

Abstract
A valid preclinical tumor model should recapitulate the tumor microenvironment. Immune and stromal components are absent in immunodeficient models of pancreatic cancer. While these components are present in genetically engineered models such as KrasG12D; Trp53R172H; Pdx-1Cre (KPC), immense variability in development of invasive disease makes them unsuitable for evaluation of novel therapies. We have generated a novel mouse model of pancreatic cancer by implanting tumor fragments from KPC mice into the pancreas of wild type mice. Three-millimeter tumor pieces from KPC mice were implanted into the pancreas of C57BL/6J mice. Four to eight weeks later, tumors were harvested, and stromal and immune components were evaluated. The efficacy of Minnelide, a novel compound which has been shown to be effective against pancreatic cancer in a number of preclinical murine models, was evaluated. In our model, consistent tumor growth and metastases were observed. Tumors demonstrated intense desmoplasia and leukocytic infiltration which was comparable to that in the genetically engineered KPC model and significantly more than that observed in KPC tumor-derived cell line implantation model. Minnelide treatment resulted in a significant decrease in the tumor weight and volume. This novel model demonstrates a consistent growth rate and tumor-associated mortality and recapitulates the tumor microenvironment. This convenient model is a valuable tool to evaluate novel therapies.
Keywords: Pancreatic cancer, Immunocompetent mouse model, Stroma, Syngeneic
Introduction
Pancreatic cancer is the 4th leading cause of cancer related deaths in the USA.1 Pancreatic ductal adenocarcinoma has the worst prognosis among all common epithelial malignancies with an overall 5-year survival of 7 %. An estimated 48, 960 people will be diagnosed with pancreatic cancer, and 40, 560 people will succumb to this deadly disease in 2015.1
Surgical resection is the treatment of choice for pancreatic cancer. However, less than 20 % of patients are eligible for surgery and, even when resected with negative margins, most patients develop recurrence.2,3 Though adjuvant and palliative chemotherapy have improved over the last decade, the overall impact of current chemotherapy continues to be small. Currently, FOLFIRINOX (combination of 5-fluoruracil, irinotecan, oxaliplatin, and leucovorin) or a combination of gemcitabine and nabpaclitaxel are considered standard treatment regimens for patients with metastatic pancreatic cancer who have a good performance status and no comorbidities.3 However, these newer regimens offer only a marginal improvement in the median survival of patients with metastatic or locally advanced unresectable pancreatic cancer. Although the poor prognosis of patients with pancreatic cancer can be attributed to the overall aggressiveness of this cancer itself and poor response to chemotherapeutic agents, a lack of relevant preclinical models to test novel therapeutic modalities contribute to the problem.4
A hallmark of pancreatic ductal adenocarcinoma is a profound fibrous inflammatory reaction composed of fibroblasts, myofibroblasts (pancreatic stellate cells), leukocytes, endothelial cells, and extracellular matrix proteins. It has been proposed that these components play a pivotal role in tumor development, tumor progression, and resistance to chemotherapy. Thus, a valid tumor model should recapitulate the tumorigenic properties as well as immune and stromal microenvironment. Current preclinical mouse models for pancreatic cancer can be broadly classified into immunodeficient and immunocompetent models. Most mouse models utilize immunodeficient hosts such as athymic nude or severe combined immunodeficiency (SCID) mice in which either resected human tumors or established human pancreatic cancer cell lines are implanted.5–7 The drawback of these models is the lack of an intact adaptive immune system and well-developed stromal components, which at least partially explains why testing new therapies in immunodeficient models fails to predict response to therapy in pancreatic cancer patients.2
Recently, a number of different genetically engineered mouse models of pancreatic cancer have been described and validated. Hingorani et al. described one such model in which expression of mutant Kras and concurrent p53 deletion, specifically in the mouse pancreas, lead to the development of pancreatic intra-epithelial neoplasia (PanIN) lesions that eventually progress to invasive ductal adenocarcinoma.8 This model was designated the KrasG12D; Trp53R172H; Pdx-1Cre or KPC model and is one of the most commonly used models for studying the pathogenesis of pancreatic cancer. While genetic models, such as KPC, recapitulate the genetic makeup, local aggressiveness, and metastatic ability of human pancreatic cancer, the immense variability in the time to development of invasive disease (47 to 355 days of life)8 is a major limitation. This inconsistency makes these models unsuitable for the evaluation of the efficacy of novel chemotherapeutic drugs since it is difficult to ensure either the presence of tumors or the uniformity of tumor size in the treatment and control groups.
To overcome the shortcomings of the immunodeficient as well as the genetically engineered mouse models of pancreatic cancer, we have developed and characterized an immunocompetent orthotopic mouse model of pancreatic cancer in which tumor pieces from KPC mice were implanted into the pancreas of immunocompetent syngeneic C57BL/6J mice. The tumors that develop in this Syngeneic Tumor Implantation Model (STIM) have a consistent and predictable growth rate and simulate the tumor microenvironment of human pancreatic cancer in terms of immune infiltration and desmoplastic reaction. The reliability and ease of this model makes it especially useful for evaluation of novel therapies.
Triptolide, a diterpene triepoxide derived from the Chinese herb Tripterygium wilfordii, has been shown to be effective against a number of different cancers. Minnelide, a water soluble prodrug of triptolide developed by our group, retains the efficacy of triptolide and is effective against pancreatic cancer in a number of different murine models.9 Minnelide is currently in phase I clinical trial against gastrointestinal malignancies. As a proof of principle, this model was utilized to evaluate the efficacy of Minnelide.
Materials and Methods
Animals
Eight-to-ten-week-old immunocompetent female wild type C57BL/6J mice were used in this study (Jackson Laboratories, Bar Harbor, ME). KRasG12D; Trp53R172H; Pdx-1Cre (KPC) animals were generated by crossing Lox Stop Lox (LSL) KRasG12D; LSL Trp53R172H animals with Pdx-1Cre animals. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Minnesota.
Isolation of Tumors from KPC Mice and Tumor Implantation
Six-month-old female KPC mice with palpable tumors were sacrificed, and their pancreatic tumors were collected. Each tumor was cut into cylindrical pieces with a diameter of 3 mm and length of 3 mm using a 3-mm punch biopsy instrument. These tumor pieces were then implanted in the pancreas of wild type C57BL/6J mice using the following operative technique. Laparotomy was performed on C57BL/6J mice, and the pancreas was identified (Fig. 1aI). One tumor piece was implanted into the pancreas of each wild type C57BL/6J mouse using a figure-of-8 stitch with a 7–0 prolene suture (Ethicon, USA) incorporating the superior and inferior borders of the pancreas thereby creating a pocket of pancreas (Fig. 1aII and aIII). After tumor implantation, the pancreas was then carefully returned to the peritoneal cavity, the abdomen was closed with a 4–0 vicryl suture (Ethicon, USA), and the skin was stapled. This model was termed the STIM.
Fig. 1.

a Operative technique for implantation of fragments of pancreatic tumors from KPC (LSL-Kras G12d/+; LSL-Trp53R172H/+; Pdx1-Cre) mice to create the Syngeneic Tumor Implantation Model (STIM), b STIM tumors demonstrate consistent growth in terms of tumor volumes at 4 and 8 weeks after implantation. c Survival analysis demonstrates that most of the STIM mice died of pancreatic cancer within 3 months post-implantation whereas in genetically engineered KPC mice, there was immense variability in mortality (57–335 days of life). d Representative picture of pancreatic tumors (number sign) and associated peritoneal metastases (arrowhead) that develop at 8 weeks post-implantation. 8 weeks after implantation, e liver metastases (arrowheads) f peritoneal and mesenteric metastases as well as g gastric outlet obstruction (arrowhead) and splenic involvement (asterisk) are observed
KPC Tumor-Derived Cell Lines Implantation Model
Cell lines were derived from primary KPC tumors and cultured in Dulbecco’s modified eagle medium (Hyclone) with 10 % fetal bovine serum (FBS) and 1 % penicillin/streptomycin.10 For animal experiments, KPC cell lines were trypsinized and resuspended in phosphate buffered saline/Matrigel in a 1:1 ratio. C57BL/6J mice were anesthetized, laparotomy was performed, and 50,000 cells were injected into the tail of the pancreas. The pancreas was then carefully returned to the peritoneal cavity, the abdomen was closed with a 4–0 vicryl suture (Ethicon, USA), and the skin was stapled. The animals were sacrificed 4 weeks after implantation, and tumor volumes and weights were measured.
Evaluation of Tumor Growth
Mice with implanted tumors were carefully followed and euthanized after 4 and 8 weeks. In a separate survival experiment, animals were sacrificed when they developed more than 20 % weight loss, anorexia, or failure to thrive. Death was confirmed by absence of heartbeat on palpation of chest and absence of blink reflex. An autopsy was performed, and abdominal and thoracic cavities of the mice were examined for presence of metastases and ascites. Tumors were harvested, and the size and weight of pancreatic tumors were measured. The tumor tissue was divided into two equal parts, one for histological analysis and the other for flow cytometry analysis.
Histological Analysis
For immunohistochemistry, tissues were placed in 10 % phosphate buffered formalin and were transferred to 70 % ethanol 24–72 h later. Subsequently, the tissues were embedded in paraffin. Four to five micrometer sections were dewaxed and rehydrated according to the standard histological procedure. Representative sections were stained with hematoxylin and eosin for light microscopy or with picrosirius red for collagen. Heat-induced antigen retrieval was performed at pH 6.0 for all antibodies.
For immunohistochemistry, peroxidase activity was blocked using 3 % H2O2 (Sigma-Aldrich). Sections were treated for 15 min with serum free protein blocker (Dako), incubated overnight with primary antibody against cytokeratin-19 (1:50 dilution, Abcam, cat. no. 52625), and subsequently incubated with HRP tagged secondary antibody for 1 h. Color was developed with DAB solution (Vector Labs). Counterstaining was performed using methyl green, and the slides were dehydrated using graded alcohols and xylene. Sections were mounted with a Permount mounting medium (Fischer Scientific).
For immunofluorescence staining (IF), primary antibodies against α-SMA (1:500 dilution; Abcam, cat. no. ab5694), vimentin (1:50 dilution, Cell Signaling Technology, cat. no. 5741), coronin-1 (1:50 dilution, Bethyl laboratories, cat. no. A300-931A), and Ki-67 (1:100 dilution, Santa Cruz, cat. no. 15402) were used in 1X Sniper (Biocare Medical) followed by incubation with secondary fluorochrome-tagged antibodies for 1 h. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay was performed using a In Situ Cell Death Detection Kit, Fluorescein (Roche, IN, USA) according to the manufacturer’s instructions. Sections were mounted using a ProLong Gold Antifade mountant with DAPI (Life Technologies).
All microscopic images were taken using a Leica DM5500B microscope (Leica Microsystems) at magnification 20× and 40×. Quantification of immunofluorescent staining was performed using ImageJ software (NIH).
Immune Cell Isolation and Flow Cytometry
Pancreatic tumors were minced using a sterile scalpel blade in RPMI medium (Hyclone). Single cell suspensions were generated using a combination of enzymatic digestion (200 U/mL Collagenase IV, Worthington) and mechanical dissociation. Tumor debris were removed using 70-μm filters (BD Falcon). Cells from all tissues were fixed using 80 % ethanol and washed with PBS. Staining was performed using a cocktail of anti-mouse antibodies against CD45 (clone: 30-F11), CD3 (clone: 145-2C11), CD4 (clone: RM4-5), CD8 (clone: 53.67), CD11b (clone: M1/70), Gr-1(clone: RB6-8C5).
All the antibodies were purchased from BD Biosciences. Isotype-matched antibodies were used as negative controls wherever appropriate. Surface marker expression was determined using multi-color flow cytometry on BD FACS Canto II and analyzed using FACS Diva (BD Biosciences) and FlowJo (Tree Star).
Evaluation of Novel Therapies
Minnelide, a novel anti-cancer drug developed by our group, was used as a model for novel therapies, and its effect on tumor growth was evaluated using the proposed STIM. For in vivo studies, mice were observed for 2 weeks after tumor implantation and then randomized into two groups: Saline and Minnelide. Minnelide was given at 0.42 mg/kg/day by intra-peritoneal injection (IP) daily.9 The mice were euthanized after 4 and 8 weeks of treatment and autopsy performed. Tumor volumes and weights were compared between the control and treatment arms.
Statistical Analysis
Values were expressed as mean±SEM. Tumor volume comparisons were analyzed by paired Student’s t test. Kaplan-Meier curve for survival was charted using the Graph Pad Prism 5.02 software. The quantification of immunofluorescent staining of tumors from different immunocompetent models was analyzed using ANOVA. A p value of less than 0.05 was considered statistically significant.
Results
Pancreatic Tumor Uptake is Uniform and Predictable Disease Progression is Observed
In our model, after implantation of tumor fragments from KPC pancreatic tumors into C57BL/6J mice, palpable tumors were noted as early as 4 weeks post-implantation. Pancreatic tumors developed in 100 % of the mice after implantation of tumor fragments. Measurement of volumes and weights of tumors obtained from the proposed STIM mice that were sacrificed at 4 and 8 weeks demonstrated a consistent and predictable progression of disease (Fig. 1b). In the survival experiment, the median survival of mice in our model (STIM) was 71 days. Most of our STIM mice died within 3 months post-implantation whereas in the genetically engineered KPC mouse model, there was immense variability in mortality (range, 2–12 months of life) (Fig. 1c).
Figure 1d demonstrates a representative photograph of a pancreatic tumor that developed in STIM mice. Liver and peritoneal metastases were frequently observed in mice 2 months after implantation and were observed in 60 and 80 % of the STIM mice at the time of death in the survival experiment (Fig. 1e, f, respectively). Gastric outlet obstruction and splenic invasion, which are events also observed clinically in pancreatic cancer patients, were observed in a number of STIM mice during the course of disease progression (Fig. 1g).
Pancreatic and Metastatic Tumor Histology in STIM is Comparable to Genetically Engineered KPC Mice
Hematoxylin and eosin staining of representative pancreatic tumors obtained from the novel STIM (Fig. 2a–b) demonstrated adenocarcinoma with high-grade nuclear features, areas of necrosis, and regions of desmoplasia (arrows, Fig. 2b) similar to that observed in tumors from KPC mice (Fig. 2c). Cytokeratin-19 (CK19), a ductal epithelial marker which is expressed in most human pancreatic adenocarcinomas, is expressed in STIM tumors as demonstrated by immunohistochemistry (Fig. 2d). Hematoxylin and eosin staining of liver and peritoneal metastases confirmed adenocarcinoma (Fig. 2e, f, respectively).
Fig. 2.

Histology of pancreatic tumors from the Syngeneic Tumor Implantation Model (STIM) confirms adenocarcinoma with a formation of glands and b regions of desmoplasia (arrowhead). The histology is comparable to that observed in c tumors from genetically engineered KPC mice. Immunohistochemistry demonstrates that pancreatic tumors from STIM mice stain for d cytokeratin-19/CK-19, a ductal epithelial marker (CK-19 in brown, methyl green counterstain, 40×). Hematoxylin and eosin staining of liver (e) and peritoneal metastases (f) from STIM mice confirms adenocarcinoma. STIM tumors are infiltrated with g distinct immune cell populations of CD4 helper T cells, CD8 cytotoxic T cell, macrophages, and myeloid-derived suppressor cells (MDSCs)
Flow Cytometry Analysis of Tumor-Infiltrating Immune Cells Reveals a Complex Immune Response
Four weeks after orthotopic implantation, single cell suspensions were generated from STIM pancreatic tumors and were analyzed using fluorescence-activated cell sorting (FACS) for detailed characterization of the intratumoral immune infiltration. Pancreatic tumors from STIM mice demonstrated leukocytic infiltration (CD45+). Further characterization of the leukocytic infiltrate demonstrated presence of helper T cells (CD45+ CD3+ CD4+), cytotoxic T cells (CD45+ CD3+ CD8+), myeloid-derived suppressor cells (MDSCs) (CD45+ CD3− Gr-1+ CD11b+), and macrophages (CD45+ CD3− Gr-1−CD11b+) in the tumors (Fig. 2g). This was similar to that observed with genetically engineered KPC mouse model of pancreatic cancer in previous studies.11
Comparison of Tumor Stromal Components Between Tumors Developing in STIM, Genetically Engineered KPC Mouse Model and KPC Tumor-derived Cell Line Implantation Model of Pancreatic Cancer
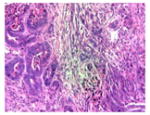
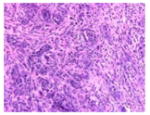
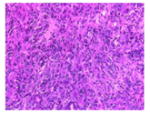
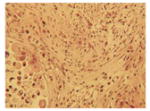
The tumor stroma was compared between the tumors that develop in the following three models of pancreatic cancer: (1) previously described KPC tumor-derived cell line implantation model of pancreatic cancer; (2) the proposed STIM; and (3) state of the art genetically engineered KPC mouse model. Immunofluorescent staining for stromal markers such as collagen, α-SMA (a marker of activated stellate cells or myofibroblasts), and vimentin (a marker of mesenchymal cells) as well as staining for coronin-1 (a marker of leukocytes) was performed on and compared between the tumors (n=3) from each group. The tumors that develop in our STIM model have collagen, α-SMA, vimentin, and coronin-1 staining that was equivalent in quantity to that observed in tumors from genetically engineered KPC mouse model (Figs. 3 and 4). Interestingly, the tumors that developed in the KPC tumor-derived cell line implantation model had significantly lower staining for collagen, α-SMA, vimentin, and coronin-1 as compared to tumors from STIM and genetically engineered KPC mice (p<0.05) (Figs. 3 and 4). This suggests that the tumor stroma in the cell line implantation model may not recapitulate the conditions observed in human disease.
Fig. 3.

Representative pictures of picrosirius red staining for stromal collagen in tumors from a KPC tumor-derived cell lines orthotopically implanted in pancreas of immunocompetent wild type C57BL/6J mice, b STIM (Syngeneic Tumor Implantation Model) and c genetically engineered KPC model. d Quantification of stromal collagen content in tumors from KPC cell lines implantation model, STIM, and genetically engineered KPC model (n=3 in each group). Representative pictures of immunofluorescent staining for vimentin in tumors from e KPC tumor-derived cell lines orthotopically implanted in pancreas of immunocompetent wild type C57BL/6J mice, f STIM (Syngeneic Tumor Implantation Model) and g genetically engineered KPC model. h Quantification of vimentin content in tumors from KPC cell lines implantation model, STIM and genetically engineered KPC model (n=3 in each group). (*p<0.05, as compared to KPC cell lines implantation model)
Fig. 4.

Representative pictures of immunofluorescent staining for α-SMA in tumors from a KPC tumor-derived cell lines orthotopically implanted in pancreas of immunocompetent wild type C57BL/6J mice, b STIM (Syngeneic Tumor Implantation Model) and c genetically engineered KPC model. d Quantification of α-SMA content in tumors from KPC cell lines implantation model, STIM and genetically engineered KPC model (n=3 in each group). Representative pictures of immunofluorescent staining for coronin-1 in tumors from e KPC tumor-derived cell lines orthotopically implanted in pancreas of immunocompetent wild type C57BL/6J mice, f STIM (Syngeneic Tumor Implantation Model) and g genetically engineered KPC model. h Quantification of coronin-1 in tumors from KPC cell lines implantation model, STIM and genetically engineered KPC model (n=3 in each group). (*p<0.05, as compared to KPC cell lines implantation model)
Effect of Minnelide on STIM Tumor Growth
Minnelide is an anti-cancer drug developed by our group at the University of Minnesota and is currently in phase I clinical trial against gastrointestinal malignancies. Minnelide has been shown to be effective in both immunocompetent and immunodeficient mouse models of pancreatic cancer.9 The proposed STIM model was used to evaluate the efficacy of Minnelide. Tumor volumes and tumor weights at both 4 and 8 weeks were significantly lower in the Minnelide group as compared to saline treated controls (tumor volume (mm3, mean±SD). 4 weeks: Minnelide 62±19 vs control 1101±347 mm3, 8 weeks: Minnelide 368±180 vs control 1985±590 mm3, p<0.05; tumor weight (grams, mean±SD. 4 weeks: Minnelide 0.27±0.02 vs control 2.27±0.6, 8 weeks: Minnelide 0.62±0.2 vs control 2.95±0.68, p<0.05) (Fig. 5a–d).
Fig. 5.

Evaluation of the efficacy of a novel chemotherapeutic drug, Minnelide, using STIM (Syngeneic Tumor Implantation Model) mice. Minnelide treatment results in significantly lower a tumor volumes and b tumor weights after 4 weeks of treatment as well as after 8 weeks c–d of treatment as compared to the saline treated group. (*p<0.05 as compared to saline treatment). Tumors from Minnelide treated STIM mice had e a significantly higher number of TUNEL positive cells as compared to tumors from saline treated STIM mice. Tumors from Minnelide treated STIM mice had f a paucity of proliferating cells, as evident by significantly lower Ki-67 staining
TUNEL staining was used as a marker of apoptosis and tumors from Minnelide-treated STIM mice had a significantly higher number of TUNEL positive cells as compared to tumors from saline treated STIM mice (Fig. 5e). Moreover, tumors from Minnelide-treated STIM mice had a paucity of proliferating cells, as evident by significantly lower Ki-67 staining, a marker of cell proliferation (Fig. 5f).
Discussion
In the current study, we have described a novel immunocompetent mouse model of pancreatic cancer which is easy to establish, has a predictable course of disease, and recapitulates the microenvironment of genetically engineered mouse models. This model also demonstrates metastatic spread and tumor-associated mortality. We believe that this model offers a convenient platform to evaluate novel therapeutics as demonstrated by our evaluation of Minnelide, a novel water soluble analog of triptolide developed by our group, which is currently in a phase I clinical trial against gastrointestinal malignancies.9
An ideal tumor model should recapitulate the tumorigenic properties and the immune and stromal microenvironment of human pancreatic cancer as well as have a predictable course of disease progression. Recent studies have demonstrated that the tumor progression as well as response to chemotherapy may be altered by pancreatic tumor stroma and components of the immune system.12–18 Therapies for pancreatic cancer have classically been evaluated using immunodeficient models. These models are created by implanting resected tumors (patient-derived xenografts or PDX)19 or injecting human pancreatic cancer-derived cell lines either subcutaneously or orthotopically in the pancreas of immunodeficient mice (athymic nude mice and severe combined immunodeficiency (SCID) mice). Although these immunodeficient mouse models are commonly the first models used to evaluate new therapies, they do not recapitulate the intricacies of the tumor microenvironment, both the immune and to a large extent the stromal components. Thus, the results obtained from these models need to be confirmed in immunocompetent models which recapitulate the tumor microenvironment more accurately (Table 1).
Table 1.
Features of the proposed and previously established mouse models of pancreatic cancer (PDAC)
| Proposed model | Existing models | ||||
|---|---|---|---|---|---|
| STIM | Genetically engineered KPC8 | KPC cell line implantation Model4,23 | Human PDAC cell line xenograft23 | Patient derived tumor xenograft (PDX)19 | |
| Mouse genetic background | C57BL/6J (wild type) | C57BL/6J (wild type) | C57BL/6J (wild type) | Athymic/SCID/NSG | Athymic/SCID/NSG |
| Immune background | Immunocompetent | Immunocompetent | Immunocompetent | Immunodeficient (lacks functional adaptive immunity) | Immunodeficient (lacks functional adaptive immunity) |
| Mutation profile | Mutations present only in implanted tumor tissue | Mutations present throughout pancreas | Mutations present only in implanted cells | Mutations present only in implanted cells | Mutations present only in implanted tumor tissue |
| Species | Mouse tumor | Mouse tumor | Mouse cell line | Human cell lines | Human tumor |
| Stromal content | High stromal content (ECM, fibroblasts and activated stellate cells) | High stromal content (ECM, fibroblasts and activated stellate cells) | Low stromal content | Absent stroma | High stromal content (ECM, fibroblasts and activated stellate cells) |
| Variability | Less variability in time to invasive disease ( within 2 weeks) | High variability in time to invasive disease (Range, 47–355 days) | Less variability in time to invasive disease (within 2 weeks) | Less variability in time to invasive disease (2–3 weeks); depends on cell line | Variability in time to tumor development (2–12 months ); depends on primary tumor |
| Predictability | Predictable course of disease progression | Unpredictable course of disease progression | Predictable course of disease progression | Predictable course of disease progression | Predictable course of disease progression and variable engraftment rates (23–75%) |
| Tumor milieu | Tumor growth is in natural milieu (pancreas) | Tumor growth is in natural milieu (pancreas) | Tumor growth is in natural milieu (pancreas) | Tumor growth is in unnatural (subcutaneous) milieu or natural (orthotopic) milieu | Tumor growth is in unnatural (subcutaneous) milieu or natural (orthotopic) milieu |
| Metastatic disease | Metastatic and loco-regional disease present after 2 months of implantation | Metastatic and loco-regional disease develops at variable time. | Metastatic and loco-regional disease present, but concerns of confounding due to cell spillage during orthotopic implantation | Metastatic and loco-regional disease absent in subcutaneous models and present in orthotopic. | Metastatic and loco-regional disease absent in subcutaneous models and present in orthotopic. |
| Tumor pathology | Well differentiated to poorly differentiated tumors (depends on parent tumor) | Well differentiated to poorly differentiated tumors | Poorly differentiated tumors | Variable | Variable |
|
|
|

|
|
|
| Tumor microenvironment | Robust desmoplastic reaction, collagen and vimentin content, immune infiltration, and stellate cells | Robust desmoplastic reaction, collagen and vimentin content, immune infiltration, and stellate cells | Paucity of desmoplastic reaction, less collagen, vimentin and stellate cells, and less immune infiltration | Paucity of desmoplastic reaction as well as infiltrating immune cells | Robust desmoplastic reaction without infiltrating adaptive immune cells |
| Surrounding pancreas | Adjacent normal/wild type pancreas | Adjacent mutant Kras-p53 pancreas | Adjacent normal/wild type pancreas | No surrounding pancreatic tissue (subcutaneous) or adjacent immunodeficient pancreas (orthotopic) | No surrounding pancreatic tissue (subcutaneous) or adjacent immunodeficient pancreas (orthotopic) |
STIM: Syngeneic Tumor Implanta3on Model; SCID: Severe combined immunodeficiency; NSG: Non-obese diabe3c (NOD) SCID gamma mice; ECM: Extracellular matrix STIM Syngeneic Tumor Implantation Model, SCID severe combined immunodeficiency, NSG non-obese diabetic (NOD) SCID gamma mice, ECM extracellular matrix
The shortcomings of the immunodeficient models of pancreatic cancer have been addressed with the advent of genetically engineered mouse models of pancreatic cancer. The development of genetically engineered spontaneous models of pancreatic cancer in immunocompetent mice has been a major breakthrough in the preclinical modeling of pancreatic cancer. The genetic landscape of pancreatic cancer is well characterized now. Human pancreatic cancer is most commonly a consequence of sporadic mutations.20 K-Ras proto-oncogene is activated in 90 %, and p53 alterations occur in 60–70 % of all pancreatic cancer patients.3,20 Apart from the KPC mice developed by Hingorani et al.,8 a number of mouse models with a combination of mutant Kras and other targeted mutations such as Ink4a/Arf and MUC1 have been used to study the pathogenesis and progression of pancreatic cancer.21–23 Genetically engineered KPC mice have an overall median survival of 5.5 months and have a chemoresistance profile comparable to that of human pancreatic cancer. This spontaneous model has complete penetrance i.e., all mice develop pancreatic tumors and a major proportion of these mice develop metastatic disease.4,8 Moreover previous studies have reported a dynamic evolution of immune response in these mice during progression of tumors which closely simulates human pancreatic carcinogenesis.11,24 Flow cytometry analysis of pancreatic tumors from KPC mice has demonstrated that leukocytes constitute a significant proportion of cells within the tumor. This leukocytic infiltration of tumors is composed of T cells (helper and cytotoxic), macrophages, and MDSCs.11,24 These immune cells play an important role in both the elimination of tumor (such as cytotoxic T cells) and in maintaining an immunosuppressive microenvironment that mitigates immune escape of pancreatic tumors (such as MDSCs).11,15
Furthermore, the genetically engineered mouse models of pancreatic cancer recapitulate the intricate stromal biology of human disease. The characteristic fibrous inflammatory microenvironment observed in pancreatic cancer has been shown to play an important role in tumor development, progression, and resistance to chemotherapy.25 The desmoplastic reaction is driven by a number of growth factors such as transforming growth factor (TGF-b), epidermal growth factor (EGF), hepatocyte growth factor (HGF), fibroblast growth factor (FGF), and insulin like growth factors (IGF). The tumor stroma is composed of myofibroblasts (pancreatic stellate cells), fibroblasts, type I collagen, extracellular matrix components as well as multiple types of inflammatory cells. This fibrous stroma can possibly affect response to chemotherapy by producing growth factors that promote the survival of tumor cells and by impairing drug delivery inside tumors due to increased interstitial pressure.25 Therefore, the stroma is an integral part of the tumor microenvironment, and its targeted ablation has been shown to improve drug delivery and induce tumor shrinkage in murine models of pancreatic cancer.26
Despite all these advantages, the variability in the time to the development of tumors in genetically engineered mice (range, 47–355 days) limits their utility in preclinical evaluation of new therapies. Another relevant shortcoming of these models is the difficulty in assessing, with certainty, the presence or absence of tumors at the beginning of an experiment. Although this is possible with the help of imaging such as ultrasonography, it is time as well as labor intensive. Orthotopic and subcutaneous models created by implanting KPC tumor-derived cell lines in immunocompetent wild type C57BL/6J mice have been utilized to overcome some of these shortcomings. However, as shown in our studies, these tumors lack a robust leukocytic infiltration and stromal content as compared to the genetically engineered models. This could be a possible explanation for the observed differences between the chemoresistance profiles of tumors from this model and tumors from genetically engineered KPC mice.4 Moreover, a false rate of metastatic disease may result from direct spillage of cells into the peritoneal cavity during injection of the cells. We observed peritoneal metastases in 90 % of mice injected with single suspensions and in 80 % of STIM mice. However, we observed liver metastases in 60 % and lung metastases in 20 % of our STIM mice as opposed to 0 % of single cell suspension injected mice during the same time period. Therefore, the peritoneal metastases observed in the cell line implantation model could possibly be a result of direct spill-age of cells in the abdominal cavity rather than true metastases. We believe that this is one of the limitations of the KPC cell line implantation model.
Although the STIM developed by us is not a spontaneous model, it addresses the shortcomings of both the genetically engineered models and immunodeficient xenograft models. The major advantage of the proposed STIM model is that it can be readily established, and the tumors display a predictable and consistent growth rate in terms of tumor volumes and weights. All the mice in our study developed palpable pancreatic tumors by 4 weeks after implantation. This is in stark contrast to the genetically engineered KPC model, in which tumor development can occur anytime between 2 and 12 months of life.8 Furthermore, we observe liver and peritoneal metastases in the majority of mice in our STIM model at 8 weeks post-implantation which confirms that our model retains the metastatic properties of pancreatic tumors. The histological examination of the tumors that develop in our STIM model demonstrated adenocarcinoma with high-grade nuclear features, regions of desmoplasia, and necrosis. Moreover, these tumors stained positively for cytokeratin-19, a marker expressed by most human pancreatic ductal adenocarcinoma. We compared the tumor microenvironment between the tumors from three separate models: (1) A previously described orthotopic model generated by implantation of KPC tumor-derived cell lines in the pancreas of immunocompetent wild type C57BL/6J mice, (2) the proposed STIM model, and (3) genetically engineered KPC mice. We observed significantly lower stromal content (collagen, α-SMA and vimentin) and immune infiltration in the tumors that developed in the KPC cell line implantation model as compared to both our STIM model and the genetically engineered KPC model. More importantly, STIM tumors at 8 weeks post-implantation had a significant amount of stromal content and immune infiltration which was comparable to that observed in tumors from genetically engineered KPC mice. A possible reason for the inadequate stroma in the cell line implantation is the fact that it accounts for only the epithelial component of pancreatic cancer. In our STIM model, we overcome this limitation by implanting a tumor piece containing both the epithelial as well as the stromal components of pancreatic cancer. We believe that preservation of the tumor epithelial and stromal components in the implanted tumor fragment results in development of robust stroma in the tumors from STIM mice which is comparable to the stroma observed in genetically engineered KPC model. This further highlights the utility of our model, which not only is quickly established and has a predictable course of disease progression but also retains the properties of genetically engineered KPC tumors.
Using immunofluorescent staining, we observed dense infiltration of the tumor and peri-tumoral regions with leukocytes in tumors from our STIM model. Moreover, flow cytometry analysis of freshly harvested pancreatic tumors from STIM mice at 4 weeks post-implantation revealed that over half of the cells in the tumors were leukocytes. Further gating on these leukocytes revealed populations of CD4+ helper T cells, CD8+ cytotoxic T cells, macrophages, and MDSCs. The immune microenvironment of tumors in our STIM mice closely resembles that found in genetically engineered models such as KPC.11,24
Triptolide, a diterpene triepoxide, is derived from the Chinese herb Trypterygium wilfordii and has been shown to downregulate heat shock genes27,28 and induce apoptotic cell death in pancreatic cancer lines.9,29,30 It has been shown to be efficacious against a number of other malignancies as well.31–37 Minnelide, the water soluble prodrug of triptolide, has been shown to induce tumor regression in a number of murine models of pancreatic cancer without causing any overt signs of toxicity9 and is currently in phase I clinical trial against advanced gastrointestinal malignancies. We therefore evaluated the efficacy of Minnelide using our novel immunocompetent mouse model. We observed significantly lower tumor volumes and weights in STIM mice treated with Minnelide as compared to saline after 4 and 8 weeks of treatment (p<0.05). This further highlights the utility of our model in assessing efficacy of new therapies.
A limitation of our model is that the tumor development does not proceed through the pre-invasive stages (PanINs), and thus, our model cannot be used for studying pancreatic carcinogenesis and pre-invasive pancreatic disease. However, our model can be used to study loco-regional as well as metastatic progression of pancreatic tumors in an immunocompetent background accompanied by tumor desmoplasia. Hence, the effect of chemotherapy on tumor stromal components can also be evaluated using the STIM model. Another limitation is that the number of cancer cells as well the stromal cells in each tumor piece were not measured before implantation. However, we believe even though the exact number of cells was not controlled for in each equal-sized tumor piece, this contributes to the heterogeneity of the tumors that develop in the STIM mice which recreates the clinical scenario of heterogeneous pancreatic tumors in different patients. Moreover, although the number of cancer cells were not measured in each equal sized tumor fragment, the tumors still grew at very similar rates as evidenced by the low variability observed in the tumor volumes and weights at 4 and 8 weeks post-implantation.
Additionally, our model can be utilized to generate mice bearing tumors with different mutations. Tumors from syngeneic genetically engineered mice such as Pdx1-Cre; K-rasG12D; Ink4a/Arf lox/lox and Pdx1-Cre; K-rasG12D; SMAD4 lox/lox can be used to generate STIM mice bearing tumors harboring different combinations of mutations which can be utilized for testing tumor genotype specific efficacy of novel chemotherapeutic drugs.
Conclusion
In summary, we have developed a reproducible and predictable orthotopic model of pancreatic cancer which retains all the tumor properties as well as the stroma-immune microenvironment of spontaneous genetic models. The histology and pattern of disease progression in our STIM mice resembles human pancreatic cancer. Additionally, our model has consistent tumor development and predictable progression that could be of great advantage in translational studies. The Syngeneic Tumor Implantation Model (STIM) could be a valuable preclinical tool for evaluation of new therapies.
Acknowledgments
This study was funded by NIH grants R01-CA170946 and CA124723 (to AKS); NIH grant R01-CA184274 (to SB); Katherine and Robert Goodale foundation support (to AKS) and Minneamrita Therapeutics LLC (to AKS).
Grant Support NIH grants R01-CA170946 and CA124723 (to AKS); NIH grant R01-CA184274 (to SB)
Footnotes
Compliance with Ethical Standards
Conflict of Interest R Chugh has ownership interest in a patent of Minnelide. AK Saluja has ownership interest (including patents) in Minneamrita therapeutics and is a consultant/advisory board member for Minneamrita Therapeutics. The remaining authors declare no conflict of interest.
Ethical Approval All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Minnesota.
References
Author names in bold designate shared first co-first authorship.
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Izeradjene K, Hingorani SR. Targets, trials, and travails in pancreas cancer. J Natl Compr Canc Netw. 2007;5:1042–1053. doi: 10.6004/jnccn.2007.0087. [DOI] [PubMed] [Google Scholar]
- 3.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–1049. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 4.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hotz HG, Reber HA, Hotz B, et al. An orthotopic nude mouse model for evaluating pathophysiology and therapy of pancreatic cancer. Pancreas. 2003;26:e89–98. doi: 10.1097/00006676-200305000-00020. [DOI] [PubMed] [Google Scholar]
- 6.Schwarz RE, McCarty TM, Peralta EA, et al. An orthotopic in vivo model of human pancreatic cancer. Surgery. 1999;126:562–567. [PubMed] [Google Scholar]
- 7.Saluja AK, Dudeja V. Relevance of animal models of pancreatic cancer and pancreatitis to human disease. Gastroenterology. 2013;144:1194–1198. doi: 10.1053/j.gastro.2013.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Chugh R, Sangwan V, Patil SP, et al. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci Transl Med. 2012;4:156ra139. doi: 10.1126/scitranslmed.3004334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sangwan V, Banerjee S, Jensen KM, et al. Primary and Liver Metastasis-Derived Cell Lines From KrasG12D; Trp53R172H; Pdx-1 Cre Animals Undergo Apoptosis in Response to Triptolide. Pancreas. 2015;44:583–589. doi: 10.1097/MPA.0000000000000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol. 2013;25:200–205. doi: 10.1016/j.coi.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabashima-Niibe A, Higuchi H, Takaishi H, et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci. 2013;104:157–164. doi: 10.1111/cas.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porembka MR, Mitchem JB, Belt BA, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61:1373–1385. doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pylayeva-Gupta Y, Lee KE, Hajdu CH, et al. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and Tcell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sideras K, Braat H, Kwekkeboom J, et al. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat Rev. 2014;40:513–522. doi: 10.1016/j.ctrv.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Hwang RF, Moore T, Arumugam T, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikenaga N, Ohuchida K, Mizumoto K, et al. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology. 2010;139:1041–1051. 1051 e1041–1048. doi: 10.1053/j.gastro.2010.05.084. [DOI] [PubMed] [Google Scholar]
- 19.Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hezel AF, Kimmelman AC, Stanger BZ, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 21.Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tinder TL, Subramani DB, Basu GD, et al. MUC1 enhances tumor progression and contributes toward immunosuppression in a mouse model of spontaneous pancreatic adenocarcinoma. J Immunol. 2008;181:3116–3125. doi: 10.4049/jimmunol.181.5.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westphalen CB, Olive KP. Genetically engineered mouse models of pancreatic cancer. Cancer J. 2012;18:502–510. doi: 10.1097/PPO.0b013e31827ab4c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 25.Feig C, Gopinathan A, Neesse A, et al. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westerheide SD, Kawahara TL, Orton K, Morimoto RI. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J Biol Chem. 2006;281:9616–9622. doi: 10.1074/jbc.M512044200. [DOI] [PubMed] [Google Scholar]
- 28.Dudeja V, Mujumdar N, Phillips P, et al. Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms. Gastroenterology. 2009;136:1772–1782. doi: 10.1053/j.gastro.2009.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips PA, Dudeja V, McCarroll JA, et al. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007;67:9407–9416. doi: 10.1158/0008-5472.CAN-07-1077. [DOI] [PubMed] [Google Scholar]
- 30.Dudeja V, Chugh RK, Sangwan V, et al. Prosurvival role of heat shock factor 1 in the pathogenesis of pancreatobiliary tumors. Am J Physiol Gastrointest Liver Physiol. 2011;300:G948–955. doi: 10.1152/ajpgi.00346.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antonoff MB, Chugh R, Borja-Cacho D, et al. Triptolide therapy for neuroblastoma decreases cell viability in vitro and inhibits tumor growth in vivo. Surgery. 2009;146:282–290. doi: 10.1016/j.surg.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 32.Banerjee S, Thayanithy V, Sangwan V, et al. Minnelide reduces tumor burden in preclinical models of osteosarcoma. Cancer Lett. 2013;335:412–420. doi: 10.1016/j.canlet.2013.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caicedo-Granados E, Lin R, Fujisawa C, et al. Wild-type p53 reactivation by small-molecule Minnelide in human papillomavirus (HPV)-positive head and neck squamous cell carcinoma. Oral Oncol. 2014;50:1149–1156. doi: 10.1016/j.oraloncology.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Pan GF, Jiang ZZ, et al. Triptolide inhibits human breast cancer MCF-7 cell growth via downregulation of the ERalpha-mediated signaling pathway. Acta Pharmacol Sin. 2015 doi: 10.1038/aps.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliveira AR, Beyer G, Chugh R, et al. Triptolide abrogates growth of colon cancer and induces cell cycle arrest by inhibiting transcriptional activation of E2F. Lab Invest. 2015 doi: 10.1038/labinvest.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rivard C, Geller M, Schnettler E, et al. Inhibition of epithelial ovarian cancer by Minnelide, a water-soluble pro-drug. Gynecol Oncol. 2014;135:318–324. doi: 10.1016/j.ygyno.2014.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang BY, Cao J, Chen JW, Liu QY. Triptolide induces apoptosis of gastric cancer cells via inhibiting the overexpression of MDM2. Med Oncol. 2014;31:270. doi: 10.1007/s12032-014-0270-7. [DOI] [PubMed] [Google Scholar]