

Abstract
Introduction
AKR1C3 is a drug target in hormonal and hormonal independent malignancies and acts as a major peripheral 17β-hydroxysteroid dehydrogenase to yield the potent androgens testosterone and dihydrotestosterone, and as a prostaglandin (PG) F synthase to produce proliferative ligands for the PG FP receptor. AKR1C3 inhibitors may have distinct advantages over existing therapeutics for the treatment of castration resistant prostate cancer, breast cancer and acute myeloid leukemia.
Area covered
This article reviews the patent literature on AKR1C3 inhibitors using SciFinder which identified inhibitors in the following chemical classes: N-phenylsulfonylindoles, N-(benzimidazoylylcarbonyl)-N-(indoylylcarbonyl)- and N-(pyridinepyrrolyl)- piperidines, N-benzimidazoles and N-benzindoles, repurposed nonsteroidal antiinflammatory drugs (indole acetic acids, N-phenylanthranilates and aryl propionic acids), isoquinolines, and nitrogen and sulfur substituted estrenes. The article evaluates inhibitor AKR potency, specificity, efficacy in cell-based and xenograft models and clinical utility. The advantage of bifunctional compounds that either competitively inhibit AKR1C3 and block its androgen receptor (AR) coactivator function or act as AKR1C3 inhibitors and direct acting AR antagonists are discussed.
Expert opinion
A large number of potent and selective inhibitors of AKR1C3 have been described however, preclinical optimization, is required before their benefit in human disease can be assessed.
Keywords: Acute myeloid leukemia, aldo-keto reductase, androgen receptor, baccharin, co-activator, estrenes, 17β-hydroxysteroid dehydrogenase, indomethacin, non-steroidal anti-inflammatory drugs, prostate cancer
1. Introduction
Aldo-keto reductase (AKR) 1C3 (type 5 17β-hydroxysteroid dehydrogenase; prostaglandin [PG] F2α synthase; and dihydrodiol dehydrogenase X) belongs to the AKR superfamily of proteins [1–4]. AKR1C3 is a drug target due to its involvement in intratumoral androgen biosynthesis in prostate and breast cancer. In breast cancer, AKR1C3 is a principal source of testosterone (T), the substrate for aromatase. The ability of AKR1C3 to regulate ligand access to the androgen receptor (AR) and estrogen receptor (ER) in a tumor-specific fashion makes it a superior drug target than either AR or ER antagonists [5–8]. By acting as a PGF2α synthase [9,10], it also deprives peroxisome proliferator activating receptor γ(PPARγ) of its putative ligand Δ15-PGJ2 and generates ligands for the PG FP receptor to stimulate the mitogen activated protein kinase cascade to promote cell proliferation [11,12]. AKR1C3 inhibitors thus offer promise for the treatment of hormonal and hormonal-independent malignancies [11,12].
AKR1C3 inhibitors have been used in clinical trials of castration-resistant prostate cancer (CPRC) [13] and acute myeloid leukemia (AML) [14]. CRPC is the fatal form of prostate cancer. This disease remains androgen dependent despite castrate levels of circulating T. Androgen dependency remains since the tumor undergoes adaptive responses to sustain AR signaling. One mechanism involves adaptive intratumoral androgen biosynthesis and a second mechanism involves changes in the AR itself [15,16]. Intratumoral androgen biosynthesis is targeted by P45017A1 (17α-hydroxylase/17,20-lyase) inhibitors to prevent the conversion of pregnenolone to dehydroepiandrosterone (DHEA) in the adrenal and hence deprive the tumor of its source of androgens. Abiraterone acetate (Abi) 1 is the P45017A1 inhibitor in clinical use and is approved by the US FDA [17–19]. The second mechanism, involving AR, is targeted with the super AR antagonist enzalutamide (ENZ) 2 [20]. To surmount the CNS side effects seen with this drug, a second-generation compound AN-509 (Apalutamide) 3 has been developed (Figure 1) [21]. These antagonists not only bind to the AR but prevent its nuclear translocation and binding to chromatin. Both Abi and ENZ increase median survival time in CRPC patients by only 3–4 months before drug resistance occurs [17–19,22,23]. Thus, a critical clinical unmet need is for better agents. While multiple mechanisms can contribute to drug resistance, both Abi and ENZ resistance can be surmounted in xenograft models using the AKR1C3 inhibitor indomethacin [24,25] first identified by Byrns et al. [26].
Figure 1.

Drugs in common use for the treatment of CRPC and some representative AKR1C3 inhibitors not covered by patents.
AKR1C3 is overexpressed in prostate cancer as part of the adaptive response to androgen deprivation therapy (ADT). AKR1C3 is overexpressed in cell lines deprived of androgens [27,28], in prostate cancer xenografts in castrate mice [24,25,28] and in CRPC patients [29–31]. AKR1C3 is involved in all pathways to T and 5α-dihydrotestosterone (DHT) in the prostate due its 17-ketosteroid reductase activity; it reduces 4-androstene-3,17-dione to T (by the canonical pathway) [32]; it reduces 5α-andros-tane-3,17-dione to DHT (by the alternative pathway) [33]; it reduces androsterone to 5α-androstane-3α,17β-diol (3α-diol) which is then oxidized by 17BHSD6 to DHT (by the backdoor pathway) [34–36]; and it reduces DHEA to 5-androstene-3β,17β-diol which is converted by 3HSDB1 to T. AKR1C3 inhibitors would block all pathways to T and DHT within the tumor and could surmount drug resistance to Abi and ENZ (Figure 2).
Figure 2.

Role of AKR1C3 in androgen biosynthesis in human prostate. The conversion of C21 steroids pregnenolone and progesterone to the corresponding C19 steroids dehydroepiandrosterone (DHEA) and 4-androstene-3,17-dione is catalyzed by CYP17 (P45017A1); and the preferred route is from pregnenolone. The evidence for intratumoral biosynthesis of C19 steroids from C21 steroids is scant and the reactions occur predominately in the adrenal. The conversion of progesterone to desoxycorticosterone is adrenal specific and is a side effect of abiraterone acetate treatment leading to mineralocorticoid excess. HSD3B1, 3β-hydroxysteroid dehydrogenase type 1, SRD5A, steroid 5α-reductase type 1 and type 2. Reproduced with permission from Adeniji AO, Twenter BM, Byrns MC, Jin, Y, et al. Development of potent and selective inhibitors of aldo-keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure-activity relationships. J Med Chem 2012;55:2311–23 Copyright American Chemical Society.
AKR1C3 is also overexpressed in ductal carcinoma in situ of the breast [37]; its overexpression is correlated with the expression of ERα [38], and with breast cancer relapse [39]. By acting as a peripheral 17β-hydroxysteroid dehydrogenase that converts 4-androstene-3,17-dione to T, AKR1C3 becomes a peripheral source of T so that aromatase can synthesize 17β-estradiol in the breast [6]. Thus, AKR1C3 inhibitors have a place in the treatment of ERα positive breast cancer and offer an advantage over aromatase inhibitors that would block estrogen biosynthesis systemically.
AKR1C3 inhibitors have been exploited in AML to alter PG signaling. In combination with PPARγ agonists, e.g. bezafibrate (BZF), the AKR1C3 inhibitor 6-medroxyprogesterone acetate (6MPA) 4 gave a superior response than was achieved by either agent alone [15]. In this treatment, BZF could stimulate PPARγ signaling and 6MPA would block the formation of PGs of the F series that would bind to the FP receptor (Figure 3). This is the first clinical example of the use of AKR1C3 inhibitors in a nonhormone-dependent malignancy.
Figure 3.

Role of AKR1C3 in prostaglandin signaling. AKR1C3 catalyzes the conversion of prostaglandin (PG) H2 and PGD2 to PGF2α and 11β-PGF2α respectively (PGF2α synthase activity). PGF2α and 11β-PGF2α are ligands for the prostaglandin FP receptor which leads to activation of mitogen activated protein kinase (MAPK) and cell proliferation, as well as activation of NFkB. AKR1C3 prevents the conversion of PGD2 to 15dPGJ2 a peroxisome proliferator activating receptor γ(PPARγ) agonist and inhibitor of NFkB signaling where the former leads to cell-differentiation and inhibition of cell growth. Reproduced with permission form Byrns MC and Penning TM. Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase (AKR1C3): Role in breast cancer and inhibition by nonsteroidal anti-inflammatory drugs. Chem Biol Inter 2009: 178: 221–7 Copyright Elsevier.
The development of AKR1C3 inhibitors that are potent and selective is challenging since it is highly related to AKR1C1, AKR1C2, and AKR1C4 that share more than 86% sequence identity and their inhibition in the context of prostate cancer would be deleterious. For example, AKR1C1 converts DHT to 5α-androstane-3β,17β-diol (3β-diol) a proapoptotic ligand for ERβ and its inhibition should be avoided [40]. Similarly, AKR1C2 inactivates DHT by forming 3α-diol and its inhibition should be avoided [41,42]. By contrast, AKR1C4 is liver specific and is required for the synthesis of bile-acids and its inhibition would lead to bile-acid deficiency [43]. Despite this challenge, both academic and industrial groups have filed patents on AKR1C3 inhibitors (Table 1).
Table 1.
Summary of patent applications reviewed on AKR1C3 inhibitors.
| Compound class | Patent number | Mode of action | Assay method | IC50 value for AKR1C3a | Specificity for AKR1C3b | Status |
|---|---|---|---|---|---|---|
| N-phenylsulfonyl indoles | CA 2644809 W02007100066 |
Enzyme inhibition | 4-AD to testosterone | 90 nM | Counter screen against 17BHSD3 selectivity > 100-fold |
Preclinical (Astellas) |
| N-(benzimidazolylcarbonyl)-piperidines | JP201202018 | Enzyme inhibition | 4-AD to testosterone | 110 nM | Counter screen against 17BHSD3 | Preclinical (Astellas) |
| N-(indolylcarbonyl)-piperidines | WO2010101127 JP2012102017 JP2010222350 |
Enzyme inhibition | 4-AD to testosterone | 3.7 nM | AKR1C3 > AKR1C1 | Lead ASP9521 (Astellas) Phase 1 clinical trial |
| N-(pyridinepyrroylylcarbonyl)-piperidines | W02010101128 | Enzyme inhibition | 4-AD to testosterone | 24 nM | Not determined | Preclinical (Astellas) |
| N-(benzimidazoles or indole) benzoic acids | CA02694216 WO2009014150 WO2010087319 |
Enzyme inhibition | 4-AD to testosterone | 40–50 nM | Counter screen against 17BHSD3 Selectivity > 40–250-fold LNCaP-AKR1C3 cells |
Preclinical |
| N-(phenylamino)-benzoates | WO2012122208 US 2014/0107085 |
COMP | Inhibits S-tetralol oxidation | 60 nM | Counter screen against AKR1C1, 1C2, 1C4 Selectivity > 250-fold |
Flufenamic acid isomer Preclinical |
| N-(naphthylamino)-benzoates | WO2012142208 US 20140107085 |
COMP; AR antagonist | Inhibits S-tetralol oxidation | 80 nM | Counter screen against AKR1C1, 1C2, 1C4 Selectivity > 100-fold |
BMT4-158 Preclinical |
| Isoquinolones | WO2013142390 WO2014039820 |
COMP; blocks AKR1C3-AR coactivator function | 4-AD to testosterone | 770 nM | AKR1C3 > AKR1C1 | GTx-560 (GTx-Therapeutics) Preclinical |
| Indomethacin analogs | WO2013/059245 US 2016/0303082 WO2015065919c |
COMP | Inhibits S-tetralol oxidation | 90 nM | Counter screen against AKR1C1, 1C2, 1C4 Selectivity > 500-fold |
Preclinical |
| Nitrogen or sulfur-substituted estrenes | WO2013045407 WO2014009274 WO2014128108 WO2016/037956 |
Enzyme inhibition | NADPH reduction of cumberone to cumberol and 4-AD to testosterone | 1 nM | HEK-293-AKR1C3 cells | Preclinical (Bayer) |
| β-Naphthylacetic acids | W02017/070448-A120170427 | COMP | Inhibits S-tetralol oxidation | 110 nM | Counter screen AKR1C2 Selectivity > 400 fold |
R-Ethyl-naproxen Preclinical |
| Baccharin | JP2015020966 | COMP | Inhibits S-tetralol oxidation | 100 nM | Counter screen | Preclinical |
| Baccharin analogs | Provisional patent | AKR1C2 Selectivity > 500-fold |
COMP: Competitive inhibition; 4-AD: 4-androstene-3,17-dione; AR: androgen receptor; ND: not determined.
IC50 value for the most potent inhibitor in the series.
Fold selectivity for most specific compound in the series in counterscreens.
Use patent.
2. Chemistry
Potent and selective AKR1C3 inhibitors that are based on non-steroidal, steroidal, and natural product scaffolds have been disclosed. Compounds reported but not claimed in patent applications include the N-benzoylanthranilates 5 [44], the 2,3-arylpropenic acids 6–9 [45], and the natural products stylopne 10 (an isoquinoline alkaloid) [46] and 2′-hydroxyflavone 11 [47]. Compound 5 was synthesized by coupling 3-hydroxybenzoate with 4-bromoaniline and the 2,3-diarylpropenic acids were synthesized by a Perkin reaction between substituted benzal-dehydes and functionalized aryl acetic acids in base (Figure 1).
The synthesis of the lead agent SN33638 12, a potent and selective inhibitor of AKR1C3 (IC50 = 13 nM), is based on an N-phenylsulfonyl-piperidine [48]. SN336381 is also related to the N-phenylsulfonyl indoles 13, developed by Astellas and claimed in patent W02007100066 (Table 1). Building on this lead, a series of N-(benzimidazolyl)-, N-(indolyl)-, and N-(-pyridinelpyroylyl)-carbonyl piperidines 14–16 were claimed in JP201202018, W02010101127, and W02010101128, respectively, by Astellas. The N-(indolyl)-carbonyl piperdines include ASP9521 15, which was taken through to a Phase 1/11b clinical trial [14,49]. A related series of N-benzimidazole or N-indole benzoic acids 17, 18 were claimed in W02009014150, WO2010087319 (Figure 4).
Figure 4.

AKR1C3 nonsteroidal inhibitors under patent.
A screen of existing drugs identified nonsteroidal antiinflammatory drugs (NSAIDs) as selective inhibitors of different AKR1C isoforms [26]. Compounds of interest include indomethacin 19 which was selective for AKR1C3; the N-phenylanthranilates (e.g. meclofenamic acid 23) were pan-AKR1C inhibitors; and aryl pro-pionic acids (e.g. naproxen 25) displayed equal AKR1C2 and AKR1C3 selectivity and potency. This led to the repurposing of these NSAIDs for AKR1C3 inhibition while eliminating inhibition of the COX-isoforms (PGH synthase I and II). Patent WO2012122208 was issued for N-phenylaminobenzoates represented by 24 and patent WO2013059245 was issued for indo-methacin analogs, represented by 20–22 [50,51]. This was subsequently followed by patent W02017070448 for β-naphthylacetic acids (R-naproxen analogs), 26 [52]. In each case, the NSAID analogs were subjected to medicinal chemistry optimization to remove structural features required for inhibition of COX-1 and COX-2 while inhibition of AKR1C3 was retained. Indomethacin gave rise to three classes of analogs: Class I analogs (retain the core structure of indomethacin, 20); Class II analogs (are des-methyl indomethacin compounds in which the 2′-methyl group has been removed, 21); and Class III analogs (are 3′-alkyl derivatives where the acetic acid side chain has been substituted with an alkyl group and the carboxylic acid side chain has been moved to the 2′-position 22 or the 3′ and 2′ positions are cyclized to yield a cyclic carboxylic acid or sulfonamide) [53,54].
Indomethacin analogs with conserved 5′-methoxy groups and p-chlorobenzoyl groups at the indole N1 position were synthesized by the method of Yamamoto [55,56]. The key reagent for the underlying Fischer indolization is 4-chloro-N-(4-methoxyphenyl)-benzohydrazide hydrochloride. Target compounds containing a 3′-propionic acid group or a 2′-des methyl-group were readily obtained from the benzohydrazide hydrochloride by using either a slight excess of 5-oxohexanoic acid (here, R1 = Me, n = 2) or 4-oxobutanoic acid (R1 = H, n = 1) in acetic acid, respectively, to give, 20–21. Use of 4-oxohexanoic acid (R1 = Me, n = 1) quantitatively yielded the reverse 2′-pro-pionic acid/3′-alkyl indole derivative, 22 [57] (Figure 4). Following the issuance of patent WO2013059245 for these indomethacin analogs, a patent claiming the use of indomethacin for CRPC was filed, WO2015065919.
For the N-phenylaminobenzoates, simple coupling chemistry involving a Buchwald–Hartwig C–N coupling reaction followed by saponification of the formed methyl ester produced an extensive library of compounds that are claimed in patent WO2012122208 and US 20140107085 [50,51]. Compounds in which the arrangement of the amine and carboxylic acid was changed from ortho-to meta- position followed by introduction of a para- electron withdrawing group on the B-ring gave compounds of mid-nanomolar potency and selectivity for AKR1C3 (Figure 4).
For the aryl propionic acids 25, β-naphthylacetic acids in which the stereochemistry at the alkyl substituent at the alpha carbon was changed from S- to R- were sufficient to abolish COX-1 and COX-2 inhibition but retain AKR1C3 inhibition; compounds such as 26 are disclosed in WO2017070448 (Figure 4) [52].
Bifunctional AKR1C3 nonsteroidal inhibitors have also been disclosed (Figure 4). Isoquinolines represented by the lead compound GTX-560 27 not only act as competitive inhibitors of AKR1C3 but also block its AR coactivator function which was previously unknown [58]. The isoquinolines were claimed in patents WO2013142390 and WO2014039820A1 filed by GTx-Therapeutics. BMT4-15828, which is a N-naphthylaminobenzoate, is covered by the patent on the N-phenylaminobenzoates and acts as a bifunctional AKR1C3 competitive inhibitor and direct acting AR antagonist [59].
Attempts have been made to develop steroidal-based inhibitors for AKR1C3 as it reduces 17-ketosteroids. Extensive nitrogen and sulfur-substituted estrenes with the core structure 30 have been claimed by Bayer in four patents WO201345407, WO2014009274, WO2014128108, and WO02016037956 (Figure 5). The key features of these steroids are the presence of either an amide, amine, or sulfone at the C3 position of the steroid coupled with the presence of either a nitrogen heterocycle or a trifluorosulfone at the C17 position.
Figure 5.

AKR1C3 steroidal inhibitors under patent.
Natural products such as baccharin analogs 29 (from the Brazilian propolis) have also be claimed as AKR1C3 inhibitors, and these derivatives contain a phenolic cinammic acid substituted with an isopropyl group and a phenylpropionic ester [60,61]. However, these compounds are likely to hydrolyze in vivo to the corresponding alcohol and acid.
3. Structure–activity relationships
Thirty-five crystal structures of AKR1C3·NADP+·inhibitor complexes exist in the PDB. Inspection of these structures shows that if the inhibitor contains a carboxylic acid, it can often form hydrogen bonds with the catalytic tetrad members Tyr55 and His117. Other portions of the inhibitor can occupy one of several subpockets (SP), e.g. SP1 Ser118, Asn167, Phe306, Phe311, and Tyr319 (e.g. occupied by the B-ring of N-phenylaminobenzoates). The SP2 sub-pocket refers to Ser129, W227, and F311 (e.g. occupied by the side-chain of PGs), and the SP3 sub-pocket which contains Y24, E192, S217, S221, Q222, Y305, and F306 [62]. While the presence of these sub-pockets can be rationalized to determine binding mode and can be used as the basis of docking studies, some important caveats exist as illustrated by the binding of indomethacin. Two different binding poses for indomethacin exist in the AKR1C3·NADP+·indomethacin depending on pH. In the AKR1C3·NADP+·indomethacin complex at pH 6.0 (PDB ID 1S2A), where indomethacin is fully protonated, the carboxylate is anchored by Q222 and Y24 in SP3, the bridge carbonyl forms a hydrogen bond with Tyr55 through an intervening water molecule, and there is no occupancy of SP1. However, in the AKR1C3·NADP+·indomethacin complex at pH 7.5 (PDB ID 3UG8), where indomethacin is deprotonated, the drug rotates so that the carboxylic acid now forms a hydrogen bond with Tyr55, the SP1 pocket is now occupied by the p-chlorobenzoyl ring, and there is interaction between W227 with the methoxyindole in the SP2 pocket (Figure 6) [54]. These structures illustrate the difficulty in performing structure-based inhibitor design for AKR1C3.
Figure 6.
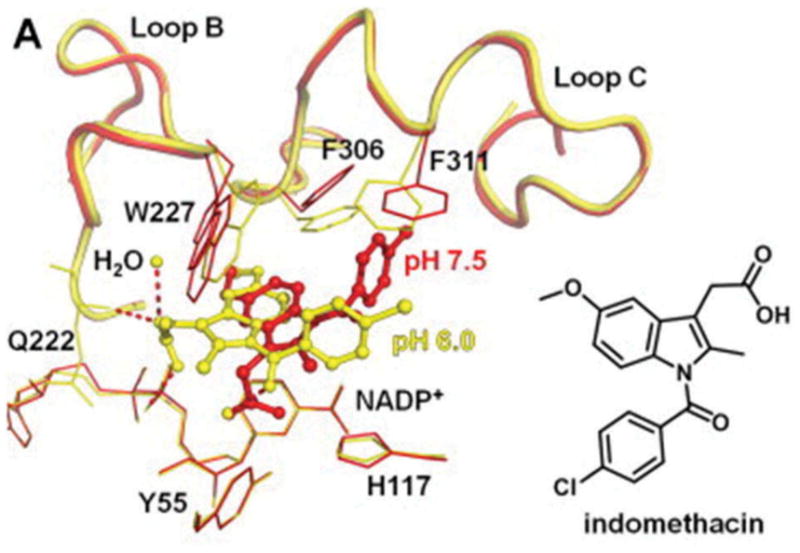
Two different binding poses for indomethacin in the AKR1C3.NADP+ complex. AKR1C3.NADP+.Indomethacin complex (yellow) at pH 6.0 (PDB ID 1S2A) and AKR1C3.NADP+.Indomethacin complex (red) at pH 7.5 (PDB ID 3UG8). Full color available online.
4. Biology and action
Tiered screening has been conducted to support patent claims. Tier 1 screening includes in vitro inhibition assays on recombinant AKR1C3 to claim compounds with mid-nanomolar affinity. Counterscreens have been performed in many instances versus either AKR1C1 or AKR1C2, to claim compounds that are 40–500-fold selective for the target (see Table 1). Many compounds have cleared this screen, but often only IC50 values are reported and the pattern of inhibition is not given. Since AKR1C3 catalyzes an ordered bi-bi mechanism, in the reduction direction, two inhibitor complexes can form e.g. E·NADPH·I (competitive complex) and E·NADP+·I (uncompetitive complex) [26]. Thus, depending on the mode of inhibition, the IC50 values may not be directly comparable.
Tier 2 screening for repurposed NSAIDs includes a subsequent counter screen against all the human AKRs, and a counter screen against COX-1 and COX-2. This level of screening was conducted for patents WO2012122208 and US 20140107085 and patents WO2013059245 and US 20160303082. In other patents, specificity was assessed by demonstrating the inability of leads to inhibit HSD17B3, the major androgenic 17β-HSD found in the testis and a member of the short-chain dehydrogenase/reductase superfamily [63].
Tier 3 screening includes cell-based assays to determine whether compounds inhibit the conversion of 4-andros-tene-3,17-dione to T in LNCaP-AKR1C3 cells or another prostate cancer cell model in which AKR1C3 is overexpressed. Often HEK-293 cells expressing AKR1C3 have been used as a substitute. These screens determine whether the inhibitor has cell bioavailability and retains potency. Claimed compounds have been shown to be effective in these models, albeit with some loss of potency. Cell-based assays using AR-reporter gene assays and AR-ligand binding assays have also been performed to determine whether compounds act as AR-antagonists or inhibit the co-activator function of AKR1C3, as is the case for GTx-560 [58]. The AR coactivator domain of AKR1C3 was located to amino-acid residues 171–237 by deletion mutagenesis [58], which is distal to the enzyme active site. This region contains a coactivator peptide consensus peptide LXXLL (LEMIL). This suggests that some small molecule competitive inhibitors may have an allosteric effect that radiates to distal portions of the protein to affect AKR1C3–AR interaction. Interestingly, indomethacin does not have this property [58].
Tier 4 screening determines whether AKR1C3 inhibitors are effective in vivo and cause a reduction in tumor volume or tumor incidence in either xenograft or patient-derived xenografts of prostate cancer. ASP9521 and indomethacin have been shown to inhibit tumor growth in xenografts ex-vivo and in vivo, respectively [24,25,49]. Similar results have been obtained with GTx-560 [58].
Some attention to the xenograft model is required. Demonstration of reduced tumor incidence and volume in SCID mice transplanted with prostate cancer tumors is not a model of CRPC. CRPC can be modeled if the recipient mouse is castrated after the transplant and the tumor then regrows under castrate conditions. This model has been rarely used. No experiments have been performed with AKR1C3 inhibitors in patient-derived xenografts. Nevertheless, proof-of-principle xenograft data indicate that AKR1C3 inhibitors are effective antitumor agents in animal models [24,25,49].
Based on preclinical data, ASP9521 15 was advanced to a Phase I/IIb clinical trial by Astellas. ASP9521 was found to be well tolerated but without efficacy [14]. In this small trial, 7/13 patients completed the regimen. Serum levels of ASP9521 reached levels that would be sufficient to inhibit AKR1C3. However, decreases in serum PSA and serum steroid hormone levels were not achieved. However, inclusion criteria did not screen for AKR1C3 expression and the authors concluded that drug failure may have been due to the exclusion of patients who had been on prior ADT, which is known to upregulate AKR1C3.
The steroid-based estrenes with substitutions at C3 and C17 have been shown to be potent competitive inhibitors in vitro using recombinant AKR1C3 and in HEK-293 cells over-expressing AKR1C3. However, counter screens against other human AKRs have not been reported. The presence of the nitrogen heterocycle at C17 is reminiscent of the heterocycle found in Abi and raises issues as to whether they inhibit P45017A1 or other steroid metabolizing P450 isoforms.
5. Expert opinion
As AKR1C3 is a major peripheral 17β-HSD required for the synthesis of T and DHT, inhibitors of the enzyme may have a place for treating endocrinological disorders associated with androgen excess in males and females, e.g. prostate cancer, benign prostatic hyperplasia, alopecia, pattern baldness, hirsutism, polycystic ovarian syndrome, etc. As T synthesized locally is also a substrate for aromatase, AKR1C3 inhibition may also be desirable in breast cancer, endometriosis, and endometrial cancer. However, the major focus has been on prostate cancer.
The majority of AKR1C3 inhibitors claimed are mono-functional agents and act downstream from Abi. Since they do not inhibit P45017A1, they do not have to be coadministered with prednisone to prevent adrenal insufficiency. The monofunctional AKR1C3 inhibitors would be superior to other P45017A1 inhibitors (orterenol and galeterone) since they target an enzyme involved in intratumoral androgen biosynthesis that is overexpressed upon ADT. Even though P45017A1 inhibitors decrease serum DHEA-SO4 and DHEA by more than 90%, the amount of DHEA-SO4 that remains leaves a substantial reservoir for intra-crine androgen biosynthesis by AKR1C3 [64,65]. Mechanisms of drug resistance to P45017A1 inhibitors also include HSD3B1 allelic variants that stabilize the enzyme responsible for the conversion of DHEA to 4-androstene-3,17-dione [66]. The properties of AKR1C3 inhibitors versus other agents that target the AR axis in prostate cancer are presented in Table 2.
Table 2.
Comparison of AKR1C3 inhibitors with other therapeutic agents for castration-resistant prostate cancer.
| Drug | Target | Resistance mechanism | Status | Company |
|---|---|---|---|---|
| Abiraterone acetate/prednisone | P45017A1 | DHEA-SO4 depot remains AR upregulation AR-SV HSDB1 variants AKR1C3 upregulation |
FDA Approval 12 December 2012 Off patent 2019 |
Johnson & Johnson |
| Orteronel (TAK-700) | P45017A1 | DHEA-SO4 depot remains AR upregulation AR-SV HSDB1 variants AKR1C3 upregulation |
Takeda terminates drug June 2014. No clinical benefit in terms of overall survival in Phase III clinical trial | Takeda |
| Galeterone (TOK-001) | P45017A1 (lyase specific) and degrades AR-SV | DHEA-SO4 depot remains AR upregulation HSDB1 variants AKR1C3 upregulation |
Phase III clinical trial. No benefit over ENZ in AR-V7 expressing CRPC. Drug terminated July 2016 | Tokai Pharmaceuticals |
| Dutasteride | SRD5A1/SRD5A2 | Intratumoral T bisosynthesis AKR1C3 upregulation |
FDA warning for use in prostate cancer | Glaxo-Smith Kline |
| Enzalutamide | AR | Adaptive tumor androgen synthesis AKR1C3 upregulation AR-SV CNS-seizures |
FDA Approval 12 September 2012 Off patent 2026 |
Medivation and now Astellas |
| Apalutamide | AR | Adaptive tumor androgen synthesis AKR1C3 upregulation AR-SV |
Aragon and now Johnson & Johnson | |
| ASP9521 | AKR1C3 | AR-SV | Phase I/IIb trial lack of efficacy in limited no of patients | Astellas |
SRD5A1/SRD5A2: Steroid 5α-reductase; AR: androgen receptor.
AKR1C3 is overexpressed in prostate cancer cells, in xenografts, and in tumors of patients that over undergone ADT [24,25,28–31,67]. But it is likely that the use of these mono-functional AKR1C3 inhibitors will require precision medicine to ensure that the target is expressed in the patient. Interestingly, steroid 5α-reductase inhibitors e.g. finasteride and dutasteride are not approved by the FDA for the treatment of prostate cancer since these may cause the appearance of a more aggressive disease (Table 2).
Few compounds claimed in the patents have undergone a complete counter screen and for many, DMPK studies have yet to be performed limiting their effective use in animal xenograft and human studies. Here, the repurposed NSAIDs hold promise since they are anticipated to retain the properties of the parent drug from which they were derived [5,68].
Monofunctional AKR1C3 inhibitors are ultimately predicted to fail in the clinic due to the issue of drug resistance. However, they could be added to existing regimens, e.g. Abi or ENZ with the prospect of achieving a synergistic effect and more durable drug response. A starting point would be to add indomethacin to patients who progress on Abi or ENZ. Both Abi and ENZ resistance are likely to involve the overexpression of AKR1C3 as a component of drug resistance [24,25].
Mechanisms of drug resistance include AR gene amplification [69], the selection of AR mutants that make it ligand promiscuous [70,71], and the appearance of AR splice variants (AR-SV) that have lost their ligand binding domain and are constitutively active [72–74].
The bifunctional AKR1C3 inhibitors, e.g. GTX-560 27, offers promise since it blocks the coactivator function of AKR1C3 on full-length AR (Table 2). Whether AKR1C3 can act as a coactivator of AR-SVs is unknown. The other bifunctional agent claimed is BMT4-158 28, which acts as a competitive inhibitor of AKR1C3 and as a direct acting AR antagonist, suggesting that single agents that target intra-tumoral androgen biosynthesis and AR signaling can be developed. Whether these single agents would be superior to a combination treatment of Abi plus prednisone plus ENZ remains to be determined.
Article highlights.
AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F2α synthase) is a drug target for hormonal and hormonal independent malignancies
Nonsteroidal inhibitors, repurposed NSAIDs and steroid based inhibitors have been claimed in multiple patent applications
Extensive steroidal-based inhibitors based on C3 and C17 substituted estrenes have been developed
AKR1C3 inhibitors have progressed to clinical trials for castration resistant prostate cancer and acute myeloid leukemia with mixed success
The majority of inhibitors require optimization for testing in animals and humans
This box summarizes key points contained in the article.
Acknowledgments
This author apologizes for the inability to describe many important studies due to space limitations.
Funding
TMP is supported by P30-ES013508 and P01-CA163227 from the National Institutes of Health.
Footnotes
Declaration of interest
Dr. Penning is founder of Penzymes, LLC, and is a consultant for Sage Pharmaceutics, Syrrix and Tokai Pharmaceuticals and has a Sponsored Research Agreement with Forendo.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1•.Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol. 2007;47:263–292. doi: 10.1146/annurev.pharmtox.47.120505.105337. This article provides a comprehensive account of the AKR protein superfamily. [DOI] [PubMed] [Google Scholar]
- 2.Penning TM, Drury JE. Human aldo-keto reductases: function, gene regulation and single nucleotide polymorphisms. Arch Biochem Biophys. 2007;464:241–250. doi: 10.1016/j.abb.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3•.Lin H-K, Jez JM, Schlegel BP, et al. Expression and characterization of recombinant type 2 3a-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3a/17b-HSD activity and cellular distribution. Mol Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. This study reports the cloning and expression of type 5 17β-HSD from a human prostate cDNA library. [DOI] [PubMed] [Google Scholar]
- 4.Penning TM, Burczynski ME, Jez JM, et al. Human 3a-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo keto reduc-tase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrns MC, Mindnich R, Duan L, et al. Overexpression of aldo-keto reductase 1C3 (AKR1C3) in LNCaP cells diverts androgen metabolism towards testosterone resulting in resistance to the 5α-reductase inhibitor finasteride. J Steroid Biochem Mol Biol. 2012;130:7–15. doi: 10.1016/j.jsbmb.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrns MC, Duan L, Lee S-H, et al. Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prosta-glandin metabolism that may explain its overexpression in breast cancer. J Steroid Biochem Mol Biol. 2010;118:177–187. doi: 10.1016/j.jsbmb.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penning TM, Steckelbroeck S, Bauman DR, et al. Aldo-keto reductase (AKR) 1C3: role in prostate disease and the development of specific inhibitors. Mol Cell Endocrinol. 2006;248:182–191. doi: 10.1016/j.mce.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Penning TM. Hydroxysteroid dehydrogenases and pre-receptor regulation of steroid hormone action. Hum Reprod Update. 2003;9:193–205. doi: 10.1093/humupd/dmg022. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki-Yamamoto T, Nishizawa M, Fukui M, et al. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett. 1999;462:335–340. doi: 10.1016/s0014-5793(99)01551-3. [DOI] [PubMed] [Google Scholar]
- 10.Matsuura K, Shirasishi H, Hara A, et al. Identification of a principal mRNA species for human 3a-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem (Tokyo) 1998;124:940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 11.Bauman DR, Rudnick S, Szewczuk L, et al. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential anti-neoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67:60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 12••.Desmond JC, Mountford JC, Drayson MT, et al. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003 Jan 15;63(2):505–512. This article develops the concept that AKR1C3 is a plausible target to account for the antineoplastic effects on NSAIDs independently of a mechanism involving inhibition of COX-1 and COX-2. [PubMed] [Google Scholar]
- 13••.Loriot Y, Fizazi K, Jones RJ, et al. Safety, tolerability and anti-tumor activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest New Drugs. 2014;32:995–1004. doi: 10.1007/s10637-014-0101-x. This report is the first description of a clinical trial with an AKR1C3 inhibitor to treat metastatic CRPC. ASP9521 was well tolerated but with limited efficacy in a small patient cohort. [DOI] [PubMed] [Google Scholar]
- 14••.Khanim F, Hayden RE, Birtwistle J, et al. Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukemia. PLoS ONE. 2009;4(12):e1847. doi: 10.1371/journal.pone.0008147. This article describes the beneficial effect of a PPRAγ receptor agonist and an AKR1C3 inhibitor in AML. It creates the precedent that AKR1C3 inhibitors can be effective in nonhormonal-dependent malignancies and that they may have superior efficacy when coadministered with a nuclear receptor ligand. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knudsen K, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15:4792–4798. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knudsen K, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Attard G, Reid AH, Yap TA, et al. Phase 1 clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;28:4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 18.Attard G, Reid AH, A’Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–37482. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Attard G, Reid AH, Auchus RJ, et al. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J Clin Endocrinol Metab. 2012;97:507–516. doi: 10.1210/jc.2011-2189. [DOI] [PubMed] [Google Scholar]
- 20••.Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. This study describes the preclinical development of MDV3100 which was later renamed as enzalutamide. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494–15 03. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Scher HI, Beer TM, Higano CS, et al. Prostate cancer foundation/department of defense prostate cancer clinical trials consortium. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;375:1437–1446. doi: 10.1016/S0140-6736(10)60172-9. This trial demonstrates the clinical benefit of MDV3100 in a phase I/II clinical trial in patients with CRPC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scher HI, Fizazi K, Saad F, et al. AFFIRM Investigators. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 24•.Liu C, Armstrong CM, Lou W, et al. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther. 2017;16:35–44. doi: 10.1158/1535-7163.MCT-16-0186. This study shows that indomethacin, an AKR1C3 inhibitor, can surmount drug resistance to abiraterone acetate in a mouse xenograft model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Liu C, Lou W, Zhu Y, et al. Intracrine androgens and AKR1C3 activation confer resistance to Enzalutamide in prostate cancer. Cancer Res. 2015;75:1413–1422. doi: 10.1158/0008-5472.CAN-14-3080. This study shows that indomethacin, an AKR1C3 inhibitor, can surmount drug resistance to enzalutamide in a mouse xenograft model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26••.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue N-(4-chlorobenzoyl)melatonin is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3a-HSD, type 5 17b-HSD and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75:484–493. doi: 10.1016/j.bcp.2007.09.008. This article is the first documentation that indomethacin can act as a potent and selective inhibitor of AKR1C3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelletier G. Expression of steroidogenic enzymes and sex-steroid receptors in human prostate. Best Pract Res Clin Endocrinol Metab. 2008;22:223–228. doi: 10.1016/j.beem.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Hofland J, van Weerden WM, Dits NFJ, et al. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70:1256–1264. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- 29••.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. This report describes the overexpression of AKR1C3 in patients with CRPC at the transcript and protein level and identifies AKR1C3 as one of the most upregulated steroidogenic enzymes in this disease. [DOI] [PubMed] [Google Scholar]
- 30.Mitsiades N, Sung CC, Schultz N, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012 Dec 1;72(23):6142–6152. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamid AR, Pfeiffer MJ, Verhaegh GW, et al. Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol Med. 2012;18:1449–1455. doi: 10.2119/molmed.2012.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fankhauser M, Tan Y, Macintyre G, et al. Canoncial androstenedione reduction is the predominat source of signaling androgens in hormone-refractory prostate cancer. Clin Cancer Res. 2014;20:5547–5557. doi: 10.1158/1078-0432.CCR-13-3483. [DOI] [PubMed] [Google Scholar]
- 33.Chang KH, Li R, Papari-Zareei M, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci USA. 2011;108:13728–13733. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohler JL, Titus MA, Bai S, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71:1486–1496. doi: 10.1158/0008-5472.CAN-10-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohler JL, Titus MA, Wilson EM. Potential prostate cancer drug target: bioactivation of androstanediol by conversion to dihydrotestosterone. Clin Cancer Res. 2011;17:5844–5849. doi: 10.1158/1078-0432.CCR-11-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab. 2004;15:432–438. doi: 10.1016/j.tem.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 37.Shibuya R, Suzuki T, Miki Y, et al. Intratumoral concentration of sex steroids and expression of sex steroid-producing enzymes in ductal carcinoma in situ of human breast. Endocr Relat Cancer. 2008 Mar;15(1):113–124. doi: 10.1677/ERC-07-0092. [DOI] [PubMed] [Google Scholar]
- 38.Oduwole OO, Li Y, Isomaa VV, et al. 17beta-hydroxysteroid dehydrogenase type 1 is an independent prognostic marker in breast cancer. Cancer Res. 2004;64:7604–7609. doi: 10.1158/0008-5472.CAN-04-0446. [DOI] [PubMed] [Google Scholar]
- 39.Jansson AK, Gunnarsson C, Cohen M, et al. 17beta-hydroxysteroid dehydrogenase 14 affects estradiol levels in breast cancer cells and is a prognostic marker in estrogen receptor-positive breast cancer. Cancer Res. 2006;66:11471–11477. doi: 10.1158/0008-5472.CAN-06-1448. [DOI] [PubMed] [Google Scholar]
- 40.Steckelbroeck S, Jin Y, Gopishetty S, Penning TM, et al. Human cytosolic 3a-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3b-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2003;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 41•.Penning TM, Bauman DR, Jin Y, et al. Identification of the molecular switch that regulates access of 5a-DHT to the androgen receptor. Mol Cell Endocrinol. 2007;265–266:77–82. doi: 10.1016/j.mce.2006.12.007. An important example of how pairs of hydroxysteroid dehydrogenases regulate ligand access to nuclear receptors, in this case the androgen receptor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bauman DR, Steckelbroeck S, Williams MV, et al. Penning, TM Identification of the major oxidative 3a-hydroxysteroid dehydrogenase in human prostate that converts 5a-andostane-3a,17b-diol to 5a-dihydrotestosterone. A potential therapeutic target for androgen dependent disease. Mol Endocrinol. 2006;20:444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- 43.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 44.Sinreih M, Sosič I, Beranič N, et al. N-Benzoyl anthranilic acid derivatives as selective inhibitors of aldo-keto reductase AKR1C3. Biorg Med Chem Lett. 2012;22:5948–5951. doi: 10.1016/j.bmcl.2012.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gazvoda M, Beranič N, Turk S, et al. 2,3-Diarylpropenoic acids as selective non-steroidal inhibitors of type-5 17β-hydroxysteroid dehydrogenase (AKR1C3) Eur J Med Chem. 2013;62:89–97. doi: 10.1016/j.ejmech.2012.12.045. [DOI] [PubMed] [Google Scholar]
- 46.Skarydova L, Hofman J, Chlebek J, et al. Isoquinoline alkaloids as a novel type of AKR1C3 inhibitors. J Steroid Biochem Mol Biol. 2014;143:2508. doi: 10.1016/j.jsbmb.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 47.Skarydová L, Zivná L, Xiong G, et al. AKR1C3 as a potential target for the inhibitory effect of dietary flavonoids. Chem Biol Inter. 2009;178:138–144. doi: 10.1016/j.cbi.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 48.Heinrich DM, Flanagan JU, Jamieson SM, et al. Synthesis and structure-activity relationships for 1-(4-(piperidin-1-ylsulfonyl)phenyl) pyrrolidin-2-ones as novel non-carboxylate inhibitors of the aldo-keto reductase enzyme AKR1C3. Eur J Med Chem. 2013;62:738–744. doi: 10.1016/j.ejmech.2013.01.047. [DOI] [PubMed] [Google Scholar]
- 49•.Kikuchi A, Furutani T, Azami H, et al. In vitro and in vivo characterization of ASP9521: a novel selective, orally bioavailable inhibitor of 17b-hydroxysteroid dehydrogenase type 5 (17b-HSD5; AKR1C3) Invest New, Drugs. 2014;32:860–870. doi: 10.1007/s10637-014-0130-5. This article describes the preclinical development of ASP9521 reported by Astellas Pharmaceuticals. [DOI] [PubMed] [Google Scholar]
- 50.Adeniji AO, Twenter BM, Byrns MC, et al. Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Bioorg Med Chem Lett. 2011;21:1464–1468. doi: 10.1016/j.bmcl.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adeniji AO, Twenter BM, Byrns MC, et al. Development of potent and selective inhibitors of aldo-keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenylaminobenzoates and their structure-activity relationships. J Med Chem. 2012;55:2311–2323. doi: 10.1021/jm201547v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adeniji A, Uddin MJ, Zang T, et al. Discovery of (R)-2-(6-Methoxynaphthalen-2-yl)butanoic acid as a potent and selective Aldo-keto reductase 1C3 inhibitor. J Med Chem. 2016 Aug 25;59(16):7431–7444. doi: 10.1021/acs.jmedchem.6b00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liedtke AJ, Kim K, Stec DF, et al. Straightforward protocol for the efficient synthesis of varied N1-acylated (aza)indole 2-/3-alkanoic acids and esters: optimization and scaleup. Tetrahedron. 2012;168:10049–10058. doi: 10.1016/j.tet.2012.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liedtke AJ, Adeniji AO, Chen M, et al. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (type 5 17b-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem. 2013 Mar 28;56(6):2429–2446. doi: 10.1021/jm3017656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamamoto H. 1-Acyl-indoles II. A new synthesis of 1-(ionchlorobenzoyl)-5-methoxy-3-indolylacetic acid and its polymorphism. Chem Pharm Bull. 1968;16:17–19. doi: 10.1248/cpb.16.17. [DOI] [PubMed] [Google Scholar]
- 56.Conn RSE, Douglas AW, Karday S, et al. An unusual Fischer indole synthesis with 4-ketoacids: an indole incorprorating the terminal hydrazine nitrogen. J Org Chem. 1990;55:2908–2913. [Google Scholar]
- 57.Allen GR., Jr Selectivity in the Fischer indolization of phenylhydrazones derived from 3-ketocyclohexanecrboxylic acid. J Heterocycl Chem. 1970;7:239–241. [Google Scholar]
- 58••.Yepuru M, Wu Z, Kyulkarni A, et al. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res. 2013;19:5613–5625. doi: 10.1158/1078-0432.CCR-13-1151. This report describes the discovery of GTx560 the first competitive inhibitor of AKR1C3 and blocker of the AR coactivator function of AKR1C3. It is the first discovery of a drug in this class. [DOI] [PubMed] [Google Scholar]
- 59••.Chen M, Adeniji AO, Twenter BM, et al. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg Med Chem Lett. 2012;22:3492–3497. doi: 10.1016/j.bmcl.2012.03.085. This report describes the discovery of BMT4-158 the first dual function competitive inhibitor of AKR1C3 and AR antagonist. It is the first discovery of a drug in this class. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zang T, Verma K, Chen M, et al. Screening baccharin analogs as selective inhibitors against type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) Chem Biol Interact. 2015 Jun 05;234:339–348. doi: 10.1016/j.cbi.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Endo S, Matsunaga T, Kanamori A, et al. Selective inhibition of human type-5 17β-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J Nat Prod. 2012;27:716–721. doi: 10.1021/np201002x. [DOI] [PubMed] [Google Scholar]
- 62.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17β-hydroxyster-oid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol. 2011;125:95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andersson S, Geissler WM, Patel S, et al. The molecular biology of androgenic 17b-hydroxysteroid dehydrogenases. J Steroid Biochem Mol Biol. 1995;53:37–39. doi: 10.1016/0960-0760(95)00039-3. [DOI] [PubMed] [Google Scholar]
- 64.Taplin ME, Montgomery B, Logothetis CJ, et al. Intense androgen-deprivation therapy with abiraterone acetate plus leuprolide acetate in patients with localized high-risk prostate cancer: results of a randomized phase II neoadjuvant study. J Clin Oncol. 2014;32:3705–3715. doi: 10.1200/JCO.2013.53.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tamae D, Mostaghel E, Montgomery B, et al. The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chem Biol Inter. 2015;234:332–338. doi: 10.1016/j.cbi.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang K-H, Li R, Kuri B, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–1084. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pfeiffer MJ, Smit FP, Sedelaar JP, et al. Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol Med. 2011;17(7–8):657–664. doi: 10.2119/molmed.2010.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson IM, Chi C, Ankerst DP, et al. Prediction of prostate cancer for patients receiving finasteride: results from the prostate cancer prevention trial. J Clin Oncol. 2007;25:3076–3081. doi: 10.1200/JCO.2006.07.6836. [DOI] [PubMed] [Google Scholar]
- 69.Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 70.Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 71.Taplin ME, Bubley GJ, Ko YJ, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–2515. [PubMed] [Google Scholar]
- 72.Yu Z, Chen S, Sowalsky AG, et al. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res. 2014;20:1590–1600. doi: 10.1158/1078-0432.CCR-13-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sprenger CCT, Plymate SR. The link between androgen receptor splice variants and castration-resistant prostate cancer. Hormones & Cancer. 2014;5:207–2014. doi: 10.1007/s12672-014-0177-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cao B, Qi Y, Zhang G, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5:1646–1656. doi: 10.18632/oncotarget.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]