

Abstract
The Asc1 protein of Saccharomyces cerevisiae is a scaffold protein at the head region of ribosomal 40S that links mRNA translation to cellular signaling. In this study, proteins that colocalize with Asc1p were identified with proximity-dependent Biotin IDentification (BioID), an in vivo labeling technique described here for the first time for yeast. Biotinylated Asc1p-birA*-proximal proteins were identified and quantitatively verified against controls applying SILAC and mass spectrometry. The mRNA-binding proteins Sro9p and Gis2p appeared together with Scp160p, each providing ribosomes with nuclear transcripts. The cap-binding protein eIF4E (Cdc33p) and the eIF3/a-subunit (Rpg1p) were identified reflecting the encounter of proteins involved in the initiation of mRNA translation at the head region of ribosomal 40S. Unexpectedly, a protein involved in ribosome preservation (the clamping factor Stm1p), the deubiquitylation complex Ubp3p-Bre5p, the RNA polymerase II degradation factor 1 (Def1p), and transcription factors (Spt5p, Mbf1p) colocalize with Asc1p in exponentially growing cells. For Asc1R38D, K40Ep, a variant considered to be deficient in binding to ribosomes, BioID revealed its predominant ribosome localization. Glucose depletion replaced most of the Asc1p colocalizing proteins for additional ribosomal proteins, suggesting a ribosome aggregation process during early nutrient limitation, possibly concomitant with ribosomal subunit clamping. Overall, the characterization of the Asc1p microenvironment with BioID confirmed and substantiated our recent findings that the β-propeller broadly contributes to signal transduction influencing phosphorylation of colocalizing proteins (e.g. of Bre5p), and by that might affect nuclear gene transcription and the fate of ribosomes.
As platforms or adaptors for protein-protein interactions, scaffold proteins dynamically organize protein proximities and thereby exert a major impact on cellular signaling (1, 2). The Gβ-like Asc1 protein from the unicellular budding yeast S. cerevisiae belongs to the WD40 family of scaffold proteins and folds into a seven-bladed β-propeller with a large surface for interactions (3–5). Asc1p is highly conserved throughout the eukaryotic kingdom and shares a high degree of structural similarity with its orthologs, e.g. the human receptor for activated C kinase 1 (RACK11; 4, 6–9). Asc1p is a core constituent of most eukaryotic ribosomes and crystallization of the S. cerevisiae 80S ribosome located Asc1p to the exposed head region of the 40S ribosome in direct proximity to the area of mRNA exit (10–12). The top surface of Asc1p is oriented to the ribosome interface, where it physically interacts with the ribosomal proteins Rps3p, Rps16p, and Rps17p (7, 10, 13). Furthermore, the ribosome facing side of Asc1p contacts helices 39 and 40 of the 18S rRNA with an area of positively charged amino acids (4, 7, 10). The bottom side of Asc1p is not involved in ribosome association and exposed for further protein-protein interactions (10, 13). Despite its ribosomal localization yeast Asc1p is not essential for viability and thus dispensable for general translation (3). Still, Asc1p affects the translational efficiency of transcripts in various ways: Asc1p physically interacts with the mRNA-binding protein Scp160p that locates specific mRNAs to polysomes and is required for its efficient ribosome association (4, 14, 15). Asc1p and Scp160p are both part of the SESA multiprotein complex (Smy2p, Eap1p, Scp160p, and Asc1p) which controls the translation of POM34 mRNA required for spindle pole body duplication (16). Furthermore, Asc1p influences the translation of specific mRNAs through their 5′UTRs (17). Asc1p/RACK1 was described as both, physical interaction partner and mediator for protein phosphorylation of various translation initiation factors (18–24), and Asc1p-depletion accordingly results in a decreased 40S binding affinity of eIF3 (21). Besides translation, Asc1p/RACK1 has a major impact on signal transduction and interacts with multiple players of signal transduction pathways including components of the cAMP/PKA and different MAPK pathways in S. cerevisiae (25, 26) and PKC, Src, and c-Jun kinases in mammals (18, 22, 27, 28). A functional requirement for ribosome association of Asc1p in MAPK signaling, however, remains elusive. An artificial asc1R38D, K40E mutant, hereafter referred to as asc1DE, has been constructed to diminish the binding affinity of Asc1DEp to the ribosome and to study possible ribosome-independent functions (4). The exchange of the two residues within the rRNA contact region to negatively charged amino acids results in repelling forces between the protein and the rRNA and to a substantial shift of the Asc1DE protein into nonribosomal fractions during sucrose density ultracentrifugation (4). However, only a minor impact on Asc1p-dependent phenotypes was observed (4). Just recently, Thompson et al. (29) revealed that the mutant is still ribosome-associated in vivo. The degree of ribosome binding of the Asc1DE protein and its exact position at the ribosome, however, remained uncertain.
Proximity-dependent Biotin IDentification, short BioID, was first described by Roux, Burke and coworkers and was developed as an in vivo screen for interacting and proximal proteins of a bait in mammalian cells (30). The emerging in vivo labeling technique uses the E. coli-derived promiscuous biotin protein ligase BirAR118Gp (hereafter referred to as BirA*p), which is fused to a bait protein, and covalently biotinylates interacting and proximal proteins of the bait if biotin is available. Thereafter, cells are lysed under denaturing conditions, and the modified proteins are purified by biotin affinity capture and finally identified by liquid chromatography mass spectrometry (LC-MS). The method was meanwhile adapted for other organisms including Trypanosoma brucei, Toxoplasma gondii, or Dictyostelium amoebae (31–33). In this study, we used BioID in combination with stable isotope labeling with amino acids in cell culture (SILAC) to quantitatively monitor the proteinaceous Asc1p-neighborhood in exponentially growing S. cerevisiae cells, but also to record alterations in response to glucose deprivation and heat stress. Furthermore, we analyzed the molecular microenvironment of the “ribosome-binding deficient” Asc1DE protein, which indicates a ribosome-associated population of Asc1DEp in vivo and suggests a distorted position of the variant at the ribosome. In direct proximity to wt-Asc1p, we identified important regulators of translation, including mRNA-binding proteins and initiation factors, as well as transcription-related proteins. Additionally, we observed major alterations in response to glucose withdrawal. In contrast, mild heat stress caused only minor effects on the Asc1p-neighborhood. Altogether, Asc1p seems to connect signal transduction not only with the translational machinery, but also with fundamental nuclear processes.
EXPERIMENTAL PROCEDURES
S. cerevisiae Strains and Their Construction
The S. cerevisiae strains used in this work are of the Σ1278b, S288c (BY strains), and W303 background and are listed in Table I. The construction of RH3494 was performed as described in Schmitt et al. (23) using RH3263 as background strain. Yeast strains were cultivated at 30 °C in yeast extract peptone dextrose medium (YEPD; 1% yeast extract, 2% peptone, 2% glucose) or yeast nitrogen base minimal medium (YNB; 0.15% YNB without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 2% glucose) with the respective supplements or in Synthetic Complete medium (SC, 0.15% YNB without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 2% glucose, 0.2 mm myo-inositol, 2 g amino acid/nitrogenous base mix). 2% agar was supplied to obtain solid medium. l-arginine (20 mg/l), l-lysine HCl (30 mg/l), l-tryptophan (20 mg/l) and uracil (20 mg/l) were supplemented if required. For differential protein labeling according to the SILAC approach 50 mg/l differentially labeled l-arginine and l-lysine were added to the growth medium (13C6-l-arginine HCl (#201203902), 13C6 15N4-l-arginine HCl (#201603902), 4,4,5,5-D4-l-lysine HCl (#211103912), 13C6-l-lysine HCl (#211203902); Silantes GmbH, München, Germany).
Table I. S. cerevisiae strains used in this work.
| Strain | Genotype | Reference |
|---|---|---|
| RH2817 | MATα, ura3-52, trp1::hisG | 40 |
| RH3263 | MATα, ura3-52, trp1::hisG, leu2::hisG, Δasc1::LEU2 | 40 |
| RH3493 | MATα, ura3-52, trp1::hisG, Δarg4::loxP, Δlys1::loxP | 23 |
| RH3494 | MATα, ura3-52, trp1::hisG, leu2::hisG, Δasc1::LEU2, Δarg4::loxP, Δlys1::loxP | This work |
| BY4741 | MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0 | Euroscarf |
| Y03444 | MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, Δsro9::kanMX | Euroscarf |
| Y06556 | MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, Δasc1::kanMX | Euroscarf |
| RH3694 RH3695 | MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, Δsro9::kanMX, Δasc1::URA3 | This work |
| RH3686 | MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, SRO9::GFP::HIS3MX | Invitrogen, 75 |
| RH3687 | MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, UBP3::GFP::HIS3MX | Invitrogen, 75 |
| RH3696 RH3697 | MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, UBP3::GFP::HIS3MX, Δasc1::URA3 | This work |
| W303-1a | W303, MATa, ura3, leu2-3 112, his3-11 15, trp1-1, ade2-1, can1-100 | 34 |
| JSY1194 | W303, MATa, ura3, leu2-3 112, his3-11 15, trp1-1, ade2-1, can1-100, eGFP-DEF1 | 34 |
| RH3698 | W303, MATa, ura3, leu2-3 112, his3-11 15, trp1-1, ade2-1, can1-100, Δasc1::URA3 | This work |
| JSY1198 | W303, MATa, ura3, leu2-3 112, his3-11 15, trp1-1, ade2-1, can1-100, 9xMyc-2xTEV-6xHis-DEF1 | 34 |
| RH3699 | W303, MATa, ura3, leu2-3 112, his3-11 15, trp1-1, ade2-1, can1-100, 9xMyc-2xTEV-6xHis-DEF1, Δasc1::URA3 | This work |
Plasmid Constructions
Plasmids used in this study are listed in Table II. Their construction is described in detail in Supplementary Experimental Procedures. A plasmid encoding an ASC1-birA* fusion gene was generated by linearizing plasmid pME2624 by PCR with a primer pair resulting in the removal of the ASC1 stop codon and the addition of a large overhang containing the linker sequence and a sequence complementary to the birA* gene. The birA* allele containing the point mutation R118G was amplified from plasmid pRS313 and supplied with a sequence complementary to the plasmid backbone by PCR. The linearized plasmid backbone and the birA* fragment were fused by homologous recombination using the In-Fusion® HD Cloning Kit (#639650, Clontech, Mountain View, CA). For the generation of a plasmid expressing the mere birA*, plasmid pME2624 was linearized by PCR with primers resulting in the removal of the ASC1 ORF and the addition of a 20 bp overhang complementary to the birA* gene. The birA* allele was amplified by PCR and fused with the linearized plasmid backbone by homologous recombination. A plasmid encoding an asc1DE-birA* fusion was generated by site directed mutagenesis using pME4478 as template. To insert the R38D and K40E substitutions within the ASC1-birA* allele, the plasmid was fully amplified by PCR with a complementary primer pair carrying the two mutations resulting in the asc1DE-birA* vector. The template DNA was removed by DpnI treatment. Plasmid pME4481 with ASC1 under control of its native promoter was constructed as described by Schmitt et al. (23) for the same plasmid with a URA3 marker for selection.
Table II. Plasmids used in this work.
| Plasmid | Description | Reference |
|---|---|---|
| pUG72 | AmpR, pUCori, loxP::URA3::loxP | 76 |
| pME2787 | MET25Prom, CYC1Term, URA3, 2 μm | 77 |
| pME2624 | pME2787 with ASC1 | Our collection |
| pRS313 | PGK1Prom, CYC1Term, HIS3, CEN/ARS, birAR118G (based on van Werven and Timmers (78)) | provided by H. D. Schmitt |
| pME4478 | MET25Prom, CYC1Term, URA3, 2 μm, ASC1-birA* | This work |
| pME4479 | MET25Prom, CYC1Term, URA3, 2 μm, asc1DE-birA* | This work |
| pME4480 | MET25Prom, CYC1Term, URA3, 2 μm, birA* | This work |
| pME2781 | MET25Prom, CYC1Term, TRP1, CEN/ARS | 77 |
| pME4481 | native promoter, CYC1Term, TRP1, CEN/ARS, ASC1 | This work |
| pPS1523 | native promoter, URA3, CEN, GFP-YRB2 | 79 |
Western Blot Analysis
50-ml yeast cultures were grown overnight to midlog phase (OD600 = 0.8) unless stated otherwise. Cells were collected by centrifugation at 3000 rpm for 3 min at 4 °C and washed in ice-cold breaking buffer (100 mm Tris-HCl pH 7.5, 200 mm NaCl, 5 mm EDTA, 20% glycerol) or 1x Strep buffer (10 mm HEPES pH 7.9, 10 mm KCl, 1.5 mm MgCl2) if protein purification with Strep-Tactin® columns was intended. Cells were lysed in the presence of glass beads in 500 μl breaking buffer containing 1 cOmpleteTM protease inhibitor mixture tablet (#11836145001, Roche Diagnostics, Mannheim, Germany) per 50 ml and 0.5% β-mercaptoethanol, or in 500 μl 1× Strep buffer with 1 cOmpleteTM protease inhibitor mixture tablet per 50 ml, 0.5 mm DTT and 0.5 mm PMSF. For denaturing cell lysis, the sample was incubated with 4% SDS at 65 °C for 5 min. After centrifugation at 13,000 rpm for 15 min at room temperature the supernatant was collected as protein extract. Protein concentrations were determined with the BCA reagent (#23224 and #23228, Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. 3× loading dye (0.25 m Tris-HCl pH 6.8, 30% glycerol, 15% β-mercaptoethanol, 7% SDS, 0.3% bromphenol blue) was added to the protein extracts and the samples were heated at 65 °C for 10 min. The proteins were separated by SDS-PAGE and blotted onto nitrocellulose membranes (Amersham BiosciencesTM Protran® Western blotting membrane, #GE10600002, Sigma-Aldrich, München, Germany). Total protein levels were visualized by staining the membrane with Ponceau Red (0.2% PonceauS, 3% trichloroacetic acid) and photographing with the FUSION-SL-4 (PEQLAB Biotechnology GmbH, Erlangen, Germany). After blocking, the membranes were incubated with polyclonal rabbit anti-Asc1p (generated from affinity purified Asc1p by Davids Biotechnologie GmbH, Regensburg, Germany), polyclonal rabbit anti-Rps3p, monoclonal mouse anti-GFP (B-2, sc-9996, Santa Cruz Biotechnology, Heidelberg, Germany) or polyclonal rabbit anti-Def1 (34) followed by incubation with peroxidase-coupled goat anti-rabbit (#G21234, MoBiTec, Göttingen, Germany) or goat anti-mouse (#115-035-003, Dianova, Hamburg, Germany) as secondary antibody. PierceTM High Sensitivity Streptavidin-HRP (#21130, Thermo Fisher Scientific) was directly added to the membrane after blocking in 1% BSA for 1 h. Chemiluminescent signals were detected with the FUSION-SL-4.
Proximity-dependent Biotin Identification
The BioID approach was modified from Roux et al. (30). For the comparison of three strains or conditions in one BioID experiment, cells were cultivated individually in 200 ml YNB medium with the required amino acids including 50 mg/l of stable isotope labeled amino acids, and with 10 μm d-biotin (#47868, Sigma-Aldrich) until an OD600 of 0.8 was reached. When comparing growth conditions all three cultures were cultivated equally until an OD600 of 0.6 or 0.7 was reached. At this point heat stress was triggered by shifting the cells to 37 °C or starvation was induced by washing and shifting the cells to YNB medium with or without glucose. Biotin was added 10 or 15 min after stress induction and the cells were cultivated for 1 h. Before gathering cells, a 20 ml aliquot was taken from each culture and used for visualization of biotinylated proteins with Streptavidin-HRP. Cells were harvested by centrifugation and resuspended in 15 ml Strep-buffer each. The three cell suspensions were combined in a 1:1:1 ratio according to the optical densities of the cultures. After denaturing cell lysis in 1× Strep-buffer the protein crude extract was split into two parts: About 60 μg were used as proteome standard and were directly separated by SDS-PAGE. The remaining crude extract was used for biotin affinity capture with Strep-Tactin® Sepharose® gravity flow columns with 1 ml column bed volume (#2–1202-001, IBA GmbH, Göttingen, Germany). The crude extract was applied to the column at room temperature, the column bed was washed with 50 ml 1x washing buffer (#2–1003-100, IBA GmbH) containing 0.4% SDS and the biotinylated proteins were eluted with 1× washing buffer containing 10 mm biotin. The proteins of the eluate were precipitated according to Wessel and Flügge (35) by chloroform-methanol extraction and separated by SDS-PAGE. This gel lane and half of the proteome gel lane were each subjected to tryptic digestion followed by LC-MS analysis. Strep-Tactin® spin columns (#2–1850-010, IBA GmbH) were used for the purification of biotinylated peptides according to the manufacturer's instructions. Peptides were eluted in three steps with each time 150 μl of 1× washing buffer containing 10 mm biotin.
Liquid Chromatography-Mass Spectrometry Analyses
Protein digestion with trypsin was performed according to Shevchenko et al. (36). The resulting peptide pellet was resolved in 20 μl sample buffer (2–5% acetonitrile, 0.1% formic acid) for further purification. Desalting of the peptide solution with the C18 stop and go extraction (stage) tips was performed according to Rappsilber et al. (37, 38). The resulting peptide solution was dried completely in the SpeedVac concentrator and resolved in the sample buffer for LC-MS analyses.
LC-MS analysis for protein identification and SILAC-based protein quantification was performed with an Orbitrap Velos ProTM Hybrid Ion Trap-Orbitrap mass spectrometer. 1–5 μl of peptide solutions were loaded and washed on an Acclaim® PepMAP 100 precolumn (#164564, 100 μm x 2 cm, C18, 3 μm, 100 Å, Thermo Fisher Scientific) with 100% loading solvent A (98% H2O, 2% acetonitrile, 0.07% trifluoroacetic acid) at a flow rate of 25 μl/min for 6 min. Peptides were separated by reverse phase chromatography on an Acclaim® PepMAP RSLC column (75 μm × 25 cm (#164536) or 50 cm (#164540), C18, 3 μm, 100 Å, Thermo Fisher Scientific) with a gradient from 98% solvent A (H2O, 0.1% formic acid) and 2% solvent B (80% acetonitrile, 20% H2O, 0.1% formic acid) to 42% solvent B for 95 min and to 65% solvent B for the following 26 min at a flow rate of 300 nl/min. Peptides eluting from the chromatographic column were on-line ionized by nanoelectrospray at 2.4 kV with the Nanospray Flex Ion Source (Thermo Fisher Scientific). Full scans of the ionized peptides were recorded within the Orbitrap FT analyzer of the mass spectrometer within a mass range of 300–1850 m/z at a resolution of 60,000. Data-dependent collision-induced dissociation (CID) fragmentation of the top-ten most intense peptides was performed with the LTQ Velos Pro linear ion trap. For data acquisition and programming the XCalibur 2.2 software (Thermo Fisher Scientific) was used.
Peptide samples from GFP-trap experiments were analyzed with a Q exactive HF mass spectrometer. Samples were loaded with 0.07% TFA on an Acclaim® PepMap 100 precolumn (100 μm × 2 cm, C18, 3 μm, 100 Å, Thermo Scientific) at a flow rate of 20 μl/min for 3 min. Analytical peptide separation by reverse phase chromatography was performed at a flow rate of 300 nl/min on an Acclaim® PepMap RSLC column (75 μm x 50 cm, C18, 3 μm, 100 Å, Thermo Scientific). A gradient from 98% solvent A (0.1% formic acid) and 2% solvent B (80% acetonitrile, 0.1% formic acid) to 55% B was applied within 30 min and was followed by 90% B for 4 min (Optima® LC/MS solvents and acids were purchased from Fisher Chemical). Chromatographically eluting peptides were on-line ionized by nano-electrospray (nESI) using the Nanospray Flex Ion Source (Thermo Scientific) at 1.5 kV (liquid junction) and continuously transferred into the mass spectrometer. Full scans within the mass range of 300–1800 m/z were taken from the Orbitrap-FT analyzer at a resolution of 30,000 with parallel data-dependent top 10 MS2-fragmentation (HCD). The resolution was set to 60,000 for tSIM scans and to 15,000 for dd-MS2 scans. The maximum ion time was 50 ms for tSIM scans (AGC target 1e6) and 1000 ms for dd-MS2 (AGC target 1e5). The loop count equaled the number of m/z values on the inclusion list. LC-MS method programming and data acquisition was performed with the software XCalibur 4.0 (Thermo Scientific).
Protein identification and SILAC-based quantification was performed with the MaxQuant 1.5.1.0 software (39). A UniProt-derived S. cerevisiae-specific database (http://www.uniprot.org, Proteome ID UP000002311, 6721 entries, download 2014) was used for database search with the Andromeda algorithm (MaxQuant version 1.5.1.0) and with the program's default parameters. As the digestion mode trypsin/P was used, maximum missed cleavage sites were set to three, carbamidomethylation of cysteins was considered as fixed modification, acetylation of the N-terminus, oxidation of methionines, and biotinylation of lysines were set as variable modifications. For SILAC-based experiments Arg6 and Lys4 or Arg10 and Lys8 were set as medium or heavy isotopic peptide labels, respectively. Match among runs, FTMS requantification, and FTMS recalibration were enabled. The mass tolerance was 4.5 ppm for precursor ions and 0.5 Da for fragment ions. The decoy mode was revert with a false discovery rate of 0.01. Subsequent data processing and statistical analysis were performed with the Perseus 1.5.1.0 software (39). For the identification of proteins and their biotinylated lysine residues also the Proteome DiscovererTM 1.4.0.288 software was used. Database searches were performed with the Mascot (versions 2.4.1 and 2.5.1) and SequestHT search algorithms (the latter implemented in Proteome DiscovererTM version 1.4.0.288) against a S. cerevisiae-specific protein database (SGD, 6110 entries including common contaminants, S288C_ORF_database release version 2011, Stanford University). The digestion mode was set to trypsin and the maximum of missed cleavage sites to three. Carbamidomethylation of cysteins was set as fixed modification, oxidation of methionines and biotinylation of lysines were set as variable modifications. The mass tolerance was 10 ppm for precursor ions and 0.6 Da for fragment ions. The decoy mode was reverse with a false discovery rate of 0.01. For GFP-trap experiments raw data from the Q Exactive HF were analyzed using MaxQuant and Perseus with phosphorylation of serine, threonine and tyrosine as variable modification, FTMS recalibration and label-free quantification. Mass tolerances of precursors and fragment ions were 4.5 ppm and 20 ppm, respectively. Raw data of tSIM scans were analyzed without calculation of LFQ-intensities.
Experimental Design and Statistical Rationale
The SILAC-BioID experiments at exponential growth, heat stress, and for the Asc1DE protein variant were performed in three biological replicates, the SILAC-BioID analysis at glucose starvation in five biological replicates. For each replicate three strains were cultivated individually and were combined in a 1:1:1 ratio according to the optical densities of the cultures. From this point on one pooled sample was handled for each replicate, thereby minimizing deviations among the three cultures of the experiment. In the SILAC-BioID experiments at exponential growth two strains were used as negative controls to differentiate between Asc1p-specific and Asc1p-unspecific biotinylation/enrichment: A wt-ASC1 strain served to reveal unspecific enrichment and naturally biotinylated proteins, and a strain expressing the mere BirA*p not fused to Asc1p to account for biotinylation independent of Asc1p. The other SILAC-BioID analyses (glucose starvation, heat stress, Asc1DEp) used the Asc1-birA* strain as a reference to account for stress- or mutation-dependent changes and the birA* strain as negative control at the respective growth condition. The protein extract of the sample without biotin affinity capture was analyzed by LC-MS to account for variations in protein expression of the different yeast strains.
In the SILAC-BioID experiments at exponential growth a significant enrichment was defined by log2 SILAC enrichment ratios ≥ 0.26 (≙ 20%) against the birA* control strain in at least four out of six biological replicates (three additional ASC1-birA*/birA* SILAC ratios were obtained from the BioID analysis with the ribosome-binding compromised Asc1DEp variant as bait), and by log2 SILAC enrichment ratios ≥ 1 (≙ 100%) against the wt-ASC1 control strain in at least two out of three biological replicates. Rpg1p was further considered as Asc1p-neighbor, because it is a subunit of eIF3, which is known to interact with Asc1p although the latter threshold was not reached. Statistical significance within the three or six repetitions was assessed with a two-sample t test (or a one sample t test in case no proteome value was available) with a p value of 0.05. t test significance is indicated in supplemental Table S1.
In the other SILAC-BioID experiments log2 SILAC ratios ≥ 0.26 or ≤ −0.26 against the ASC1-birA* reference strain were considered as stress-specific or DE-specific changes in the Asc1p-neighborhood. A log2 SILAC ratio ≥ 0.26 against the birA* negative control strain was required as threshold in either case in at least three out of five biological replicates (glucose starvation) or two out of three replicates (heat stress or Asc1DEp). Statistical significance was again assessed with a two-sample t test (or a one sample t test in case no proteome value was available) with a p value of 0.05. t test significance is indicated in supplemental Tables S3, S4, and S6.
The rationale for the value 0.26 as threshold for log2 SILAC ratios derived from the known interacting partner Eap1p, one of four subunits of the SESA complex (Smy2p, Eap1p, Scp160p, Asc1p; 16). From several known Asc1p interacting proteins found in our BioID study including all SESA subunits, Eap1p displayed the lowest enrichment of 20% and with that defined the minimum enrichment factor required for a candidate being considered as Asc1p-proximal.
Sucrose Density Gradient Centrifugation
For sucrose density gradient centrifugation S. cerevisiae cells were cultivated overnight in 200 ml YNB medium until midlog phase. Cells were incubated with 100 μg/ml cycloheximide for 15 min on ice and collected by centrifugation at 3,000 rpm for 3 min at 4 °C. The cell sediment was washed in washing buffer (20 mm HEPES-KOH, pH 7.5, 10 mm KCl, 2.5 mm MgCl2, 1 mm EGTA) and lysed in lysis buffer (20 mm HEPES-KOH, pH 7.5, 10 mm KCl, 2.5 mm MgCl2, 1 mm EGTA, 1 mm DTT, 100 μg/ml cycloheximide, 1 cOmpleteTM EDTA-free protease inhibitor mixture tablet (#4693132001, Roche Diagnostics) per 50 ml). Mechanical cell lysis in the presence of glass beads was performed with the Fast-Prep-24 (MP Biomedicals, Illkirch, France). 15 OD260 units were loaded onto a 7–47% sucrose gradient, generated with the Gradient Master 108 (BioComp Instruments, Fredericton, Canada). The sucrose gradients were centrifuged at 40,000 rpm for 2 h 50 min at 4 °C in a TH-641 rotor in a Sorvall WX80 ultracentrifuge (Thermo Fisher Scientific) and fractions were collected with the Foxy Jr. Fraction Collector (Optical Unit Type 11, Absorbance detector UA-6, Teledyne Isco, Lincoln, NE). The absorbance was measured at 254 nm for polysome profiling. Proteins from the collected fractions were precipitated with 10% trichloroacetic acid for subsequent Western blot analyses.
BioID Experiments with Strep-Tactin® Spin Columns
For BioID experiments with subsequent Western blot analysis the Strep-Tactin® spin columns (#2–1850-010, IBA GmbH) were used according to the manufacturer's instructions. Cells were cultivated in 450 ml YNB medium as described in the “Proximity-dependent biotin identification” section. Cell lysis was performed under denaturing conditions. The columns were washed four times with 500 μl 1x washing buffer (#2–1003-100, IBA GmbH) containing 4% SDS. Elution of proteins was performed using 50 μl 1× washing buffer comprising 2 mm biotin.
GFP-trap Experiments
The enrichment of GFP-tagged proteins was performed as described before (23). Strains were cultivated to mid-log phase in 150 ml SC medium.
Drop Dilution Assay
Yeast cells were grown to midlog phase, diluted in YNB or SC medium with the respective amino acids to an OD600 of 0.1, and 10-fold dilution series were prepared. 20 μl of each dilution were dropped on YNB or SC agar plates and 10 μl on YEPD plates. For the analysis of cell wall integrity YNB medium was supplemented with 125 μg/ml congo red. Respiratory capacity was investigated on YNB medium containing 2% glycerol as sole nonfermentable carbon source. Osmotic stress was induced on YNB medium containing 75 mm NaCl or YEPD medium containing 1 m NaCl. The translational fitness was analyzed on YNB medium with the protein misfolding agent canavanine (600 ng/ml) or the translation inhibitor cycloheximide (0.15 μg/ml). After 3 to 5 days of incubation at 30 °C the plates were photographed with the Gel iX20 Imager (Intas Science Imaging Instruments, Göttingen, Germany).
Adhesion Assay
Yeast cells were patched on YNB agar plates containing 10 mm 3-amino-1,2,4-triazole (3-AT) and incubated at 30 °C for 3 days. Nonadhesive cells were removed by gentle washing under a constant stream of water. Plates were photographed before and after washing.
RESULTS
Scaffold proteins contribute to proximity among proteins and, thereby, allowing for functional relations between them. The Gβ-like protein Asc1p is involved in signal transduction and affects the intricate process of protein biosynthesis. It is implicated in translational regulation by the spatiotemporal organization of its microenvironment, and thus the coordination of functional protein interactions at the site of translation initiation. These proximities or microenvironments are difficult to map as many colocated proteins are only inefficiently enriched by classical pull-down strategies but require novel in vivo imaging techniques for their discovery. To identify proteins located within the Asc1p-microenvironment in vivo, we applied the emerging technique of proximity-dependent Biotin Identification (BioID; 30) and established the method for yeast. The promiscuous biotin protein ligase BirA*p, originated from E. coli, was fused to the C-terminus of the Asc1 protein bridged by four Gly-Ser-Ser repeats as a flexible linker (Fig. 1A). The ligase moiety covalently labels lysine residues of proteins in the direct proximity to Asc1p with biotin, which can afterward be captured with Strep-Tactin® and identified with LC-MS. Beyond the Asc1p-neighborhood at exponential growth, this approach was also used to monitor changes in its proximity at challenging growth conditions or because of amino acid exchanges. The Asc1R38D, K40Ep (short Asc1DEp) variant shows a decreased ribosome-binding affinity during ultracentrifugation of cell extracts in sucrose density gradients (4). To get a comprehensive view on the in vivo localization and the proteinaceous neighbors of this protein variant, it was also fused to BirA*p in this work. Expression of the Asc1-BirA* and Asc1DE-BirA* fusion proteins from a high copy number plasmid in a Δasc1 strain background yielded in wildtype-like Asc1 protein levels (Fig. 1B) and growth of both the ASC1-birA* and the ASC1DE-birA* strain in the presence of elevated biotin levels resulted in an increase of biotinylated proteins when compared with the unfused wt-ASC1 strain (Fig. 1C).
Fig. 1.

Scheme of Asc1-BirA*p at the head of the 40S ribosome. A, The Asc1 protein is a constituent of the 40S ribosomal subunit and interacts physically with the ribosomal proteins Rps3p, Rps16p and Rps17p. Amino acid residues Arg38 and Lys40 contribute to ribosome-binding and their exchange to Asp or Glu (Asc1DEp) weakens ribosome-binding. The BirA* protein is fused to the C-terminus of Asc1p and Asc1DEp via four repeats of a Gly-Ser-Ser linker sequence indicated as letter sequence. Because of the ribosome averted orientation of the Asc1p C-terminus, ribosome binding of the fusion proteins should not be sterically compromised. The crystal structure data of the S. cerevisiae 80S ribosome and the E. coli BirA protein derive from the PDB entries 4V88 (10) and 1BIB (73) and were combined to model the fusion protein with the PyMOL Molecular Graphics System software. B, Expression of the Asc1-BirA* and the Asc1DE-BirA* fusion proteins (∼70 kDa) from a high copy number plasmid in the Δasc1 strain background provides wild-type-like Asc1p levels. Proteins were detected in Western experiments using an Asc1p-specific antibody. The Ponceau staining of the lane is shown for a small part of the lanes. C, Protein biotinylation, detected with horseradish peroxidase (HRP)-coupled streptavidin, was elevated in strains expressing Asc1-BirA*p, Asc1DE-BirA*p or the mere BirA* protein cultivated in the presence of biotin. Parts of the Ponceau stained lanes are shown.
The Asc1-BirA* Fusion Protein Functionally Complements Asc1p Depletion
The Asc1 protein affects a variety of cellular processes including signal transduction, which manifests in obvious growth phenotypes of asc1− cells. They are impaired in respiration, are osmotically frail and show a defect in the cell wall integrity pathway as apparent from slow growth in the presence of glycerol as sole and nonfermentable carbon source, at elevated concentrations of NaCl and with the cell wall perturbing agent congo red, respectively (17, 40, 41). Additionally, Asc1p-depletion results in the inability of yeast cells to grow adhesively on agar surfaces during starvation further indicating a cell wall defect (41). In accordance with its role in translation, an asc1− strain additionally shows increased sensitivity against protein misfolding agents and translational inhibitors like canavanine and cycloheximide (23, 42). The ASC1 coding sequence is interrupted by an intron bearing the SNR24 gene that encodes the small nucleolar RNA (snoRNA) U24 (3). Therefore, an ASC1 deletion simultaneously results in the loss of U24 snoRNA. Rachfall et al. (17) as well as Schmitt et al. (23) however showed that the phenotypes mentioned above are U24-independent and clearly a consequence of Asc1p depletion. Accordingly, expression of a plasmid-borne Asc1-BirA* fusion protein in the Δasc1 strain rescued these Δasc1 phenotypes (Fig. 2A). The sensitivity against the translational inhibitor cycloheximide and protein misfolding agent canavanine was at least partially complemented by ASC1-birA*. Thus, Asc1-BirA*p is generally functional and takes over Asc1p's role within Δasc1 cells, implicating that it locates at its native cellular position. Consistently, the ribosome averted orientation of the Asc1p C-terminus suggests that ribosome association of the fusion protein should not be sterically hindered. Indeed, sucrose density gradient centrifugation of wt-ASC1 and ASC1-birA* cell extracts followed by the visualization of Asc1p/Asc1-BirA*p distribution within the sucrose gradient by Western blot analysis confirmed that not only wt-Asc1p, but also the Asc1-BirA* fusion protein is present within the 40S, monosomal and polysomal fractions (Figs. 2B and C). An increase of Asc1-BirA*p within the ribosome-free fraction and a slight decrease within the 80S monosomal fraction was, however, observed. The latter observation was even more pronounced for Rps3p, a direct interactor of Asc1p. Maybe these changes reflect a shift in the in vivo distribution of Asc1-BirA*p and Rps3p, or they reflect an increase of in vitro disintegration caused by forces during ultracentrifugation, possibly increased for Asc1p through its fusion with the BirA* moiety. The fact that the Rps3 protein of Asc1DE-BirA*p-expressing cells remained stably associated with ribosomes during ultracentrifugation when Asc1DE-BirA*p readily left the ribosomes hints to a weakened interaction of Asc1DE-BirA*p with Rps3p, leading to the stabilization of Rps3p at the ribosome. To generally meet concerns of misinterpreting proximal proteins of ribosome-free Asc1-BirA*p as ribosomal Asc1p-neighbors we integrated a yeast strain into the BioID study that expresses the mere BirA* protein not fused to Asc1p and, therefore, not located to ribosomes as a negative control.
Fig. 2.

ASC1-birA* and asc1DE-birA* complement asc1− phenotypes and the fusion proteins associate with translating ribosomes. A, For drop dilution assays 10-fold dilution series of wt-ASC1, Δasc1, ASC1-birA* and asc1DE-birA* cell suspensions were spotted on YNB agar plates with the protein misfolding agent canavanine (600 ng/ml) or the translation inhibitor cycloheximide (0.15 μg/ml), or with NaCl (75 mm). Additionally, cells were dropped on YNB plates with 2% glycerol instead of glucose and on medium with congo red (125 μg/ml). A YNB agar plate was used for the growth control, and all plates were photographed after 3 to 4 days of growth. Adhesive growth was induced with 1 mm 3-AT and scored after 3 days of growth followed by gentle washing. ASC1-birA* and asc1DE-birA* were expressed from high copy number plasmids in the Δasc1 strain background. The wt-ASC1 and Δasc1 strains were transformed with an empty vector (pME2787). B, The ribosome-binding ability of the Asc1-BirA* and Asc1DE-BirA* fusion proteins in comparison to Asc1p was analyzed with sucrose gradient centrifugation and Western blot analysis with an Asc1p-specific antibody. The abundances of the different Asc1p variants in the nonribosomal fraction, the 40S and 60S subunits as well as the 80S monosome and the polysomes revealed ribosome-binding of the Asc1-BirA* protein, but ribosome dissociation of Asc1DE-BirA*p during ultracentrifugation. The distribution of Rps3p within these fractions was visualized with an Rps3p-specific antibody as marker for the migration of the small ribosomal subunit. C, Input loading control verified by Western blotting and detection with the Asc1p- and Rps3p-specific antibodies.
A Quantitative SILAC Approach Enables the Accurate Integration of Controls into the BioID Experiment
After confirming Asc1-BirA*p's functionality and authentic positioning we next identified its proteinaceous neighbors in vivo with the BioID approach. ASC1-birA*-expressing cells were grown in the presence of elevated levels of biotin resulting in the covalent biotinylation of interacting and proximal proteins. This covalent modification enabled the enrichment of the labeled proteins after denaturing cell lysis with a stationary chromatographic Strep-Tactin® phase. Affinity captured proteins were digested with trypsin for bottom-up protein identification with LC-MS analysis (Fig. 3A). Additionally, the sites of biotinylation within the proteins were identified by mass spectrometry in case the peptides carrying this modification were covered. To increase the coverage of the biotinylated peptides, half of the digested and enriched sample was used for a second Strep-Tactin® peptide enrichment followed by another LC-MS analysis (Fig. 3A).
Fig. 3.

SILAC-BioID: Enrichment quantified against controls. A, Supplemented biotin was used by the Asc1-BirA* fusion protein to covalently biotinylate proteins in reach (up to ∼35 Å). After cell lysis in the presence of 4% SDS biotinylated proteins were enriched via biotin affinity capture and afterwards digested with trypsin. The resulting peptides were subjected to LC-MS analysis for protein identification and for the determination of biotinylated lysine residues. SILAC quantification was performed with the MaxQuant software (39). Additional database search with the Proteome Discoverer software enhanced the identification of biotinylated sites. To improve the coverage of biotinylated peptides, half of the peptide solution was subjected to additional biotin affinity capture and LC-MS analysis. Peptide samples without biotin affinity capture were also analyzed by LC-MS. The resulting proteome values were used to normalize affinity capture SILAC ratios against total protein abundances. Modified from Smolinski and Valerius (74). B, All four BioID-experiments were performed with triple SILAC to quantitatively compare cells of three different cultures in one batch. The birA* and wt-ASC1 strains served as negative controls. Alterations in the Asc1p-neighborhood on glucose starvation and heat stress (37 °C) were determined against ASC1-birA* at exponential growth (2% glucose, 30 °C) as reference and birA* at stress as negative control. The neighborhood of Asc1DE-BirA*p was compared with ASC1-birA* as reference and birA* as negative control. Yeast strains were individually cultivated in the presence of 10 μm biotin and were labeled with lysine and arginine isotope variants as indicated. All genes were expressed from high copy number plasmids in a Δarg4 Δlys1 strain background to ensure exclusive incorporation of the labeled amino acids into all proteins. The number of biological replicates is indicated for each experiment. °One replicate of the “heat stress” BioID experiment was performed with a label swap: ASC1-birA*, 37 °C (heavy), ASC1-birA*, 30 °C (medium), birA*, 37 °C (light).
Coenrichment experiments coupled to sensitive LC-MS analysis are generally prone to an unspecific capture of hundreds of proteins emerging even from negative controls. The combination of affinity-capture strategies with the relative quantification against essential controls enables to identify false positive candidates and to exclude them from further consideration. Asc1-BirA*p-dependent capture of biotinylated proteins was quantitatively monitored against two negative control strains using the stable isotope labeling with amino acids in cell culture (SILAC) technique (43). By applying a triple SILAC approach, proteins of these three strains were differentially labeled in culture with light, medium, or heavy arginine and lysine variants, respectively, to enable a direct quantitative comparison in one cell culture pool. To differentiate between Asc1p-specific and Asc1p-unspecific biotinylated/enriched proteins, the following strains were used as negative controls: (1) A wt-ASC1 strain to reveal unspecific enrichments and naturally biotinylated proteins and (2) a strain expressing the mere BirA*p not fused to Asc1p to account for BirA*p-dependent biotinylations independent of Asc1p (Fig. 3B). Following this SILAC strategy, a similar number of cells from the respective strains were pooled directly after cultivation and further processed as one batch according to the described BioID workflow (Fig. 3A). The finally obtained LC-MS/MS2 raw data files were subjected to database searches using the MaxQuant software (39). The downstream analysis with the Perseus software (39) resulted in quantitative ratios giving the relative enrichment of proteins from one strain compared with the others. Also, an aliquot of the total protein extract, taken before the first biotin affinity capture, was processed and analyzed by LC-MS (Fig. 3A). The resulting proteome data were used to account for variations in protein expression within the different yeast strains. In total, four different BioID analyses were conducted: We investigated the Asc1p-neighborhood in (1) exponentially growing cells, we monitored dynamic alterations on (2) glucose depletion and (3) mild heat stress, and (4) we studied the effect of the Asc1p R38D K40E amino acid exchanges on the Asc1p-microenvironment. The corresponding negative control/reference strains and the number of replicates are indicated in Fig. 3B. In the following chapters the term “SILAC ratio” refers to protein ratios with the value of the respective negative control or reference strains in the denominator.
Def1p, Stm1p, and mRNA-binding Proteins are in Close Proximity to Asc1p
BioID-captured proteins were considered as at least temporal Asc1p-neighbors when the following two criteria were fulfilled: (1) at least 20% enrichment (≙ log2 SILAC ratio ≥ 0.26) compared with the birA* negative control in at least four out of six biological replicates (three additional ASC1-birA*/birA* SILAC ratios were available from the BioID analysis investigating the Asc1DEp neighborhood) and (2) at least 100% enrichment (≙ log2 SILAC ratio ≥ 1) compared with the wt-ASC1 control strain in at least two out of three biological replicates (Table III). With these thresholds our SILAC-BioID setup revealed 40 proteins that were Asc1p-specifically enriched and further regarded as putative Asc1p-neighbors. We distinguish between candidates with or without proteome value for normalization and with or without at least one identified biotinylated lysine residue as second proof of proximity (Table III; for detailed information see supplemental Table S1). Within the group of the top candidates with proteome value and biotin site, expected and well known Asc1p interaction partners were found like the mRNA-binding protein Scp160p and the ribosomal protein Rps3p (14, 16, 10). Evidence for physical interaction of Asc1p with Rps26p, Ubp3p, Bre5p and Nob1p was also previously reported (44–46). The proteins with the highest enrichment factors relative to the birA* control strain were the RNA polymerase II degradation factor Def1p, the ribosomal protein Rps26p and the mRNA-binding proteins Scp160p and Sro9p. Def1p and Sro9p were so far not reported as Asc1p interaction partners. Also, the Pbp1p-binding protein Pbp4p was highly enriched in the Asc1p-neighborhood and was biotinylated at five sites. Def1p, Asc1p itself, and the ribosome clamping factor Stm1p showed the strongest biotinylation with 10, 15, and 14 identified biotinylated lysine residues, respectively (Table III). The nonexclusive assignment of the candidates to functional groups revealed an enrichment of proteins with distinct molecular functions (Fig. 4): the largest group contains proteins with mRNA-binding activity including Scp160p, Sro9p, and Gis2p. Additionally, constituents and regulators of mRNP granules, like Pbp1p, Pbp4p, and Lsm12p were identified in the Asc1p-neighborhood as well as regulators of protein ubiquitylation (e.g. Def1p, Ubp3p, and Ubp2p). Other Asc1p-neighbors are involved in translational regulation including the translation initiation factors Cdc33p (eIF4E) and Rpg1p (eIF3/a) or they even function in mRNA transcription like e.g. the transcription elongation factor Spt5p. Further groups of molecular functions are listed in Fig. 4. In summary, our data reveal an accumulation of proteins involved in mRNA fate and functionality, ribosome homeostasis and mRNA translation, and nuclear gene transcription within the microenvironment of Asc1p.
Table III. Asc1p-neighbors during exponential growth. The ratios were normalized against the respective proteome value (if available). A two-sample t-test (or one-sample t-test if no proteome value was available) with a p value of 0.05 indicated the enrichment as statistically significant. Standard deviations and the number of identified biotinylation sites are listed. Exceptions of the described thresholds are indicated as follows: 2/6 only two valid values available from six replicates; T elevated p value in the t-test; o one clear outlier was not considered for the calculation of mean and standard deviation. * Although the threshold for the wt-Asc1p control was not reached, Rpg1p was further considered as Asc1p-neighbor, because it is a subunit of eIF3, which is known to interact with Asc1p (21).
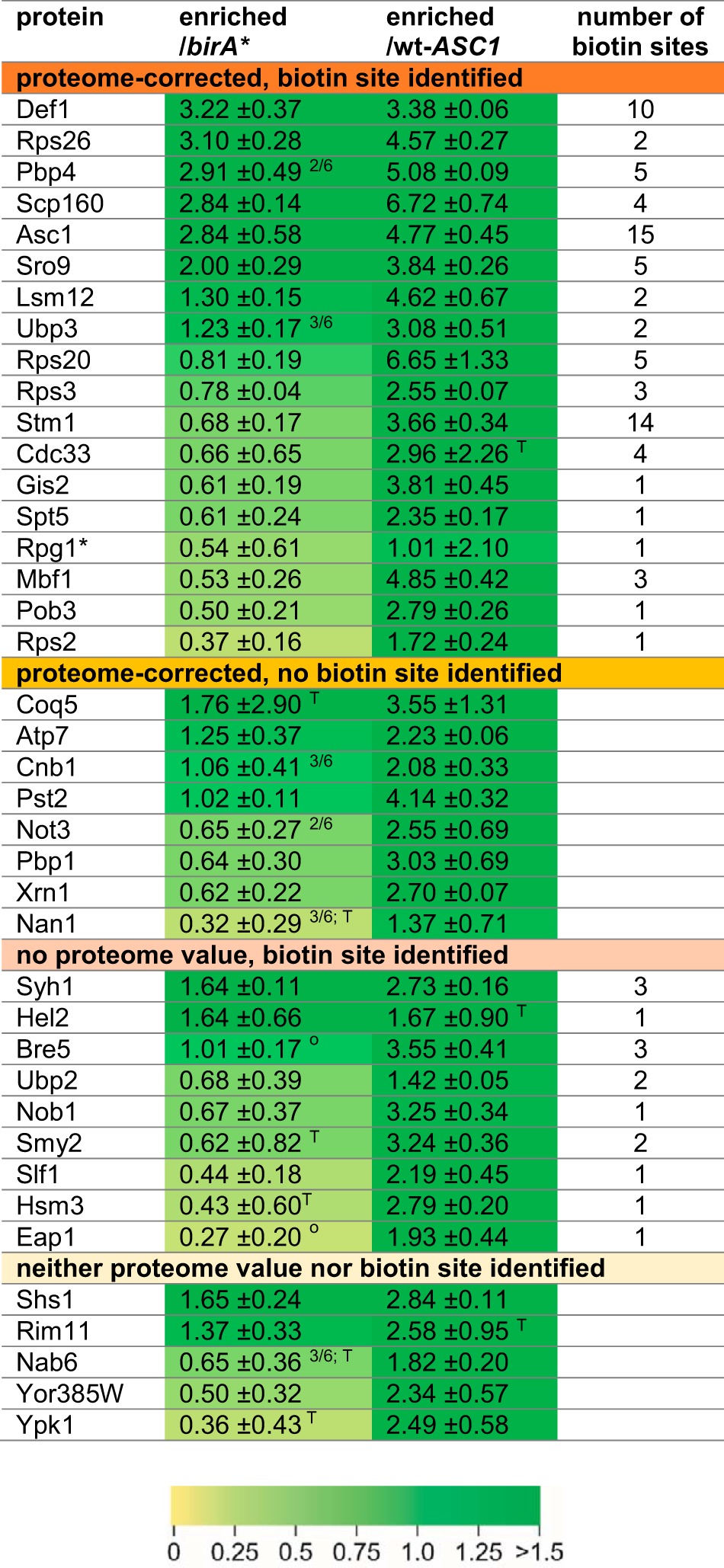
Fig. 4.

Functional grouping of Asc1p-neighbors. 40 proteins identified in the Asc1p-vicinity were assigned to groups reflecting molecular functions in a nonexclusive manner. The number of proteins per group is given by the x axis. The number of potential Asc1p-neighbors per group with or without proteome value, and with or without identified biotin site is reflected by the respective colors. Corresponding candidates are listed.
Functional Validation of Selected BioID Candidates
SILAC/LC-MS-based BioID experiments (BioID proteomics) generate complex subproteome data sets that require highly reliable relative quantification of the StrepTactin®-enriched proteins against appropriate controls and an efficient filtering workflow to sort out false positive proteins. To validate selected proteomics-derived BioID candidates, we applied alternative techniques: 1. Coimmunoprecipitation by GFP-pulldown experiments for three selected BioID candidates: We verified the coenrichment of Asc1p with Sro9p-GFP and Ubp3p-GFP, however, failed to significantly coenrich Asc1p with GFP-Def1p (Fig. 5A). This experiment gives evidence that the β-propeller Asc1p physically binds to Sro9p and Ubp3p. 2. To confirm the proteomics-based identification of Def1p as Asc1p-proximal protein, we performed BioID without SILAC/LC-MS analysis but subjected the StrepTactin® eluates to Western blot analysis (BioID Western). We successfully validated the enrichment of Def1p from extracts of Asc1-BirA*p expressing cells with a Def1p-specific antibody (Fig. 5B). Furthermore, we observed a slow growth phenotype for cells expressing N-terminally myc-tagged Def1p in a Δasc1 background (Fig. 5C1). This growth defect can be complemented through transformation with plasmid-borne ASC1 (Fig. 5C2). Thus, Asc1p and Def1p are not only proximal to each other, but also genetically interact. We further observed a synthetic genetic interaction between ASC1 and SRO9. Deletion of ASC1 rescues the growth defect of Δsro9 cells on osmotic stress induced by high concentrations of NaCl (Fig. 5E).
Fig. 5.

Validation of selected BioID candidates. A, GFP-traps. GFP-tagged Def1p, Sro9p, Ubp3p, and Yrb2p (as a negative control) were enriched from the respective strains expressing the fusion-proteins. The wt strain BY4741 expresses no GFP and was used for a second negative control. Input and eluate fractions were subjected to Western blot experiments for the subsequent detection of Asc1p and GFP-fusion proteins with the respective antibodies. B, BioID Western. birA* and ASC1-birA* were expressed from high copy number plasmids in the wt-ASC1 and Δasc1 strains, respectively. The strains were cultivated in the presence of biotin, and biotinylated proteins were enriched from cell lysates of these cultures. The abundance of Def1p in the input control samples and the eluate fractions was analyzed with Western blot experiments using a Def1p-specific antibody. C, Genetic interaction of ASC1 and DEF1. (C1) Synthetic growth defect. Ten-fold dilution series of W303 wt-ASC1, Δasc1, myc-DEF1 ASC1, and myc-DEF1 Δasc1 cell suspensions were spotted on YEPD and SC medium. (C2) Complementation of the synthetic growth defect. myc-DEF1 ASC1 and myc-DEF1 Δasc1 cells were transformed with centromere plasmids either expressing ASC1 from its own promoter (pME4481) or without any ASC1 gene (EV, pME2781) as control. Cell suspensions were spotted onto SC medium without tryptophan (-Trp) for selection. D, Asc1p-dependent phosphorylation of Ubp3p-associated Bre5p. (D1) Bre5p phospho-peptides identified by mass spectrometry. Bre5p phospho-peptides with phosphorylation (ph) at S282 identified from tryptic digests of proteins enriched with Ubp3p-GFP. Both peptides cover the amino acid sequence 280 to 298. Because of a missed cleavage site C-terminal of K279, the upper peptide starts at T278. See supplemental Figs. S2 and S3 for the corresponding fragmentation spectra. The b- and y-ions found in these spectra are indicated. Highest phospho-site probabilities for S282: 100% (short peptide) and 93% (long peptide). (D2) Label-free quantification (LFQ) of proteins/peptides copurified with Ubp3p-GFP in the absence and presence of Asc1p. The experiment was performed with two biological replicates (1 and 2). LFQ intensities and MS/MS counts for Ubp3p-GFP, Asc1p, Bre5p, and the Bre5p phospho-site S282 are depicted. MS/MS counts for Bre5p phosphorylation at S282 derived from peptides depicted in D1 including tSIM analyses. E, Genetic interaction of ASC1 and SRO9. 10-fold dilution series of BY wt-ASC1, Δasc1, Δsro9, and Δasc1 Δsro9 cell suspensions were spotted on YEPD plates with and without 1 m NaCl. Two Δasc1 Δsro9 clones were tested. Strains of the S288c background are less osmosensitive then strains of the Σ1278b background; therefore, an increased osmosensitivity of the Δasc1 strain was not observed here (compare with Fig. 2).
In a previous study, we analyzed the impact of an ASC1 deletion on the yeast phospho-proteome and observed strongly reduced phosphorylation of the Ubp3p-interacting protein Bre5p at S282 (23). Here, we enriched Ubp3p-GFP from wt-ASC1 and Δasc1 cells and analyzed proteins with mass spectrometry. Label-free quantification (LFQ) with MaxQuant showed that Ubp3p-GFP as well as Bre5p was equally enriched from all samples (Fig. 5D2). However, two unique Bre5p peptides phosphorylated at S282 (Fig. 5D1) were exclusively identified in samples derived from wt-ASC1 cells (Fig. 5D2). Even with targeted single ion monitoring (tSIM), these peptides were not identified in samples of Δasc1 cells. A phosphorylated control peptide of Bre5p (E329-K359) for the tSIM analyses was found in all samples. Thus, phosphorylation of Bre5p at S282 seems to occur Asc1p-dependently in a common microenvironment of Asc1p, Bre5p, and Ubp3p. We conclude that new physical interacting proteins of Asc1p (e.g. Sro9p and Ubp3p) as well as additional proximal proteins (e.g. Def1p) have been discovered with our BioID experiment in yeast, and that the combination of BioID with state-of-the-art proteomics is a powerful tool that enables the description of microenvironments near target proteins.
Glucose Limitation Depletes the Asc1p-microenvironment in Favor of Ribosomal Proteins
We next monitored changes in the Asc1p-neighborhood on glucose deprivation. ASC1-birA* cells deprived of glucose were compared with cells of the very same strain grown at 2% glucose. The birA* strain was again used as negative control (Fig. 3B). In five biological replicates all strains were cultivated to the exponential growth phase (OD600 of 0.7). Cells were then shifted to fresh media, either with or without glucose. After 15 min of adjustment, biotin was added to label starvation-induced changes in the Asc1p-proximity with biotin during one further hour of cultivation. Proteins that were at least 20% enriched compared with the reference culture were considered as starvation-related Asc1p-neighbors, and proteins that were at least 20% less enriched were considered as starvation-dependently displaced from the direct Asc1p-neighborhood. We observed a significant change in the molecular microenvironment of Asc1-BirA*p in glucose depleted cells: 15 proteins identified as Asc1p-neighbors at exponential growth were less enriched during glucose deprivation (depicted for a selection in Fig 5A). These included the mRNA-binding proteins Scp160p, Sro9p or Gis2p, as well as Def1p, Stm1p, Rpg1p or Mbf1p. The Asc1-BirA* protein itself is also less efficiently enriched with BioID from cells at glucose deprivation. The enrichment of naturally biotinylated proteins like the acetyl-CoA carboxylase Acc1p, the pyruvate carboxylases Pyc1p/Pyc2p or the mitochondrial acetyl-CoA carboxylase Hfa1p, was not altered on glucose starvation (supplemental Table S2). This indicates unchanged general biotinylation efficiency at glucose limitation. 29 proteins specifically appeared as Asc1p-neighbors on glucose limitation, many of them ribosomal proteins of both the small and large ribosomal subunits, but also proteins involved in amino acid metabolism (Fig. 6B; for detailed information on starvation-dependent changes see supplemental Table S3). This implicates a rearrangement of ribosomes during glucose starvation when translation in general is not favored and adaptation to low energy supply occurs. The decreased biotinylation and subsequent decreased enrichment of Asc1-BirA*p itself hints to an impeded accessibility of Asc1p for self-biotinylation, which might result from a modified arrangement of ribosomes at glucose deprivation. Coq5p, a protein of the multimeric Coq-complex required for CoenzymeQ synthesis essential for aerobic respiration, displays a strong enrichment in the Asc1p-neighborhood on glucose deprivation. This protein also appeared as Asc1p-neighbor during exponential growth with glucose, however to a far lesser extent (Table III). Our data reveal a significant remodeling of the molecular microenvironment at the ribosomal scaffold protein Asc1p on glucose deprivation, including an accumulation of ribosomal proteins, and a decrease of translational and transcriptional regulators hinting to local rearrangements of ribosomes on nutrient stress.
Fig. 6.

Changes within the proteinaceous neighborhood of Asc1p. A, The bar chart depicts changes in the Asc1-BirA*p neighborhood in cells during glucose deprivation and heat stress (37 °C), and because of the R38D K40E exchanges relative to the neighborhood of Asc1-BirA*p during exponential growth. 19 proteins identified as Asc1p-neighbors during exponential growth are listed on the left. The bars represent log2 SILAC ratios/enrichment values of the ASC1-birA*-strain starved from glucose (green), cultivated at 37 °C (red) or of the ASC1DE-birA*-strain (blue). Proteins were considered as dynamic Asc1p-neighbors if log2 SILAC ratios were outside −0.26 and 0.26. Stable Asc1p-neighbors, independent of the tested growth conditions or the DE-exchange, are listed at the top within the gray box (log2 SILAC ratio between −0.26 and 0.26). Missing SILAC ratios are indicated by asterisks. Mean values that do not reach the respective threshold because of outlier values are indicated by triangles. B–D, Proteins specifically enriched in the Asc1p-neighborhood at (B) glucose starvation, (C) heat-stress, or (D) of the Asc1DEp-variant were assigned to functional groups in a nonexclusive manner. Candidates of the respective groups are listed within the bar chart, those that are exclusively enriched in these specific experiments are listed in bold. The number of proteins per group is given by the x axis. Further proteins that do not belong to the chosen groups are listed below the diagrams.
Mild Heat Treatment Emphasizes the Proximity of mRNA-binding Proteins to Asc1p
As an important player in signal transduction the Asc1 protein is required for yeast cells to cope with environmental changes manifesting in a variety of Asc1p-dependent phenotypes. Among others, Asc1p is required to cope with heat evident by slowed growth of Asc1p-depleted cells at 37 °C (47, 48). In three biological replicates of further BioID-experiments, alterations in the Asc1-BirA*p neighborhood caused by mild heat during growth at 37 °C were monitored. The ASC1-birA* strain was exposed to the elevated temperature and was directly compared with the very same strain cultivated at 30 °C and to the birA* strain grown at 37 °C as negative control. All three strains were initially cultivated at 30 °C until they reached an OD600 of 0.6. The two cultures meant for heat stress were shifted to 37 °C, whereas the ASC1-birA* reference strain remained at 30 °C. After 10 min of adjustment biotin was added to the cultures followed by further growth at the respective temperatures for 1 h. One replicate was performed with a label swap. In contrast to the depiction in Fig. 3B, the ASC1-birA* strain cultivated at 37 °C was in this case labeled with heavy isotopic amino acids, the ASC1-birA* reference strain with medium and the birA* negative control strain with light lysine and arginine. It was reported that the E. coli derived biotin ligase BirA*p is increasingly active during growth at 37 °C (49), so that protein enrichments could be a consequence of an increased biotinylation activity. However, the visualization of total protein biotinylation with HRP-coupled streptavidin of our protein extracts derived from cells cultivated at 30 °C or 37 °C revealed no obvious difference (supplemental Fig. S1). Additionally, natural biotinylation targets were similarly enriched at 30 °C and at 37 °C (supplemental Table S5). Eight proteins increasingly accumulated in the neighborhood, namely Scp160p, Stm1p, Xrn1p, Rpg1p, Sro9p, Rps20p, Pst2p, and Def1p with more than 20% enrichment (Fig. 6A and supplemental Table S4). Three further mRNA-binding proteins, namely Hek2p (also designated as Khd1p), Psp2p and New1p, were as well specifically enriched at elevated temperature. Thus, in total eleven proteins were heat-dependently enriched from the Asc1p-neighborhood, among them eight proteins with mRNA-binding activity (Fig. 6C). These data imply that during growth at increased temperature mRNA-binding proteins congregate at the scaffold protein Asc1p.
Exchange of Residues Arg38 and Lys40 at Asc1p's rRNA Contact Site for Aspartate and Glutamate Does Not Efficiently Release the Protein from the Ribosome
The Asc1 protein is part of the head of the 40S ribosomal subunit and was suggested to function as a central hub connecting cellular signaling and translation at this exposed site. Coyle et al. (4) described an Asc1p variant with two amino acid exchanges that showed decreased binding affinity to ribosomes on sucrose density gradient ultracentrifugation. Exchange of Arg38 and Lys40 to negatively charged Asp (D) and Glu (E), respectively, both sites located at the interface of Asc1p to the 18S rRNA, was meant to budge Asc1p off the ribosome (Fig. 1A). Strikingly, this asc1DE mutant strain revealed almost no asc1− phenotypes (4, 23). Recently published experiments using formaldehyde cross-linking before ultracentrifugation gave evidence that Asc1DEp still localizes to the ribosome in vivo (29). We generated an asc1DE-birA* fusion to quantitatively study changes in the Asc1p-neighborhood of the Asc1DEp variant. Also, the asc1DE-birA* fusion complemented the asc1−phenotypes with the exceptions of an impaired growth on NaCl and on cycloheximide (Fig. 2A). The latter phenotype is compatible with the increased sensitivity against cycloheximide of a strain expressing the unfused Asc1DEp (23). During ultracentrifugation in sucrose gradients the Asc1DE-BirA* fusion protein behaved like Asc1DEp (4), as it was mainly found in the nonribosomal fractions of the ribosome profile (Figs. 2B and 2C). This confirmed its decreased anchorage at the ribosome during ultracentrifugation. The SILAC-BioID experiment with the Asc1DE-BirA* protein was performed with the ASC1-birA* strain as reference and the birA* strain as negative control in three biological replicates (Fig. 3B). Proteins were considered as DE-specific neighbors when they were at least 20% enriched compared with the ASC1-birA* reference strain or as removed from the Asc1DEp neighborhood in case of 20% less enrichment. The BioID-analysis indicates that Asc1DEp is present at the ribosome because most of the proteins identified in vicinity of wt-Asc1p, e.g. the ribosomal proteins Rps3p, Rps26p, or Rps2p as well as Def1p, Scp160p, and Sro9p, were equally enriched from the both strains as depicted in Fig. 6A. Still, five proteins displayed changes relative to wt-Asc1p, namely Cdc33p, Pst2p, Atp7p, and Lsm12p as they were less frequently captured from the asc1DE-birA* strain. The Asc1DE protein itself was even more enriched from the asc1DE-birA* strain in comparison to the ASC1-birA* strain, although its protein abundance in general was not affected. This is evidence for neighboring Asc1DEp molecules, possibly even to Asc1p-dimer formation, or to a more efficient self-biotinylation because of a sterical relief of BirA*. Strikingly, additional 16 proteins were found as neighbors exclusively for the Asc1DEp variant (Fig. 6D and supplemental Table S6). These include further ribosome binding proteins, NTPases, and proteins involved in nucleotide metabolism hinting to spatial reorganization or flexibility at the ribosome. Altogether, these data suggest that the R38D K40E exchanges within Asc1p do not lead to a ubiquitous release from the ribosome in vivo, but rather to a more flexible spatial positioning at the ribosome.
DISCUSSION
Scaffold proteins organize protein-protein interactions and are required for proximity organization of proteins and protein complexes thereby realizing cellular signaling. The highly conserved Gβ-like Asc1 protein serves as scaffold at the exposed head region of the small 40S ribosomal subunit and was proposed to function as a central hub for cellular signaling into the translational machinery (3, 4, 10, 13). With a combination of BioID and SILAC the molecular microenvironment of Asc1p was studied in vivo taking essential control strains quantitatively into account. This combined BioID-SILAC approach was validated by the identification of previously reported physical Asc1p interaction partners (14, 16, 45) and of four ribosomal proteins known to colocalize with Asc1p according to the 80S crystal structure. Beyond these established Asc1p-neighbors further 32 candidate proteins were identified to colocalize with Asc1p at exponential growth including translational and transcriptional regulators as well as mRNA-binding proteins. At glucose starvation, ribosomal proteins of the small and the large ribosomal subunit accumulated in the Asc1p proximity. The Asc1DE protein variant was shown to bind ribosomes in vivo, possibly with an increased spatial flexibility. Asc1p-proximal proteins as well as changes within the Asc1p-microenvironment are discussed in the following sections.
Translation Initiation Factors and mRNA-binding Proteins Colocalize with the Asc1p β-propeller
At exponential growth, a major group of Asc1p-neighbors consists of proteins with mRNA-binding activity. The delivery of mRNAs to the ribosomal translational initiation site depends on mRNA-binding proteins that also influence mRNA stability and translational fate (50). The BioID experiments confirmed the formation of the SESA network consisting of Asc1p, the mRNA-binding protein Scp160p, Smy2p and Eap1p together required for POM34 mRNA regulation (16). Furthermore, the two paralogous mRNA-binding proteins Sro9p and Slf1p as well as Gis2p were identified proximal to Asc1p. Evidence for functional interaction between Asc1p and Sro9p is given by the genetic suppression of the growth defect of Δsro9 cells on high-salt medium through deletion of ASC1. Additionally, the increased abundance of Sro9p in Asc1p-depleted cells implies an interplay with Asc1p (23). Regulating the proximities of mRNA-binding proteins at the head region of the 40S ribosomal subunit seems suitable to control mRNA translation in a spatiotemporal manner probably in coordination with further regulatory components at the ribosome.
Further proteins required for the initiation of mRNA translation colocalize within the Asc1p-microenvironment during exponential growth. The cap-binding protein eIF4E encoded by CDC33 in S. cerevisiae was found proximal to Asc1p. Also in mammals, RACK1 interacts with eIF4E and regulates the phosphorylation of the initiation factor by PKCβII (22). Furthermore, a physical interaction of Asc1p with the multisubunit initiation factor eIF3 was reported, specifically with the b- and c-subunits Prt1p and Nip1p (13, 21). Here, the eIF3/a-subunit Rpg1p was shown to colocalize with Asc1p supporting an eIF3 arrangement as described from the crystal structure by Llácer et al. (51). We showed recently that these three subunits of this initiation factor get Asc1p-dependently phosphorylated (23) suggesting a regulatory impact of Asc1p on the translation initiation process. Together, these data reveal that Asc1p occupies a key site within the area of translation initiation.
The RNA Polymerase II Degradation Factor Def1p and Ubp3p-Bre5p Cooperate with Asc1p
A strongly biotinylated Asc1-BirA*p neighbor identified with BioID was the RNA polymerase (RNAP) II degradation factor Def1p. Within the cytosol, ubiquitylation of Def1p through Rsp5p in response to transcription stress leads to its C-terminal truncation close to amino acid 530 (Fig. 7C) in a proteasome-dependent manner (34). The remaining N-terminal moiety of Def1p translocates into the nucleus where it associates to the RNAPII subunit Rbp1p of stalled elongation complexes. It mediates the poly-ubiquitylation of Rbp1p through the Elongin-Cullin ubiquitin ligase complex and its subsequent degradation (34, 56). A C-terminal Def1p peptide ranging from N631 to K661 similarly enriched with other Def1p peptides of the N-terminal moiety suggesting that Asc1p and Def1p colocalize already within the cytoplasm before Def1p processing (Fig. 7C). Our recent phospho-proteome analyses showed that Def1p is Asc1p-dependently phosphorylated at T258 and S260 (23). Hence, Asc1p seems to be involved in signal transduction to Def1p, thereby possibly affecting its processing and migration into the nucleus. We failed to obtain Δasc1 Δdef1 double deletion strains with different approaches. Deletion of ASC1 in myc-def1 strains already led to synthetic growth impairment. Ubp3p reverses ubiquitylation of RNAPII in the nucleus and is considered as Def1p antagonist. It copurifies with Def1p, both RNAPII subunits (Rbp1p and Rbp2p), its cofactor Bre5p and the transcription elongation factor Spt5p (57). Strikingly, Ubp3p, Bre5p and Spt5p were also identified as Asc1p-neighboring proteins in this study. We showed that phosphorylation of Ubp3p-bound Bre5p at serine 282 occurs Asc1p-dependently in a common microenvironment. Thus, a group of proteins involved in nuclear RNA polymerase turnover are target of Asc1p-mediated modification.
Fig. 7.

Stm1p and Def1p are biotinylated by the Asc1-BirA* fusion protein at several sites. A, At glucose deprivation the ribosomal clamping factor Stm1p is located in between the 40S and 60S subunits and prevents mRNA translation. At this condition, Asc1p forms a tightly connected network with Rps3p and Stm1p at the ribosome. B, Stm1p appears as a protein with almost no intrinsic structure during glucose starvation. Biotinylated lysine residues are indicated by red dots. The crystal structure data derive from the PDB entry 4V88 (10) and were used for visualization with the PyMOL Molecular Graphics System software. C, Def1p was biotinylated mainly in the N-terminal half of the protein. Proteasome-dependent processing of Def1p in response to transcription stress occurs before amino acid 530, and the presence of a C-terminal peptide confirms cytoplasmic proximity of Def1p to Asc1p. Biotinylated lysine residues are colored in red. The area of Def1p-processing is indicated in blue and peptides identified by LC-MS analysis are highlighted in green.
The Ribosome-clamping Factor Stm1p Colocalizes with Asc1p During Exponential Growth
Translation attenuation and re-initiation in response to nutritional stimuli is tightly regulated. During poor growth conditions global translation is massively slowed down, and nontranslating 80S ribosomes are kept together in readiness to facilitate translation re-initiation as soon as nutrients get available again (52). Stm1p locates into nontranslating 80S ribosomes and is required to allow for efficient re-initiation of translation after nutritional constraints (53). It functions as a clamping factor that prevents ribosome subunit disassembly in the absence of mRNA translation by occupying partially the path that is normally occupied by mRNA during translation (Fig. 7A; 10, 52). Thus, mRNA decoding at the ribosome and Stm1p placement within the decoding center are mutually exclusive events. The BioID analysis of cells during exponential growth revealed Stm1p as an Asc1p-proximal protein with several biotinylated lysine residues (Fig. 7B). None of the identified biotinylated residues within Stm1p is exposed toward Asc1p according to the 80S ribosome crystal structure obtained from starved yeast cells (Fig. 7B; 10). The BioID analyses suggest that Stm1p is deposited close to Asc1p within the 40S ribosomal head region when mRNAs are actively translated and might be kept there in standby until starvation signaling moves it into its clamping position. For Hela cells, a physical interaction of RACK1 with the Stm1p-homologous part of the putative human ortholog SERBP1 was reported just recently (55).
Transcription Factors Colocalize with Asc1p
Several transcriptional regulators were identified in the proximity to Asc1p. Spt5p is a universally conserved transcription elongation factor (58). It also affects cotranscriptional mRNA-processing while binding the nascent transcript and recruiting the respective processing factors (59, 60). Spt5p was described as a binding platform at the RNAPII for the recruitment of further transcriptional regulators (60). Proximity of this nuclear scaffold protein to the ribosomal scaffold protein Asc1p connects the two fundamental processes of cytosolic mRNA translation and nuclear mRNA synthesis.
Further transcriptional regulators were identified in the proximity of Asc1p: Pob3p, which is part of the facilitates chromatin transcription (FACT) complex involved in nucleosome remodeling and thus implicated in transcription initiation and elongation (61–64); Not3p, part of the Ccr4-NOT complex that functions as transcriptional regulator (65–67); and Mbf1p, a transcriptional coactivator that bridges the interaction of Gcn4p with the TATA-binding protein Spt15p to promote Gcn4p-dependent transcriptional activation of amino acid biosynthesis-related genes (68). In conclusion, Asc1p might not only connect cellular signaling to ribosomal activity for protein biosynthesis, but might also synchronize cytoplasmic mRNA translation with aspects of mRNA synthesis in the nucleus.
Glucose Depletion Triggers Rearrangements in the Asc1p Microenvironment
Poor growth conditions like glucose shortage lead to major adaptation processes in the cell. Changes in the microenvironment of Asc1p at the head region of the 40S ribosomal subunit were traced with BioID in this work. Stm1p, proximal to Asc1p during exponential growth, probably relocates in its clamping position within the 80S complex of nontranslating monosomes on glucose deprivation (10). Many of the proteins identified in the proximity of Asc1p during exponential growth, especially proteins implicated in mRNA translation and gene transcription, disappeared during glucose starvation.
Ribosomal proteins from both the small and large ribosomal subunit accumulate within the Asc1p-microenvironment during glucose shortage suggesting a process of ribosome aggregation. In analogy hibernating 70S dimers were observed in the stationary growth phase of E. coli cells (69). Translationally repressed 80S dimers as well as 80S-60S heterodimers were reported for rat glioma cells in response to different stresses (70). In yeast cells, the level of polysomes rapidly decreases on glucose deprivation whereas inactive monosomes accumulate (71). These nontranslating monosomes might dimerize/aggregate and thereby provide proximity to Asc1p to proteins of the 60S subunit and further 40S subunit constituents. This remodeled Asc1p-surrounding with ribosomal proteins close by might shield Asc1-BirA*p against self-biotinylation leading to the observed reduction in Asc1-BirA*p enrichment from glucose starved cells with BioID.
Coq5p, a mitochondrial protein implicated in ubiquinone biosynthesis, is highly enriched in the proximity of Asc1p during glucose starvation. The protein was identified as Asc1p-neighbor already for exponential growth, but showed increased enrichment on glucose deprivation. Coq5p is a methyltransferase that locates to the matrix site of the inner mitochondrial membrane where it is involved in the biosynthesis of ubiquinone (coenzyme Q; 72). Its 19 N-terminal amino acids are required for the mitochondrial import of the protein and are processed after the import. As a constituent of the electron transfer chain, ubiquinone is required for cellular respiration. Coq5p seems to be of special importance during glucose deprivation, possibly by the enforcement of increased ubiquinone production for respiration to efficiently generate energy. The strong BioID enrichment of Coq5p on glucose starvation suggests that Coq5p together with ribosomes accumulates at the cytosolic face of mitochondria. In summary, the Asc1p-BioID analysis of glucose-starved yeast cells indicates an extensive rearrangement within the head region of the 40S ribosomal subunit that reflects adaptation processes taking place during starvation.
Asc1DE-BirA*p Binds to Ribosomes During Exponential Growth
As an integral constituent of the mRNA translational machinery the functionality of Asc1p is expected to depend on its binding to the ribosome. Surprisingly, the Asc1DEp variant, which has an obvious ribosome-binding defect during ultracentrifugation of cell extracts in sucrose density gradients, caused only very mild phenotypes (4, 23). The quantitative BioID analyses of the Asc1DE-BirA* fusion protein provided insight in the in vivo microenvironment of this mutated version of the scaffold protein. Most of the wt-Asc1-BirA*p neighbors were also found proximal to Asc1DE-BirA*p suggesting that the mutated protein is still positioned at the ribosome. Hence, our observation supports the result of qualitative formaldehyde crosslink experiments reported by Thompson et al. (29). However, further proteins were enriched and identified in the proximity of Asc1DEp including ribosome associated proteins (e.g. Gcd1p and Tif6p) and ribosomal core constituents, namely Rps11p and Rpl10p. Therefore, we conclude that Asc1DEp might not be as tightly incorporated into ribosomes as the wt-Asc1p but engages a more flexible position. The stronger self-biotinylation of Asc1DE-BirA*p in comparison to wt-Asc1-BirA*p might be a consequence of this increased spatial flexibility. It remains to be shown whether the artificial Asc1DEp version resembles any physiological status of an Asc1 protein with altered ribosome-binding characteristics, e.g. as consequence of post-translational modifications or structural dynamics within the ribosome.
SILAC-based BioID in Yeast - An In Vivo Tool to Study Dynamic Microenvironments in the Unicellular Model Organism Saccharomyces cerevisiae
The BioID approach was for the first time performed quantitatively with SILAC labeling enabling a direct comparison of the ASC1-birA* strain to fundamental controls and among differential growth conditions. This approach will be further exploited to study dynamic changes of the Asc1p-microenvironment in response to environmental stimuli and in dependence of post-translational Asc1p modifications. The use of a smaller and optimized biotin ligase will be considered for further experimental improvement (49). The BioID experiments performed in this work provide insight into the proteinaceous microenvironment of Asc1p and demonstrate the applicability and worth of the method for S. cerevisiae.
DATA AVAILABILITY
The raw mass spectrometric data and search output files are deposited at ProteomeXchange via the PRIDE database with the accession numbers “PXD005612” (BioID experiments) and “PXD007858” (GFP-trap experiments).
Supplementary Material
Acknowledgments
We thank Hans Dieter Schmitt and Sabrina Knape (Department of Neurobiology, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany) for inspiring us to apply BioID and for sharing the respective birA* plasmid with us. We thank Miriam Kolog Gulko for her support and expertise in protein purification for antibody production. We thank Jesper Svejstrup and Michelle Harreman Lehner (Mechanisms of Transcription Laboratory, The Francis Crick Institute, London, UK) for providing us with the Def1p-specific antibody and yeast strains.
Footnotes
Author contributions: N.O., K.S., and O.V. designed research; N.O., K.S., V.H., B.N., H.K., G.H.B., and O.V. performed research; N.O., K.S., and O.V. analyzed data; N.O., K.S., and O.V. wrote the paper.
* This work was supported by the Deutsche Forschungsgemeinschaft (DFG) to OV (Grant VA352/2–1 and Grant GSC 226/2 of the Göttingen Graduate School for Neurosciences and Molecular Biosciences), and to GHB (SFB860).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- RACK1
- Receptor for Activated C Kinase 1
- BioID
- proximity-dependent Biotin Identification
- HRP
- horseradish peroxidase
- MAPK
- mitogen-activated protein kinase
- PKA
- protein kinase A
- PKC
- protein kinase C
- SILAC
- stable isotope labeling with amino acids in cell culture
- stage tips
- stop and go extraction tips
- YEPD
- yeast extract peptone dextrose
- YNB
- yeast nitrogen base
- LFQ
- label-free quantification
- tSIM
- targeted single ion monitoring.
REFERENCES
- 1. Pan C. Q., Sudol M., Sheetz M., and Low B. C. (2012) Modularity and functional plasticity of scaffold proteins as p(l) acemakers in cell signaling. Cell Signal 24, 2143–2165 [DOI] [PubMed] [Google Scholar]
- 2. Vondriska T. M., Pass J. M., and Ping P. (2004) Scaffold proteins and assembly of multiprotein signaling complexes. J. Mol. Cell Cardiol 37, 391–397 [DOI] [PubMed] [Google Scholar]
- 3. Chantrel Y., Gaisne M., Lions C., and Verdière J. (1998) The transcriptional regulator hap1p (cyp1p) is essential for anaerobic or heme-deficient growth of saccharomyces cerevisiae: genetic and molecular characterization of an extragenic suppressor that encodes a wd repeat protein. Genetics 148, 559–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coyle S. M., Gilbert W. V., and Doudna J. A. (2009) Direct link between rack1 function and localization at the ribosome in vivo. Mol. Cell. Biol. 29, 1626–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tarnowski K., Fituch K., Szczepanowski R. H., Dadlez M., and Kaus-Drobek M. (2014) Patterns of structural dynamics in rack1 protein retained throughout evolution: a hydrogen-deuterium exchange study of three orthologs. Protein Sci. Publ. Protein. Soc. 23, 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ero R., Dimitrova V. T., Chen Y., Bu W., Feng S., Liu T., Wang P., Xue C., Tan S. M., and Gao Y.-G. (2015) Crystal structure of gib2, a signal-transducing protein scaffold associated with ribosomes in cryptococcus neoformans. Sci. Rep 5, 8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rabl J., Leibundgut M., Ataide S. F., Haag A., and Ban N. (2011) Crystal structure of the eukaryotic 40s ribosomal subunit in complex with initiation factor 1. Science 331, 730–736 [DOI] [PubMed] [Google Scholar]
- 8. Ruiz Carrillo D., Chandrasekaran R., Nilsson M., Cornvik T., Liew C. W., Tan S. M., and Lescar J. (2012) Structure of human rack1 protein at a resolution of 2.45 å. Acta Crystallograph Sect F Struct Biol. Cryst. Commun. 68, 867–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ullah H., Scappini E. L., Moon A. F., Williams L. V., Armstrong D. L., and Pedersen L. C. (2008) Structure of a signal transduction regulator, rack1, from arabidopsis thaliana. Protein Sci. Publ. Protein. Soc. 17, 1771–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ben-Shem A., Garreau de Loubresse N., Melnikov S., Jenner L., Yusupova G., and Yusupov M. (2011) The structure of the eukaryotic ribosome at 3.0 å resolution. Science 334, 1524–1529 [DOI] [PubMed] [Google Scholar]
- 11. Inada T., Winstall E., Tarun S. Z., Yates J. R., Schieltz D., and Sachs A. B. (2002) One-step affinity purification of the yeast ribosome and its associated proteins and mrnas. RNA N Y N 8, 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Link A. J., Eng J., Schieltz D. M., Carmack E., Mize G. J., Morris D. R., Garvik B. M., and Yates J. R. (1999) Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 17, 676–682 [DOI] [PubMed] [Google Scholar]
- 13. Sengupta J., Nilsson J., Gursky R., Spahn C. M. T., Nissen P., and Frank J. (2004) Identification of the versatile scaffold protein rack1 on the eukaryotic ribosome by cryo-em. Nat. Struct. Mol. Biol. 11, 957–962 [DOI] [PubMed] [Google Scholar]
- 14. Baum S., Bittins M., Frey S., and Seedorf M. (2004) Asc1p, a wd40-domain containing adaptor protein, is required for the interaction of the rna-binding protein scp160p with polysomes. Biochem. J. 380, 823–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirschmann W. D., Westendorf H., Mayer A., Cannarozzi G., Cramer P., and Jansen R.-P. (2014) Scp160p is required for translational efficiency of codon-optimized mrnas in yeast. Nucleic Acids Res. 42, 4043–4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sezen B., Seedorf M., and Schiebel E. (2009) The sesa network links duplication of the yeast centrosome with the protein translation machinery. Genes Dev. 23, 1559–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rachfall N., Schmitt K., Bandau S., Smolinski N., Ehrenreich A., Valerius O., and Braus G. H. (2013) RACK1/asc1p, a ribosomal node in cellular signaling. Mol. Cell. Proteomics MCP 12, 87–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ceci M., Gaviraghi C., Gorrini C., Sala L. A., Offenhäuser N., Marchisio P. C., and Biffo S. (2003) Release of eif6 (p27bbp) from the 60s subunit allows 80s ribosome assembly. Nature 426, 579–584 [DOI] [PubMed] [Google Scholar]
- 19. Gavin A.-C., Bösche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J. M., Michon A.-M., Cruciat C.-M., Remor M., Höfert C., Schelder M., Brajenovic M., Ruffner H., Merino A., Klein K., Hudak M., Dickson D., Rudi T., Gnau V., Bauch A., Bastuck S., Huhse B., Leutwein C., Heurtier M.-A., Copley R. R., Edelmann A., Querfurth E., Rybin V., Drewes G., Raida M., Bouwmeester T., Bork P., Seraphin B., Kuster B., Neubauer G., and Superti-Furga G. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147 [DOI] [PubMed] [Google Scholar]
- 20. Guo J., Wang S., Valerius O., Hall H., Zeng Q., Li J.-F., Weston D. J., Ellis B. E., and Chen J.-G. (2011) Involvement of arabidopsis rack1 in protein translation and its regulation by abscisic acid. Plant Physiol. 155, 370–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kouba T., Rutkai E., Karásková M., and Valášek L. S. (2012) The eif3c/nip1 pci domain interacts with rna and rack1/asc1 and promotes assembly of translation preinitiation complexes. Nucleic Acids Res. 40, 2683–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruan Y., Sun L., Hao Y., Wang L., Xu J., Zhang W., Xie J., Guo L., Zhou L., Yun X., Zhu H., Shen A., and Gu J. (2012) Ribosomal rack1 promotes chemoresistance and growth in human hepatocellular carcinoma. J. Clin. Invest. 122, 2554–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmitt K., Smolinski N., Neumann P., Schmaul S., Hofer-Pretz V., Braus G. H., and Valerius O. (2017) Asc1p/rack1 connects ribosomes to eukaryotic phospho-signaling. Mol. Cell. Biol. 37, e00279–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y., Shen G., Gong J., Shen D., Whittington A., Qing J., Treloar J., Boisvert S., Zhang Z., Yang C., and Wang P. (2014) Noncanonical gβ gib2 is a scaffolding protein promoting camp signaling through functions of ras1 and cac1 proteins in cryptococcus neoformans. J. Biol. Chem. 289, 12202–12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Breitkreutz A., Choi H., Sharom J. R., Boucher L., Neduva V., Larsen B., Lin Z.-Y., Breitkreutz B.-J., Stark C., Liu G., Ahn J., Dewar-Darch D., Reguly T., Tang X., Almeida R., Qin Z. S., Pawson T., Gingras A.-C., Nesvizhskii A. I., and Tyers M. (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zeller C. E., Parnell S. C., and Dohlman H. G. (2007) The rack1 ortholog asc1 functions as a g-protein beta subunit coupled to glucose responsiveness in yeast. J. Biol. Chem. 282, 25168–25176 [DOI] [PubMed] [Google Scholar]
- 27. Chang B. Y., Chiang M., and Cartwright C. A. (2001) The interaction of src and rack1 is enhanced by activation of protein kinase c and tyrosine phosphorylation of rack1. J. Biol. Chem. 276, 20346–20356 [DOI] [PubMed] [Google Scholar]
- 28. Gandin V., Gutierrez G. J., Brill L. M., Varsano T., Feng Y., Aza-Blanc P., Au Q., McLaughlan S., Ferreira T. A., Alain T., Sonenberg N., Topisirovic I., and Ronai Z. A. (2013) Degradation of newly synthesized polypeptides by ribosome-associated rack1/c-jun n-terminal kinase/eukaryotic elongation factor 1a2 complex. Mol. Cell. Biol. 33, 2510–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thompson M. K., Rojas-Duran M. F., Gangaramani P., and Gilbert W. V. (2016) The ribosomal protein asc1/rack1 is required for efficient translation of short mrnas. eLife 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Batsios P., Meyer I., and Gräf R. (2016) Proximity-dependent biotin identification (bioid) in dictyostelium amoebae. Methods Enzymol. 569, 23–42 [DOI] [PubMed] [Google Scholar]
- 32. Chen A. L., Kim E. W., Toh J. Y., Vashisht A. A., Rashoff A. Q., Van C., Huang A. S., Moon A. S., Bell H. N., Bentolila L. A., Wohlschlegel J. A., and Bradley P. J. (2015) Novel components of the toxoplasma inner membrane complex revealed by bioid. mBio 6, e02357–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morriswood B., Havlicek K., Demmel L., Yavuz S., Sealey-Cardona M., Vidilaseris K., Anrather D., Kostan J., Djinovic-Carugo K., Roux K. J., and Warren G. (2013) Novel bilobe components in trypanosoma brucei identified using proximity-dependent biotinylation. Eukaryot. Cell 12, 356–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson M. D., Harreman M., Taschner M., Reid J., Walker J., Erdjument-Bromage H., Tempst P., and Svejstrup J. Q. (2013) Proteasome-mediated processing of def1, a critical step in the cellular response to transcription stress. Cell 154, 983–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wessel D., and Flügge U. I. (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138, 141–143 [DOI] [PubMed] [Google Scholar]
- 36. Shevchenko A., Wilm M., Vorm O., and Mann M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 37. Rappsilber J., Ishihama Y., and Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and lc/ms sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 38. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using stagetips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 39. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 40. Melamed D., Bar-Ziv L., Truzman Y., and Arava Y. (2010) Asc1 supports cell-wall integrity near bud sites by a pkc1 independent mechanism. PloS One 5, e11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valerius O., Kleinschmidt M., Rachfall N., Schulze F., López Marín S., Hoppert M., Streckfuss-Bömeke K., Fischer C., and Braus G. H. (2007) The saccharomyces homolog of mammalian rack1, cpc2/asc1p, is required for flo11-dependent adhesive growth and dimorphism. Mol. Cell. Proteomics 6, 1968–1979 [DOI] [PubMed] [Google Scholar]
- 42. Parsons A. B., Brost R. L., Ding H., Li Z., Zhang C., Sheikh B., Brown G. W., Kane P. M., Hughes T. R., and Boone C. (2004) Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 22, 62–69 [DOI] [PubMed] [Google Scholar]
- 43. Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, silac, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 44. Ho Y., Gruhler A., Heilbut A., Bader G. D., Moore L., Adams S.-L., Millar A., Taylor P., Bennett K., Boutilier K., Yang L., Wolting C., Donaldson I., Schandorff S., Shewnarane J., Vo M., Taggart J., Goudreault M., Muskat B., Alfarano C., Dewar D., Lin Z., Michalickova K., Willems A. R., Sassi H., Nielsen P. A., Rasmussen K. J., Andersen J. R., Johansen L. E., Hansen L. H., Jespersen H., Podtelejnikov A., Nielsen E., Crawford J., Poulsen V., Sørensen B. D., Matthiesen J., Hendrickson R. C., Gleeson F., Pawson T., Moran M. F., Durocher D., Mann M., Hogue C. W. V., Figeys D., and Tyers M. (2002) Systematic identification of protein complexes in saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183 [DOI] [PubMed] [Google Scholar]
- 45. Ossareh-Nazari B., Bonizec M., Cohen M., Dokudovskaya S., Delalande F., Schaeffer C., Van Dorsselaer A., and Dargemont C. (2010) Cdc48 and ufd3, new partners of the ubiquitin protease ubp3, are required for ribophagy. EMBO Rep. 11, 548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schütz S., Fischer U., Altvater M., Nerurkar P., Peña C., Gerber M., Chang Y., Caesar S., Schubert O. T., Schlenstedt G., and Panse V. G. (2014) A rangtp-independent mechanism allows ribosomal protein nuclear import for ribosome assembly. eLife 3, e03473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Auesukaree C., Damnernsawad A., Kruatrachue M., Pokethitiyook P., Boonchird C., Kaneko Y., and Harashima S. (2009) Genome-wide identification of genes involved in tolerance to various environmental stresses in saccharomyces cerevisiae. J. Appl. Genet. 50, 301–310 [DOI] [PubMed] [Google Scholar]
- 48. Sinha H., David L., Pascon R. C., Clauder-Münster S., Krishnakumar S., Nguyen M., Shi G., Dean J., Davis R. W., Oefner P. J., McCusker J. H., and Steinmetz L. M. (2008) Sequential elimination of major-effect contributors identifies additional quantitative trait loci conditioning high-temperature growth in yeast. Genetics 180, 1661–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim D. I., Jensen S. C., Noble K. A., Kc B., Roux K. H., Motamedchaboki K., and Roux K. J. (2016) An improved smaller biotin ligase for bioid proximity labeling. Mol. Biol. Cell 27, 1188–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Angenstein F., Evans A. M., Settlage R. E., Moran S. T., Ling S.-C., Klintsova A. Y., Shabanowitz J., Hunt D. F., and Greenough W. T. (2002) A receptor for activated c kinase is part of messenger ribonucleoprotein complexes associated with polya-mrnas in neurons. J. Neurosci. 22, 8827–8837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Llácer J. L., Hussain T., Marler L., Aitken C. E., Thakur A., Lorsch J. R., Hinnebusch A. G., and Ramakrishnan V. (2015) Conformational differences between open and closed states of the eukaryotic translation initiation complex. Mol. Cell 59, 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van den Elzen A. M. G., Schuller A., Green R., and Séraphin B. (2014) Dom34-hbs1 mediated dissociation of inactive 80s ribosomes promotes restart of translation after stress. EMBO J. 33, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Van Dyke N., Baby J., and Van Dyke M. W. (2006) Stm1p, a ribosome-associated protein, is important for protein synthesis in saccharomyces cerevisiae under nutritional stress conditions. J. Mol. Biol. 358, 1023–1031 [DOI] [PubMed] [Google Scholar]
- 54. Van Dyke N., Chanchorn E., and Van Dyke M. W. (2013) The saccharomyces cerevisiae protein stm1p facilitates ribosome preservation during quiescence. Biochem. Biophys. Res. Commun. 430, 745–750 [DOI] [PubMed] [Google Scholar]
- 55. Bolger G. B. (2017) The rna-binding protein serbp1 interacts selectively with the signaling protein rack1. Cell Signal 35, 256–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Woudstra E. C., Gilbert C., Fellows J., Jansen L., Brouwer J., Erdjument-Bromage H., Tempst P., and Svejstrup J. Q. (2002) A rad26-def1 complex coordinates repair and rna pol ii proteolysis in response to dna damage. Nature 415, 929–933 [DOI] [PubMed] [Google Scholar]
- 57. Kvint K., Uhler J. P., Taschner M. J., Sigurdsson S., Erdjument-Bromage H., Tempst P., and Svejstrup J. Q. (2008) Reversal of rna polymerase ii ubiquitylation by the ubiquitin protease ubp3. Mol. Cell 30, 498–506 [DOI] [PubMed] [Google Scholar]
- 58. Harris J. K., Kelley S. T., Spiegelman G. B., and Pace N. R. (2003) The genetic core of the universal ancestor. Genome Res. 13, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lindstrom D. L., Squazzo S. L., Muster N., Burckin T. A., Wachter K. C., Emigh C. A., McCleery J. A., Yates J. R., and Hartzog G. A. (2003) Dual roles for spt5 in pre-mrna processing and transcription elongation revealed by identification of spt5-associated proteins. Mol. Cell. Biol. 23, 1368–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mayer A., Schreieck A., Lidschreiber M., Leike K., Martin D. E., and Cramer P. (2012) The spt5 c-terminal region recruits yeast 3′ rna cleavage factor i. Mol. Cell. Biol. 32, 1321–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brewster N. K., Johnston G. C., and Singer R. A. (2001) A bipartite yeast ssrp1 analog comprised of pob3 and nhp6 proteins modulates transcription. Mol. Cell. Biol. 21, 3491–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Formosa T., Eriksson P., Wittmeyer J., Ginn J., Yu Y., and Stillman D. J. (2001) Spt16-pob3 and the hmg protein nhp6 combine to form the nucleosome-binding factor spn. EMBO J. 20, 3506–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schlesinger M. B., and Formosa T. (2000) POB3 is required for both transcription and replication in the yeast saccharomyces cerevisiae. Genetics 155, 1593–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schwabish M. A., and Struhl K. (2004) Evidence for eviction and rapid deposition of histones upon transcriptional elongation by rna polymerase ii. Mol. Cell. Biol. 24, 10111–10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Basquin J., Roudko V. V., Rode M., Basquin C., Séraphin B., and Conti E. (2012) Architecture of the nuclease module of the yeast ccr4-not complex: the not1-caf1-ccr4 interaction. Mol. Cell 48, 207–218 [DOI] [PubMed] [Google Scholar]
- 66. Collart M. A., Panasenko O. O., and Nikolaev S. I. (2013) The not3/5 subunit of the ccr4-not complex: a central regulator of gene expression that integrates signals between the cytoplasm and the nucleus in eukaryotic cells. Cell Signal. 25, 743–751 [DOI] [PubMed] [Google Scholar]
- 67. Panasenko O. O., and Collart M. A. (2012) Presence of not5 and ubiquitinated rps7a in polysome fractions depends upon the not4 e3 ligase. Mol. Microbiol. 83, 640–653 [DOI] [PubMed] [Google Scholar]
- 68. Takemaru K., Harashima S., Ueda H., and Hirose S. (1998) Yeast coactivator mbf1 mediates gcn4-dependent transcriptional activation. Mol. Cell. Biol. 18, 4971–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wada A., Yamazaki Y., Fujita N., and Ishihama A. (1990) Structure and probable genetic location of a “ribosome modulation factor” associated with 100s ribosomes in stationary-phase escherichia coli cells. Proc. Natl. Acad. Sci. U.S.A. 87, 2657–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krokowski D., Gaccioli F., Majumder M., Mullins M. R., Yuan C. L., Papadopoulou B., Merrick W. C., Komar A. A., Taylor D., and Hatzoglou M. (2011) Characterization of hibernating ribosomes in mammalian cells. Cell Cycle Georget Tex 10, 2691–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ashe M. P., De Long S. K., and Sachs A. B. (2000) Glucose depletion rapidly inhibits translation initiation in yeast. Mol. Biol. Cell 11, 833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dibrov E., Robinson K. M., and Lemire B. D. (1997) The coq5 gene encodes a yeast mitochondrial protein necessary for ubiquinone biosynthesis and the assembly of the respiratory chain. J. Biol. Chem. 272, 9175–9181 [DOI] [PubMed] [Google Scholar]
- 73. Wilson K. P., Shewchuk L. M., Brennan R. G., Otsuka A. J., and Matthews B. W. (1992) Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and dna-binding domains. Proc. Natl. Acad. Sci. U.S.A. 89, 9257–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Smolinski N., and Valerius O. (2016) Nachbarschaftsstudien am ribosom: proximity dependent biotin identification. BIOspektrum 22, 134–136 [Google Scholar]
- 75. Huh W.-K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., and O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 76. Gueldener U., Heinisch J., Koehler G. J., Voss D., and Hegemann J. H. (2002) A second set of loxp marker cassettes for cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mumberg D., Müller R., and Funk M. (1994) Regulatable promoters of saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 22, 5767–5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. van Werven F. J., and Timmers H. T. M. (2006) The use of biotin tagging in saccharomyces cerevisiae improves the sensitivity of chromatin immunoprecipitation. Nucleic Acids Res. 34, e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Taura T., Krebber H., and Silver P. A. (1998) A member of the ran-binding protein family, yrb2p, is involved in nuclear protein export. Proc. Natl. Acad. Sci. U.S.A. 95, 7427–7432 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw mass spectrometric data and search output files are deposited at ProteomeXchange via the PRIDE database with the accession numbers “PXD005612” (BioID experiments) and “PXD007858” (GFP-trap experiments).