

Abstract
Purpose
The cure rate for patients with advanced head and neck squamous cell carcinoma (HNSCC) remains poor due to resistance to standard therapy primarily consisting of chemoradiation. Since mutation of TP53 in HNSCC occurs in 60-80% of non-HPV associated cases and is in turn associated with resistance to these treatments, more effective therapies are needed. In this study, we evaluated the efficacy of a regimen combining vorinostat and AZD1775 in HNSCC cells with a variety of p53 mutations.
Experimental Design
Clonogenic survival assays and an orthotopic mouse model of oral cancer were used to examine the in vitro and in vivo sensitivity of high-risk mutant p53 HNSCC cell lines to vorinostat in combination with AZD1775. Cell cycle, replication stress, homologous recombination (HR), live cell imaging, RNA-sequencing, and apoptosis analyses were performed to dissect molecular mechanisms.
Results
We found that vorinostat synergizes with AZD1775 in vitro to inhibit growth of HNSCC cells harboring high-risk mutp53. These drugs interact synergistically to induce DNA damage, replication stress associated with impaired Rad51-mediated homologous recombination through activation of CDK1 and inhibition of Chk1 phosphorylation, culminating in an early apoptotic cell death during the S-phase of the cell cycle. The combination of vorinostat and AZD1775 inhibits tumor growth and angiogenesis in vivo in an orthotopic mouse model of oral cancer and prolongs animal survival.
Conclusions
Vorinostat synergizes with AZD1775 in HNSCC cells with mutant p53 in vitro and in vivo. A strategy combining HDAC and WEE1 inhibition deserves further clinical investigation in patients with advanced HNSCC.
Keywords: HNSCC cells, high-risk mutant p53, WEE1, AZD1775, vorinostat, DNA damage, CDK1, Chk1, RRM2, replication stress, Rad51, homologous recombination, CD31, apoptosis
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the seventh most common cancer worldwide (1). The cure rate for patients with advanced HNSCC remains poor due to resistance to standard therapy primarily consisting of cisplatin and radiation. Mutation of TP53 in HNSCC occurs in 60-80% of HPV-negative cases (2,3) and is associated with resistance to these treatments. Recently, we developed a novel computational approach termed evolutionary action (EAp53), which can stratify patients with tumors harboring TP53 mutations as high or low risk. Patients with high-risk TP53 mutations had the poorest survival outcomes, shortest time to the development of distant metastases, and increased resistance to cisplatin-based therapy (4,5). Since the group of tumors with this high-risk mutations show resistance to cisplatin or radiation, we are interested in the development of novel therapeutic approaches to overcome this resistance and improve treatment outcomes. WEE1 is a kinase that has been linked to DNA damage-induced S-phase replication stress and G2/M arrest, owing to its ability to inactivate cyclin dependent kinase 1 (CDK1) through phosphorylation of the Tyr15 residue (6–10). Recently, we demonstrated that the WEE1 kinase inhibitor, AZD1775 sensitizes HNSCC cells harboring high-risk TP53 mutations to cisplatin both in vitro and in vivo through induction of persistent DNA damage response associated with mitotic delay and subsequent senescence (11).
Modulation of the acetylation status of histones and transcription factors is an essential mechanism for regulating gene expression (12,13). Histone acetylation is generally associated with elevated transcription, whereas deacetylated histones are often linked to repressed transcription (14). Histone deacetylases (HDACs) act enzymatically to remove the acetyl group from histones and silence gene expression (14). Elevated activities of histone deacetylases (HDACs) have been observed in several human malignancies, including HNSCC, and their overexpression is associated with poorer prognosis in oral cancer patients (2,15,16). Collectively, these findings indicate that histone deacetylation may represent a potential therapeutic target in HNSCC.
Recent reports have shown that HDAC inhibitors (HDACIs) induce growth arrest, differentiation, and apoptosis in various cancer cell lines in vitro and suppress tumor growth in animal xenograft models, including HNSCC (12,17,18). Additionally, several studies have demonstrated that vorinostat, a small molecule inhibitor of HDAC displays preferential cytotoxicity in vitro and in vivo in cancer cells harboring TP53 mutations (19–21). Although recent evidence suggests that defects in DNA damage repair processes contribute to the selective cytotoxic effects of HDAC inhibitors in tumor cells, the detailed molecular mechanism is not well understood (22,23). The HDAC and WEE1 inhibitors are now emerging as attractive classes of antitumor agents being tested clinically either as single agents or in combination with conventional chemotherapeutics or targeted agents (24,25). Taken together, these preclinical results and the ongoing clinical trials have prompted us to evaluate the combination of WEE1 and HDAC inhibitors in HNSCC with mutant TP53.
In this study, we demonstrate that vorinostat interacts synergistically with AZD1775 in vitro and in vivo in HNSCC tumor cells expressing high-risk mutant p53 (mutp53). Notably, vorinostat alone or in combination with AZD1775 results in increased markers of replication stress, DNA damage response, and impaired Rad51-mediated homologous recombination, leading to an early apoptotic cell death during the S-phase and subsequently in the G2/M cell cycle phase. Using live cell imaging, RNA-seq analyses and RPPA proteomic profiling, we further provide evidence that the mechanism of the synergistic interaction between these two drugs may be in part due to vorinostat’s ability to epigenetically modulate expression of a transcript-signature containing genes involved in regulating replication stress, mitosis, and the cell cycle checkpoints in p53 mutant HNSCC cells. Taken together, our findings support a strategy including a combination of WEE1 and HDAC inhibition, which is a novel therapeutic regimen warranting investigation in patients with advanced HNSCC.
Materials and Methods
Tissue culture, reagents and generation of stable cell lines
The HNSCC cell line PCI13 lacking endogenous expression of p53 was obtained from the laboratory of Dr. Jennifer Grandis (University of Pittsburgh, Pittsburgh, PA) in August 2008 and engineered to stably express constructs containing wild-type p53 (wtp53), high-risk EA score mutant p53 (C238F and G245D), as described previously (4). The HNSCC cell lines, HN30 expressing wtp53 and HN31 expressing mutp53 were obtained in December 2008 from the laboratory of Dr. John Ensley (Wayne State University, Detroit, MI). OSC-19 was obtained from Health Science Research Resource Bank (HSRRB, Japan) in 2010. Detroit562 was purchased from ATCC in 2009. HN5 was obtained from Dr. D. M. Easty (Ludwig Institute for Cancer Research, London, UK) in 2003. The cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% FBS, L-glutamine, sodium pyruvate, nonessential amino acids, and vitamin solution, and incubated at 37°C in 5% CO2 and 95% Air. The identity of all cell lines was authenticated using short tandem repeat testing within 6 months of cell use. The WEE1 inhibitor, AZD1775 was provided by AstraZeneca through a collaborative agreement arrange by NCI-CTEP. Vorinostat [formerly known as suberoylanide hydroxamic acid (SAHA)] was obtained from Merck Corp. For in vitro studies, AZD1775 and vorinostat were prepared as 10 mmol/L stock solution in DMSO and stored at -20⁰C. AZD1775 and vorinostat were diluted in culture medium and used at 0.25 μmol/L and 4 μmol/L final concentration respectively before use. The staurosporine was purchased from Sigma (St Louis, MO) and used at 1 μmol/L final concentration.
Clonogenic survival assay
The HNSCC cells were seeded in 6-well plates at predetermined densities, concurrently exposed to different fixed-ratio combinations of vorinostat (dose range, 0.01-20 μmol/L) and AZD1775 (dose range, 0.01-1 μmol/L) for 48 hours and the clonogenic cell survival was determined as previously described (11, 26).
Analysis of combined drug effects
Drug synergy between vorinostat and AZD1775 was determined by the combination-index and isobologram analyses, which were generated according to the median-effect method of Chou and Talalay (27) using the CalcuSyn software (Biosoft, Ferguson, MO). For details, see Supplementary Materials and Methods section.
Antibodies
Antibodies used for western blotting were phospho-H2AX (Ser139; #2577), phospho-CDC2 (CDK1)-Tyr15 (#9111), CDC2 (CDK1) (#9112), cyclin B1 (#4138), PARP-1 (#9542), Survivin (71G4B7; #2808), Rad51 (#8875), RB (Ser807/811; #9308), FOXM1 (D12D5; #5436S), p-HH3 (#9701), CHK1 (#2345), pCHK1 (Ser345; #2341), Acetyl-Histone H3 (Lys9/Lys14; #9677) and PLK1 (208G4; #4513S); all from Cell Signaling Technology; β-actin (#A5316; Sigma-Aldrich); p53(DO-1) (#sc-126), R2 (N-M; sc-10844), and RB (C-15; sc-50), were from Santa Cruz; p21WAFI (Ab-1) (#OP64; Calbiochem); RPA32 (clone RPA-34-20; #MABE285; EMD Millipore); EMA (2F6; #ab156947), and CD31 (#ab28364), were from Abcam.
Western blot analysis
Cells grown on 10-cm plates were treated with minimally toxic and clinically relevant concentrations of vorinostat (i.e., 0.5-4 μmol/L) and slightly toxic AZD1775 (i.e., 0.25-0.5 μmol/L) either alone or in combination for 16 or 48 hours. Whole cell lysates were prepared and western blot analysis was conducted with indicated antibodies as described previously (11).
Cell cycle analysis and apoptosis detection
Cells were seeded in 60-mm dishes, treated the next day with vorinostat (4 μmol/L), AZD1775 (0.25 μmol/L) either alone or in combination and then harvested at 24, 48, or 72 hours. The cell cycle analysis and apoptosis detection were performed as previously described (11).
Live cell imaging and EdU labeling
For live cell imaging studies, HNSCC cell lines (PCI13-G245D) were stably transfected with histone H2B-RFP lentiviral vector (Addgene), selected in 2 μg/mL blasticidin, and sorted by flow cytometry to enrich for highly expressing cells according to published protocol (26). The histone H2B-RFP-tagged cells were then plated at a predetermined density followed by treatment with 0.01% DMSO alone, 0.25 μmol/L AZD1775 alone, 4 μmol/L vorinostat alone, or in combination. Live video imaging, EdU labeling, and DNA content analyses were carried out as described in the Supplementary Materials and Methods.
Immunofluorescence
Cells were plated on glass coverslips and treated with drugs the following day as previously indicated. Cells were then fixed in 2% paraformaldehyde for 1 hour, washed, permeabilized in 0.2% Triton X-100 in PBS for 20 minutes, washed, and blocked for 1 hour at room temperature in PBS buffer containing 2% normal goat serum, and 0.3% Triton X-100. Then, glass coverslips were incubated with primary Rad51 (MA5-14419; Thermo Scientific) antibody overnight at 4°C followed by secondary Alexa Fluor-conjugated antibody 594 as appropriate and Rad51 foci images were acquired on a Leica confocal microscope as described previously (11). Detailed description is included in the Supplementary Materials and Methods.
Orthotopic mouse model of oral tongue cancer and tumor growth delay
All animal experimentation was approved by the Institutional Animal Care and use Committee (IACUC) of the University of Texas MD Anderson Cancer Center. Our orthotopic nude mouse tongue model has been previously described in the literature (11). The high-risk mutp53 HNSCC cells (PCI13-G245D) were injected into the tongues of the male athymic nude mice and they were randomized into different groups 8–10 days after injection. Treatment protocol and tumor growth delay measurement are described in the Supplementary Materials and Methods.
In vivo TUNEL assay
Apoptosis was assessed in mice tissue sections with terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay with DeadEnd™ Fluorometric TUNEL System (Promega) according to the manufacturer’s protocol with some modifications. A detailed description is included in the Supplementary Materials and Methods.
RNA-Seq profiling
HNSCC cell lines (PCI-13) stably expressing high-risk mutp53 (G245D) were treated with vorinostat (4 μmol/L), AZD1775 (0.25 μmol/L) or in combination. Total RNA was isolated 36 hours after treatment using the RNeasy mini kit reagents (QIAGEN) according to the manufacturer’s instructions. Cells treated with 0.1% DMSO served as untreated controls. For RNA-Seq, total RNA was submitted to the Sequencing and Microarray Facility at MD Anderson Cancer Center for next-generation sequencing. Detailed description of the RNA-seq is included in the Supplementary Materials and Methods.
Reverse phase protein array (RPPA)
PCI13-G245D mutant cells were treated with vorinostat (4 μmol/L), AZD1775 (0.25 μmol/L) or in combination. Protein lysate was collected 48 hour after treatment from each cell line under full-serum (10% FBS) conditions and subjected to RPPA analysis as described previously (28). Detailed description of the RPPA is included in the Supplementary Materials and Methods.
Immunohistochemistry
Sections were prepared from formalin-fixed paraffin embedded mice tumor tissues and subjected to immunohistochemistry with indicated antibodies according to the protocol as described in Supplementary Materials and Methods.
Statistical analysis
The Student’s t and one-way ANOVA tests were carried out to analyze in vitro data. For mouse studies, a two-way ANOVA test was used to compare tumor volumes between control and treatment groups. For immunohistochemical analyses, a one-way ANOVA and a chi-square test were used to compare control and treatment groups. Survival following drug treatment was analyzed by the Kaplan–Meier method and compared with log-rank test. All data were expressed as mean ± standard error and P-values of 0.05 or less were considered to indicate nominal statistical significance.
Results
Vorinostat synergizes with AZD1775 to inhibit the in vitro clonogenic survival of HNSCC cells expressing high-risk mutp53
To explore the degree of sensitivity of high-risk mutp53 HNSCC cells to the HDAC inhibitor vorinostat as a single agent, we performed dose-response studies with the drug in isogenic HNSCC PCI-13 cells with wildtype p53, lacking p53 expression (pBabe null), or expressing high-risk mutp53 (C238F, G245D) using standard clonogenic survival assays. As shown in Fig. 1A and B, the PCI-13 isogenic cells displayed differential sensitivity to vorinostat as a single agent. The cell lines expressing high-risk mutp53 were more sensitive to vorinostat as a single agent (IC50; 0.5-0.7 μmol/L) than the cells expressing wtp53 (IC50; 1.2 μmol/L) or pBabe vector control-p53 null cell lines (1.1 μmol/L), consistent with previous publications (13,19,20).
Figure 1. Vorinostat synergizes with AZD1775 to inhibit in vitro clonogenic survival of HNSCC cells expressing high-risk mutp53.
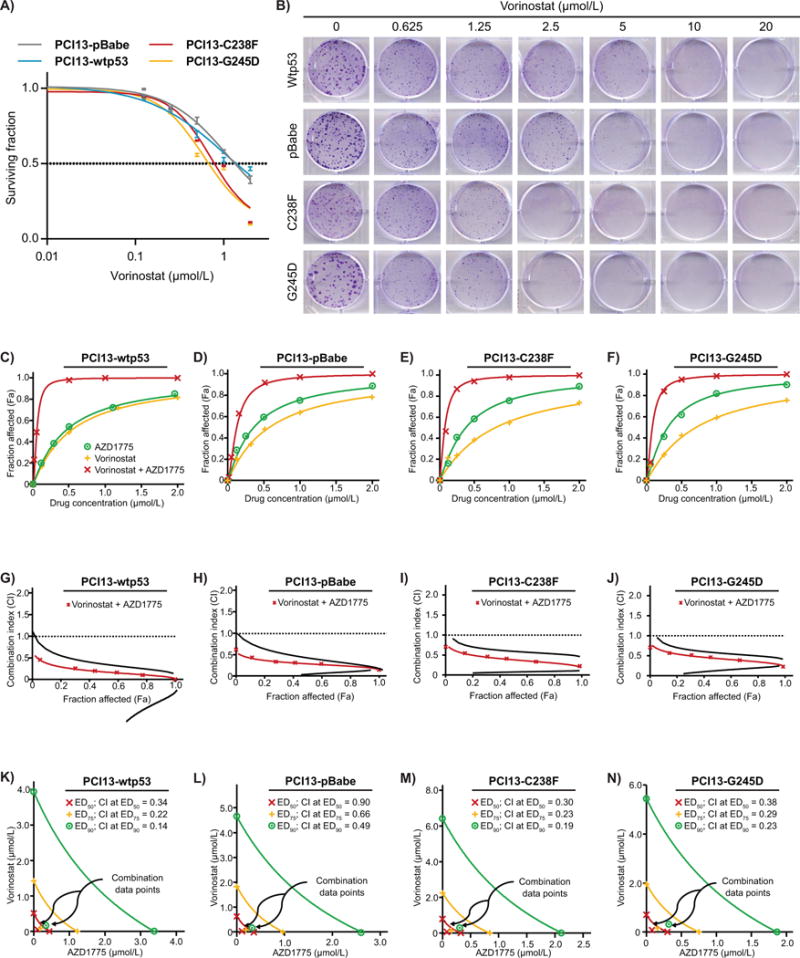
A, clonogenic survival curves for isogenic HNSCC PCI-13 cells lacking p53 (pBabe null), expressing wtp53 or high-risk mutp53 (C238F, G245D) and treated for 48 hours with a range of vorinostat concentrations (0.01-20 μmol/L) to determine the IC50. B, representative images of the results of clonogenic survival assays. C-F, assessment of the degree of synergy between vorinostat and AZD1775 in PCI13-Wtp53, PCI13-pBabe, PCI13-C238F, and PCI13-G245D, using the Chou and Talalay method (median dose-effect analysis). Vorinostat and AZD1775 were used at constant ratios (1:1 and 2:1 respectively). G-J, Fa-CI plots generated to determine the CI values (< 1.0 indicate synergism). The CI values for combination of effective drug doses (ED) that result in clonogenic survival inhibition of 50% (ED50; fa = 0.5), 75% (ED75; fa = 0.75), and 90% (ED90; fa = 0.90) were generated from the conservative isobolograms. The ED50 (red X), ED75 (green crosses) and ED90 (blue circles) graphed against fractional concentrations of vorinostat and AZD1775 on the y and x axis, respectively are indicated. K-N, conservative isobologram plots demonstrate that vorinostat and AZD1775 acts synergistically to inhibit in vitro clonogenic survival of HNSCC cells. All treatments were performed in triplicate and each experiment was repeated at least three times.
We next examined whether vorinostat was synergistic with AZD1775 treatment of the isogenic PCI-13 cell lines, using the combination index (CI) and fraction affected (Fa) method of Chou and Talalay (27). Treatment of these cells with the combination of vorinostat and AZD1775 revealed strong synergism manifested by the shift of vorinostat response curves and the CI values (Fa 0.5) of 0.90, 0.30, 0.38, and 0.34 (Fig. 1C–F, top plots and Supplementary Table S1) for PCI13-pBabe (null), PCI13-C238F, PCI13-G245D, and PCI13-Wtp53 respectively. Furthermore, the CI was less than 1.0 in all cell tested representing strong synergism at the more relevant Fa values (≥50%) (Fig. 1G–J, middle plots). Conservative isobologram plots of effective dose ED50, ED75, and ED90 were also generated and further confirmed strong synergism (Fig. 1K–N, bottom plots and Supplementary Table S1). The degree of synergy between vorinostat and AZD1775 was also confirmed in other human HNSCC cell lines with different TP53 status (Supplementary Fig. S1A–S1H, S2A–S2H, and Supplementary Table S1). The data clearly demonstrated that vorinostat and AZD1775 interacted synergistically to inhibit in vitro clonogenic survival in various HNSCC irrespective of the TP53 status.
Multiple cell cycle perturbations leading to significant apoptosis in HNSCC cells expressing high-risk mutp53 treated with combination of vorinostat and AZD1775
To further characterize the synergistic response of HNSCC cells expressing mutp53 to vorinostat and AZD1775, we conducted live cell imaging in PCI13-G245D cells expressing a histone H2B-RFP construct. The results of the live cell imaging studies are shown in Figure 2A as event charts, where the history of each cell depicted as a line over time, the color of the line indicates whether the cell is in interphase (gray) or mitosis (blue), and the time of cell apoptosis is marked by a red symbol. The majority of control PCI13-G245D cells entered mitosis within 15 hours and reached the second mitosis with an average interphase time of 15.6 hours (Fig. 2A). While AZD1775 treatment (Fig. 2A and B) slightly increased the initial rate of mitotic entry (during the first 4 hours) and mitotic duration (from an average of 37 minutes for the control to 53 minutes for AZD1775 treatment), the mitotic entry rate then decreased for the next 7 hours. In addition, AZD1775 treatment alone decreased the fraction of cells capable of reaching the second mitosis and increased the fraction of apoptotic cells, especially in cells that had undergone a first mitosis in the presence of AZD1775 (Fig. 2A and B and Supplementary Video S1). In contrast, vorinostat treatment was associated with a slower progression rate to mitosis, a decreased fraction of cells capable of reaching the first mitosis, a slightly longer mitotic duration in those cells reaching mitosis (average, 61 minutes), and resulted in a further increased apoptotic fraction both in cells that did and did not reach a first mitosis (Fig. 2A and B and Supplementary Video S2). Interestingly, when vorinostat and AZD775 were combined, the initial rate of mitotic entry again increased compared to cells treated with vorinostat alone, however the average mitotic duration increased to 119 minutes and an increased fraction of apoptotic cells (Fig. 2A and B and Supplementary Video S3).
Figure 2. Multiple cell cycle perturbations lead to significant apoptosis in HNSCC cells expressing high-risk mutp53 treated with combination of vorinostat and AZD1775.
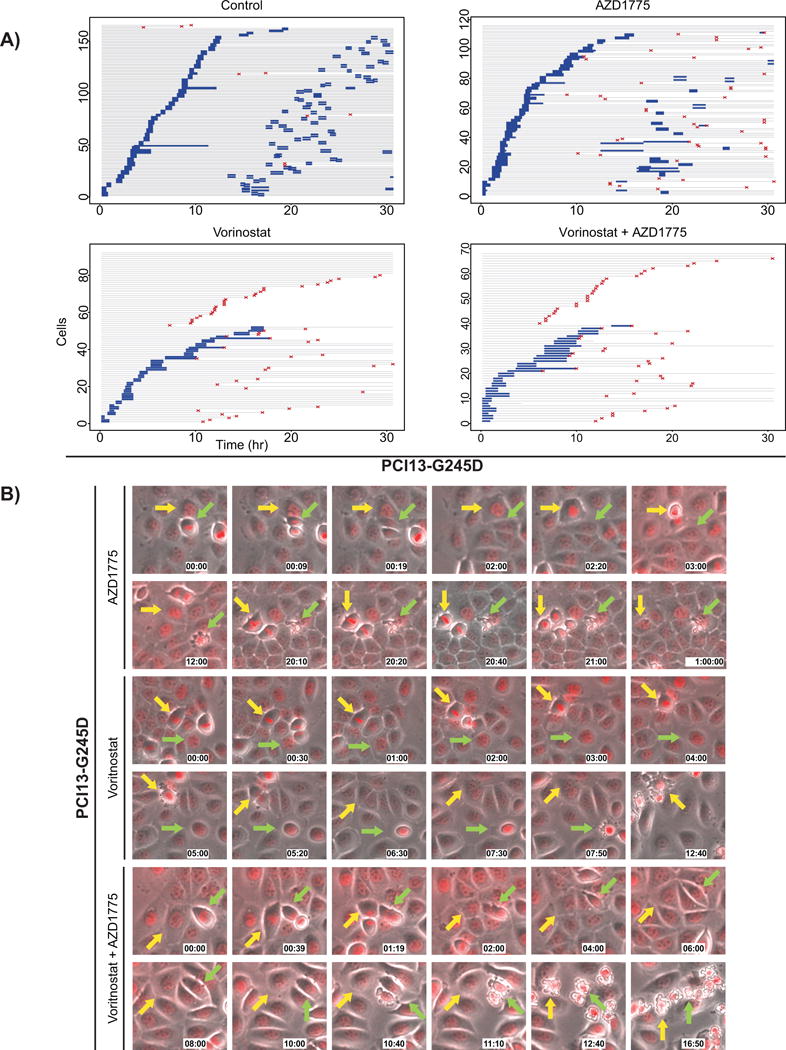
Histone-H2B-RFP expressing PC13-G245D cells were treated with the drugs as indicated and followed by live cell imaging. A, live cell imaging results depicted as event charts, where each line represents one cell where the cell’s time in interphase is labeled gray, its time in mitosis is labeled blue, and the time of apoptosis is marked by a red symbol. B, Representative image frames from the live cell imaging movies illustrating mitotic and interphase events leading to apoptosis.
Apoptosis occurs in early S-phase in HNSCC cells expressing high-risk mutp53 treated with combination of vorinostat and AZD1775
The prior live cell imaging studies (Fig. 2) showed that cells began undergoing apoptosis at increased rates after 7–9 hours of treatment with vorinostat and AZD1775. To better understand the underlying causes and timing of apoptosis, PCI13-G245D cells expressing a histone H2B-RFP construct were treated for various durations and were subjected to pulse EdU labeling and DNA content measurements. As shown in Figure 3A (9 hour treatment), AZD1775 alone slightly increased the rate of entry into S-phase (increased fraction of replicating cells at the G1/S boundary) and S-phase fraction (59.3%) compared to untreated control (48.7%). Vorinostat alone also caused S-phase accumulation (66.1%) (especially early S-phase) (Fig. 3A), slowed DNA replication (Fig. 3B), and increased the G2 fraction compared to untreated control (Fig. 3A and B). Furthermore, the combination of vorinostat and AZD1775 resulted in a more profound S-phase accumulation (77.3%) due to enhanced entry into S phase but profound replication slowing with fewer cells reaching G2 phase compared to all treatments (Fig. 3A and 3B). Combining these findings with the live cell imaging data, we observed that the mitotic rate in the combination treatment was slowed by 2–3 hours of treatment due to AZD1775 effect and cells stopped reaching mitosis by 12 hours. Moreover, we observed increasing rates of apoptosis starting at around 8 hours, mostly in the subpopulation that had not reached its first mitosis or reached mitosis later during treatment.
Figure 3. Apoptosis occurs in early S-phase in HNSCC cells expressing high-risk mutp53 treated with combination of vorinostat and AZD1775.
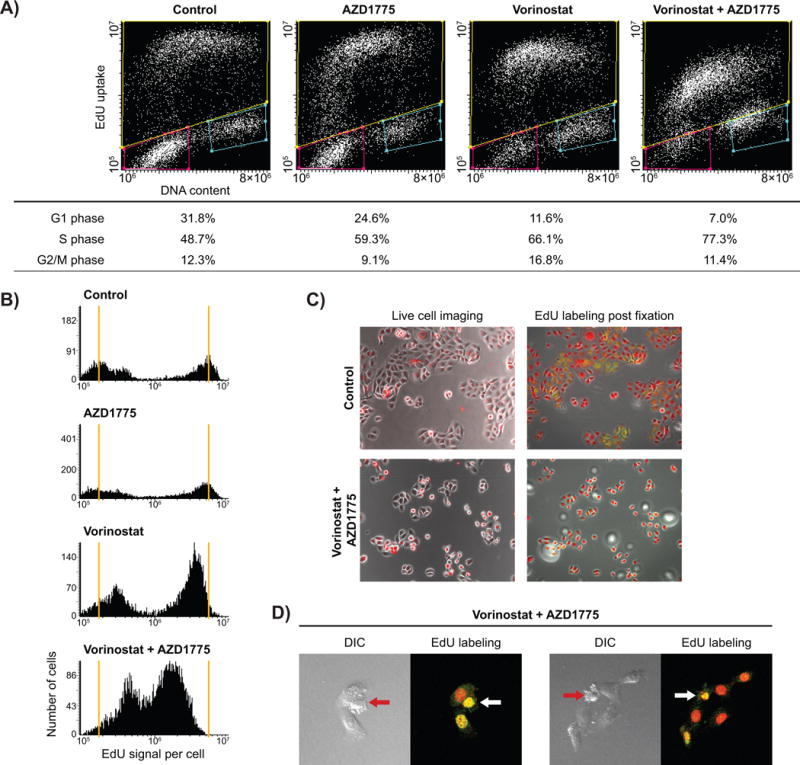
A, cell cycle analyses at 9 hour time point of drug treatment based on DNA content and EdU labeling. B, EdU uptake (log scale) into cells at 9 hour time point following treatment with drugs. C, comparison of live cell imaging and same field after EdU (green) and DNA content (red) staining. D, laser scanning confocal images of combination treated samples pulsed with EdU prior to fixation for cellular morphology (DIC, left), EdU uptake (green), nuclear morphology (TOPRO-3 DNA staining, red), (fluorescence, right). The arrows indicate cells showing a stressed morphology along with the associated EdU and nuclear images, suggestive of cells about to undergo apoptosis during an impeded S-phase.
The above data suggest that cells treated with the combination of drugs experienced replication stress and eventually underwent apoptosis within S-phase. If this were the case, one might expect to detect EdU-labeled cells undergoing apoptosis after treatment. The live cell imaging studies showed that the cells underwent distinct morphological changes (stress-induced blebbing) just prior to apoptosis (Fig. 3C). We therefore imaged populations of cells that were pulsed with EdU for 1 hour prior to fixation (9 hour sample) for morphology (DIC), EdU staining, and nuclear morphology (TOPRO-3 staining) using laser scanning confocal microscopy (Fig. 3D and Supplementary Fig. S3). The arrows indicate cells showing the stressed morphology (DIC image, left side) along with the associated EdU and nuclear images (right side), suggestive of cells about to undergo apoptosis during an impeded S-phase.
To better understand the longer term effects of drug treatment, PCI13-G245D cells were drug treated and examined at 48 hours. Treatment with AZD1775 alone, or vorinostat alone were associated with increased G2 fractions (25% and 38.3 %, respectively) (Supplementary Fig. S4A and S4B), and the combination of drugs was associated with and even higher G2/M fraction (57.4%) (Supplementary Fig. S4A and S4B). Moreover, a sub G1 peak indicative of apoptosis was increased in PCI13-G245D cells after 48 hours of combination treatment (Supplementary Fig. S4A and S4B). Apoptosis in these cells was further confirmed by the presence of positive APO-BrdU tunnel staining (Supplementary Fig. S4C).
Vorinostat synergizes with AZD1775 in HNSCC mutp53 cells to induce replication stress associated with impaired Rad51-mediated homologous recombination and apoptosis
The cell cycle kinase inhibitor p21 (CDKN1A), acetylated histone H3 and H4 are known targets for induced effects of HDACIs in various transformed cells (29,30,31). Significant increases in p21 and acetylated H3 protein levels were observed in all the cells tested by western blotting following treatment for 48 hours with vorinostat alone or in combination with AZD1775 (Fig. 4A and B and Supplementary Figure S5A-C). These results suggest that vorinostat increases p21 level independent of its upstream activator, p53, in HNSCC cells. The increase in acetylated histone 3 further confirmed that the concentrations of vorinostat used in vitro in our study were inhibiting the activity of HDACs.
Figure 4. The synergism between vorinostat and AZD1775 in HNSCC mutp53 cells is mediated by increased replication stress associated with impaired Rad51-mediated homologous recombination and apoptosis.
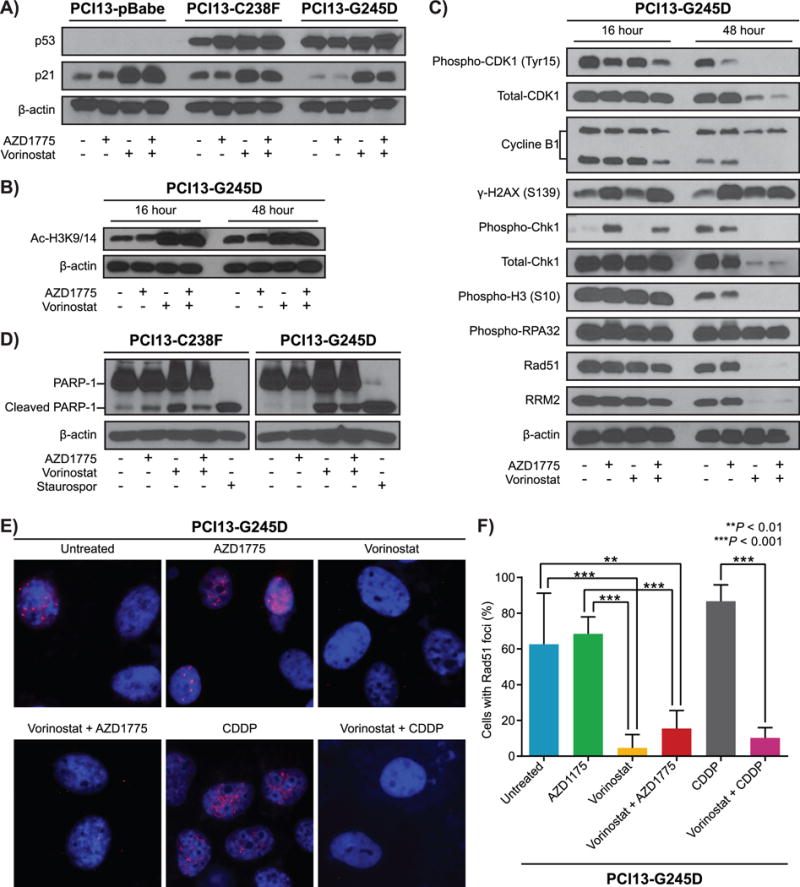
PCI13-pBabe and PCI13 with mutant TP53 (C238F, G245D) cells treated with either vorinostat, AZD1775 alone or in combination for 16 and/or 48 hours and subjected to immunoblot analysis using antibodies as indicated. A and B, protein expression levels of p53, p21 and acetylated histone 3 respectively. C, levels of phosphorylation of H2AX (S139), Chk1 (S345), total Chk1, hyperphosphorylation of RPA32, Rad51, and RRM2. The mitotic entry marker histone H3 S10 phosphorylation, cyclin B1 total protein, CDK1 (CDC2) tyr15 phosphorylation, and total protein levels were also analyzed. D, western blots for HNSCC cells with mutant TP53 (PCI13-C238F, and PCI13-G245D) treated with either vorinostat, AZD1775 alone or in combination as indicated and analyzed for the presence of PARP-1 cleavage as marker of apoptosis. Lysates from staurosporine-treated (1 μmol/L) cells were used as positive controls for apoptosis. β-actin served as loading control .E, vorinostat treatment lead to reduced Rad51 focus formation, indicating impaired Rad51-mediated homologous recombination in HNSCC cells with high-risk TP53 mutation. F, quantification of Rad51 foci images shown in Figure 4E.
HDAC and WEE1 inhibitors have been reported to interact synergistically to induce DNA damage and replication stress through modulation of checkpoint kinase 1 (Chk1) and CDK1 phosphorylation levels in leukemia and glioblastoma cancer cells (22,31–33). Therefore, we evaluated whether vorinostat and AZD1775 similarly affected phosphorylation and protein levels of CDK1 and Chk1 in PCI13-G245D cells and various HNSCC cell lines. The combination of vorinostat and AZD1775 markedly decreased the inhibitory phosphorylation of Tyr15 on CDK1, total CDK1 and its partner cyclin B1 protein levels in a time dependent manner (Fig. 4C and Supplementary Figure S5D–F), suggesting a shift in cells stalled during S-phase. Additionally, the levels of the mitotic marker, phospho-histone 3 (S10), were dramatically reduced within 48 hours following vorinostat treatment and in combination with AZD1775, indicating that a fraction of cells were either arrested at the G2 phase or lost during S-phase at this time point (Fig. 4C). Vorinostat sharply abrogated AZD1775-induced Chk1 phosphorylation and further decreased the total Chk1 protein levels in a time dependent manner (Fig. 4C and Supplementary Figure S5D–F). Reduced Chk1 activation was associated with induction of DNA damage, manifested by the marked increase in the double-strand break marker, phospho-γH2AX following treatment with the drugs in these cells (Fig. 4C and Supplementary Figure S5D–F). These findings raise the possibility that WEE1 and HDAC inhibition induce lethal replicative stress in HNSCC cells expressing mutp53. To test this possibility, the effects of vorinostat and AZD1775 on replication stress markers were examined in PCI13-G245D and HNSCC cells with different TP53 status. Hyperphosphorylation of the replication stress marker, RPA32 associated with reduced levels of the ribonucleotide reductase enzyme, RRM2 were observed in a time dependent manner consistent with the induction of replication stress in these cells following treatment with drugs (Fig. 4C and Supplementary Figure S5D–F). Western blot analyses provided evidence that treatment of HNSCC cells with vorinostat, or in combination with AZD1775 for 48 hours induced apoptosis as indicated by PARP1 cleavage (Figure 4D and Supplementary Figure S5D–F).
One of the pleiotropic actions of vorinostat includes its effect on Rad51 expression and homologous recombination (HR) in cancer cells (34,35). On the basis of this finding, we examined alterations in the expression levels of the HR competent marker, Rad51 in PCI13-G245D cells and various HNSCC cell lines following treatment with the drugs. Vorinostat or in combination with AZD1775 is associated with decreased Rad51 protein levels within 48 hours post treatment (Fig. 4C and Supplementary Figure S5D–F). Correspondingly, Rad51 focus formation under the same treatment conditions was markedly decreased in these cells (Fig. 4E and F). Taken together, these data suggest that vorinostat and AZD1775 exert their synergistic effects in HNSCC cells expressing high-risk mutp53 through induction of replication stress associated with insufficient Rad51-mediated homologous recombination repair.
HDAC and WEE1 inhibition affects key genes involved in DNA replication and G2/M cell cycle checkpoint in TP53 mutant HNSCC
To better understand the cellular phenotype associated with the synergistic interaction between vorinostat and AZD1775 in HNSCC cells with mutp53, cells were treated for 48 hours with the drugs and differentially expressed genes (DEGs) were examined by RNA sequencing (RNA-seq) as described in Materials and Methods. A linear regression model (false discovery rate (FDR) cutoff = 0.05) and filtering criteria (i.e. DEG with an adjusted P-value of < 0.01, genes with 50 average reads among the comparison groups, exclusion of noncoding RNAs) were used. Principal clustering component analysis (PCA) revealed that DEGs among vorinostat and vorinostat plus AZD1775 treatment groups are alike. Approximately 1374 genes that are significantly different between vorinostat and untreated cells with at least a four-fold difference in expression were identified (Supplementary Table S2). Because our in vitro data presented here suggested that vorinostat and AZD1775 resulted in replicative stress and specifically affected the cell cycle at multiple checkpoints and induced cell killing (Fig. 2, 3 and 4), we focused our attention on the dysregulation of genes that are typically involved in these cellular processes. To examine this possibility, the 1374 genes were evaluated by GO enrichment and IPA analysis. Not surprisingly, 5 major pathways significantly associated with the top filtered 139 DEGs regulated by vorinostat or in combination with AZD1775 were identified (Fig. 5 and Supplementary Table S3), including chromatid segregation, G1/S phase transition, mitotic anaphase DNA replication, mitotic M phase and cell cycle checkpoint. All of these processes were deregulated in cancer and a heatmap associated with the target GO terms representing the distribution of the 139 DEGs is shown in Figure 5. Many of the 139 DEGs were associated with multiple cellular cell functions. Expression of top target genes involved in induction of replication stress, homologous recombination, regulation of the S/G1 and G2/M checkpoints, including CHK1, CDK1, PLK1, CCNB1 (cyclin B1), RRM2, RAD51, and BIRC5 (survivin), were significantly decreased following treatment with vorinostat or in combination with AZD1775 (Fig. 5). The expression level of p21 (CDKN1A), the immediate downstream target of p53, was increased by vorinostat (Fig. 5). Furthermore, decreased mRNA expression levels of 130 genes out of the 139 DEGs trended toward almost significant correlation with better survival in the TCGA HPV-negative HNSCC cohort (Supplementary Fig. S6). Change in protein expression levels of some of these key genes were confirmed by western blotting (Fig. 4 and Supplementary Fig. S7B) providing partial validation for the RNA-seq data.
Figure 5. HDAC and WEE1 inhibition affects key genes involved in DNA replication and G2/M cell cycle checkpoint in TP53 mutant HNSCC.
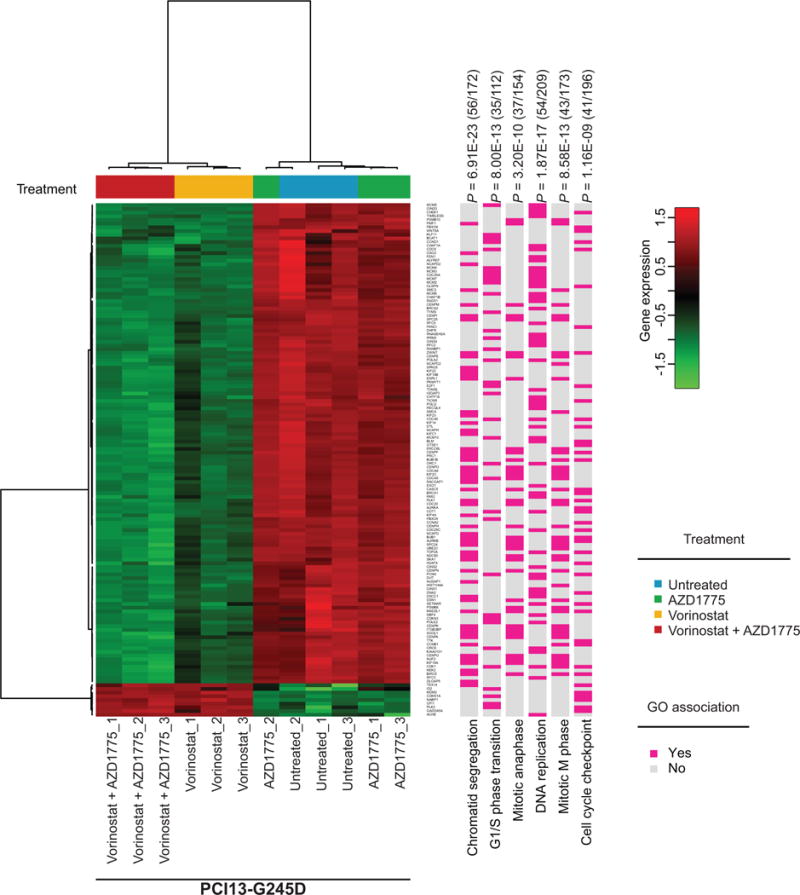
TP53 mutant PCI13-G245D cells were treated with either vorinostat, AZD1775 alone or in combination for 48 hours and subjected to RNA-seq analysis as described in Methods. Shown is a hierarchical clustering heatmap associated with the target GO terms representing the distribution of the significant 139 differentially expressed genes (DEGs) identified by RNA-seq profiling and their specific cell functions. Red and green colors indicate increased or decreased gene expression levels, respectively. P-values are shown and fractional numbers (in parenthesis) on top indicate the DEGs from the list of 139 DEGs that are linked to a cell function (bottom and pink boxes) divided by the total number of genes listed for the function by GO enrichment.
To identify the signaling proteins mediating the effects of vorinostat alone or in combination with AZD1775, a reverse phase protein array (RPPA) was performed using PCI13-G245D cells treated with the drugs as indicated. A total of 118 proteins were significantly expressed (P-value < 0.05), and were similar among vorinostat and vorinostat plus AZD1775 treatment groups. GO enrichment analysis identified two significant pathways involving G1/S phase transition and cell cycle checkpoint as annotated in the heatmap (Supplementary Fig. S7A and Supplementary Table S4). To focus our investigation, we limited our analysis to protein level changes of > 1.5 fold, with a false discovery rate of < 1% and P-value of < 0.05 (28). RPPA identified a number of proteins involved in cell cycle regulation that were significantly downregulated in PCI13-G245D cells, including CDK1, cyclin B1, PLK1, EMA (mucin), FoxM1 and Rb_pS807_S811 (Supplementary Fig. S7A), suggesting decreased progression to mitosis as seen with the live cell imaging and cell cycle analyses. Downregulation of these proteins was further confirmed by western blotting (Supplementary Fig. S7B).
The combination of vorinostat and AZD1775 inhibits tumor growth in an orthotopic mouse model of oral cancer and prolongs animal survival
Based on the in vitro results, the impact of vorinostat and AZD1775 either alone or in combination was evaluated in an orthotopic nude mouse model implanted with PCI13-G245D cells in the tongue. While vorinostat (65 mg/kg) or AZD1775 (45 mg/kg) alone had little effects on tumor growth in vivo at these doses, the combination of vorinostat and AZD1775 significantly inhibited tumor growth (P < 0.001, when compared with AZD1775 alone and P < 0.0001 when compared with vehicle or vorinostat treated group, respectively; Fig. 6A and Supplementary Fig. S8). Furthermore, combined treatment significantly prolonged animal survival compared to animals in the untreated control group (P < 0.022; Fig. 6B). To confirm that the enhancement of antitumor efficacy by the combination drug treatment was associated with engagement of downstream targets in vivo, phosphorylation of CDK1 (Y15) and expression of p21 were evaluated in tissue sections obtained from tongue tumor xenografts. Consistent with the in vitro results, treatment with vorinostat, AZD1775 or in combination was associated with decreased levels of phospho-CDK1 in vivo in orthotopic tongue tumors compared to untreated tumors (Fig. 6C–D). Increased p21 and decreased PCNA, RRM2 and survivin immunostaining levels were also evident in these tumor sections following treatment with vorinostat and in combination with AZD1775 (Supplementary Fig. S9). During the study, mice in the combination treatment arm show no more than 10% body weight loss or any signs of drug toxicity (Supplementary Figure S10).
Figure 6. The combination of vorinostat and AZD1775 inhibits tumor growth in an orthotopic mouse model of oral cancer and prolongs animal survival.
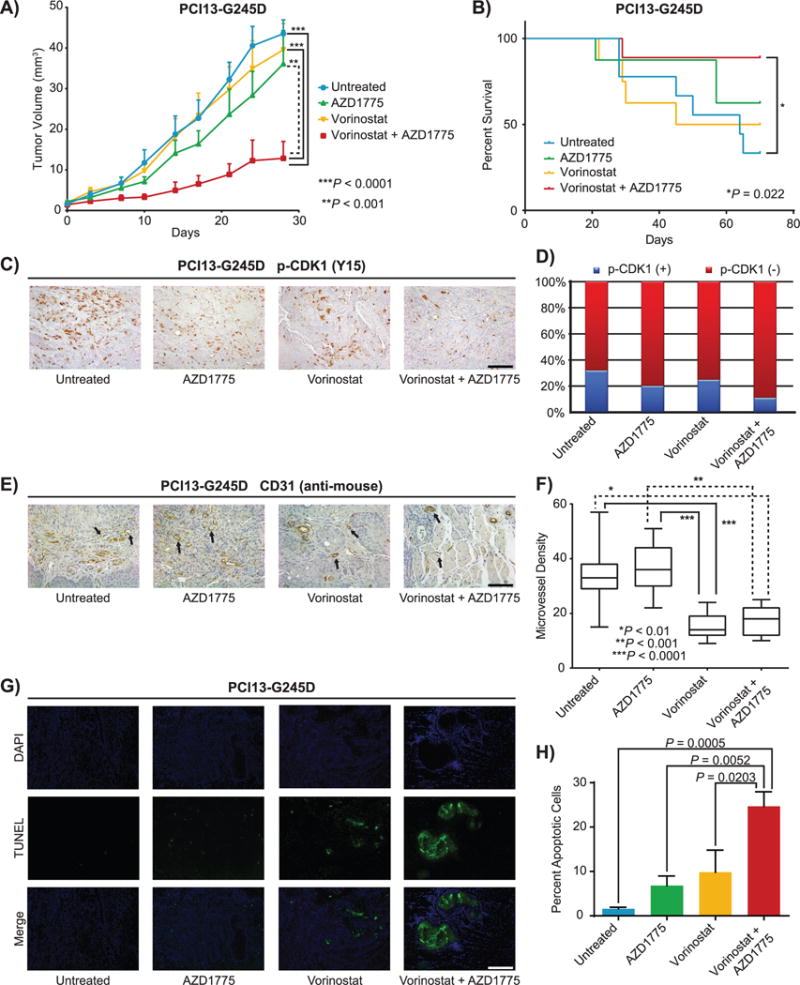
A, tumor growth curves in orthotopic mouse model oral tongues bearing PC13-G245D cells with high-risk mutant TP53 following treatment with either vorinostat (65 mg/kg), AZD1775 alone (45 mg/kg) or in combination. B, Kaplan-Meier analysis for overall survival. C, phospho-CDK1 (tyr15) expression levels in the orthotopic tongue tumors. D, Quantification of phospho-CDK1 levels shown in Figure 5C. E and F, CD31 immunohistochemistry and quantification of microvessel density. G and H, TUNEL-positive apoptotic cells in tissue sections obtained from tumors of mice treated with drugs as indicated.
Angiogenesis is a critical component of tumor progression that is regarded as an important target in cancer therapy. Since vorinostat can inhibit vascularization (36,37), we evaluated the effects of these drugs administered alone or in combination on angiogenesis by measuring intra-tumor vasculature using CD31 as an endothelial cell marker. A significant decrease in the fraction of CD31-positive cells in vivo in mice tumor tongue sections was observed (Fig. 6E and F), suggesting that disruption of microvessel density or lack of new vessel formation may contribute to the efficacy of vorinostat and AZD1775 combination in vivo. The occurrence of apoptosis in vivo in tongue tumor xenografts bearing PCI13-G245D cells was also examined using the TUNEL assay. TUNEL-positive apoptotic cells were significantly increased in tissue sections obtained from tumors of mice treated with combination of vorinostat and AZD1775 compared to either drug alone or untreated control (Fig. 6G and H) indicative of apoptosis.
Discussion
Since HPV-negative HNSCC has a high burden of TP53 mutations, contributing to low cure rate and resistance to standard therapy consisting of cisplatin and radiation, novel therapeutic approaches are needed for patients with advanced HNSCC. In this study, we investigated the antitumor effects of a regimen combining the HDAC inhibitor, vorinostat and the WEE1 kinase inhibitor, AZD1775, in HNSCC tumor cells harboring high-risk TP53 mutations. Consistent with previous reports, we show that vorinostat-induced cell death in HNSCC is independent of TP53 status (22,38). However, HNSCC cells expressing mutp53 are more sensitive to vorinostat than isogenic counterparts with wtp53 or p53 null, in agreement with data showing that vorinostat preferentially kills mutp53 cancer cells, established from various tumor types, in vitro (19–21). This difference in sensitivity towards cells with different TP53 status could be due to the pleiotropic nature of vorinostat primarily acting at the epigenetic transcriptional level (39,40). One significant finding of our study is that vorinostat is highly synergistic with AZD1775 in vitro in HNSCC cells with high-risk TP53 mutation. Additionally, our in vitro experiments guided by live cell imaging, demonstrate for the first time that, vorinostat not only arrests cells at the G2 cell cycle phase but also causes accumulation of cells in early S-phase and the combination of drugs results in significant replication stress and an early apoptotic death within S-phase, perhaps in response to stalled and collapsed replication forks. Taxanes are drugs used in the regimen for treating HNSCC and their effects on radiosensitization have been attributed to their abilities to block tumor cells at the mitotic (M) phase. Therefore, combining taxanes with HDAC inhibitors may seem an attractive combination to improved HNSCC cell killing compared to Wee1 inhibitor. However, this is unlikely since vorinostat is expected to arrest cells at the S and G2-phases and cause mitotic delay.
Mechanistically, combination of vorinostat and AZD1775 significantly increases the DNA damage marker, γ-H2AX (S139) levels, associated with replication stress followed subsequently by apoptosis in all TP53 mutant HNSCC cells tested. This mechanistic possibility is supported by our finding that vorinostat alone or in combination with AZD1775 resulted in activated CDK1 and increased hyperphosphorylation of the replication stress marker, RPA32 associated with reduced protein levels of RRM2. Moreover, the concomitant downregulation of Chk1 activation and Rad51 likely contribute to the HNSCC cells inability to deal with replication stress, thereby leading to cell death.
For the first time, we show that vorinostat treatment alone or in combination with AZD1775 impacts regulation of nearly 1400 genes in TP53 mutant HNSCC, and based on significant fold change in expression, key pathway genes implicated in cancer are modulated. Consistent with published reports, vorinostat downregulates essential genes involved in regulation of DNA synthesis, homologous recombination, and regulation of the G2/M checkpoints, including CHK1, CDK1, PLK1, CCNB1 (cyclin B1), RRM2, RAD51, FOXM1, and BIRC5 (survivin) (41–45). Based upon their well-established functions in regulating replication stress and G2/M cell cycle phase in cancer cells (41–45), it is likely that these genes play an important mechanistic role in synergism between vorinostat and AZD1775 in TP53 mutant HNSCC cells. In addition, HDAC inhibitors act through prevention of the de-acetylation of histones thereby increasing histone acetylation favoring a relaxed chromatin confirmation favoring transcription, our RNA seq data indicates that transcription levels of 9 genes were increased and 130 genes were decreased upon vorinostat addition and when combined with AZD1775 compared to untreated control or AZD1775 alone.
As a preclinical evaluation, we demonstrated that oral administration of vorinostat combined with AZD1775 markedly inhibited in vivo growth of high-risk TP53 mutant HNSCC tongue xenografts and prolonged animal survival. Furthermore, tumor growth inhibition was associated with increased apoptosis in vorinostat alone or in combination with AZD1775 compared with the control groups consistent with the in vitro results. Moreover, vorinostat, AZD1775 or in combination significantly lead to CDK1 activation in vivo in orthotopic tongue tumors compared to untreated tumors. While few studies have shown that vorinostat alone suppresses tumor growth of myeloid leukemia and B cell lymphoma cells in vivo (22,46), we observed little single agent activity of this drug in vivo in mice injected with TP53 mutant HNSCC cells and treated with similar reported pharmacologic doses. Angiogenesis plays an important role in a broad array of physiological and pathological processes, including tumor growth. Consistent with published results (36,37), we show that the fraction of cells with vasculature marker, CD31 is decreased following vorinostat alone or in combination with AZD1775, supporting our previous finding that disruption of the tumor microvasculature can contribute to antitumor efficacy in HNSCC (47). While similar findings were reported previously in myeloid leukemia (22), our data revealed the underlying causes, timing of apoptosis and identified key pathway genes that possibly regulate the synergy between vorinostat and AZD1775 in HNSCC cells. Furthermore, we provide evidence that low expression of these genes trended toward almost significant correlation with better survival in the TCGA HPV-negative HNSCC cohort, suggesting that vorinostat synergizes with AZD1775 to downregulate basal transcriptional levels of prosurvival and proliferative genes in HNSCC.
In summary, we demonstrate that combination of vorinostat and AZD1775 is highly synergistic in HNSCC tumors bearing high-risk TP53 mutation in vitro and in vivo. One hypothesis for this synergism is that WEE1 inhibition promotes progression of cells in different phases of the cycle into the next phase, and that vorinostat decreases the expression of genes that the cells need to adapt in next phase of the cell cycle and this leads to cell death. More specifically, WEE1 inhibition enhances CDK1 activity and this leads to either cells in G1 to be pushed prematurely into S-phase causing replication stress, or cells in G2 phase to be pushed prematurely into mitosis, leading to mitotic dysfunction. The loss of important replication stabilizing and homologous recombination proteins Chk1, and Rad51, and likely others leaves unrepaired DNA which in S-phase cells causes replication fork destabilization and more DNA damage that cannot be repaired, leading to apoptosis. Taken together, our findings argue that a strategy aimed at simultaneous interruption of WEE1 and HDAC function is a novel therapeutic regimen and deserves investigation in patients with advanced HNSCC.
Supplementary Material
Translational Relevance.
Although the treatment of locally advanced head and neck cancer has evolved recently, the cure rate for patients with aggressive tumors and high-risk of failure remains poor due to resistance to standard chemoradiotherapy. Since TP53, the gene that encodes the p53 protein, is by far the most commonly mutated gene found in head and neck cancer and is, in turn, associated with treatment failure and poor survival outcomes, more effective therapies are urgently needed. In this study, we evaluate the efficacy of combining the HDAC inhibitor, vorinostat, and the WEE1 inhibitor, AZD1775, in head and neck tumor cells expressing high-risk mutant p53. Our data demonstrate the vulnerability of HNSCC to vorinostat and AZD1775 through induction of replication stress associated with decreased Rad51-mediated homologous recombination. Thus, we provide preclinical foundation for initiation of clinical trials using combination of HDACs and WEE1 inhibitors particularly for patients with advanced and recurrent metastatic HNSCC tumors.
Acknowledgments
Funding: This work was supported by the National Institute of Health/NIDCR R01DE024601 (A.A. Osman and J.N. Myers), and The German Research Foundation (A. Lindemann).
Abbreviations
- HNSCC
head and neck squamous cell carcinoma
- CDK1
cyclin dependent kinase 1
- HDACs
Histone deacetylases
- RRM2
ribonucleotide reductase enzyme-2
- EdU
5-ethynyl-2-deoxyuridine
- DIC
differential interference contrast
- HR
homologous recombination
- RPPA
Reverse phase protein array
Footnotes
Conflicts of Interest: The authors have no potential conflicts of interest to disclose
References
- 1.Clayman GL, Ebihara S, Terada M, Mukai K, Goepfert H. Report of the Tenth International Symposium of the Foundation for Promotion of Cancer Research: basic and clinical research in head and neck cancer. Jap J Clin Oncol. 1997;27:361–368. doi: 10.1093/jjco/27.5.361. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neskey DM, Osman AA, Ow TJ, Katsonis P, McDonald T, Hicks SC, et al. Evolutionary action score of TP53 identifies high-risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Cancer Res. 2015;75(7):1527–36. doi: 10.1158/0008-5472.CAN-14-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Osman AA, Neskey DM, Katsonis P, Patel AA, Ward AM, Hsu TK, et al. Evolutionary Action Score of TP53 Coding Variants is Predictive of Platinum Response in Head and Neck Cancer Patients. Cancer Res. 2015;75(7):1205–15. doi: 10.1158/0008-5472.CAN-14-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Squire CJ, Dickson JM, Ivanovic I, Baker EN. Structure and inhibition of the human cell cycle checkpoint kinase, wee1a kinase: an atypical tyrosine kinase with a key role in CDK1 regulation. Structure. 2005;13:541–50. doi: 10.1016/j.str.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 7.Portugal J, Mansilla S, Bataller M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr Pharm Des. 2010;16:69–78. doi: 10.2174/138161210789941801. [DOI] [PubMed] [Google Scholar]
- 8.Beck H, Nähse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck H, Nähse V, Larsen MS, Groth P, Clancy T, Lees M, et al. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol. 2010;188:629–638. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forced Mitotic Entry of S-Phase Cells as a Therapeutic Strategy Induced by Inhibition of WEE1. Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Cancer Discov. 2012;2(6):524–39. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 11.Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, Neskey DM, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther. 2015;14:608–619. doi: 10.1158/1535-7163.MCT-14-0735-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 13.Gillenwater AM, Zhong M, Lotan R. Histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis through both mitochondrial and Fas (Cd95) signaling in head and neck squamous carcinoma cells. Mol Cancer Ther. 2007;6:2967–2975. doi: 10.1158/1535-7163.MCT-04-0344. [DOI] [PubMed] [Google Scholar]
- 14.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 15.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–176. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 16.Chang HH, Chiang CP, Hung HC, Lin CY, Deng YT, Kuo MY. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009;45:610–614. doi: 10.1016/j.oraloncology.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 17.Prystowsky MB, Adomako A, Smith RV, Kawachi N, McKimpson W, Atadja P, Chen Q, Schlecht NF, Parish JL, Childs G, Belbin TJ. The histone deacetylase inhibitor LBH589 inhibits expression of mitotic genes causing G2/M arrest and cell death in head and neck squamous cell carcinoma cell lines. J Pathol. 2009;218:467–477. doi: 10.1002/path.2554. [DOI] [PubMed] [Google Scholar]
- 18.Iglesias-Linares A, Yanez-Vico RM, Gonzalez-Moles MA. Potential role of HDAC inhibitors in cancer therapy: Insights into oral squamous cell carcinoma. Oral Oncol. 2010;46:323–329. doi: 10.1016/j.oraloncology.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904–1913. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan W, Liu S, Xu E, Zhang J, Zhang Y, Chen X, Chen X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene. 2013;32:599–609. doi: 10.1038/onc.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blagosklonny MV, Trostel S, Kayastha G, Demidenko ZN, Vassilev LT, Romanova LY, et al. Depletion of mutant p53 and cytotoxicity of histone deacetylase inhibitors. Cancer Res. 2005;65:7386–7392. doi: 10.1158/0008-5472.CAN-04-3433. [DOI] [PubMed] [Google Scholar]
- 22.Zhou L, Zhang Y, Chen S, Kmieciak M, Leng Y, Lin H, et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia. 2015;29(4):807–18. doi: 10.1038/leu.2014.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson AJ, Holson E, Wagner F, Zhang YL, Fass DM, Haggarty SJ, et al. The DNA damage mark pH2AX differentiates the cytotoxic effects of small molecule HDAC inhibitors in ovarian cancer cells. Cancer Biol Ther. 2011;12(6):484–93. doi: 10.4161/cbt.12.6.15956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 25.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol. 2015;33(30):3409–15. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012;18(1):290–300. doi: 10.1158/1078-0432.CCR-11-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 28.Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2:798–811. doi: 10.1158/2159-8290.CD-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci. 2004;101(5):1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci. 2000;97(18):10014–9. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cornago M, Garcia-Alberich C, Blasco-Angulo N, Vall-Llaura N, Nager M, Herreros J, Comella JX, 3, et al. Histone deacetylase inhibitors promote glioma cell death by G2 checkpoint abrogation leading to mitotic catastrophe. Cell Death Dis. 2014;5:e1435. doi: 10.1038/cddis.2014.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010;70(11):4470–80. doi: 10.1158/0008-5472.CAN-09-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Domínguez-Kelly R, Martín Y, Koundrioukoff S, Tanenbaum ME, Smits VAJ, Medema RH, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–579. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A. 2007;104(49):19482–7. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konstantinopoulos PA, Wilson AJ, Saskowski J, Wass E, Khabele D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol Oncol. 2014;133(3):599–606. doi: 10.1016/j.ygyno.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N, et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 2002;21(3):427–36. doi: 10.1038/sj.onc.1205108. [DOI] [PubMed] [Google Scholar]
- 37.Mahalingam D, Medina EC, Esquivel JA, Espitia CM, Smith S, Oberheu K, et al. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res. 2010;16(1):141–53. doi: 10.1158/1078-0432.CCR-09-1385. [DOI] [PubMed] [Google Scholar]
- 38.Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci USA. 2007;104:8071–8076. doi: 10.1073/pnas.0702294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 40.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 41.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 42.Bhatla T, Wang J, Morrison DJ, Raetz EA, Burke MJ, Brown P, Carroll WL. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood. 2012;119(22):5201–10. doi: 10.1182/blood-2012-01-401687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dasmahapatra G, Patel H, Nguyen T, Attkisson E, Grant S. PLK1 inhibitors synergistically potentiate HDAC inhibitor lethality in imatinib mesylate-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin Cancer Res. 2013;19(2):404–14. doi: 10.1158/1078-0432.CCR-12-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dibb M, Han N, Choudhury J, Hayes S, Valentine H, West C, et al. The FOXM1-PLK1 axis is commonly upregulated in oesophageal adenocarcinoma. Br J Cancer. 2012;107(10):1766–75. doi: 10.1038/bjc.2012.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, et al. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol. 2008;10(9):1076–82. doi: 10.1038/ncb1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang B, YU D, Liu J, Yang K, Wu G, Liu H. Antitumor activity of SAHA, a novel histone deacetylase inhibitor, against murine B cell lymphoma A20 cells in vitro and in vivo. Tumor Biol. 2015;36:5051–5061. doi: 10.1007/s13277-015-3156-1. [DOI] [PubMed] [Google Scholar]
- 47.Choi S, Sano D, Cheung M, Zhao M, Jasser SA, Ryan AJ, Mao L, Chen WT, El-Naggar AK, Myers JN. Vandetanib inhibits growth of adenoid cystic carcinoma in an orthotopic nude mouse model. Clin Cancer Res. 2008;14(16):5081–9. doi: 10.1158/1078-0432.CCR-08-0245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.