosoma brucei causes African trypanosomiasis and contains three full-length oligosaccharyltransferase (OST) genes; two of which, TbSTT3A and TbSTT3B, are expressed in the bloodstream form of the parasite. These OSTs have different peptide acceptor and lipid-linked oligosaccharide donor specificities, and trypanosomes do not follow many of the canonical rules developed for other eukaryotic N-glycosylation pathways, raising questions as to the basic architecture and detailed function of trypanosome OSTs. Here, we show by blue-native gel electrophoresis and stable isotope labeling in cell culture proteomics that the TbSTT3A and TbSTT3B proteins associate with each other in large complexes that contain no other detectable protein subunits. We probed the peptide acceptor specificities of the OSTs in vivo using a transgenic glycoprotein reporter system and performed glycoproteomics on endogenous parasite glycoproteins using sequential endoglycosidase H and peptide:N-glycosidase-F digestions. This allowed us to assess the relative occupancies of numerous N-glycosylation sites by endoglycosidase H-resistant N-glycans originating from Man5GlcNAc2-PP-dolichol transferred by TbSTT3A, and endoglycosidase H-sensitive N-glycans originating from Man9GlcNAc2-PP-dolichol transferred by TbSTT3B. Using machine learning, we assessed the features that best define TbSTT3A and TbSTT3B substrates in vivo and built an algorithm to predict the types of N-glycan most likely to predominate at all the putative N-glycosylation sites in the parasite proteome. Finally, molecular modeling was used to suggest why TbSTT3A has a distinct preference for sequons containing and/or flanked by acidic amino acid residues. Together, these studies provide insights into how a highly divergent eukaryote has re-wired protein N-glycosylation to provide protein sequence-specific N-glycan modifications. Data are available via ProteomeXchange with identifiers PXD007236, PXD007267, and PXD007268.
Keywords: glycoprotein biosynthesis, N-linked glycosylation, oligosaccharyltransferase, parasitology, Trypanosoma brucei, STT3, glycoproteomics, machine learning
Introduction
The tsetse-fly–transmitted protozoan parasite Trypanosoma brucei and its close relatives are responsible for human and animal African trypanosomiasis. The animal-infecting bloodstream forms of these organisms depend on surface coats made of glycosylphosphatidylinositol (GPI)3-anchored and N-glycosylated variant surface glycoprotein (VSG) to evade the innate host immune system (1) and the acquired immune system through antigenic variation (2). Furthermore, they express many less abundant glycoproteins such as their novel transferrin receptors (3–5), a novel lysosomal/endosomal protein called p67 (6), the so-called invariant surface glycoproteins (ISGs) (7), and invariant endoplasmic reticulum glycoproteins (8), the Golgi/lysosomal glycoprotein tGLP-1 (9), the membrane-bound histidine acid phosphatase TbMBAP1 (10), the flagellar adhesion zone glycoproteins Fla1 and Fla2 (11), and others. Whereas some of these are bloodstream-form specific glycoproteins (VSGs, ISGs, TbMBAP1, and transferrin receptors), others are common to the tsetse midgut-dwelling procyclic form of the parasite. Furthermore, procyclic form parasites also express unique glycoproteins, notably the abundant GPI-anchored procyclins, some of which are N-glycosylated (12, 13), and the partially characterized high-molecular-weight glycoconjugate (14, 15). Many of the N-glycan structures expressed by T. brucei have been solved, and these include conventional oligomannose and biantennary complex structures as well as paucimannose and extremely unusual “giant” poly-N-acetyl-lactosamine (poly-LacNAc) containing complex structures in the bloodstream form of the parasite (16–19). In contrast, only oligomannose N-glycans have been structurally described in wild-type procyclic trypanosomes (12, 20). The unusual repertoire of the T. brucei bloodstream-form N-glycans and the original observation by Bangs et al. (21) that Endo H-resistant N-glycans appear immediately following protein synthesis, and not following transport to the Golgi apparatus, has stimulated our group to study the fundamentals of protein N-glycosylation in this divergent eukaryotic pathogen.
Protein N-glycosylation is believed to be a ubiquitous post-translational modification among the eukaryotes, with the canonical model based primarily on extensive studies in mammalian cells and the yeast Saccharomyces cerevisiae (22, 23). In this canonical model, there are a number of tenets that include: (i) The mature lipid-linked oligosaccharide (LLO) donor, and the preferred substrate for oligosaccharyltransferases (OSTs) is Glc3Man9GlcNA2-PP-dolichol. (ii) The OSTs are hetero-oligomers of 8 or 9 distinct subunits. (iii) The OSTs may fall into two classes (A and B) according to their subunit composition with different peptide acceptor specificities. (iv) The ER enzyme UDP-glucose–glycoprotein glucosyltransferase (UGGT) operates on tri-antennary Man9–7GlcNA2 structures with a complete a-branch, but not on bi-antennary structures. (v) The action of Golgi mannosidase II is a pre-requisite for the conversion of oligomannose to complex N-glycans. (vi) The enzymes GnTI and GnTII have strict acceptor substrate specificities, operating on Man5GlcNA2 and GlcNAcMan3GlcNA2, respectively. However, none of these tenets apply to N-glycosylation in the protozoan parasite T. brucei where the following points have been noted. (i) The largest LLO is Man9GlcNA2 (24, 25) and different OSTs preferentially transfer either this structure or bi-antennary Man5GlcNA2 (25–28). (ii) There is no evidence for OST subunits other than the catalytic SST3 subunits in T. brucei or the related parasites Trypanosoma cruzi and the Leishmania (26, 28–32). (iii) OST subclasses and their peptide acceptor specificities (which are more disparate in T. brucei than for other eukaryotes) are defined only by their STT3 components (28). (iv) The parasite UGGT works efficiently on all structures (bi- and tri-antennary) with an intact a-branch (28). (v) Golgi-mannosidase II is absent, preventing the conversion form the oligomannose series to the complex series of N-glycans (25). (vi) The parasite GnTI and GnTII βGlcNAc-transferases have different specificities to canonical GnTIs and GnTIIs and belong to a different glucosyltransferase family (33, 34).
Here, we present the following. (i) We directly address the oligomeric states of T. brucei STT3 subunits. (ii) We look for evidence for any non-canonical OST subunits in addition to the TbSTT3s. (iii) We probe the peptide acceptor specificities of TbSTT3A (Tb927.5.890) and TbSTT3B (Tb927.5.900) using a reporter glycoprotein expression system and by glycoproteomics. (iv) We use machine learning to predict which putative N-glycosylation sites in bloodstream-form T. brucei will be modified by TbSTT3A or TbSTT3B.
Results
Blue native gel electrophoresis of in situ tagged TbSTT3A suggests it is present in high molecular weight complexes
To enable immunoprecipitation of TbSTT3A, the 3′-end of the endogenous gene in a heterozygote cell line (TbSTT3A/B/C+/−) was tagged in situ with a sequence encoding a C-terminal HA3 epitope. Transfected cells were cloned and analyzed by Southern blotting to confirm correct insertion of the tag (supplemental Fig. S1A). To check that the tag did not impair the function of TbSTT3A, the glycosylation of VSG221 was analyzed in these cells. VSG221 receives different types of glycan at the two N-glycosylation sequons in the protein: Endo H-resistant Man5GlcNAc2 at Asn-263 and Endo H-sensitive Man9GlcNAc2 at Asn-428 (17, 28). Following PNGaseF and Endo H treatment, the typical digestion pattern for wild-type VSG221 was seen also for the transgenic cells (supplemental Fig. S1B), showing that C-terminally tagged TbSTT3A-HA3 is functional. Subsequently, cells were lysed under mild conditions (0.5% digitonin on ice for 30 min), and the clarified cell lysates were incubated with anti-HA mouse antibody, followed by magnetic beads coupled to protein G. The pull-out eluates were analyzed by SDS-PAGE and Western blotting using an anti-HA antibody. As expected, epitope-tagged TbSTT3A-HA3 was detected running just below the position of the 75-kDa molecular mass marker in the TbSTT3A-HA3 pull-out, whereas no band was seen in the wild-type cell pull-out (Fig. 1A). However, when the same eluates were analyzed by blue native gel electrophoresis and anti-HA Western blotting, a smear (specific for the TbSTT3A-HA3 cell line) was detected between 700 and 1200 kDa, suggesting that TbSTT3A is present in large complexes (Fig. 1B).
Figure 1.
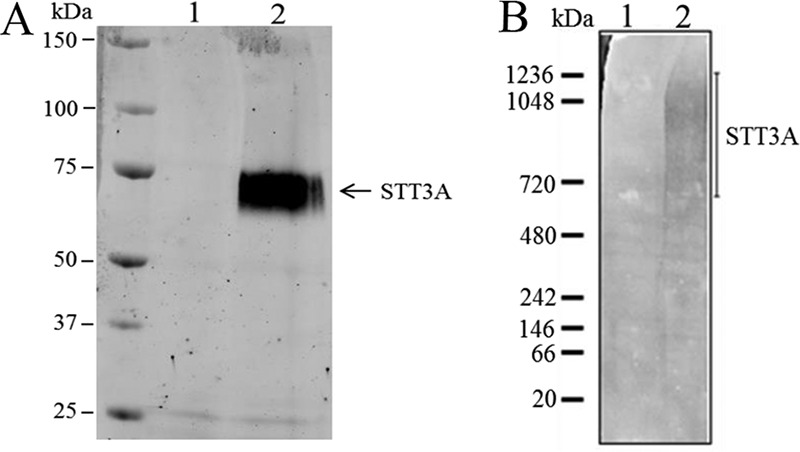
Denaturing and blue native gel electrophoresis of TbSTT3A-HA3. A and B, SDS-PAGE and anti-HA Western blotting (A) or blue native gel electrophoresis and anti-HA Western blotting (B) of anti-HA/protein G magnetic bead pull-outs from digitonin lysates of wild-type (lane 1) and in situ TbSTT3A-HA3-tagged (lane 2) bloodstream-form trypanosomes.
SILAC proteomics shows TbSTT3A and TbSTT3B form hetero-oligomeric complexes without other subunits
Because the results from the blue native gel electrophoresis suggested TbSTT3A is present in high molecular weight complexes, we carried out pull-out experiments using SILAC. For this experiment, wild-type and transgenic parasites (expressing TbSTT3A-HA3) were grown under identical conditions for eight cell divisions, except that the transgenic TbSTT3A-HA3 cell line was grown in “heavy medium” containing stable isotope-labeled Lys and Arg (R6K4), whereas the wild-type cells were grown in “light medium” containing unlabeled Lys and Arg (R0K0). The transgenic TbSTT3A-HA3 and the wild-type cells were harvested, washed, counted, mixed together in a 1:1 ratio, and lysed in 0.5% digitonin buffer. Anti-HA antibodies and protein G magnetic beads were used to pull-out the TbSTT3A-HA3–tagged protein and any binding partners, and the bead eluate was processed to tryptic peptides for LC-MS/MS analysis. In this kind of SILAC experiment, TbSTT3A-HA3 and any proteins specifically associated with it can be distinguished from non-specific contaminant proteins by the isotope ratios of their tryptic peptides. Thus, TbSTT3A-HA3 and true associated protein peptides will have high heavy/light isotope ratios, whereas contaminant proteins will have approximately equal heavy/light isotope ratios (Fig. 2A). The data set from the experiment was used to search a T. brucei predicted protein database using MaxQuant software. Each protein was displayed on a plot of the log10 value of the intensities of the unique peptides of that protein (y axis) and the log2 value of the heavy to light isotope ratios of the same peptides (x axis) (Fig. 2B). The TbSTT3A-HA3 (bait) protein had the highest heavy/light ratio (14:1), closely followed by TbSTT3B (10:1). Only three other proteins were significantly enriched (orange crosses, Fig. 2B). However, these were only marginally (1.5-fold) enriched hits that are not known to localize to the ER.
Figure 2.

Overview of the SILAC pull-out experiment and plot of proteomics data. A, overview of the SILAC experiment. Wild-type bloodstream-form cells were grown in light (R0K0) medium, and cells expressing in situ-tagged TbSTT3A-HA3 were labeled with heavy (R6K4) medium. The cells were mixed 1:1 and lysed with digitonin, and TbSTT3A-HA3 was enriched by affinity selection on anti-HA magnetic beads. Peptides from TbSTT3A-HA3 and genuine binding proteins have high heavy/light isotope ratios, whereas those from contaminants will have ratios close to 1:1 (log2 = 0). B, results from the TbSTT3A SILAC pull-out experiment. The plot shows the log2 of the heavy-to-light isotope ratio (x axis) versus the log10 value of the intensities of the peptides belonging to each protein that was detected (y axis). The black curves (marked sigma 3) represent three standard deviations from the mean. Proteins plotted in orange have a heavy-to-light ratio above the sigma 3 cut off and are significantly enriched. TbSTT3AHA3 (bait) and TbSTT3B (both annotated) were shown to be highly enriched and are highlighted in red. C, results from the TbSTT3B-Myc3 SILAC pull-out experiment. The plot is the same as in B except that in situ-tagged TbSTT3B-Myc3 was used as bait. Again, TbSTT3A and TbSTT3A (both annotated and highlighted in red) were significantly enriched.
The TbSTT3A-HA3 cell line was further modified by the in situ tagging of the remaining TbSTT3B allele, to yield a cell line expressing C-terminally Myc3-tagged TbSTT3B-Myc3. A complementary SILAC experiment, using in situ TbSTT3B-Myc3-tagged bait and an anti-Myc pull-out, produced similar results to the TbSTT3A-HA3 pull-out, with TbSTT3A being the only obvious binding partner for TbSTT3B-Myc3 (Fig. 2C). In this case, there was one other significant protein hit (orange cross, Fig. 2C), corresponding to a glucose transporter, but this was different from those seen in Fig. 2B, and also unlikely to be an ER component.
Taken together, these data suggest that TbSTT3A and TbSTT3A form hetero-oligomeric complexes, with no other candidate subunits, although we cannot rule out the presence of low-affinity subunits that might be lost during immunoprecipitation. The data for the SILAC proteomics experiments can be found at ProteomeXchange under entry PXD007236.
Co-immunoprecipitation of TbSTT3A-HA3 and TbSTT3B-Myc3
The results from the SILAC pull-out experiments suggested that TbSTT3A is in a complex with TbSTT3B, and to further test this hypothesis, immunoprecipitation (IP) experiments were performed. Cells from the double-tagged cell line were harvested, washed, and lysed in 0.5% digitonin, and TbSTT3A-HA3 or TbSTT3B-Myc3 was captured from the lysate using anti-HA or anti-Myc magnetic beads. Subsequently, the tagged proteins were detected by Western blotting using anti-HA and anti-Myc antibodies. Wild-type cell lysates (containing no HA- or Myc-tagged genes) were used as a control. The results from the co-IP experiments are shown in (Fig. 3). As expected, no bands in the region of TbSTT3A-HA3 or TbSTT3B-Myc3 were seen in the IPs from the control wild-type lysates (Fig. 3, lanes 1, 3, 5, and 7). Also, as expected, TbSTT3A-HA3 and TbSTT3B-Myc3 were detected in the homologous anti-HA IP/anti-HA blot (Fig. 3, lane 2) and anti-Myc IP/anti-Myc blot (Fig. 3, lane 8). Significantly, TbSTT3B-Myc3 can be seen to co-IP with TbSTT3A-HA3 (Fig. 3, lane 6), and TbSTT3A-HA3 can be seen to co-IP with TbSTT3B-Myc3 (Fig. 3, lane 4), confirming their physical association predicted by the SILAC experiment.
Figure 3.
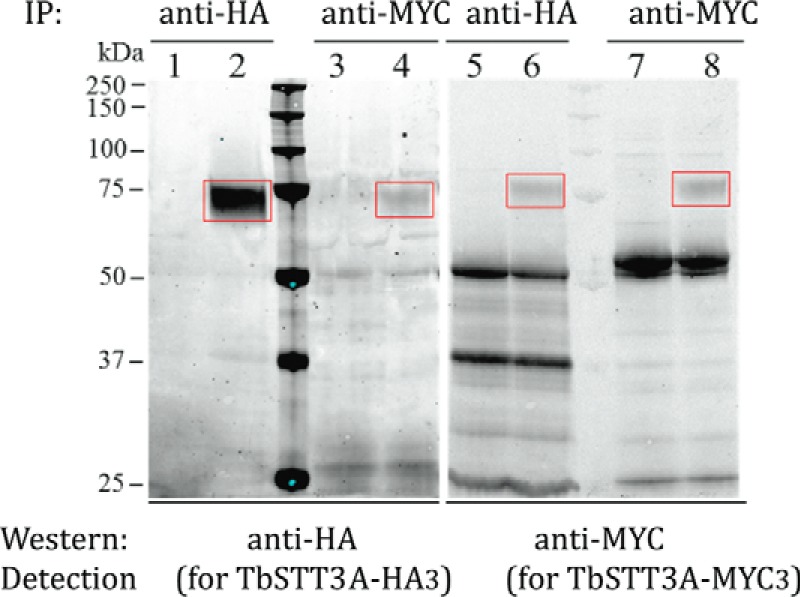
Co-immunoprecipitation of TbSTT3A and TbSTT3B. Digitonin lysates from wild-type cells (lanes 1, 3, 5, and 7) and TbSTT3A-HA3 and TbSTT3B-Myc3 double in situ tagged cells (lanes 2, 4, 6, and 8) were subjected to IP and Western blotted with anti-HA or anti-Myc antibodies, as indicated. The red rectangles highlight the bands corresponding to TbSTT3AHA3 (lanes 2 and 4) and TbSTT3B-Myc3 (lanes 6 and 8).
In these experiments, the anti-HA IP/anti-HA Western blot signal for TbSTT3A-HA3 is much stronger than the anti-Myc IP/anti-Myc Western blot signal for TbSTT3B-Myc3. Whereas some of this difference may be due to relative antibody affinities, it is also consistent with the higher expression of TbSTT3A in wild-type bloodstream-form trypanosomes at both the mRNA and protein levels (28, 35). The co-IP data suggest that a significant proportion of the total TbSTT3B-Myc3 appears in the TbSTT3A-HA3 IP (Fig. 3, compare lanes 6 and 8), whereas only a minority of TbSTT3A-HA3 appears in the TbSTT3B-Myc3 IP (Fig. 3, compare lanes 2 and 4). These data suggest that there may be some high molecular weight complexes made exclusively, or almost exclusively, of TbSTT3A, whereas all, or most, of the TbSTT3B is present in complexes containing TbSTT3A.
Probing peptide acceptor substrate specificities of TbSTT3A and TbSTT3A using a reporter glycoprotein expression system
TbSTT3A is responsible for co-translational transfer of biantennary Man5GlcNAc2 predominantly to N-glycosylation sequons containing and/or flanked by acidic amino acids, whereas TbSTT3B catalyzes post-translational transfer of triantennary Man9GlcNAc2 to the remaining sterically accessible sequons (28). To improve our understanding of the acceptor peptide specificity in T. brucei, an in vivo assay was established using an artificial reporter glycoprotein, based on the TbBiPN system described previously (36). TbBiPN is a non-glycosylated truncated version of TbBiP, retaining its N-terminal signal peptide but lacking its C-terminal ER retention peptide, which enters the ER and is eventually secreted out of the cell via the Golgi apparatus. A pLEW82 expression plasmid (37) was modified to contain the TbBiPN open reading frame fused to a 3′-sequence into which we could insert additional sequences, via AvrII and MfeI restriction sites, immediately upstream of a C-terminal HA3 epitope tag. This construct was used to introduce sequences encoding a single reporter N-glycosylation sequon, flanked by five amino acid residues on each side. These TbBiPN-(XXXXXNXTXXXXX)-HA3 constructs were transformed into bloodstream-form trypanosomes to express the reporter glycoprotein.
We first validated the in vivo reporter assay by introducing TEGLLNATDEIAL and TILKSNYTAEPVR into the TbBiPN construct and expressing them in T. brucei. The former sequence (with a pI of 3.42) is found in VSG MITat1.8 and is known, in that context, to receive exclusively biantennary Man5GlcNAc2 from TbSTT3A (38), whereas the latter (with a pI of 8.3) is found in the ESAG6 subunit of the transferrin receptor and is known not to be recognized and modified by TbSTT3A, and therefore, it receives exclusively triantennary Man9GlcNAc2 from TbSTT3B (5). Aliquots of trypanosome lysates expressing these constructs were treated with and without Endo H or PNGaseF, followed by SDS-polyacrylamide gel and Western blotting with anti-HA antibodies. The endoglycosidase Endo H can only digest triantennary Man9GlcNAc2 glycans transferred by TbSTT3B, whereas PNGaseF can digest both Man9GlcNAc2 and biantennary Man5GlcNAc2 transferred by TbSTT3A. Thus, distinct digestion patterns, depending on what type(s) of glycan(s) are bound to the sequon asparagine, can be visualized by Western blotting. The in vivo reporter assay faithfully recapitulated the experimental data for the VSG MITat1.8 and ESAG6 glycosylation sites, with TEGLLNATDEIAL- and TILKSNYTAEPVR-containing TbBipN glycoproteins occupied predominantly by Endo H-resistant and Endo H-sensitive glycans, respectively (supplemental Fig. S2).
Next, we investigated how each position flanking and within the sequon affects TbSTT3A recognition and transfer. First, the neutral sequence AAAAANATAAAAA (pI 6.01) was introduced into the reporter glycoprotein. For this construct, the majority (about 93% as measured by quantitative Licor imaging of the upper and lower bands of the Endo H digests) of the anti-HA binding signal was sensitive to Endo H (Fig. 4A, 1st lane; Table 1). One aspartic acid was then introduced in all 11 possible positions, yielding peptides with the same pI value (3.10), and the proportion of the reporter glycoprotein processed by TbSTT3A (and therefore resistant to Endo H) was measured (Fig. 4A, 2nd and 3rd lanes; Table 1). The quantitative data, derived from two technical replicates of three biological replicates (Table 1), are summarized in Fig. 4B. From these data, it can be seen that the aspartic acid scan across the different positions leads to variation in recognition and glycan transfer by TbSTT3A, with the two positions immediately flanking the Asn residue apparently having the greatest influence and with residues N-terminal to the glycosylation sequon having greater influence to those C-terminal to the sequon.
Figure 4.
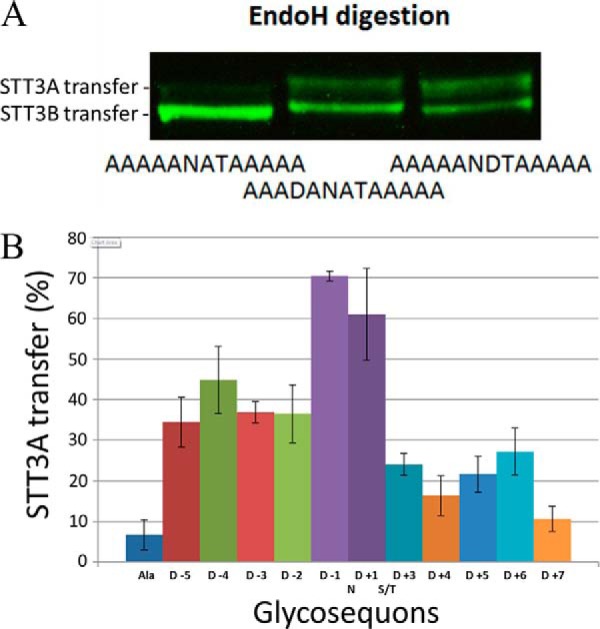
In vivo assay of TbSTT3A substrate specificity. A, constructs described in Table 1 were expressed in bloodstream-form trypanosomes, and the resulting TbBiPN-(XXXXXNXTXXXXX)-HA3 reporter glycoproteins were visualized in cell lysates by SDS-PAGE and anti-HA Western blotting. Representative examples are shown for the sequences indicated. The proportions of the anti-HA signals that were sensitive (lower bands) and resistant (upper bands) to Endo H were quantified and are reported in Table 1. B, summary of the quantitative data from Table 1 showing the influence of replacing single neutral Ala residues with an acidic Asp residue at each possible position.
Table 1.
Effect of aspartic acid on glycosite recognition by TbSTT3A
| Name | Sequence | pI | % EndoH-resistant (% STT3A transfer)a |
|---|---|---|---|
| Alanine control (Ala) | AAAAANATAAAAA | 6.01 | 6.8 ± 3.7 |
| D −5 | DAAAANATAAAAA | 3.10 | 34.4 ± 6.1 |
| D −4 | ADAAANATAAAAA | 3.10 | 44.8 ± 8.9 |
| D −3 | AADAANATAAAAA | 3.10 | 36.9 ± 2.6 |
| D −2 | AAADANATAAAAA | 3.10 | 36.5 ± 7.2 |
| D −1 | AAAADNATAAAAA | 3.10 | 70.5 ± 1.1 |
| D +1 | AAAAANDTAAAAA | 3.10 | 61.0 ± 11.3 |
| D +3 | AAAAANATDAAAA | 3.10 | 24.0 ± 2.7 |
| D +4 | AAAAANATADAAA | 3.10 | 16.4 ± 4.8 |
| D +5 | AAAAANATAADAA | 3.10 | 21.6 ± 4.4 |
| D +6 | AAAAANATAAADA | 3.10 | 27.2 ± 5.8 |
| D +7 | AAAAANATAAAAD | 3.10 | 10.7 ± 3.1 |
a The mean and standard deviations of the mean figures are based on n = 8 from two technical replicates of three biological replicates.
Probing endogenous peptide acceptor substrate specificities of TbSTT3A and TbSTT3B by glycoproteomics
The glycoproteomics data from Ref. 28 were reprocessed and significantly augmented by combining them with data derived from the experiments outlined under “Experimental procedures.” These experiments assess whether the endogenous trypanosome N-glycosylation sites are occupied by Endo H-sensitive oligomannose glycans (originating from the action of TbSTT3B) or by Endo H-resistant paucimannose and/or complex glycans (originating from the action of TbSTT3A).
Parasites were osmotically lysed to release >90% of the VSG coat as soluble form VSG, through the action of the endogenous GPI-specific phospholipase C (39, 40). The recovered cell ghosts, containing the majority of the non-VSG cellular glycoproteins, were solubilized, denatured, and S-alkylated. This preparation was then processed in two ways. In one approach, the intact glycoproteins were first affinity-purified using immobilized ricin (RCA120) and concanavalin-A (ConA) lectins. The enriched glycoproteins were then sequentially digested with Endo H and PNGaseF (the latter in the presence of H2[18O]), and digested with Lys-C and trypsin. In the other approach the denatured and S-alkylated proteins were first digested with Lys-C and trypsin, and the glycopeptides were trapped with ricin (RCA120) and ConA and subsequently digested with Endo H and PNGaseF (the latter in the presence of H2[18O]). In both cases, the resulting peptides were analyzed by LC-MS/MS, and the data were used to search the T. brucei predicted protein database allowing for the possible presence of Asn-N-GlcNAc residues, the product of Endo H cleavage, and/or for the conversion of Asn residues into [18O]Asp residues, the product of PNGaseF cleavage. These data and reprocessed data from Izquierdo et al. (28), are shown in supplemental Table S1. Peptides containing Asn-N-GlcNAc and/or [18O]Asp within an Asn-Xaa-Ser/Thr sequon that were detected ≥3 times were assigned as being predominantly TbSTT3A or TbSTT3B substrates when the proportion of the [18O]Asp feature was ≥0.8 and ≤0.4, respectively (supplemental Table S2). We then analyzed the amino acid frequencies immediately adjacent to N-glycosylation sequons of the assigned TbTT3A and TbTT3B substrates using WebLogo (41). The enrichment of negatively charged residues is the most striking feature of the TbSTT3A substrates (Fig. 5A) along with a bias toward Thr over Ser in the sequon +2 position. Conversely, the TbSTT3B substrates are relatively enriched for positively charged residues upstream of the sequon but show no preference for Thr over Ser in the +2 position (Fig. 5B). The data were also analyzed by the two-sample logo visualization method (42), which compares two input peptide sequence lists against each other, highlighting features that predominate in each. This suggests enrichment for negatively charged amino acids, especially at positions −6, −3, and +7 for the TbSTT3A substrates and a preference for hydrophobic and positively charge amino acids in the −1 to −5 positions of the TbSTT3B substrates (Fig. 5C). The reprocessed data of Ref. 19 can be found at ProteomeXchange under entry PXD007237 and the new glycoproteomics data under PXD007238.
Figure 5.

Glycoproteomic data logos and predictor. The amino acid frequencies of the preferentially deamidated (A) or preferentially HexNAc-modified peptides (B) identified by mass spectrometry are visualized with the WebLogo web service. The Two Sample logo web service was used to visualize the amino acids enriched (upper part) or depleted (lower part) in the sequences of the preferentially deamidated peptides identified by mass spectrometry (C) or predicted by the machine learning algorithm (D) by using the preferentially HexNAc-modified peptides as negative sample.
Building a glycosylation site predictor for T. brucei using machine learning
Machine learning has been successfully used in biological research to infer the peptide recognition specificities of, for example, protein kinases, phosphatases, and Src homology 2 domains (43). The first step of machine learning consists of transforming peptide sequences into biochemical features such as charge, hydrophobicity, and relative positions. These features, organized in a machine-readable template, are then evaluated by artificial intelligence algorithms to highlight which amino acid properties of a peptide sequence are the most important in determining substrate recognition. We therefore decided to apply a machine learning approach to further leverage our glycoproteomics and glycoprotein reporter data and to build an ensemble of prediction algorithms to assign putative N-glycosylation sites in the predicted T. brucei proteome. This ensemble algorithm (Voting Classifier) averages the outputs of a Random Forest classifier (RF), an Extra Tree Classifier (ETC), and a Support Vector Machine (SVM) classifier to predict which putative N-glycosylation sites will more likely be modified by TbSTT3A or TbSTT3B. The RF, ETC, and SVM classifiers all weigh the features derived from the upstream (N-terminal) side of the glycosylation sequon more than the features extracted from the downstream (C-terminal) side (supplemental Fig. S3, A–C). Moreover, the RF, ETC, and SVM classifiers all preferentially use the cumulative negative charge upstream of the glycosylated asparagine, and the hydrophobicity of the peptide upstream and downstream, to discriminate between TbSTT3A and TbSTT3B substrates (supplemental Fig. S4). We could detect a core of five important features shared by the three classifiers, namely the charge at pH 7.3 and the isoelectric point for the sequon ±10 amino acid residues, the cumulative charge upstream from the modified asparagine with a window of 13 and 16 amino acids, and the bonus score derived from the aspartic acid scanning experiment described in Fig. 4, see under “Experimental procedures.” A list of putative N-glycosylation sites and the Voting Classifier predictions are shown in supplemental Table S3. We performed a two-sample logo visualization on the output (Fig. 5D). As expected, the trends in this plot are similar to those generated from the glycoproteomics data alone (Fig. 5C).
Molecular modeling
Although TbSTT3C (Tb927.5.910) has not been studied in this paper because it is not expressed at detectable levels in bloodstream-form T. brucei, data from its heterologous expression in yeast suggest that its peptide acceptor specificity is much more similar to TbSTT3A than TbSTT3B (28, 44). To try to rationalize the preferences of TbSTT3A and TbSTT3C for acceptor sequons containing and/or flanked by negatively charged amino acid residues, we built molecular models of TbSTT3A, TbSTT3B, and TbSTT3C using Phyre2 (45) based on the Campylobacter lari PglB structure (46). The predicted models were aligned with the PglB structure using PDBeFold (47), and the binding pockets of TbSTT3A and TbSTT3B were visualized with Chimera (48). Next, we looked for basic amino acid residues that were conserved in TbSTT3A and TbSTT3C that were different in TbSTT3B (supplemental Fig. S6). Of these, the active-site proximal residue 397 is particularly interesting as it contains a His residue in TbSTT3B but an Arg residue in TbSTT3A and TbSTT3C. Arginine has a flexible and strongly basic guanidinium cation side chain that could conceivably interact with acidic amino acid residues at or close to the NX(S/T) glycosylation sequon in the active site (Fig. 6). Position 406 contains an Arg residue in TbSTT3A and TbSTT3C (in place of a neutral Gly residues in TbSTT3B) that could also conceivably interact with sequon-adjacent anionic residues in the acceptor peptide (Fig. 6). Such ionic interactions between the acceptor peptide and the enzyme surface might increase the efficiency of substrate recognition and glycosylation of sequons containing and/or flanked by acidic amino acids.
Figure 6.
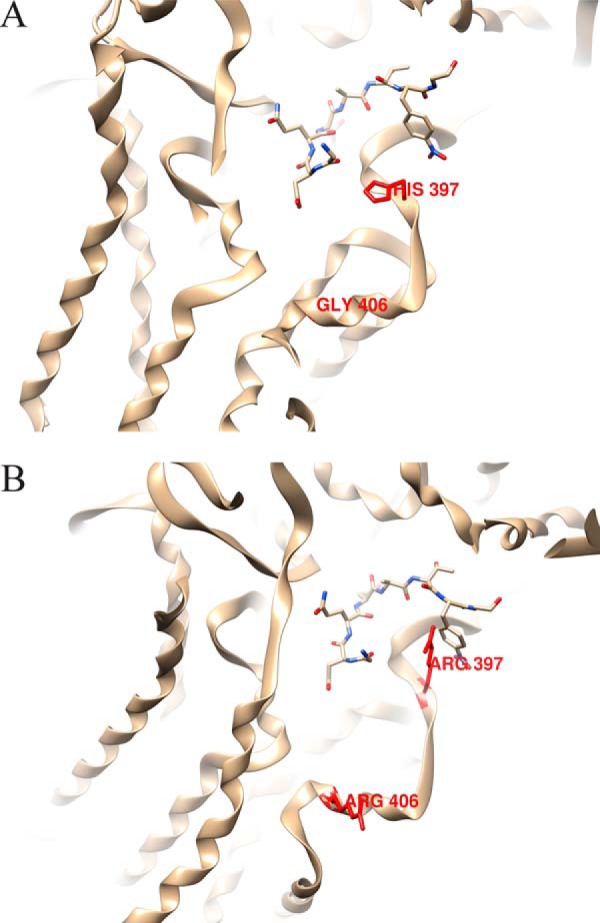
Molecular models of the active sites of TbSTT3A and TbSTT3B. A and B, molecular models of the predicted active sites of TbSTT3A (A) and TbSTT3B (B) with an acceptor peptide (GDQNAT) based on the crystal structure of C. lari PglB (46). Of note are the residues in red: Arg-397 in TbSTT3A (His-397 in TbSTT3B) and Arg-406 in TbSTT3A (Gly-406 in TbSTT3B), where the guanidinium cations of the Arg side chains could interact with acidic residues at or close to the acceptor peptide sequon.
Discussion
Although the T. brucei genome does not encode for any identifiable OST subunits, other than three intact and one truncated STT3, we decided to investigate whether there might be novel non-canonical T. brucei OST subunits. Precedents for kinetoplastid-specific subunits of otherwise conserved cellular machineries include clathrin-associating proteins and endocytic components (49, 50), exocyst components (50), nuclear pore complex and nuclear lamina components (51), and subunits of the GPI transamidase (52). Blue native gel electrophoresis of gently solubilized epitope-tagged endogenous TbSTT3A showed that it is present in high-molecular-weight complexes, but quantitative SILAC proteomics of tagged TbSTT3A and TbSTT3B pull-outs showed that although these are mutual binding partners (confirmed by co-immunoprecipitation), no other subunits could be found by these methods. The lack of non-canonical or canonical T. brucei OST subunits (other than the STT3 catalytic subunits) is consistent with the ease with which T. brucei and other kinetoplastid STT3s can be functionally expressed in other eukaryotes, like S. cerevisiae and Pichia pastoris (29, 31, 32, 53). Nevertheless, the blue native gel and co-immunoprecipitation experiments show that, at least in the native environment of a bloodstream-form trypanosome, TbSTT3A associates with itself and with the less-abundant TbSTT3B to form complexes with apparent molecular masses between 600 kDa and 1.2 MDa. The nature of these complexes remains to be determined but has implications for how and whether TbSTT3s associate with the parasite translocon complex and how they access nascent glycoprotein sequons during and/or following protein translocation. The dimer/oligomer nature of yeast OST subunits, including Stt3, has been previously described (54).
To probe TbSTT3 peptide acceptor substrate specificities, we developed an artificial glycoprotein reporter system, based on a truncated version of TbBiP (36) fused to a single glycosylation sequon flanked by five variable residues on either side. Constructs were expressed in bloodstream-form T. brucei, and their products were assayed for the relative proportions of N-glycosylation by TbSTT3A and TbSTT3B. With this, we were able to recapitulate the preferential N-glycosylations of native peptide acceptor sequences. We then applied the system to analyze the N-glycosylation of an artificial 13-mer sequence (AAAAANATAAAAA) into which we sequentially introduced a single Asp residue in all 11 possible Ala sites. The data clearly confirmed that the presence of an acidic amino acid proximal to the sequon significantly increased its N-glycosylation by TbSTT3A, with the −1 and +1 positions relative to the N-glycosylated Asn residue having the greatest effect and the positions N-terminal to the sequon having a greater effect than those C-terminal to the sequon.
We then created a richer glycoproteomics dataset than we previously reported (28) by combining two alternative approaches: (i) glycoprotein enrichment by lectin affinity chromatography, followed by trypsin digestion and sequential Endo H and PNGaseF digestion; and (ii) tryptic glycopeptide enrichment by lectin affinity chromatography, followed by sequential Endo H and PNGaseF digestion. In both cases, the PNGaseF digestion step was performed in H2[18O] to distinguish between PNGaseF-mediated Asn deamidation and non-enzymatic deamidation during sample preparation and handling. These data were combined with reprocessed raw data from Ref. 28 to provide quantitative data on TbSTT3A and TbSTT3B N-glycosylation of 141 unique N-glycosylation sites. Logo plots confirmed the enrichment of acidic amino acid residues (Asp and Glu) surrounding TbSTT3A N-glycosylated sequons, the general depletion of hydrophobic residues, and the selective depletion of basic residues (Arg and Lys) N-terminal to the sequon.
We also used hypothesis-free machine-learning techniques to identify features that predispose sequons to be preferentially modified by TbSTT3A or TbSTT3B, and finally, we combined these features and parameters derived from the experimental reporter glycoprotein data to develop a Voting Classifier prediction algorithm. This predictor was then applied to all the putative N-glycosylation sequons in the T. brucei proteome to predict those sites preferentially modified by TbSTT3A (leading to paucimannose and/or complex N-glycan occupancy) or TbSTT3B (leading to oligomannose N-glycan occupancy) in bloodstream-form trypanosomes. Two-sample logo plot analysis of the output (a total of 1291 predicted occupied N-glycosylation sites) largely echoes the experimental glycoproteomic and reporter glycoprotein data and implies that the TbSTT3A N-glycosylation sites are enriched for acidic residues and depleted of basic and hydrophobic residues, with the effects of these features more profound to the N-terminal side and within the sequon than to the C-terminal side of the sequon.
It is important to note that available pulse-chase data (21, 55) suggest that TbSTT3A modifies VSG glycoproteins co-translationally, whereas TbSTT3B can act post-translationally (25), and that TbSTT3A and TbSTT3B knockdown data suggest that TbSTT3B is able to modify TbSTT3A sites, but not vice versa (28). Thus, all or most of the aforementioned TbSTT3A versus TbSTT3B sequon selectivity features are dictated by the peptide/sequon acceptor specificity of TbSTT3A and not TbSTT3B, which appears to be able to utilize sequons in almost any amino acid sequence context. This property of TbSTT3B could be particularly useful from a biotechnological point of view to boost the efficient N-glycosylation of recombinant glycoproteins in eukaryotic expression systems. It also nicely explains why trypanosomes can transition from expressing a rich mixture of oligomannose and paucimannose/complex N-glycans in the bloodstream-form parasite (16, 18, 19) to predominantly oligomannose N-glycans in the procyclic form of the parasite (12, 20) by simply down-regulating the expression of TbSTT3A, as observed at both the mRNA (28, 56) and protein levels (35, 57, 58).
The results reported here and in Ref. 44 are consistent with the mechanisms of resistance in T. brucei to certain toxic lectins and carbohydrate-binding small molecules reported in an interesting series of studies (59–61). These workers demonstrated that the parasites could escape the effects of these trypanocidal agents, all of which bind principally to oligomannose N-glycans, by either switching to the expression of a VSG type that naturally does not carry oligomannose N-glycans or by suppressing the expression of TbSTT3B. In this way, the parasites effectively exchange oligomannose for puacimannose and complex N-glycans that are poor ligands for the trypanocides.
Interestingly, TbSTT3A and TbSTT3B are found in tandem array in the trypanosome genome, together with a preceding truncated TbSTT3 pseudogene and followed by a full-length TbSTT3C gene that is more similar to TbSTT3B than to TbSTT3A. However, TbSTT3C is not significantly expressed in bloodstream-form or procyclic form of the parasite (28, 35, 56–58). Nevertheless, transgenic expression of TbSTT3C in S. cerevisiae clearly shows that it is a functional OST with a similar preference for sequons flanked by acidic amino acids to TbSTT3A but an LLO donor specificity like TbSTT3B (28, 44). Amino acid sequence alignment of TbSTT3A, -B, and -C and molecular model building, based on the C. lari PglB structure (46) was performed. The models suggest that the presence of a large, flexible and highly positively charged Arg residue side chain (Arg-397) very close to the active site of the enzyme in TbSTT3A and TbSTT3C, compared with a His residue in TbSTT3B, may play a role in the selectivity of TbSTT3A and TbSTT3C for sequons containing and flanked by acidic Asp and Glu residues. TbSTT3A and TbSTT3C also contain Arg residues in place of neutral Gln-567 and Gly-406 residues in TbSTT3B, locations close enough to the active site to potentially interact with sequon-flanking anionic residues. The accompanying paper (44) elegantly addresses the issues of peptide acceptor and LLO donor specificities of all three TbSTT3s by heterologous expression of each and chimeras thereof in various yeast mutants. The accompanying paper concludes that the region containing Arg-397 and Arg-406 in TbSTT3A and TbSTT3C controls peptide acceptor specificity, and this is consistent with our suggestions from molecular modeling.
There are similarities and differences between the multisubunit mammalian STT3A- and STT3B-based OSTs and the single subunit TbSTT3A and TbSTT3B OSTs of T. brucei. (i) In both, the STT3A OSTs operate co-translationally and get the first option to glycosylate a given sequon, whereas the STT3B OSTs can operate post-translationally on what is left (25, 62). (ii) In both, the OSTs show differences in peptide acceptor substrate specificity. However, in T. brucei this is controlled by the physicochemical properties of the amino acids surrounding the acceptor sequon, whereas in mammalian cells this is controlled by the position of the sequon relative to the C terminus of the protein (63) or proximity to the signal peptide–cleaved N terminus of the protein and to cysteine residues (23, 63, 64). The latter appears to relate to the presence of the mutually redundant MagT1 or TUSC3 thioredoxin-like oxidoreductase subunits (equivalent to the yeast Ost3 and Ost6 subunits) in the STT3B OST that may form mixed disulfides with the sequon-proximal cysteine residues and thus increase residence time with the STT3B OST (64, 65). A role for oxidoreductase activities of Ost3 and Ost6 in yeast OST acceptor site specificity was also previously shown (66). In this regard, it is worth noting that TbSTT3B and TbSTT3C contain a CXC motif (absent in TbSTT3A) that is predicted from the C. lari OST structure to be proximal to the acceptor peptide (46). Such CXC sequences can have a disulfide isomerase activity (67) that might conceivably increase the acceptor substrate range of TbSTT3B and TbSTT3C. (iii) Whereas both STT3A and STT3B OSTs prefer the mature Glc3Man9GlcNAc2-PP-dolichol LLO donor, the T. brucei OSTs have distinct LLO donor specificities such that the presence of the ALG12-dependent c-branch of the conventional (but glucose-free) triantennary Man9GlcNAc2-PP-dolichol LLO is required by TbSTT3B but not tolerated by TbSTT3A (27, 44). An important consequence of this differential LLO specificity is that, because Golgi mannosidase II activity is also absent in T. brucei, N-glycans derived from TbSTT3B glycosylation cannot be processed to paucimannose or complex structures, which must instead be derived exclusively from TbSTT3A glycosylation.
In summary, the two simultaneously operating acceptor substrate-specific and donor substrate-specific N-glycosylation systems of bloodstream-form T. brucei have been further characterized in this paper. Whereas no canonical OST subunits, other than catalytic STT3 subunits, could be found in the parasite genome, we can now confirm that there are no non-canonical subunits either. Instead, TbSTT3A and TbSTT3B appear to form multimeric high-molecular-weight complexes containing either TbSTT3A alone or TbSTT3A and TbSTT3B. Further insights into the peptide acceptor specificity of TbSTT3A have been provided, and an algorithm has been generated to predict, proteome-wide, which OST will likely operate on which putative N-glycosylation site. Taken together with the unusual specificities of T. brucei UGGT, GnTI, and GnTII enzymes described in the Introduction (19, 33, 34), and the apparent absence of a regulated ER unfolded protein response (19, 68), we may conclude that protein N-glycosylation and downstream processing in this divergent eukaryote is worthy of note and that its unusual features may provide therapeutic possibilities.
Experimental procedures
Cultivation of trypanosomes
Bloodstream-form T. brucei, genetically modified to express T7 polymerase, and the tetracycline repressor protein (37) were cultured in HMI-9T medium (69) supplemented with 10% fetal calf serum, 2 mm GlutamaxTM (Invitrogen), 56 μm 1-thioglycerol (in place of 2-mercaptoethanol), and 2.5 μg/ml G418 antibiotic at 37 °C in a 5% CO2 incubator. Other antibiotics used, as appropriate, were hygromycin (4 μg/ml), puromycin (2.5 μg/ml), phleomycin (0.1 μg/ml), and tetracycline (0.5 μg/ml). SILAC labeling, using dialyzed fetal calf serum, was performed in HMI11-SILAC media, as described previously (35). l-Arginine U-13C6 and l-lysine 4,4,5,5–2H4 (R6K4) were purchased from Cambridge Isotope Labs.
Generation of genetically modified trypanosomes with in situ tagged TbSTT3A-HA3 and TbSTT3B-Myc3
The TbSTT3A,B,C−/+ heterozygote described in Ref. 28 was used for C-terminal HA3 in situ tagging of the remaining TbSTT3A allele using a pMOTagH4 plasmid and C-terminal Myc3 in situ tagging of the remaining TbSTT3B allele using a pMOTag4M4 plasmid (70). For the pMOTagH4 plasmid, the 1328 bp from the C terminus of TbSTT3A and the 1057 bp 3′-UTR downstream of the gene ORF were PCR-amplified from genomic DNA using Kod Hot Start polymerase with primers 5′-ataagtatctcgagcaagtttgcttgccccgttcg-3′ and 5′-ataagtaactcgagctcgctctgaaaatacaggttttcgacttcgtaatggaaccgcttcgct-3′ and 5′-ataagtatggatccccacatcgtttcaatcgccgc-3′ and 5′-ataagtaaggatccactcacaatcgtgcttacagcc-3′ as forward and reverse primers, respectively. The PCR products were cloned into the plasmid using the XhoI and BamHI (underlined). A tobacco etch virus restriction site was included downstream of the ORF of TbSTT3A (italics). The construct was linearized before being transfected into the TbSTT3A,B,C−/+ heterozygote cell line, and transfected cells were selected by addition of hygromycin. The pMOTag4M4 plasmid was ordered from Genescript. It included 1032 bp from the C terminus of TbSTT3B, located upstream of the Myc3 epitope in the plasmid. The plasmid also included 835 bp of the 3′ UTR of TbSTT3B, which were located downstream of the blasticidin resistance gene in the plasmid. The construct was linearized before being transfected into the TbSTT3A/B/C−/+ TbSTT3A-HA3 heterozygote cell line, and transfected cells were selected by addition of blasticidin.
SDS-PAGE and Western blotting
Reducing SDS-PAGE was run using pre-cast Novex BisTris gels with MOPS running buffer (Invitrogen). Proteins were transferred to nitrocellulose using an iBlot system (Invitrogen) and stained with Ponceau S (Sigma) before being blocked in 50 mm Tris-HCl, 0.15 m NaCl, 0.05% Tween 20, 0.25% BSA, 0.05% sodium, and 2% fish skin gelatin, pH 7.4, for 20 min. The membrane was then incubated for 30 min with primary antibody in a 50-ml Falcon tube followed by washing using a SnapID system (Millipore). Subsequently, the labeled secondary antibody was incubated for 30 min followed by a washing step. The blots were imaged using an ODYSSEY® SA near infrared imager (LI-COR Biosciences). Secondary LI-COR antibodies (IRDye-800CW goat anti-mouse 1:15,000 or IRDye-680RD donkey anti-mouse 1:20,000) were used to bind the primary mouse anti-HA and anti-Myc antibodies.
Blue native gels and Western blotting
Blue native gel electrophoresis was run using components from the native PAGE kit (Invitrogen). The protocol followed the manufacturer's instructions except that no G-250 was added to the sample buffer and the 1× native PAGE Light Cathode buffer was diluted 1:4 in 1× running buffer to reduce Coomassie interference of the post-blotting LiCor imaging.
Immunoprecipitation
Cells cultures (100 ml) were grown to log phase (∼2.5 × 106 cells/ml) and lysed for 30 min on ice in 0.5% digitonin, 50 mm Tris-HCl, pH 6.8, 20 mm EDTA plus the protease inhibitors 0.8 mm PMSF, 0.1 mm TLCK, and 1× EDTA-free protease inhibitor mixture (Roche Applied Science). Subsequently, the lysate was centrifuged (4 °C, 11,000 × g, 15 min), and the supernatant was moved to a new tube. Protein G magnetic beads, pre-washed in lysis buffer, were added to the lysate (30 min, 4 °C) and captured to absorb non-specific binding components. The lysate was moved to a new tube followed by incubation for 1 h with anti-HA or anti-Myc antibody (1 μg/ml) at 4 °C followed by fresh pre-washed magnetic beads. The beads were captured and washed twice with 1 ml of lysis buffer and once with 10 mm Tris-HCl, pH 6.8, 4 mm EDTA, 0.1% digitonin containing the same protease inhibitors. Proteins were eluted from the beads in 30 μl of reducing SDS-sample buffer with heating (100 °C for 10 min). The eluted proteins were subsequently separated by SDS-PAGE.
SILAC proteomics
Heavy and light labeled cells were harvested separately (15 min, 800 × g, 4 °C) and washed and resuspended in trypanosome dilution buffer (20 mm Na2HPO4, 2 mm NaH2PO4, 80 mm NaCl, 5 mm KCl, 20 mm glucose) for cell counting. The cells were mixed 1:1 before undergoing immunoprecipitation, as described above. The eluted proteins in 25 μl of reducing SDS sample buffer were S-alkylated with 5 μl of 300 mm iodoacetamide (30 min, dark) and loaded on Novex NUPAGE 4–12% BisTris gel and run at 200 V using MOPS buffer until the proteins had migrated about 2 cm into the gel (visualized by Simply Blue Safe Stain, Thermo Fisher Scientific). The protein-containing region of the gel was excised and subjected to in-gel trypsin digestion, and aliquots of the extracted peptides were analyzed on an LTQ-Orbitrap Velos Pro mass spectrometer coupled with a Dionex Ultimate 3000 RS HPLC system (Thermo Fisher Scientific). The sample peptides were loaded at 5 μl/min onto a trap column (100 μm × 2 cm, PepMap nanoViper C18 column, 5 μm, 100 Å, Thermo Fisher Scientific) equilibrated in 98% buffer A (2% acetonitrile and 0.1% formic acid (v/v)) and 2% buffer B (80% acetonitrile and 0.08% formic acid (v/v)). The trap column was washed for 3 min at the same flow rate and then switched in-line with a Thermo Fisher Scientific resolving C18 column (75 μm × 50 cm, PepMap RSLC C18 column, 2 μm, 100 Å). The peptides were eluted from the column at a constant flow rate of 300 nl/min with a linear gradient from 98% buffer A to 40% buffer B in 128 min and then to 98% buffer B by 130 min. LTQ-Orbitrap Velos Pro was used in data-dependent mode. A scan cycle comprised an MS1 scan (m/z range from 335 to 1800) in the Orbitrap (resolution 60,000) followed by 15 sequential data-depandant collision-induced dissociation MS2 scans (the threshold value was set at 5000 and the minimum injection time was set at 200 ms).
Glycoproteomics
The protein-based approach was based on the methodology described previously (28). Cells were harvested by centrifugation and osmotically lysed at 3.5 × 108 cells/ml for 5 min at 37 °C in the presence of 0.1 μm TLCK, 1 mm benzamidine, 1 mm phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml leupeptin, 1 μg/ml aprotinin, and Phosphatase Inhibitor Mixture II (Calbiochem). Cell ghosts from a total of 3.5 × 109 trypanosomes, enriched for non-VSG cellular glycoproteins, were collected by centrifugation (16,000 × g, 15 min, 4 °C) and solubilized in 250 μl of detergent buffer (4% SDS, 0.1 m DTT, 0.1 m Tris-HCl, pH 7.5) using probe sonication for 30 s before and after heating to 85 °C for 20 min. S-Alkylation was performed by mixing with 250 μl of 8 m urea, 0.5 m iodoacetamide, 0.1 m Tris-HCl, pH 8.5, for 1 h, room temperature, in the dark. Unreacted iodoacetamide was quenched by the addition of 10 μl of the detergent buffer. After centrifugation (16,000 × g, 15 min), the supernatant was transferred to a filtration device with a 30-kDa molecular cutoff (Sartorius), and the majority of the detergent was removed by diafiltration with 8 m urea, 0.1 m Tris-HCl, pH 8.5, followed by lectin-binding buffer (1 mm CaCl2, 1 mm MnCl2, 150 mm NaCl in 40 mm Tris-HCl, pH 7.4). An aliquot (400 μg of protein) was mixed with 150-μl packed volume of ricin (RCA120)-agarose beads (Vector Laboratories) and rotated gently for 2 h. The beads were washed with lectin-binding buffer and eluted with 300 μl of 30 mg/ml lactose and 30 mg/ml galactose (Sigma) in 40 mm Tris-HCl, pH 7.4. After overnight incubation, the supernatant containing the eluted glycoproteins was collected by centrifugation (10,000 × g for 10 min). ConA-coupled agarose beads (0.15 ml packed volume, Vector Laboratories) was added to the recovered supernatant from the RCA120 pulldown and incubated at 4 °C overnight. The beads were washed with lectin-binding buffer and eluted with 300 μl of 0.5 m methyl-α-d-mannopyranoside (Sigma) in 40 mm Tris-HCl, pH 7.4. After gentle rotation for 2 h, the supernatant containing glycoproteins was recovered by centrifugation. The RCA120 and ConA-eluted glycoproteins were mixed, transferred to a 30-kDa filter, the buffer exchanged with 25 mm ammonium acetate, pH 5.5, and subjected to digestion with 180 milliunits of Endo H (Roche Applied Sciences) overnight at 37 °C. The Endo H-released glycans were removed by centrifugation, and the remaining material was exchanged into 40 mm ammonium bicarbonate buffer in H2[18O] (Sigma), and subsequently digested with 100 units of N-glycosidase F (PNGaseF, Roche Applied Sciences) dissolved in H2[18O]. PNGaseF in the presence of H2[18O] converts Asn to [18O]Asp with a mass increment of 2.9890 Da that can be readily distinguished from spontaneous deamidation (mass increment of 0.9858 Da). After overnight incubation at 37 °C, the PNGaseF-released glycans were removed by centrifugation, and the deglycosylated proteins were diafiltered into 40 mm ammonium bicarbonate and subsequently digested with a mixture of 1:100 (enzyme/substrate) Lys-C and 1:20 trypsin (Roche Applied Sciences) at 37 °C for 48 h. The peptides were collected by centrifugation through the filter, dried in a Speedvac, and desalted using a ZipTip C18 micro-column (10 μl, Merck Millipore) prior to liquid chromatography tandem mass spectrometry (LC-MS/MS).
An alternative (glyco)peptide-based approach was also performed, based on the method of Ref. 71. An aliquot of the denatured and S-alkylated sample (400 μg) was first digested on a 10-kDa filter with a mix of Lys-C and trypsin, as described above. The (glyco)peptides were collected by centrifugation through the filter in lectin-binding buffer and were mixed with lectin solution containing a mixture of ConA and RCA120 resulting in mixtures of (glyco)peptides and lectins with a mass proportion of 1:2. After gentle rotation for 2 h, the mixtures were transferred to a 30-kDa filter, and the lectin-captured glycopeptides were diafiltered into 25 mm ammonium acetate, pH 5.5, and subjected to Endo H digestion. After overnight incubation at 37 °C, the Endo H-released peptides (Asn N-GlcNAc residue) were collected by gentle centrifugation. The peptides containing Endo H-resistant glycans still bound to lectins were diafiltered into 40 mm ammonium bicarbonate buffer in H2[18O] and digested with PNGaseF overnight at 37 °C. The de-glycosylated peptides were collected by centrifugation, mixed with the Endo H released fraction, dried in a Speedvac, and desalted using ZipTip C18 prior LC-MS/MS, which was performed as described above.
LC-MS/MS analysis and data processing
For glycoproteomics, the LC was performed on a fully automated Ultimate U3000 Nano LC System (Dionex) fitted with a C18 trap (PepMap nanoViper, Thermo Fisher Scientific) and resolving columns (PepMap RSLC) with inner diameters of 100 and 75 μm and lengths of 2 and 50 cm, respectively. Mobile phases consisted of 0.1% formic acid (Sigma) in 2% acetonitrile (Merck, Darmstadt Germany) for solvent A and 0.08% formic acid in 80% acetonitrile for solvent B. Samples were loaded in solvent A. A linear gradient was set as follows: 0% B for 5 min and then a gradient up to 40% B in 122 min and to 98% B in 10 min. A 20-min wash at 98% B was used to prevent carryover, and a 20-min equilibration with 2% B completed the gradient. The LC system was coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) equipped with an Easy spray ion source and operated in positive ion mode. The spray voltage was set to 2 kV and the ion transfer tube at 250 °C. The full scans were acquired in a Fourier transform MS mass analyzer that covered an m/z range of 335–1800 at a resolution of 60,000. The MS/MS analysis was performed under data-dependent mode to fragment the top 15 precursors using collision-induced dissociation. A normalized collision energy of −35 eV, an isolation width of m/z 2.0, an activation Q value of 0.250, and a time of 100 ms were used. The raw files were converted to mgf format by MSConvert software from ProteoWizard (proteowizard.sourceforge.net).4 The searches were carried out against the T. brucei 927 annotated proteins database (version 8.0, downloaded from TriTrypDB (72), www.tritrypdb.org/)4 using Mascot software (version 2.4.0, Matrix Science Inc., Boston). The search parameters for Mascot software were set as follows: peptide tolerance, 5 ppm; MS/MS tolerance, 0.5 Da; enzyme, trypsin; one missed cleavage allowed; and fixed carbamidomethyl modifications of cysteines. Oxidation of methionine, N-acetylglucosamine modification of Asn, and deamidation of Asn to Asp containing a single 18O atom (2.9890 Da mass increase) were used as variable modifications.
Dataset extraction
We extracted from the Mascot result files all the peptides with an ion score of >20. From this we selected 350 glycosylated sites with a deamidation (Asn to [18O]Asp conversion) or Asn–N-HexNAc modification embedded in the N[caret]P(ST) consensus. The list was used to count the number of times (≥ 3) that these changes were detected for each peptide. To increase the number of modified peptides we decided to re-process a previously published work in our laboratory (28). This dataset was re-searched with the same Mascot parameters and database used for this publication. This made it possible to include 14 new peptides preferentially HexNAc-modified and to compile a list of 186 peptides used for the next phase of machine learning implemented in Python with the scikit-learn package (73).
The data set reported here and that from Ref. 28 identified 170 common and 180 and 155 unique glycosylation sites, respectively (supplemental Fig. S1A). To check the consistency of the two datasets, we selected 92 peptides that were observed ≥ 4 times in both datasets and plotted the frequencies of the Asn–N-HexNAc and Asn to [18O]Asp modifications. The two datasets had a good correlation (r2 = 0.83) (supplemental Fig. S1B). However, the experimental procedures used to generate the 2009 dataset (i.e. without the use of H218O) cannot discriminate between the spontaneous non-enzymatic deamidation of asparagine to aspartic acid versus the PNGaseF-mediated deamidation produced during the cleavage of the N-glycan. From the linear regression, we could deduce that non-enzymatic deamidation contributed a significant amount (about 18%) of the total deamidation seen in the 2009 dataset (supplemental Fig. S1B). For this reason, we only used Asn–N-HexNAc containing (TbSTT3B substrate) peptides (with a frequency of ≥ 0.6) from this dataset to augment our machine-learning training set.
Machine learning
The deamidation proportion was computed for each peptide as (DC/DC + HC), where DC is the Deamidation Count (i.e. the number of times a peptide with [18O]Asp is detected), and HC is the HexNAc Count (i.e. the number of times a peptide with Asn–N-HexNAc is detected). This score was used to classify each peptide as preferentially deamidated (score >0.8 n = 70) or preferentially HexNAc-modified (score <0.3% n = 56). This dataset was used to extract a sequence-based feature from 10 amino acids before and after the glycosylated asparagine with the ASAP package in Python (74). We also added some in-house features derived from the knowledge of the TbSTT3A recognition and transfer experiment reported in Fig. 4B and Table 1. To this end we created “Bonus Features” for each residue position probed in that experiment based on the increased transfer efficiency observed when that site is occupied by an Asp (or, by inference, a Glu residue). We also included a “Bonus All” feature that summed all of the bonus scores for the peptide when it contained more than one Asp and/or Glu residue and a “Bonus Max” feature that selected only the highest bonus score in such cases. Finally, we also created “Bonus Presence D” and “Bonus Presence E” features that simply recorded the presence or absence of Asp or Glu, respectively, at each residue location. We then developed three machine learning algorithms: an RFC, an ETC, and a support vector machine classifier (SVM). The predictors were used to extract the importance of all the features and to rank the features with recursive feature elimination and cross-validated selection of the best number of features (RFECV methodology). The selected features were used to train the three machine learning algorithms (RFC, ETC, and SVM) that were further optimized using a Bayesian global optimization with Gaussian processes (https://github.com/fmfn/BayesianOptimization).4 The optimized machine learning algorithms were combined in a voting classifier to produce our final predictor. The ability of the developed classifiers (RFC, ETC, SVM, and Voting Classifier) to discriminate between the deamidated or HexNAc-modified peptides was assessed with the area under the curve of the receiver-operating characteristic curve. The area under the curve score was computed 100 times with a 5-fold cross-validation, using each time a different random split of the original dataset (supplemental Fig. S5).
Author contributions
M. A. J. F. conceived and coordinated the study. A. J., L. A., and M. A. J. F. designed the experiments. A. J. and M. L. S. G. performed the biochemical and SILAC proteomic experiments. L. A. performed the glycoproteomic experiments. A. J. and M. T. performed the molecular modeling. M. T. performed the bioinformatic analyses and designed the machine-learning experiments.
Supplementary Material
Acknowledgments
We thank the Dundee Proteomics facility team for their help and Amy Tavendale for assistance with processing the SILAC data. We thank Angela Mehlert, Mick Urbaniak, and Jim Proctor for helpful advice and comments and Markus Aebi for helpful discussions and kindly sharing information prior to publication. Wellcome Trust grant 097045/B/11/Z provided mass spectrometry and proteomics infrastructure support.
This work was supported in part by Wellcome Trust Investigator Award 101842/Z13/Z (to M. A. J. F.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S6 and Tables S1–S3.
The mass spectrometric raw data and spectral libraries associated with this manuscript are available from ProteomeXchange with the accession numbers PXD007236, PXD007267, and PXD007268.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- GPI
- glycosylphosphatidylinositol
- OST
- oligosaccharyltransferase
- VSG
- variant surface glycoprotein
- ISG
- invariant surface glycoprotein
- RFC
- random forest classifier
- ETC
- Extra Tree Classifier
- SVM
- Support Vector Machine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- TLCK
- 1-chloro-3-tosylamide-7-amino-2-heptanone
- Endo H
- endoglycosidase H
- PNGaseF
- peptide:N-glycosidase-F
- ER
- endoplasmic reticulum
- IP
- immunoprecipitation
- SILAC
- stable isotope labeling in cell culture
- UGGT
- UDP-glucose–glycoprotein glucosyltransferase
- LLO
- lipid-linked oligosaccharide
- ConA
- concanavalin-A.
References
- 1. Schwede A., Macleod O. J., MacGregor P., and Carrington M. (2015) How does the VSG coat of bloodstream form African trypanosomes interact with external proteins? PLoS Pathog. 11, e1005259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horn D. (2014) Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 195, 123–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salmon D., Geuskens M., Hanocq F., Hanocq-Quertier J., Nolan D., Ruben L., and Pays E. (1994) A novel heterodimeric transferrin receptor encoded by a pair of VSG expression site-associated genes in T. brucei. Cell 78, 75–86 [DOI] [PubMed] [Google Scholar]
- 4. Steverding D. (2000) The transferrin receptor of Trypanosoma brucei. Parasitol. Int. 48, 191–198 [DOI] [PubMed] [Google Scholar]
- 5. Mehlert A., Wormald M. R., and Ferguson M. A. (2012) Modeling of the N-glycosylated transferrin receptor suggests how transferrin binding can occur within the surface coat of Trypanosoma brucei. PLoS Pathog. 8, e1002618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peck R. F., Shiflett A. M., Schwartz K. J., McCann A., Hajduk S. L., and Bangs J. D. (2008) The LAMP-like protein p67 plays an essential role in the lysosome of African trypanosomes. Mol. Microbiol. 68, 933–946 [DOI] [PubMed] [Google Scholar]
- 7. Jackson A. P., Allison H. C., Barry J. D., Field M. C., Hertz-Fowler C., and Berriman M. (2013) A cell-surface phylome for African trypanosomes. PLoS Negl. Trop. Dis. 7, e2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allison H., O'Reilly A. J., Sternberg J., and Field M. C. (2014) An extensive endoplasmic reticulum-localised glycoprotein family in trypanosomatids. Microb. Cell 1, 325–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lingnau A., Zufferey R., Lingnau M., and Russell D. G. (1999) Characterization of tGLP-1, a Golgi and lysosome-associated, transmembrane glycoprotein of African trypanosomes. J. Cell Sci. 112, 3061–3070 [DOI] [PubMed] [Google Scholar]
- 10. Engstler M., Weise F., Bopp K., Grünfelder C. G., Günzel M., Heddergott N., and Overath P. (2005) The membrane-bound histidine acid phosphatase TbMBAP1 is essential for endocytosis and membrane recycling in Trypanosoma brucei. J. Cell Sci. 118, 2105–2118 [DOI] [PubMed] [Google Scholar]
- 11. LaCount D. J., Barrett B., and Donelson J. E. (2002) Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J. Biol. Chem. 277, 17580–17588 [DOI] [PubMed] [Google Scholar]
- 12. Treumann A., Zitzmann N., Hülsmeier A., Prescott A. R., Almond A., Sheehan J., and Ferguson M. A. (1997) Structural characterisation of two forms of procyclic acidic repetitive protein expressed by procyclic forms of Trypanosoma brucei. J. Mol. Biol. 269, 529–547 [DOI] [PubMed] [Google Scholar]
- 13. Acosta-Serrano A., Cole R. N., Mehlert A., Lee M. G., Ferguson M. A., and Englund P. T. (1999) The procyclin repertoire of Trypanosoma brucei. Identification and structural characterization of the Glu-Pro-rich polypeptides. J. Biol. Chem. 274, 29763–29771 [DOI] [PubMed] [Google Scholar]
- 14. Güther M. L., Lee S., Tetley L., Acosta-Serrano A., and Ferguson M. A. (2006) GPI-anchored proteins and free GPI glycolipids of procyclic form Trypanosoma brucei are nonessential for growth, are required for colonization of the tsetse fly, and are not the only components of the surface coat. Mol. Biol. Cell 17, 5265–5274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Güther M. L., Beattie K., Lamont D. J., James J., Prescott A. R., and Ferguson M. A. (2009) Fate of glycosylphosphatidylinositol (GPI)-less procyclin and characterization of sialylated non-GPI-anchored surface coat molecules of procyclic-form Trypanosoma brucei. Eukaryot. Cell 8, 1407–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zamze S. E., Wooten E. W., Ashford D. A., Ferguson M. A., Dwek R. A., and Rademacher T. W. (1990) Characterisation of the asparagine-linked oligosaccharides from Trypanosoma brucei type-I variant surface glycoproteins. Eur. J. Biochem. 187, 657–663 [DOI] [PubMed] [Google Scholar]
- 17. Zamze S. E., Ashford D. A., Wooten E. W., Rademacher T. W., and Dwek R. A. (1991) Structural characterization of the asparagine-linked oligosaccharides from Trypanosoma brucei type II and type III variant surface glycoproteins. J. Biol. Chem. 266, 20244–20261 [PubMed] [Google Scholar]
- 18. Atrih A., Richardson J. M., Prescott A. R., and Ferguson M. A. (2005) Trypanosoma brucei glycoproteins contain novel giant poly-N-acetyllactosamine carbohydrate chains. J. Biol. Chem. 280, 865–871 [DOI] [PubMed] [Google Scholar]
- 19. Izquierdo L., Atrih A., Rodrigues J. A., Jones D. C., and Ferguson M. A. (2009) Trypanosoma brucei UDP-glucose:glycoprotein glucosyltransferase has unusual substrate specificity and protects the parasite from stress. Eukaryot. Cell 8, 230–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bandini G., Mariño K., Güther M. L., Wernimont A. K., Kuettel S., Qiu W., Afzal S., Kelner A., Hui R., and Ferguson M. A. (2012) Phosphoglucomutase is absent in Trypanosoma brucei and redundantly substituted by phosphomannomutase and phospho-N-acetylglucosamine mutase. Mol. Microbiol. 85, 513–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bangs J. D., Doering T. L., Englund P. T., and Hart G. W. (1988) Biosynthesis of a variant surface glycoprotein of Trypanosoma brucei. Processing of the glycolipid membrane anchor and N-linked oligosaccharides. J. Biol. Chem. 263, 17697–17705 [PubMed] [Google Scholar]
- 22. Aebi M. (2013) N-Linked protein glycosylation in the ER. Biochim. Biophys. Acta 1833, 2430–2437 [DOI] [PubMed] [Google Scholar]
- 23. Cherepanova N., Shrimal S., and Gilmore R. (2016) N-Linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 41, 57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Acosta-Serrano A., O'Rear J., Quellhorst G., Lee S. H., Hwa K. Y., Krag S. S., and Englund P. T. (2004) Defects in the N-linked oligosaccharide biosynthetic pathway in a Trypanosoma brucei glycosylation mutant. Eukaryot. Cell 3, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manthri S., Güther M. L., Izquierdo L., Acosta-Serrano A., and Ferguson M. A. (2008) Deletion of the TbALG3 gene demonstrates site-specific N-glycosylation and N-glycan processing in Trypanosoma brucei. Glycobiology 18, 367–383 [DOI] [PubMed] [Google Scholar]
- 26. Jones D. C., Mehlert A., Güther M. L., and Ferguson M. A. (2005) Deletion of the glucosidase II gene in Trypanosoma brucei reveals novel N-glycosylation mechanisms in the biosynthesis of variant surface glycoprotein. J. Biol. Chem. 280, 35929–35942 [DOI] [PubMed] [Google Scholar]
- 27. Izquierdo L., Mehlert A., and Ferguson M. A. (2012) The lipid-linked oligosaccharide donor specificities of Trypanosoma brucei oligosaccharyltransferases. Glycobiology 22, 696–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Izquierdo L., Schulz B. L., Rodrigues J. A., Güther M. L., Procter J. B., Barton G. J., Aebi M., and Ferguson M. A. (2009) Distinct donor and acceptor specificities of Trypanosoma brucei oligosaccharyltransferases. EMBO J. 28, 2650–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castro O., Movsichoff F., and Parodi A. J. (2006) Preferential transfer of the complete glycan is determined by the oligosaccharyltransferase complex and not by the catalytic subunit. Proc. Natl. Acad. Sci. U.S.A. 103, 14756–14760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kelleher D. J., Banerjee S., Cura A. J., Samuelson J., and Gilmore R. (2007) Dolichol-linked oligosaccharide selection by the oligosaccharyltransferase in protist and fungal organisms. J. Cell Biol. 177, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nasab F. P., Schulz B. L., Gamarro F., Parodi A. J., and Aebi M. (2008) All in one: Leishmania major STT3 proteins substitute for the whole oligosaccharyltransferase complex in Saccharomyces cerevisiae. Mol. Biol. Cell 19, 3758–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hese K., Otto C., Routier F. H., and Lehle L. (2009) The yeast oligosaccharyltransferase complex can be replaced by STT3 from Leishmania major. Glycobiology 19, 160–171 [DOI] [PubMed] [Google Scholar]
- 33. Damerow M., Rodrigues J. A., Wu D., Güther M. L., Mehlert A., and Ferguson M. A. (2014) Identification and functional characterization of a highly divergent N-acetylglucosaminyltransferase I (TbGnTI) in Trypanosoma brucei. J. Biol. Chem. 289, 9328–9339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Damerow M., Graalfs F., Güther M. L., Mehlert A., Izquierdo L., and Ferguson M. A. (2016) A gene of the β3-glycosyltransferase family encodes N-acetylglucosaminyltransferase II function in Trypanosoma brucei. J. Biol. Chem. 291, 13834–13845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Urbaniak M. D., Martin D. M., and Ferguson M. A. (2013) Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei. J. Proteome Res. 12, 2233–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bangs J. D., Brouch E. M., Ransom D. M., and Roggy J. L. (1996) A soluble secretory reporter system in Trypanosoma brucei. Studies on endoplasmic reticulum targeting. J. Biol. Chem. 271, 18387–18393 [DOI] [PubMed] [Google Scholar]
- 37. Wirtz E., Leal S., Ochatt C., and Cross G. A. (1999) A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89–101 [DOI] [PubMed] [Google Scholar]
- 38. Mehlert A., Sullivan L., and Ferguson M. A. (2010) Glycotyping of Trypanosoma brucei variant surface glycoprotein MITat1.8. Mol. Biochem. Parasitol. 174, 74–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cardoso de Almeida M. L., and Turner M. J. (1983) The membrane form of variant surface glycoproteins of Trypanosoma brucei. Nature 302, 349–352 [DOI] [PubMed] [Google Scholar]
- 40. Ferguson M. A., Haldar K., and Cross G. A. (1985) Trypanosoma brucei variant surface glycoprotein has a sn-1,2-dimyristyl glycerol membrane anchor at its COOH terminus. J. Biol. Chem. 260, 4963–4968 [PubMed] [Google Scholar]
- 41. Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vacic V., Iakoucheva L. M., and Radivojac P. (2006) Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics 22, 1536–1537 [DOI] [PubMed] [Google Scholar]
- 43. Miller M. L., Jensen L. J., Diella F., Jørgensen C., Tinti M., Li L., Hsiung M., Parker S. A., Bordeaux J., Sicheritz-Ponten T., Olhovsky M., Pasculescu A., Alexander J., Knapp S., Blom N., et al. (2008) Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poljak K., Breitling J., Gauss R., Rugarabamu G., Pellanda M., and Aebi M. (2017) Analysis of substrate specificity of Trypanosoma brucei oligosaccharyltransferases (OSTs) by functional expression of domain-swapped chimeras in yeast. J. Biol. Chem. 292, 20342–20352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lizak C., Gerber S., Numao S., Aebi M., and Locher K. P. (2011) X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 [DOI] [PubMed] [Google Scholar]
- 47. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 48. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 49. Kalb L. C., Frederico Y. C., Boehm C., Moreira C. M., Soares M. J., and Field M. C. (2016) Conservation and divergence within the clathrin interactome of Trypanosoma cruzi. Sci. Rep. 6, 31212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boehm C. M., Obado S., Gadelha C., Kaupisch A., Manna P. T., Gould G. W., Munson M., Chait B. T., Rout M. P., and Field M. C. (2017) The trypanosome exocyst: a conserved structure revealing a new role in endocytosis. PLoS Pathog. 13, e1006063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maishman L., Obado S. O., Alsford S., Bart J. M., Chen W. M., Ratushny A. V., Navarro M., Horn D., Aitchison J. D., Chait B. T., Rout M. P., and Field M. C. (2016) Co-dependence between trypanosome nuclear lamina components in nuclear stability and control of gene expression. Nucleic Acids Res. 44, 10554–10570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nagamune K., Ohishi K., Ashida H., Hong Y., Hino J., Kangawa K., Inoue N., Maeda Y., and Kinoshita T. (2003) GPI transamidase of Trypanosoma brucei has two previously uncharacterized (trypanosomatid transamidase 1 and 2) and three common subunits. Proc. Natl. Acad. Sci. U.S.A. 100, 10682–10687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Choi B. K., Warburton S., Lin H., Patel R., Boldogh I., Meehl M., Meehl M., d'Anjou M., Pon L., Stadheim T. A., and Sethuraman N. (2012) Improvement of N-glycan site occupancy of therapeutic glycoproteins produced in Pichia pastoris. Appl. Microbiol. Biotechnol. 95, 671–682 [DOI] [PubMed] [Google Scholar]
- 54. Yan A., Wu E., and Lennarz W. J. (2005) Studies of yeast oligosaccharyl transferase subunits using the split-ubiquitin system: topological features and in vivo interactions. Proc. Natl. Acad. Sci. U.S.A. 102, 7121–7126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ferguson M. A., Duszenko M., Lamont G. S., Overath P., and Cross G. A. (1986) Biosynthesis of Trypanosoma brucei variant surface glycoproteins. N-Glycosylation and addition of a phosphatidylinositol membrane anchor. J. Biol. Chem. 261, 356–362 [PubMed] [Google Scholar]
- 56. Siegel T. N., Hekstra D. R., Wang X., Dewell S., and Cross G. A. (2010) Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 38, 4946–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Butter F., Bucerius F., Michel M., Cicova Z., Mann M., and Janzen C. J. (2013) Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite's host adaptation machinery. Mol. Cell. Proteomics 12, 172–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Urbaniak M. D., Guther M. L., and Ferguson M. A. (2012) Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS One 7, e36619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Castillo-Acosta V. M., Ruiz-Pérez L. M., Etxebarria J., Reichardt N. C., Navarro M., Igarashi Y., Liekens S., Balzarini J., and González-Pacanowska D. (2016) Carbohydrate-binding non-peptidic pradimicins for the treatment of acute sleeping sickness in murine models. PLoS Pathog. 12, e1005851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Castillo-Acosta V. M., Ruiz-Pérez L. M., Van Damme E. J., Balzarini J., and González-Pacanowska D. (2015) Exposure of Trypanosoma brucei to an N-acetylglucosamine-binding lectin induces VSG switching and glycosylation defects resulting in reduced infectivity. PLoS Negl. Trop. Dis. 9, e0003612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Castillo-Acosta V. M., Vidal A. E., Ruiz-Pérez L. M., Van Damme E. J., Igarashi Y., Balzarini J., and González-Pacanowska D. (2013) Carbohydrate-binding agents act as potent trypanocidals that elicit modifications in VSG glycosylation and reduced virulence in Trypanosoma brucei. Mol. Microbiol. 90, 665–679 [DOI] [PubMed] [Google Scholar]
- 62. Ruiz-Canada C., Kelleher D. J., and Gilmore R. (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shrimal S., Cherepanova N. A., and Gilmore R. (2015) Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin. Cell Dev. Biol. 41, 71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cherepanova N. A., Shrimal S., and Gilmore R. (2014) Oxidoreductase activity is necessary for N-glycosylation of cysteine-proximal acceptor sites in glycoproteins. J. Cell Biol. 206, 525–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mohorko E., Owen R. L., Maloǰcić G., Brozzo M. S., Aebi M., and Glockshuber R. (2014) Structural basis of substrate specificity of human oligosaccharyl transferase subunit N33/Tusc3 and its role in regulating protein N-glycosylation. Structure 22, 590–601 [DOI] [PubMed] [Google Scholar]
- 66. Schulz B. L., and Aebi M. (2009) Analysis of glycosylation site occupancy reveals a role for Ost3p and Ost6p in site-specific N-glycosylation efficiency. Mol. Cell. Proteomics 8, 357–364 [DOI] [PubMed] [Google Scholar]
- 67. Woycechowsky K. J., and Raines R. T. (2003) The CXC motif: a functional mimic of protein disulfide isomerase. Biochemistry 42, 5387–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tiengwe C., Muratore K. A., and Bangs J. D. (2016) Surface proteins, ERAD and antigenic variation in Trypanosoma brucei. Cell Microbiol. 18, 1673–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hirumi H., and Hirumi K. (1989) Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75, 985–989 [PubMed] [Google Scholar]
- 70. Oberholzer M., Morand S., Kunz S., and Seebeck T. (2006) A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol. Biochem. Parasitol. 145, 117–120 [DOI] [PubMed] [Google Scholar]
- 71. Zielinska D. F., Gnad F., Wiśniewski J. R., and Mann M. (2010) Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141, 897–907 [DOI] [PubMed] [Google Scholar]
- 72. Aslett M., Aurrecoechea C., Berriman M., Brestelli J., Brunk B. P., Carrington M., Depledge D. P., Fischer S., Gajria B., Gao X., Gardner M. J., Gingle A., Grant G., Harb O. S., Heiges M., et al. (2010) TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 38, D457–D462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Abraham A., Pedregosa F., Eickenberg M., Gervais P., Mueller A., Kossaifi J., Gramfort A., Thirion B., and Varoquaux G. (2014) Machine learning for neuroimaging with scikit-learn. Front. Neuroinform. 8, 1424600388 [Google Scholar]
- 74. Brandes N., Ofer D., and Linial M. (2016) ASAP: a machine learning framework for local protein properties. Database 2016, baw133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.