

Abstract
UV-irradiation of a self-assembled benzophenone bis-urea macrocycle generates µM amounts of radicals that persist for weeks under ambient conditions. High-Field EPR and variable temperature X-band EPR studies suggest a resonance stabilized radical pair through H-abstraction. These endogenous radicals were applied as a polarizing agent for magic angle spinning (MAS) dynamic nuclear polarization (DNP) NMR enhancement. The field-stepped DNP enhancement profile exhibits a sharp peak with a maximum enhancement of εon/off = 4 superimposed on a nearly constant DNP enhancement of εon/off = 2 over a broad field range. This maximum coincides with the high field EPR absorption spectrum, consistent with an Overhauser effect mechanism. DNP enhancement was observed for both the host and guests, suggesting that even low levels of endogenous radicals can facilitate the study of host-guest relationships in the solid-state.
Keywords: benzophenone, DNP, EPR spectroscopy, macrocycle, MAS
FULL PAPER
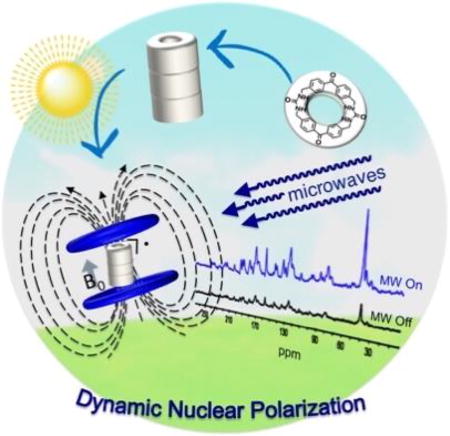
Persistent organic radicals are generated upon UV-irradiation as a consequence of their self-assembled structure. The endogenous host framework radicals, present in ~µM quantities, are characterized and applied as a polarizing agent for magic angle spinning (MAS) dynamic nuclear polarization (DNP) NMR spectroscopy where they displayed a maximum enhancement of four for both host and guest.
Introduction
Dynamic Nuclear Polarization (DNP) has gained widespread use as a means to improve the sensitivity of nuclear magnetic resonance (NMR) signals.1–3 In material science, solid-state DNP methods primarily rely on exogenous radicals, such as TOTAPOL and AMUPol, which are typically introduced by incipient wetness impregnation in mM concentrations.4 Recent work suggests that high field DNP enhancement may also be observed with endogenous radicals.5 This manuscript probes the structure of an unusually persistent endogenous radical in bis-urea macrocycle 1 and its use as a polarizing agent for DNP enhancement. The photogenerated radical was first noted when investigating the applications of benzophenone bis-urea macrocycle 1 to facilitate selective photooxidations.6, 7 The prolonged stability of these endogenous radicals at room temperature appears to be a consequence of the columnar assembly and crystal packing of the porous organic crystals, where as no evidence of radical formation is observed in solution.6 Herein, we probe the structure of the radical, estimate its quantity, and evaluate its lifetime by EPR spectroscopy. Finally, we demonstrate that the low levels of endogenous radicals in 1 can be applied to hyperpolarize nuclei and enhance the NMR signals of both the host and its encapsulated DMSO guest (Figure 1).
Figure 1.

A benzophenone bis-urea macrocycle self-assembles from DMSO to form host 1 with encapsulated DMSO. A persistent paramagnetic species is generated when 1 is UV irradiated at 350 nm with a Hanovia 450 W medium pressure mercury arc lamp. This long-lived intermediate was used to create DNP enhancement over a broad field range in MAS NMR experiments.
Solutions of exogenous stable radicals such as AMUPol, known as “DNP juice”, are typically used in mM concentration as polarizing agents for solid-state DNP MAS NMR at ~100 K.1, 4 Under microwave irradiation, the exogenous radical transfers its spin polarization to neighbouring protons. The large spin polarization of the protons generates a spin polarization gradient leading to spin diffusion to nearby protons resulting in a uniform proton hyperpolarization throughout the sample.8 The proton spin polarization can be transferred to other nuclei, such as 13C, using a Cross-Polarization (CP) pulse sequence. Recently, Eichorn et al. demonstrated DNP in pyruvic acid following low temperature UV-irradiation without any exogenous radical.9,10 Instead, the quasi-stable, short-lived UV-induced radicals were shown to afford sizeable DNP enhancements at low temperatures.9, 10 Larger DNP enhancements are possible in solution and have been observed in frozen media under constant irradiation (photochemically induced DNP).11 With exogenous radicals, optimum concentration is key, as high radical concentration can result in excessive paramagnetic relaxation and line broadening.4, 12 Few studies have examined DNP using endogenous radicals in the solid-state or single crystals.5, 9, 10
Recently, the Shimizu group found columnar supramolecular assembly of benzophenone altered its photophysics and lead to the formation of stable radicals as an emergent property. Benzophenone is an extensively studied chromophore with promising applications ranging from photosensitization to genome sequencing and materials chemistry.13 Its photochemistry affords a triplet state as a result of fast intersystem crossing, which can rapidly undergo H-abstraction to yield a ketyl radical. The radical is only observed at low temperatures (77 K) as a doublet with a g value of 2.0061 or as a radical anion via 1-electron reductions.14
Compound 1 preorganizes two benzophenones within a macrocycle. Upon recrystallization from hot DMSO, 1 assembles in needle-like crystals through predictable bifurcated urea hydrogen-bonding interactions to afford columns (Figure 1).7 The crystals are robust and contain accessible channels that are filled with DMSO guests (Figure 3b). DMSO guests can be removed by heating and other solvents and substrates can be readily loaded in the channels.7 The assembled structure enforces close contacts between the benzophenone groups to the methylene H’s on neighbouring columns (2.44 – 2.81 Å, Figure S4) and orients individual benzophenone units close in space.6 Molecular self-assembly and crystal packing in 1 dramatically quenches the phosphorescence lifetime from µs to < 1 ns, likely through a non-radiative pathway.6 Mechanistic investigations suggest that columnar assembly and packing stabilizes some type of photogenerated radical that is stable for weeks in the dark at room temperature. No evidence of radical formation is observed in solution where the molecule only exists in monomer form. Our hypothesis is that the solid-state structure may facilitate an H-abstraction reaction to form the ground state triplet radical pair (RP) shown in Figure 2A.
Figure 3.

Steady-state EPR studies of 1. (A) Variable temperature X-band EPR study of 1 at 293 K (black line), 100 K (red line) and 10 K (blue line) (B) CW high-field (240 GHz) EPR absorption curve of 1 at 6 K vs. ppm (black line, see ESI for plot in mT). An EasySpin simulation of the high field EPR (red line) for two S=1/2 electron spins, one weighted x4. Inset: Plot of DNP enhancement vs. ppm observed in a field-step study.
Figure 2.

(A) Proposed photochemistry of 1, which suggests the known photoreaction of benzophenone in the presence of an H donor. (B) X–band EPR signal, pre- and post-UV exposure (simulation inset) centered at an average g value of 2.006. (C) Dark-decay study of 1 (5 mg) after 1 h of UV irradiation. (D) Radical generation study shows that radical signal reaches a maximum intensity after 5h.
Results and Discussion
We probed the structure of the photogenerated radicals of 1 through solid–state X-band EPR studies on 1 and on a fully 15N-labeled derivative. Both samples exhibited nearly identical broad peaks (g = 2.006, Figures 2B and S10). A simulation using parameters for a weakly exchange–coupled RP (see inset Figure 2B and S13 for more details and parameters) shows that the overall spectral width and main features of the experimental spectrum can be accounted for with such a model. This also rules out the presence of a photochemically excited (or thermally relaxed) molecular triplet state, which would be expected to exhibit much broader line widths > 1000 G. The similar broad peak observed for the 15N-labeled derivative suggests that the triplet RP, drawn in Figure 2A, adopts a conformation where the 15N hyperfine is less than half of the natural line width (~14 G, Figure S13), which is reasonable for the benzylic radical structure shown.
The stability of the photoinduced RP was investigated through dark decay studies, which were performed by UV irradiating 1 (1 h, rt) and recording the EPR spectra over time (0 h – 26 days) while storing the sample in the dark. The double integration of the EPR signal is plotted vs. time after UV-irradiation in Figure 2C. Little to no loss of signal intensity is observed, suggesting that once generated, the radicals created in 1 are stable for weeks. The small fluctuations in the observed EPR signal intensity are likely due to variations in the orientation of the crystalline sample with respect to the magnetic field.15 The stability of the photogenerated radical is likely a consequence of two effects. First, it is probable that once generated, the radicals are unable to terminate as benzophenone is known to do in solution, due to the rigid structure of the macrocycles. Second, the proposed photochemistry suggests that the radicals are generated in positions that allow resonance stabilization into the neighbouring benzene rings resulting in enhanced stabilization of the radicals.
The concentration dependence of the radicals generated by UV-irradiation of 1 was monitored for exposures from 30 min to 7 h. Figure 2D plots the double integration of the EPR signal vs. irradiation time and indicates that the number of radicals reaches a maximum after 5–7 h. After 7 h of UV-irradiation, 1 was slightly yellow in colour but still suitable for single crystal X-ray diffraction. No changes were observed in the X-ray structure (Figure S14) or in the 1H NMR, indicating that 1 is stable and the absolute radical concentration is low. The maximum concentration was approximated by calibration with standard solutions of TEMPO in benzene under identical conditions (Figure S15).10, 16 The number of radicals generated by 1 (4.5 mg) is similar to a 5.4 µM stock solution (0.1 mL), which is equivalent to a radical forming in ~0.01% of the macrocycles.
Variable temperature EPR spectra were recorded to resolve hyperfine couplings that may not be observed at rt. First, spectra were recorded at high temperatures (293, 348 and 398K, Figure S16) for 1 and the 15N labelled 1. No change in the g-factor or the coupling pattern was observed, although the intensity of the signal decreased with increasing temperature. Cooling the sample to 100K did not markedly change the g-factor, although a slight anisotropy was observed at g = 2.001 (Figure 3A, red spectra). Further cooling to 10 K resulted in a change in the EPR spectrum, leading to a powder pattern shape with an overall shift in g-factor to 2.001 and a slight anisotropy. This can be explained by the orientation-dependent dipolar contribution of rigid radical pairs, which are often further complicated by hyperfine interactions ultimately resulting in line broadening due to motional averaging. By cooling the sample, we were able to overcome the Boltzmann distribution and rebuild the g-factor matrix leading to the significant over population of the lower energy states. The lack of hyperfine interactions at low temperatures is consistent with delocalized radicals, since hyperfine interactions are described by the probability of finding an electron at the site of a nucleus (i.e. Fermi contact interaction). For a delocalized radical the probability is small resulting in an averaged effect.17
The high field EPR shown in Fig. 3B was acquired on the NHMFL’s 240 GHz spectrometer and converted to a ppm scale to assist in our interpretation of the field-stepped DNP data. The solid-state high-field CW absorbance EPR spectra of UV-irradiated 1 (25 mg) at 6K shows a broad baseline component at g = 2.006 with a sharper transition at g = 2.003. Spectral simulations carried out using the EasySpin software are in agreement with two S=½ radical species with a sharp isotropic signal at g = 2.003 as well as a second broad anisotropic signal. An extrapolated simulation to X-band indicates that the lines would overlap at low field, consistent with the observations in Figure 3. Variable temperature EPR and high-field experiments both suggest the assignment as two radicals, possibly a delocalized RP.
The thermally polarized CP MAS NMR spectra were recorded at a spinning speed of 11.3 kHz after 0h, 2h and 4h of UV-irradiation (Figure 4). The 13C NMR peaks are surprisingly sharp for both the host and included DMSO. The spectra before and after UV-irradiation are nearly identical and no paramagnetic broadening is observed, consistent with the low estimated radical concentration and a well-ordered structure.12 The THF loaded host crystals, also show a sharp thermally polarized CP MAS NMR spectra under similar conditions (Figure S24).
Figure 4.

Top: (A) Peak assignment of the benzophenone bis-urea macrocycle, which self-assembles to form 1. (B) XRD structure of 1 depicting the encapsulated DMSO (space fill), the assembly of 1 promotes the formation of a stable radical upon UV irradiation. Bottom: CP MAS NMR spectra of thermally polarized 1 recorded at a spinning speed of 11.3 kHz. (I) no UV, (II) 2 h. UV and (III) 4 h. UV.
Solid-state MAS DNP experiments gave a maximum enhancement factor εon/off = 4. The DNP enhancement profile, shown in the inset of Figure 3B, demonstrates that nearly constant DNP enhancement εon/off = 2 is obtained over a broad field range of approximately 120 mT with a sharp peak in the enhancement factor of εon/off = 4. DNP enhancement profiles are more typically limited to a much smaller field range of 30–40 mT compared to our experimentally observed 120 mT range for 1.2, 18 Given the broad EPR signal observed by 1, this profile is not surprising. Figure 5 depicts the optimized DNP CP-MAS NMR spectrum at the magnetic field that optimized the DNP enhancement, where εon/off = 4 was recorded at 112 K and a spinning speed of 7.0 kHz. Similar enhancement factors were observed for all NMR peaks including the encapsulated DMSO. The spinning side bands are a result of the low spinning speed required to keep the sample stabilized at the low temperatures employed. The non-irradiated sample showed no DNP enhancement, which indicates that the enhancement results from irradiation of the sample and is not a microwave induced heating artifact (Figure S25). The constant and positive sign of this field-stepped study suggests an Overhauser mechanism.19
Figure 5.

Optimized DNP CP-MAS NMR enhancement observed at 14.085 T under CP DNP MAS NMR conditions demonstrating a total enhancement of εon/off = 4. Recorded at 112 K and a spinning speed of 7.0 kHz, microwave on (red line) vs. microwave off (black line). *Indicates spinning sidebands
Much larger DNP enhancements can be observed through traditional impregnation methods; therefore, the UV-irradiated sample was impregnated with AMUPol (10 mM, 10–12 µL of a 6:3:1 glycerol-d8, D2O,H2O solution) and the DNP CP-MAS NMR spectrum acquired using the optimal field conditions for 1 (Figure S27). A DNP enhancement of εon/off ~ 6 was observed for all peaks with the exception of the glycerol peaks which were enhanced by a factor of εon/off ~ 20. This higher observed enhancement is likely due to the dual effect of the endogenous and exogenous radicals in this sample. The larger observed enhancement for glycerol is expected because the exogenous radical is in more direct contact with glycerol molecules present in the bulk DNP juice. It should be noted that impregnating the sample did not yield much higher polarization levels while introducing solvent signals. These results suggest that in these porous organic crystals, it may be more fruitful to use the endogenous radicals formed by 1 to enhance the NMR signals of the host:guest materials as opposed to traditional impregnation methods. Such DNP enhancement may be observable in other structures.
Conclusions
We have demonstrated that UV-irradiation of assembled benzophenone bis-urea macrocycles can generate low ~µM concentrations of long-lived RPs that persist for weeks at room temperature in the dark. Labelling experiments ruled out nitrogen-centered radicals. High field EPR data and variable temperature X-band EPR studies suggest the formation of two radicals. Our hypothesis is that the columnar assembled structure of 1 facilitates an H-abstraction reaction and significantly stabilizes the triplet RP. Stable and persistent organic radicals are rare and typically belong to four structural classes.20 These results suggest that additional organic radicals may be stabilized by similar supramolecular assembly. Thus, we are currently exploring building blocks that contain other known sensitizers to investigate if their crystalline solids also afford stable radicals upon photolysis.
In summary, we have demonstrated that the photo-induced radical species generated by 1 can be utilized as a polarizing agent to significantly enhance NMR signals for both the host and its encapsulated guest DMSO, even though the concentration of endogenous radicals in 1 is orders of magnitudes below typical quantities of exogenous agents used for DNP NMR. UV-irradiated 1 showed a surprisingly broad DNP enhancement profile of over 120 mT, demonstrating that it is not necessary to tune the microwave frequency in order to observe DNP enhancement. The contribution of the cross-effect mechanism appears to be insignificant, since the field-stepped DNP profile exhibits only a single maximum with εon/off > 0. These results suggest that the design and incorporation of low levels of endogenous radicals into host frameworks may be broadly applied for NMR signal enhancement. Future DNP studies will focus on investigating a variety of guest molecules to see if such systems can be widely applied to study host: guest interactions in the solid-state.
Experimental
EPR Sample Preparation
Neat crystals of 1 were added to a Norell quartz EPR tube, purged under Argon gas, sealed under Parafilm, and capped. The samples were UV-irradiated at 350 nm at rt with a Hanovia 450 W medium pressure mercury arc lamp cooled in a quartz immersion well. EPR signals were doubly integrated three times and averaged from the 3305 G to 3370 G range using Xenon software (v 1.1b.66).
MAS DNP NMR Sample Preparation
UV irradiated sample
The sample was prepared by UV-irradiating a crystalline sample of 1 (25 mg) for 7 hours using a medium pressure Hanovia Hg lamp using Corning glass filters to isolate the 366 nm Hg line. After UV irradiation, the sample was packed neat into a 3.2 mm sapphire rotor and DNP experiments were performed.
Non-irradiated sample
The sample was prepared by packing a neat unirradiated crystalline sample of 1 (25 mg) into a 3.2 mm sapphire rotor and DNP experiments were performed.
AMUPol impregnated sample
The previously packed UV-irradiated host 1 sample (25 mg) was unpacked from the sapphire rotor and impregnated with AMUPol (10 mM, 10–12 µL of a 6:3:1 glycerol-d8, D2O, H2O solution) and the DNP CP-MAS NMR spectrum acquired using the optimal field conditions for 1.
MAS DNP NMR Experimental
All 13C NMR spectra were acquired using a ramped CP-MAS pulse sequence on a 600 MHz Bruker DNP spectrometer (3.2 mm sapphire rotor) at the National High Magnetic Field Laboratory. DNP experiments were conducted with a high-powered Cryomagnetics 394 GHz gyrotron with an output of 24 mW. The output was guided via quasi-optics to a corrugated waveguide and into the probe head. Experiments were conducted at 112 K with spinning speeds at 7.0 kHz.
Supplementary Material
Acknowledgments
Thanks to Zhehong Gan, Ivan Hung, Jose Amaya, Steven Ratigan, and Job Grant. This work was supported by the National Science Foundation (CHE-1608874, CHE-1305136, and CHE-1464817). The National High Magnetic Field Laboratory is supported by the NSF (DMR-1157490) and by the State of Florida. The DNP system is funded in part by NIH S10 OD018519 (magnet and console), and NSF CHE-1229170 (gyrotron).
References
- 1.Lee, Hediger S, De Paëpe G. Solid State Nucl. Magn. 2015;66–67:6–20. doi: 10.1016/j.ssnmr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Smith AN, Long JR. Anal. Chem. 2016;88:122–132. doi: 10.1021/acs.analchem.5b04376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossini AJ, Zagdoun A, Lelli M, Lesage A, Copéret C, Emsley L. Acc. Chem. Res. 2013;46:1942–1951. doi: 10.1021/ar300322x. [DOI] [PubMed] [Google Scholar]
- 4.Akbey U, Franks WT, Linden A, Orwick-Rydmark M, Lange S, Oschkinat H. In: Topics in Current Chemistry. Kuhn LT, editor. Vol. 6. Springer; New York: 2013. pp. 181–228. 338. [DOI] [PubMed] [Google Scholar]
- 5.(a) Maly T, Cui D, Griffin RG, Miller A-F. J. Phys. Chem. B. 2012;116:7055–7065. doi: 10.1021/jp300539j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Riikonen J, Rigolet S, Marichal C, Aussenac F, Lalevée J, Morlet-Savary F, Fioux P, Dietlin C, Bonne M, Lebeau B, Lehto V-P. J. Phys. Chem. C. 2015;119:19272–19278. [Google Scholar]
- 6.Geer M, Walla M, Solntsev K, Strassert C, Shimizu L. J. Org. Chem. 2013;78:5568–5578. doi: 10.1021/jo400685u. [DOI] [PubMed] [Google Scholar]
- 7.Dewal M, Xu Y, Yang J, Mohammed F, Smith M, Shimizu LS. Chem. Commun. 2008:3909–3911. doi: 10.1039/b805895d. [DOI] [PubMed] [Google Scholar]
- 8.(a) Thurber KR, Tycko R. J. Chem. Phys. 2012;137:84508. doi: 10.1063/1.4747449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mentink-Vigier F, Akbey U, Hovav Y, Vega S, Oschkinat H, Feintuch AJ. Magn. Reson. 2012;224:13–21. doi: 10.1016/j.jmr.2012.08.013. [DOI] [PubMed] [Google Scholar]; (c) Mentink-Vigier F, Paul S, Lee D, Feintuch A, Hediger S, Vega S, De Paëpe G. Phys. Chem. Chem. Phys. 2015;17:21824–21836. doi: 10.1039/c5cp03457d. [DOI] [PubMed] [Google Scholar]
- 9.Eichhorn TR, Takado Y, Salameh N, Capozzi A, Cheng T, Hyacinthe J–N, Mishkovsky M, Roussel C, Comment AJ. Proc. Natl. Acad. Sci. U.S.A. 2013;110:18064–18069. doi: 10.1073/pnas.1314928110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capozzi A, Hyacinthe J–N, Cheng T, Eichhorn TR, Boero G, Roussel C, van der Klink JJ, Comment AJ. Phys. Chem. C. 2015;119:22632–22639. [Google Scholar]
- 11.(a) Liu G, Levien M, Karchin N, Parigi G, Luchinat C, Bennati M. Nature Chem. 2017 doi: 10.1038/NCHEM.2723. [DOI] [PubMed] [Google Scholar]; (b) Daviso E, Jacoba Janssen G, Alia A, Jeschke G, Matysik J, Tessari M. J. Am. Chem. Soc. 2011;133:16754–16757. doi: 10.1021/ja206689t. [DOI] [PubMed] [Google Scholar]
- 12.Langea S, Linden AH, Akbey U, Franks WT, Loening NM, van Rossum B–J, Oschkinat HJ. Magn. Reson. 2012;216:209–212. doi: 10.1016/j.jmr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 13.(a) Baldovi HG, Kruger M, Reinsch H, Alvaro M, Stock N, Garcia HJ. Mater. Chem. C. 2015;3:3607–3613. [Google Scholar]; (b) Dormán G, Nakamura H, Pulsipher A, Prestwich GD. Chem. Rev. 2016;116:15284–15398. doi: 10.1021/acs.chemrev.6b00342. [DOI] [PubMed] [Google Scholar]; (c) Marazzi M, Mai S, Roca-Sanjuán D, Delcey MG, Lindh R, González L, Monari AJ. Phys. Chem. Lett. 2016;7:622–626. doi: 10.1021/acs.jpclett.5b02792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Barash L, Wasserman E, Yager WA. J. Am. Chem. Soc. 1967;89:3931. [Google Scholar]; (b) Lin T–S. J. Chem. Phys. 1972;57:2260. [Google Scholar]; (c) Murai H, Imamura T, Obi K. Chem. Phys. Lett. 1982;87:295. [Google Scholar]; (d) Jones C, McDyre L, Murphy DM, Stasch A. Chem. Commun. 2010;46:1511–1513. doi: 10.1039/b922002j. [DOI] [PubMed] [Google Scholar]
- 15.(a) Gast P, Gottschalk A, Norris JR, Closs GL. FEBS Lett. 1989;243:1–4. [Google Scholar]; (b) Žilić D, Rakvin B, Dalal NS. J. Appl. Phys. 2011;110:1–5. [Google Scholar]
- 16.Prasad A, Ferretti U, Sedlářová M, Pospíšil P. Sci. Rep. 2016;6:1–13. doi: 10.1038/srep20094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyakonov MI. Spin Physics in Semiconductors. Springer; Verlag Berlin Heidelberg: 2008. [Google Scholar]
- 18.Kaushik M, Bahrenberg T, Can TV, Caporini MA, Silvers R, Heiliger J, Smith AA, Schwalbe H, Griffin RG, Corzilius B. Phys. Chem. Chem. Phys. 2016;18:27205–27218. doi: 10.1039/c6cp04623a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Can TV, Caporini MA, Mentink-Vigier F, Corzilius B, Walish JJ, Rosay M, Maas WE, Baldus M, Vega S, Swager TM, Griffin RG. J. Chem. Phys. 2014;141:1–8. doi: 10.1063/1.4891866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Hicks RG. Org. Biomol. Chem. 2007;5:1321–1338. doi: 10.1039/b617142g. [DOI] [PubMed] [Google Scholar]; (b) Rajca A. Chem. Rev. 1994;94:871. [Google Scholar]; (c) Rawson JM, Alberola A, Whalley AE. J. Mater. Chem. 2006;16:2560. [Google Scholar]; (d) Nakatsuji S, Anzai HJ. Mater. Chem. 1997;7:2161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.