

Abstract
Fungi and mammals share a co-evolutionary history and are involved in a complex web of interactions. Studies focused on commensal bacteria suggest that pathologic changes in the microbiota, historically termed as ‘dysbiosis’, are at the root of many inflammatory diseases of non-infectious origin. Yet, the importance of dysbiosis in the fungal community— the mycobiota — was only recently acknowledged to have a pathological role as novel findings suggest that mycobiota disruption can have detrimental effects on host immunity. Fungal dysbiosis and homeostasis are dynamic processes that are probably more common than actual fungal infections, and therefore constantly shape the immune response. In this Review, we summarize specific patterns associated with fungal dysbiosis, and discuss how mucosal immunity has evolved to distinguish fungal infection from dysbiosis and how it responds to these different conditions. We propose that gut microbiota dysbiosis is a collective feature of complex interactions between prokaryotic and eukaryotic microbial communities that can affect immunity and influence health and disease.
Introduction
Fungi are omnipresent in our living environment and are an integral part of all ecosystems1. They have played a central role in the evolution of life2. Molecular evidence suggests that animals and fungi have coevolved since diverging from plants over 1 billion years ago3 This has provided plentiful opportunities for fungi to strongly influence the evolution of animals3 and their immune system4,5. The immune system has been crucial in establishing a close relationship between host, bacteria and fungi, by keeping the peace at the highly colonized barrier surfaces. Although this relationship often results in symbiosis6–8 (BOX 1), it can lead to diseases that have devastating socioeconomic impact9–11.
Box 1. Ecological relationships between fungi and their host.
There are numerous examples of oppositional and symbiotic relationships between fungi and animals. While some fungi such as members of Pleurotaceae family have become predators using elaborated strategies to catch and digest nematodes living in the soil, other fungi such Harposporium spp. and Ophiocordyceps spp. have adapted a parasitic lifestyle128. Ophiocordyceps fungal species have gone as far as manipulating the host behaviour to ensure their own survival and dispersal128. Other fungi such as Blastomyces dermatitidis and some Aspergillus spp. have developed mechanisms to mimic mammalian regulatory molecules and suppress host immunity129,130. The skin and the mucosal surfaces of animals and humans are rich on nutrients and provide temperate environment for commensal fungal symbionts to thrive. Malassezia spp. are well adapted to sebaceous microenvironments and are abundantly present on the skin9,60. The feet are home of a diverse fungal community dominated by saprophytic fungi that feed from the dead cells, which are continuously shed at this skin site9. Symbiotic relationships between fungi and their host are well established in the gastrointestinal tract of different orders of animals from insects to mammals10,11. Whereas some fungal symbionts are crucial for processing indigestible lignocellulose associated with the herbivores diet, other symbionts produce enzymes necessary for the neutralization of dietary toxic compounds11. The host, on the other hand, provides its commensal mycobiota with an optimal environment to grow, feed and procreate. In some cases the fungal symbiont are even vertically transmitted through generations131, suggesting that similar to bacterial symbiosis, some fungal commensals might be inherited from the mother. Recently, high throughput sequencing approaches revealed that mouse and human intestines are home to diverse fungal community which might play an important role in host physiology and immunity13,15–17,21.
The rapid development of deep sequencing and computational technologies has provided great opportunities to explore the structure and functionality of microbial communities associated with our body surfaces. Extensive microbial data generated worldwide has enabled the scientific community to uncover the functions of the human microbiota12. The bacterial communities have been the primary focus of these sequencing efforts and less data is currently available characterizing the fungal microbiota — the mycobiota. Nevertheless, recent studies reveal that host-associated fungal populations are also dynamic and responsive to environmental and pathophysiological changes9,13–17.
A century of studies focused on commensal bacteria in the gut suggest that ‘dysbiosis’18 is in the root of several diseases of complex etiology involving genetic polymorphisms, immune mechanisms and the microbiota19,20,21. Although the term dysbiosis is imperfect, it describes conditions that are distinct from and is widely used to describe altered bacterial communities as both a cause and a consequence of pathologies (BOX 2).. Nonetheless, infections and dysbiosis can prelude to each other (homeostasis-dysbiosis-infection vs homeostasis-infection-dysbiosis) in a dynamic continuum. Several studies indicate that a similar process involving fungal communities — ‘fungal dysbiosis’ — could affect the host mycobiota (Figure 1)9,16,17,21–24. Since fungi are common inhabitants of all barrier surfaces, changes in fungal communities might have substantial effects on the host immune responses.
Box 2. Uncoupling infection from dysbiosis.
Early in the 16th century Girolamo Francastoro proposed that minute invisible seeds — ‘seminaria’ — transmit diseases from person to person132. This revolutionary theory of the microbial origin of diseases was proven about 100 years later with the discovery of the microscope by Antony van Leeuwenhoek133. It took another 200 years for the theory to fully ripe. In the second half of the 19th century Luis Pasteur and Friedrich Jakob Henle independently published their ideas of the microbial origin of diseases134,135. The idea was further developed by Robert Koch112, who eventually developed guidelines known as “Koch’s postulates” intending to move the field forward from observations to evidence proved causative relationship between microbes and disease136. Over the years Koch’s postulates underwent several modifications incorporating application to acute and chronic diseases with diverse etiologies137 and comprising the advances that genomics-based approaches have had on clinical microbiology and virology138,139. Although Koch’s postulates framed the idea of the microbial origin of infectious diseases, they cannot explain situations where microbes indirectly shape a pro-inflammatory or anti-inflammatory environment to affect immunity. During the second half of the 19th century, while working on causes of longevity, Ilya Metchnikoff theorized that intestinal microbes can “auto-intoxicate and auto-infect” their host18. He and his colleagues Stamen Grigoroff and Leon Massol proposed that lactic acid producing bacteria, which they called Bulgarian bacillus, can decelerate this process18. Together with the concept of beneficial bacteria, Metchnikoff coined the term ‘dysbiosis’ (initially dysbacteriosis) to define pathologic changes of intestinal microflora18. However, dysbiosis is a generalizing and wide definition, which can be applied to either cause or effect. Some have criticized these shortcomings140 or have attempted to define dysbiosis within several conditions141. Similar to the modifications that Koch’s postulates underwent over time, the concept of ‘dysbiosis’ will probably evolve to reflect the modern advances in microbiome science.
Figure 1. The mycobiota during health and in dysbiosis.

A) During homeostasis, diverse fungal communities reside all human barrier surfaces during homeostasis, such as the mouth32; lung53; skin9, gut22 and vagina57 (left hand side). The pie charts represent the relative abundance of the observed taxa at the phylum and genus levels (inner and outer circles, respectively). Of note, the data for the vagina are estimates that are based on culture-dependent studies, due to a lack of sequencing-based studies related to disease conditions (indicated with an asterisk). ‘Other’ refers to sequences with <5% relative abundance. During diseases, those communities are perturbed (right hand side). Dysbiotic fungal communities are observed in the oral cavity32 and the vagina58 of HIV patients; in the lung of cystic fibrosis patients51; on the skin of primary immunodeficiency61 and chronic wounds63 patients; in the gut of Crohn’s disease22 patients. B) Factors contributing to fungal dysbiosis at different barrier surfaces. AF, antecubital fossa; HF, hind foot; STAT3, signal transducer and activator of transcription 3.
In this Review article, we discuss the mycobiota in homeostasis and dysbiosis, and how mucosal immunity distinguishes between fungal infections and fungal dysbiosis and responds to these conditions. We define specific patterns associated with fungal dysbiosis and changes in mycobiota diversity at several body sites. Finally, we discuss the impact of fungal dysbiosis on commensal bacteria and how these two fundamentally different communities influence each other to affect the outcome of the immune response.
Mycobiota at steady state and in dysbiosis
Fungi have been traditionally studied using culture-dependent methods. Only recently, advances in deep sequencing technologies have provided opportunities to unveil the complexity of the fungal communities colonizing the mammalian barrier surfaces. Most approaches have relayed on sequencing the fungal internal transcribed spacer 1 (ITS1) and ITS2 regions and using computational algorithms to align operational taxonomic units (OTUs) to respective fungal databases9,16,25,26. Whereas fungal identification remains challenging in environmental and soil samples due to the enormous fungal diversity and the presence of many unknown fungal species27, the development of several databases designed to specifically target host-associated fungal communities have made it possible to identify over 80% of the fungal ITS sequences derived from fecal or mucosal samples9,28–30. Despite this progress, each step of this identification approach can contribute to biases26,28 and complicate the comparison of mycobiota sequencing results from different studies. Additional bias might be introduced by therapeutic and dietary interventions that can contribute to further changes in fungal communities15. Despite these confounding factors, several studies have revealed similarities in how fungal communities adapt to environmental perturbations and these will be summarized in this section. Keeping in mind the limitations that the term “dysbiosis” imposes (BOX 2), we will further define specific conditions associated with fungal community disruption in the context of inflammatory diseases, host immunity and drug-triggered microbial alterations.
Mycobiota in the oral cavity
The oral cavity is one of the first mucosal sites where asymptomatic carriage of fungi was described 31. Diversity in the oral mycobiota is low and dominated by members of the Ascomycota phylum, mainly Candida spp.32. Culture independent methods have revealed the presence of several members of the Saccharomycetaceae family (Candida albicans, Candida dubliniensis, Candida rugosa Candida pararugosa, Saccharomyces cerevisiae, Hanseniaspora uvarum, and Pichia spp.), and Fusarium spp. that represent the majority of fungal species found in the oral cavity32. Dysbiosis in the oral mucosa is poorly characterized and most studies have focused on the overgrowth of Candida albicans upon immunosuppression and diabetes32 (Figure 1B). The first culture-independent assessment of the oral mycobiota in patients with HIV revealed that in addition to overgrowth of C. albicans, the presence of other species belonging to the Ascomycota phylum, including Dothideomycetes spp. and Leotiomycetes spp., was also increased whereas Tremellomycetes spp. were reduced32. Notably, this study did not observe any alteration of the bacterial microbiota, indicating that the observed fungal dysbiosis is probably mediated by a direct effect of the compromised immune system and not by an indirect effect mediated by changes in the bacterial microbiota.
Mycobiota in the gastrointestinal tract
The fungal community in the lower gastrointestinal tract is more rich and diverse than in the upper gastrointestinal tract. A recent review of 36 published gut mycobiota articles concluded that only 15 out of the 267 identified species were detected in more than 5 studies33. Among these 15 species, 13 can grow at 37°C and thus have the potential to permanently inhabit the intestinal niche22,34. These species belong to the genera Candida, Saccharomyces, Aspergillus, Cryptococcus, Malassezia, Cladosporium, Galactomyces and Trichosporon33. Nevertheless, commensalism is not the only way fungi can influence host immunity. For instance, although Histoplasma spp., Blastomyces spp. and Coccidioides spp. cannot colonize the mucosal surfaces they can cause severe infections in the lung35. Similarly, non-commensal fungi present in the diet, the air or other environmental sources, might also activate immune responses in the gut 15,24 (Figure 1B).
The high fungal variability observed in different studies has been interpreted as a sign of the temporal instability of the intestinal mycobiota13,34. Yet, most of the studies rely on the sampling of fecal material. Although additional data are needed to make definitive conclusions, a recent study of the mucosa-associated fungi suggests that a more stable community is associated to the gut mucosa36 and this can be perturbed during intestinal disease37. These results are interesting since fungi that are in close proximity to the intestinal mucosa might have increased potential to interact locally with the intestinal epithelium and the mucosal immune system.
Inflammatory conditions that target the lower gastrointestinal tract have been known to promote bacterial dysbiosis for a long time and this has highlighted the important contribution of bacteria in the etiology of inflammatory bowel disease (IBD)38,39. In light of these findings, several studies have investigated the fungal dysbiosis in patients with IBD and revealed a general increase of fungal burden in the colonic mucosa during both Crohn’s disease21,22 and ulcerative colitis40. Consistent with these results, the fungal burden increases and pronounced fungal translocation occurs in the mucosa during the chronic stage of colitis41 and during the acute stage of colitis in mice deficient in the fungal receptor dectin-1(clec7a, see below)16. Together, these studies suggest that genetic factors, extensive tissue damage and the presence of inflammatory environment can cause fungal dysbiosis in the gut.
Several fungal taxa appear to be consistently altered during chronic intestinal inflammation throughout multiple studies. Patients with IBD have an increased occurrence and abundance of cultivable C. albicans in the feces, which was further confirmed by culture-independent studies17,21–23. In patients with Crohn’s disease, the presence of Candida albicans and Candida parapsilosis increased in the inflamed mucosa37. Increased abundance of Candida tropicalis in Crohn’s disease correlated with inflammation and increased levels of anti-S. cerevisiae antibody (ASCA)23. Consistently, intestinal inflammation is sufficient to promote Candida spp. colonization and increased abundance in the mouse gut16,41. Although it is clear that Candida spp. expand during intestinal disease (Figure 1), its effect on other gut fungi remains unclear. Some studies suggest that the presence of the Ascomycota phylum decrease in favour of expansion of Basydiomycota spp.17,37, whereas another study report a general increase of five Ascomycota species in IBD21.
Clone libraries approaches based on 18S rDNA denaturing gradient gel electrophoresis (DGGE) show that patients with Crohn’s disease have increased fungal diversity and richness in the faeces and the mucosa, compared with healthy individuals or individuals with other forms of intestinal inflammation such as patients with infectious colitis 37,40. By contrast, ITS-sequencing approaches showed either no changes in the mycobiota composition in fecal samples 17 or colonic mucosa22 during Crohn’s disease, or a reduction in fungal diversity in a paediatric IBD cohort that mainly included patients with Crohn’s disease42. A further study showed a decreased species richness in the feces from patients with Crohn’s disease and their healthy relatives when compared with unrelated healthy controls, but no change in fungal diversity was reported23. Interestingly, one study shows that patients with active ulcerative colitis have a clear reduction in fungal diversity17. The differences in the gut mycobiota observed among different studies suggest that in addition to inflammation, other factors, such as immunosuppressive therapy, disease type (Crohn’s disease or ulcerative colitis) or disease-specific diet might influence the structure of the mycobiota in patients with IBD.
Patients with Crohn’s disease have increased levels of antifungal antibodies, which show that an immune response to intestinal fungi is elicited during IBD43. These systemic ASCA IgG and IgA are widely recognized as reliable diagnostic markers for Crohn’s disease and are good predictors of the disease course43. Recent studies show that C. albicans can act as immunogen for ASCA production44 and that ASCA are cross-reactive to cell wall mannans isolated from other yeasts including commensal Candida species45. Despite this link with intestinal fungi, the exact mechanism behind ASCA generation and their role in antifungal immunity is yet to be understood.
The frequent co-occurrence of gastrointestinal symptoms and neurodevelopmental disorders has uncovered the presence of bacterial dysbiosis in autism-spectrum disorders (ASD), schizophrenia and other neurodevelopmental disorders46,47. Patients with ASD and Rett syndrome were recently reported to have alterated intestinal mycobiota47,48; however whether fungi also contribute to the development of gastrointestinal symptoms in these patient groups is currently unknown.
Mycobiota in the lung, vagina- and skin
Disease-mediated fungal dysbiosis has been described at other barrier sites with lower fungal burden than the gut. The healthy lung has long been considered an aseptic organ and bacterial colonization was thought to occur only during disease20. Similarly, the fungal burden is generally low in healthy individuals whose lung mycobiota appears to be largely composed of environmental fungi or fungi disseminating from the oral cavity, including species belonging to the Cladosporium, Aspergillus, Penicillium and Candida genera 25,32,49. By contrast, more stable fungal communities can colonize the lung when its physiology is altered (Figure 1B). In the majority of patients with cystic fibrosis, for example, the fungal burden in the lung appears increased, whereas alpha diversity is reduced and correlate with disease severity50,51. Despite the high inter-individual variability of mycobiota composition, fungal genera such as Candida and Aspergillus, have been linked to morbidity in individuals with cystic fibrosis50,51. Notably, once established, lung dysbiotic fungal communities seem to stabilize and persist even in the presence of antibiotic or immunosuppressant therapy51,52. Fungi have also been associated with the severity of asthma20,53, however it is unclear whether the effect is local or influenced by the intestinal mycobiota as suggested by experimental studies24,54,55.
Vaginal candidiasis is by far one of the most common mucosal fungal infections in humans. Yet, fungal communities are found in healthy women without active infections110. Similar to the oral mucosa, the vaginal mycobiota is less diverse and is mainly colonized by C.albicans, C glabrata, C. krusei, C. parapsilosis and S. cerevisiae56,57,58. Several factors, such as antibiotic use, pregnancy, HIV infection and recurrent vulvovaginal candidiasis have been associated with alterations of the vaginal fungal community structure (Figure 1B)56,58,59. Vaginal Candida spp. colonization increases up to 40% during pregnancy, due to the increased estrogen levels and glycogen production by vaginal epithelial cells, and this might have important consequences on pregnant women, including preterm delivery56.
In contrast to the other surfaces of the body, the skin is cold, dry and has higher availability of lipids and proteins (such as keratin), which constitute the major nutrients for the colonizing fungal flora including lipophilic species such as Propionibacterium spp. and Malassezia spp.9,60. The skin microbiota shows temporal stability and can be influenced by primary immunodeficiencies60,61,62. When skin integrity is compromised, fungi rapidly populate the wound site. A recent study demonstrated that fungi are present in 80% of the chronic wounds in patients with type 1 and type 2 diabetes (Figure 1B)63. Despite the high interpersonal variability of the wound mycobiota the study showed an association between Candida spp., Trichosporon spp. and Rhodotorula spp. with non-healing wounds and amputation while temporally unstable fungal and bacterial communities were associated with a positive healing outcome63. Therefore, in addition to bacteria, a specific signature of wound fungal community can have a negative outcome of the skin healing process.
Altogether, these studies suggest that disease-induced dysbiotic fungal communities are often well adapted to the changes induced by pathological conditions and can contribute to the disease course. Further studies are required in treatment-naive individuals, including paediatric patients and newly diagnosed patients, to make critical insight into a purely disease-driven dysbiosis. Mouse studies are an essential tool to analyze the effect of fungal dysbiosis under such conditions and have already provided some evidence for fungal involvement in several pathologies as discussed below.
Influence of age, sex, diet and environment
Several studies show a high variability of the mycobiota between different individuals, which complicates the distinction between healthy and dysbiotic fungal communities. This variability highlights the need for a more strict definition of dysbiosis (BOX 2) and the need of experimental studies that define the consequences of an altered mycobiota. The mycobiota composition is substantially influenced by diet, gender, age and geographical locations64. Aspergillus spp. and Tremellomycetes spp. are more abundant in male individuals whereas Candida spp. are more abundant in females possibly due to spreading from the vagina64. Genera such as Aspergillus, Tremellomycetes and Penicillium are abundant in infants whereas the overall diversity of fungal community decreases with age42,64. Similarly, the diversity of the skin mycobiota decreases with age, probably due to the increasingly sebaceous skin that favours colonization by Malassezia species65.
The diet can influence not only bacterial but also fungal communities, mainly as a constant source of fungal species that are associated with vegetables, fruits and dairy products15. Individuals eating a controlled animal-derived diet that was rich in cheese were associated with an increased fungal burden and fungal species detected in their feces corresponded to those isolated from the food they had eaten15,34. Diet might be one of the factors contributing to the differences observed in the fungal community across different ethnicities21. Altogether, these studies suggest that dietary fungal intake contributes substantially to the gut mycobiome and that specific dietary nutrients can sustain specific fungal community members (Figure 1B). In support for this, a recent study reported that Candida spp. associated positively with carbohydrates and negatively with saturated fatty acids, whereas Aspergillus spp. correlated negatively with dietary short chain fatty acids14.
Mucosal immunity to fungi
The immune system has evolved to tolerate fungi and respond to them upon injury or infection, and the presence of fungi at mammalian barrier surfaces might constantly shape immunity. Mucosal immunity to fungi has been explored largely in the context of fungal infections8,35,66–72 (Figure 2A). Yet very little is known about the role of fungi in influencing immunity during steady state or during dysbiosis (Figure 2B). In this section, we discuss mechanisms of antifungal immunity using the lower gastrointestinal tract as a model barrier surface, where despite their high abundance, fungi rarely cause infection whereas fungal dysbiosis occurs frequently16,17,21–24.
Figure 2. Examples of mucosal immune responses to fungal infection and dysbiosis.

The host barrier surfaces are inhabited by mycobiota. Several sites, such as the skin, lung, oral cavity and the vagina are prone to fungal infections and fungal dysbiosis upon compromised host immunity, whereas the gastrointestinal tract is resistant to fungal infections, but susceptible to fungal dysbiosis. Host mucosal immunity has evolved to distinguish fungal infection from fungal dysbiosis. A) Fungal infection leads to breaches in the epithelial surfaces due to fungal activity, which in turn leads to rapid infiltration of neutrophils and monocytes at the site of infection. The production of cytokines and chemokines by epithelial and innate immune cells leads to further influx and recruitment monocytes, neutrophils and T cells among others. Resident phagocytes such as macrophages and dendritic cells (DCs) recognize and process fungal antigens and promote fungal specific T cell responses and polarization. Innate lymphoid cells (ILC3) can respond directly to fungi or to the inflammatory environment during infection with production of IL-22 and IL-17. Epithelial cells produce antimicrobial peptides that directly affect fungal survival. B) By contrast, fungal dysbiosis is characterized by alterations in the mycobiota composition that might be both the cause and/or consequence of changes in the tissue environment or changes in the intestinal lumen. Fungal dysbiosis can influence both local and systemic immunity through several mechanisms including the modulation of cytokine milieu, the activation of different cell types and the release of metabolites. Fungal dysbiosis in the gut can influence immunity at distant sites such as the lung and contribute to allergy. Intestinal inflammation and breaches of the intestinal epithelial barrier caused by non-fungal triggers can lead to direct exposure to fungal antigens coming from the intestinal lumen and development of systemic IgG and IgA anti-Saccharomyces cerevisiae antibodies (ASCA). Mucosal immunity to gut fungi during dysbiosis is poorly explored and only a few molecules have been studied in this context. At the barrier surfaces, fungi and bacteria can affect each other; thus, microbiota dysbiosis is probably a collective feature of the complex crosstalk between the fungal and bacterial microbiota, and the host. CCR2, CC-chemokine receptor 2; CXCL2, CXC-chemokine ligand 2; IDO1, indoleamine 2,3-dioxygenase 1; IFNγ, interferon-γ; iTreg, induced T regulatory; NK, natural killer; PGE2, prostaglandin E2; TNF, tumour necrosis factor; TH, T helper.
The role of cell-mediated immunity
Cell types involved in innate and adaptive immune responses to fungi during infection have been well characterized at several barrier surfaces such as the oral mucosa, the lungs, the vaginal mucosa and the skin (Figure 2A)66,68,69,72–76. Innate immune cells are important for the clearance of fungi and for the initiation of adaptive immune responses during fungal infections8,35,66–72. Neutrophils, CCR2+Ly6C+ monocytes, CX3CR1+ mononuclear phagocytes, CD11b+CD103+ dendritic cells, natural killer cells and epithelial cells have important functions in antifungal immunity by having a role in recognition, phagocytosis and killing of fungi as well as through their indirect activation by cell-cell crosstalk66,68,69,72–78. The activation of specific cell types or cell-cell crosstalk is further dependent on the specific barrier surface affected by fungal infection (Figure 2A). Whereas some fungi, such as Candida spp., are widely present and have the ability to infect several barrier sites, the distribution of other fungi is tissue-dependent 29. As a central hub of mucosal immunity, the gastrointestinal tract is naturally equipped with a cellular machinery to recognize and interact with the microbiota79. The intestine harbours several subsets of phagocytes, which are known to respond to bacterial infections or to fluctuations in the bacterial communities79. Although phagocytic cell populations probably sense mycobiota in the intestine, their role during fungal dysbiosis remains unknown.
In addition to cellular innate and adaptive immunity, humoral immune mechanisms such as the complement and antifungal antibodies play an important role in antifungal immunity80 and might be involved in shaping the mycobiota at the mucosal surfaces. Innate immune cells are further equipped with an array of receptors necessary to sense and interact with fungi8.
The role of C type lectin receptors
Genetic evidence and experimental studies have underlined a central role for C-type lectin receptors (CLRs) in antifungal immunity, whereas Toll-like receptors (TLRs) and NOD-like receptors (NLRs) mostly play a secondary role5,81. CLRs, such as dectin-1, dectin-2, dectin-3, mincle and the mannose receptor, recognize several molecules present in the fungal cell wall5,81. Spleen tyrosine kinase (syk) is the primary signal transduction molecule used by several of these receptors, and activation requires an immunoreceptor tyrosine-based activation motif (ITAM) or ITAM-like motif present within the receptor tail (in the case of dectin-1) or within an associated FcRγ adapter molecule (in the case of dectin-2, dectin-3, mincle)5,81,62. Upon receptor engagement, ITAM motifs are phosphorylated by Src family kinases, leading to the recruitment and phospohorilation of syk and activation of a downstream signalling cascade including phospholipase Cγ2 (PLCγ2), protein kinase Cδ (PKCδ), the CARD9-Bcl10-MALT1 complex and the IκB kinase (Figure 3). The mannose receptor is an exception and does not activate this pathway5,81.
Figure 3. C-type lectin receptor (CLR) recognition and signaling.
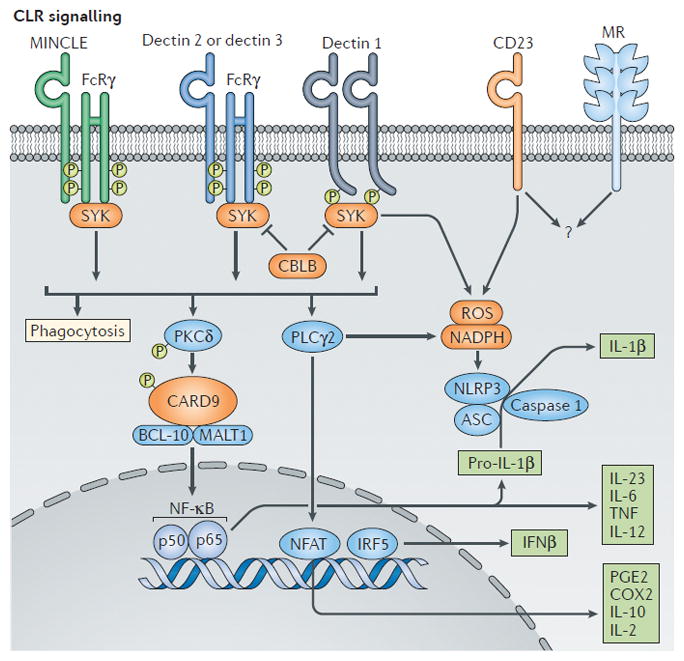
Fungal polysaccharides are recognized by C type lectin receptors (CLRs) such as macrophage-inducible C-type lectin (MINCLE), dectin 1, dectin 2and dectin 2, resulting in the activation of spleen tyrosine kinase (SYK). The E3 ubiquitin ligase CBLB, which ubiquitylates SYK, regulates antifungal immune responses downstream of dectin 1 and dectin 2. CLRs trigger phagocytosis and respiratory burst (reactive oxygen species production, ROS) through the SYK-dependent phospholipase Cγ2 (PLCγ2) activation of NADPH phagocyte oxidase and killing of fungi. Engagement of protein kinase Cδ (PKCδ) followed by activation of the CARD9–BCL-10–MALT1 complex leads to nuclear factor-κB (NF κB) activation, caspase 1 activity and the production of cytokines and other mediators that are crucial for host defence against fungi. Dectin-SYK induction promotes IRF5-dependent IFN-γ production. CD23 is a newly identified CLR that is upregulated upon dectin 1 activation and that leads to the production of ROS. The signaling events downstream of CD23 and the mannose receptor (MR) remain unknown (indicated by a question mark). ASC, apoptosis- associated speck-like protein containing a CARD; CARD9, caspase recruitment domain-containing protein 9; CBLB, casitas B-lineage lymphoma-b; COX2, cyclooxygenase 2;FcRγ, Fc receptor common γ-chain; IFNβ, interferon-β; IRF5, interferon-regulatory factor 5; MALT1, mucosa-associated lymphoid tissue lymphoma translocation protein 1; NFAT, nuclear factor of activated T cells; NLRP3, NOD, LRR and Pyrin domain-containing protein 3; PGE2, prostaglandin E2; TNF, tumor necrosis factor.
Dectin-1
The fungal β-glucan receptor dectin-1has been widely studied as a crucial receptor for fungal phagocytosis, killing and cytokine response82. Mouse studies have revealed a protective role of dectin-1 in systemic candidiasis82 and in the control of fungi at several mucosal surfaces, such as the skin69, the oral mucosa67 and the lungs72. The role of dectin-1 in protection against Candida spp. infection appears to depend on specific fungal strains83, suggesting that some fungi can mask their cell wall β-glucans and escape recognition. In humans, a mutation in CLEC7A, the gene encoding dectin-1, leads to poor dectin-1 expression and impaired cytokine production upon fungal re-stimulation70. As a result, patients carrying this mutation suffer from recurrent vulvovaginal infections and onychomycosis, partially due to defective IL-17 production in response to fungi70.
Dectin-1 might also control antifungal immunity during fungal dysbiosis. We have shown that dectin-1-deficient mice developed more severe colitis accompanied by mycobiota changes and overgrowth of opportunistic genera such as Candida and Trichosporon16. This phenotype was dependent on the inability of Clec7a−/− mice to control Candida spp. during intestinal inflammation and treatment with the antifungal drug fluconazole ameliorated colitis16,84. In situ examination of mouse colons revealed absence of hyphal growth in the lower gastrointestinal tract during Candida spp. colonization of wild-type mice and mice with defects in antifungal immunity16 (and unpublished observations; I.L., I.D.I.). By contrast, hyphal growth is commonly observed during Candida spp. infections at other mucosal sites66,69 and factors involved in hyphal elongation or yeast to hyphae transition have been shown to either aid fungal invasion (which is mediated by cell elongation 1 gene (ECE1))85 or commensalism (which is mediated by Efg1p) 86. This suggests that colitis in Clec7a−/− mice is not aggravated by a typical fungal infection, but that other aspects of host–fungal interactions affecting the mycobiota might be involved. Consistent with the mouse models, patients carrying a two-marker CLEC7A haplotype developed more severe refractory ulcerative colitis supporting a possible role of dectin-1 in severity16, but not susceptibility to intestinal disease87. Furthermore, dectin-1 deficiency is also associated with increased colonization of Candida spp. in the gut and the development of graft-versus-host disease in patients who have received transplants88,89. Altogether, these studies suggest that impaired antifungal immunity, Candida spp. overgrowth and fungal dysbiosis might be involved in several intestinal diseases.
The role of dectin-2
Dectin-2 has been implicated in the induction of T helper 17 (Th17) responses and protection from systemic candidiasis90; however, its role in mucosal immunity is less explored. Dectin-2 mediated immunity might be important for the generation of fungi-specific Th17 responses to Fonsecaea pedrosoi and resolution of skin infection in a mouse model of chromoblastomycosis 91. Immunity to the skin commensal Malassezia spp. is also partially controlled by dectin-292. In the lung, dectin-2 might be involved in recognition of fungal and mite allergens93, yet dectin-2 knockout mice do not develop more severe lung inflammation upon infection with fungi such as Cryptococcus neoformans94. The role of dectin-2 in human antifungal mucosal defense is even less clear. A recent study described a polymorphism in CLEC6A (encoding dectin-2) that is associated with susceptibility to pulmonary cryptococcosis95. However, it is uncertain whether dectin-2 is directly responsible for this phenotype.
The role of other CLRs
Other CLRs, such as mincle and dectin-3, that induce Syk-CARD9-Bcl10-MALT1- dependent signaling are also involved in mucosal immunity. Dectin-3 induces the expression of mincle upon specific stimulation96 and forms heterodimers with dectin-2 to induce signalling presumably through FcRγ coupling (Figure 3)97, suggesting that these receptors are expressed in an interdependent manner. Dectin-3- and mincle-deficient mice are all susceptible to Candida spp. systemic infections81,90,97; however it is less clear whether these receptors are directly involved in mucosal immunity to fungi. Mincle, for instance, can recognize a lipophilic component of Malassezia spp. cell wall in vitro92, but whether such recognition of commensal Malassezia spp. occurs in the skin has not been determined.
Dectin-3, which is encoded by Clec4d, was recently shown to control intestinal inflammation in mice. Clec4d −/− mice developed more severe colitis, which was associated with increased C. tropicalis burden and efficiently treated with fluconazole98. Decreased numbers of Th17 cells, impaired phagocytosis by dectin-3-deficient macrophages and defective intestinal epithelial cell barrier in these mice can be partially responsible for C. tropicalis overgrowth. However, the mechanisms behind increased susceptibility to colitis remain unclear as several pro-inflammatory cytokines (including tumour necrosis factor) were reduced in the colons of Clec4d −/− mice98. Although the structure of the microbiota was not assessed in this study, augmented fungal burden and the increased relative abundance of C. tropicalis in the intestines of Clec4d −/− mice indicate changes in the fungal community. Finally, the relevance of dectin-3 to human intestinal disease remains uncertain due to the lack of reports on genetic associations between CLEC4D polymorphisms and IBD.
The role of CARD9
CARD9 holds a key downstream position to several antifungal receptors such as the CLRs discussed above (Figure 3)81. As such, CARD9 is crucial for the induction of cytokine production and triggering of cellular immunity to fungi at several mucosal sites8,71,99,100. Thus, biallelic mutations in CARD9 have important consequences on the control of mucosal and systemic fungal infections8,71,101. Although CARD9 might be involved in signalling via receptors that sense stimuli other than fungi, mice and humans deficient in CARD9 appear to be specifically susceptible to fungal infections71,99–101. CARD9-deficient patients often develop chronic mucocutaneous candidiasis characterized by persistent and recurrent Candida spp. infections of the mouth, skin, vagina and other mucosal surfaces8,71. CARD9 mutations can also lead to severe onychomycosis, phaeohyphomycosis and deep-seated dermatophytosis, affecting deeper subcutaneous tissue8,102. Fungal infection in these patients are caused by Trichophyton spp. and Phialophora spp. and can expand to affect lymph nodes and bones8,102,103. These rare, but severe effects of CARD9 deficiency suggest a central role for this adaptor protein in controlling mucosal and systemic immunity to fungal infections in humans.
Additionally, CARD9 has been implicated in IBD. A nonsynonymous single nucleotide polymorphism in CARD9 has been detected with high frequency in both Caucasian (at a frequency of 53%) and African (at a frequency of 25%) populations104,105. This polymorphism is strongly associated with the risk of developing both Crohn’s disease and ulcerative colitis106. Using experimental models of colitis, it was shown that CARD9 signaling can be protective against fungi that come in contact with the intestinal mucosa during intestinal barrier damage107. CARD9−/− mice had increased antifungal antibodies in serum and colitis could be partially ameliorated by antifungal treatment, suggesting fungal involvement in the observed phenotype107. Furthermore, both the fungal and bacterial gut communities in CARD9−/− mice were altered108. Interestingly, Candida spp. and Aspergillus spp. were hardly detectible in CARD9 −/− mice, whereas Sporobolomyces genus was the most prevalent group of fungi108, suggesting that C. tropicalis, which expands in dectin-1 and dectin-3 deficient animals16,98, was not the main driver of dysbiosis during CARD9 deficiency. Consistently, human mycobiota studies failed to establish a positive correlation between CARD9 polymorphism and Candida spp. overgrowth, which are otherwise present in patients with IBD107. Finally, besides the CARD9 polymorphism reported above104,105 other CARD9 deficiencies in humans have not been associated with predisposition to IBD.
While some CARD9 mutations are accompanied with a defect in the production of interleukin-17 (IL-17)71, other mutations do not affect IL-17 mediated immunity109. Consistently, mechanistic studies in mice demonstrated that a subtle tuning and timing of CLRs-CARD9-IL-17 mediated response to fungal infections might be taking place100,74. Although CARD9 has a clear role in adaptive Th17 cell responses to oral C. albicans infection, it was not required for IL-17-dependent innate immunity in this model100. In addition, the role of CARD9 in the late stages of mucosal immunity to fungi has been described during Aspergillus fumigatus lung infection74. Of note, CARD9 has a redundant role in the induction of Th17 immunity during systemic infection with other Candida species, such as C. tropicalis110. Whether CARD9 can influence Th17 cell responses during mucosal infection with other non-Candida opportunistic fungi remains to be elucidated.
The role of IL-17 and IL-22
While more studies are needed to define CARD9-dependent and CARD9-independent regulation of mucosal IL-17 responses to fungi, the role of this cytokine during mucosal fungal infections has been evidenced by multiple human genetic and mouse experimental studies summarized in several recent reviews80,81,111. Despite the excellent body of experimental work, it is still unclear whether the IL-17 pathway has an effect on commensal fungal communities. A proof-of-concept clinical study exploring IL-17A blockage with secukinumab in patients with Crohn’s disease identified an unexpected higher rate of fungal infections among the treated individuals112. This adverse outcome correlated with augmented intestinal pathology and suggests a possible role of IL-17 in controlling fungal communities; IL-17 could mediate the production of antimicrobial peptides (AMPs) by epithelial cells, which has been shown to clear Candida spp. infection in the oral mucosa 78. Similar to IL-17, IL-22 acts on epithelial cells as a potent inducer of AMPs and is protective against mucosal Candida spp. and Aspergillus spp. infections66,68,72,73. These barrier enhancing properties might explain the indispensable role of IL-17 and IL-22 in the protection against fungal infection at the mucosal surfaces and provide strong rational to explore the role of these pathways in relation to fungal communities in the gut.
Other contributors
Several other molecules, including the cellular machineries such as NLRP3, NLRC4, and Caspase 1 — which are involved in the processing of pro-IL-1β into its biologically active form — and IL-1β, have been explored in the context of mucosal immunity to fungal infections67,80,113. NLRP3 and NLRC4 inflammasomes are protective in mouse models of vaginal Candida spp. infection67,80,114. Furthermore, genetic polymorphisms in NLRP3 are associated with susceptibility to recurrent vulvovaginal candidiasis, increased IL-1β production and hyper-inflammation115,116. Fungal sensing by NLRP3 inflammasomes seems to be morphotype dependent with a preferential activation by Candida spp. hyphae80. However, whether any of these molecules is involved in control of the mycobiota during steady state or upon dysbiosis remains unclear.
Despite our growing knowledge of antifungal immunity and available antifungal drugs, a rapid rise of fungal infections has prompted the search for novel immunomodulatory strategies to induce immunity to fungi. A recent study demonstrated that inhibition of JNK1 promotes expression of CD23 (a recently discovered CLR), production for nitric oxide and a potent antifungal effect both in vitro and in vivo117. E3 ubiquitin ligase CBLB that ubiquitinates SYK, was shown to regulate antifungal immune responses downstream of dectin-1 and dectin-2 (Figure 3)118,119. Genetic deletion or peptide-based inhibition of CBLB promoted strong antifungal response, enhanced fungal killing and protected mice during systemic and cutaneous C. albicans infections118. Because of their downstream position of several antifungal receptors, CBLB or JNK1 blockage might be a therapeutic avenue to pursue in individuals with increased risk of fungal infections and gastrointestinal complications, such as patients who are immunosuppressed or are infected with HIV. Finally, several other CLRs, chemokine receptors and pathways have been studied in the context of antifungal immunity, however their role in mucosal immunity to fungi has not been explored.
Interplay between bacterial and fungal communities
Fungi and bacteria share similar niches on the mucosal surfaces and can undoubtedly influence each other. Interactions between fungi and bacteria can occur directly by physical contact and through secreted molecules, or indirectly through alteration of the host immune response. Several studies have attempted to identify inter- and intra-kingdom correlations between fungi and bacteria using next generation sequencing14,17,23. However, this approach needs to be validated experimentally since correlations between relative abundances are at high risk of identifying false associations120.
Experimental studies suggest that fungal colonization is modulated by bacterial communities, both clinically21 and in mouse models13,54. Short term antibiotic treatment is sufficient to promote a persistent Candida spp. colonization in the mouse gut13,54 with immunological effects at distant sites such as the lung54,55. Although antibiotic treatment rarely promotes the outgrowth of Candida spp. in the oral mucosa121, antibiotic-induced vaginal candidiasis is relatively common, suggesting a mucosal site-specific nature of bacterial–fungal interplay. Lactobacilli-induced reduction of pH or production of bacteriocin-like compound can inhibit the growth of Candida spp. and are both an essential barrier for the colonization of several fungal species in the vaginal mucosa31,122. In addition to lactobacilli, other bacterial species including Pseudomonas and Burkholderia, as well as bacterial metabolites such as butyrate, can inhibit Candida yeast-to-hypha transition; a process needed for the formation of resistant biofilms of fungi and bacteria123,124. However, fungal communities can adapt to the selective pressure exerted by the host and their bacterial neighbours. Analysis of the relatively stable fungal communities in the cystic fibrosis lung, suggests that Candida spp. clones were resistant to the filamentation-repressive effects of Pseudomonas aeruginosa through a mutation in the repressive gene NRG151. Although the formation of biofilms might contribute to the morbidity of both oral and vaginal candidiasis32,124 such biofilms have not been detected in the gut16,125, which is probably due to inability of Candida to switch to hyphal growth in this environment. According to a recent study, C. albicans adopts a third ‘GUT’ (gastrointestinally induced transition) morphology in the intestines that cannot induce systemic disease and is metabolically accustomed to the intestinal environment125.
In addition to Candida spp., other fungal species can compete for the establishment in an environmental niche. Compounds secreted by Pichia spp. can inhibit the in vitro growth of Candida spp., Aspergillus spp. and Fusarium spp., the formation of biofilms by Candida spp. and reduce the fungal burden in a mouse model of oral candidiasis32. Similarly, bacteria can affect fungal colonization indirectly through the modulation of the host immune response. For example, in mice, Bacteroidetes thetaiotamicron prevents C. albicans colonization of the gut via the HIF-1α mediated production of the antimicrobial peptide CRAMP126.
The host immune response to fungi might in turn indirectly influence bacteria in the gut. A recent study demonstrated that in the absence of commensal Candida spp., Clec7a−/− mice are protected from colitis through a mechanism involving intestinal bacteria84. The authors found that in the absence of Candida spp., antimicrobial peptides targeting gram-positive bacteria were reduced in the colons of Clec7a−/− mice, leading to an increase in commensal Lactobacillus murinus that induced regulatory T cell expansion and protection from colitis. However, upon colonization with C. tropicalis, this potentially beneficial effect was lost and Clec7a−/− mice developed more severe intestinal inflammation84. Similarly, CARD9−/− mice that are susceptible to fungi99,107, also carry altered populations of tryptophan metabolizing bacteria including reduced lactobacilli108. This correlated with decreased aryl hydrocarbon receptor (AHR) ligands and defective Il22, Reg3g, and Reg3b expression in the colons of CARD9−/− mice that could be rescued by supplementation with lactobacilli108. The protective role of lactobacilli through the production of AHR ligands and stimulation of IL-22 release by ILCs has been previously described as a mechanism promoting Candida spp. colonization resistance in the gut 127. Whether fungal and bacterial communities influence each other during these cases of apparent interkingdom dysbiosis in genetically susceptible host remains unclear. Nevertheless, such studies suggest a more complex microbial interkingdom relationship and possibly a dual role of antifungal immunity pathways in the control of bacterial and fungal populations in the gut.
Finally, specific disruption of the healthy gut fungal community can also have an adverse effect to the host health. We and others have recently demonstrated that prolonged antifungal treatment of healthy wild-type mice exacerbated immune-mediated disease in several experimental models of colitis and lung allergy, leading to the expansion of neutrophils, monocytes, Th1 cells, Th17 cells (which was shown in a colitis model) or eosinophils, Th2 cells and IgE-producing B cells (which was shown in a lung allergy model, Figure 2B)24,55. Analysis of the mycobiota from mice treated with fluconazole and amphotericin B revealed dramatic changes in the composition and diversity of the gut mycobiota, suggesting the induction of gut fungal community dysbiosis24. Oral supplementation with Aspergillus amstelodami, Epicoccum nigrum and Wallemia sebi, which expanded during fungal dysbiosis, was sufficient to recapitulate the detrimental effects of antifungal drugs on inflammatory disease. In addition to the mycobiota, drug-induced fungal dysbiosis may affect bacterial community structure and genera such as Bacteroides, Clostridium and Lactobacillus, which are also decreased during fungal dysbiosis25,.
Concluding remarks
A fast growing number of studies are detailing the complexity of antifungal immune responses, yet most evidence focuses on fungal infections. Increasing evidence indicates that human barrier surfaces harbour diverse communities of fungi that cohabitate with the host throughout its life. Although opportunistic fungi within those communities can cause pathologies in susceptible individuals, the majority of host–fungal interactions at the body surfaces are non-infectious. This raises several questions: Do commensal fungi, similar to bacteria, contribute to shaping and maintaining immune homeostasis? Which pathways and intercellular networks are involved in sensing commensal fungi during the steady state? The characterization of immune responses in presence of dysbiotic and steady state fungal communities might provide key answers. Future studies on the crosstalk between the mycobiota, fungal metabolites, and epithelium and mucosa resident immune cells will propel the field toward understanding how mycobiota interacts with host immunity at the barrier surfaces. Understanding the mechanisms behind fungal dysbiosis might provide diagnostic and therapeutic targets by identifying fungi that could or should not be targeted during inflammatory conditions.
Interkingdom microbiome interactions involving fungi can also modulate immunity indirectly by affecting microbiome function and metabolism. The application of metagenomics, metatranscriptomics and metabolomics holds promise to begin identifying the mediators of interkingdom interactions; however, the use of experimental approaches to validate the mediators remains crucial. The fast growing field of antifungal immunity has entered a new age in which a focus on complex microbial communities rather than single fungal species will shed light on the role of mycobiota in homeostasis and inflammation. Collectively, an increasing body of evidence suggests a crosstalk between gut fungi, bacteria and the host mucosal immunity. Disruption of this network can contribute to interkingdom microbial community alterations with detrimental consequences to the host and thus position fungi in the crossfire between mucosal immunity and commensal bacteria.
Acknowledgments
The authors would like to thank members of the Iliev laboratory and the New York Host-Mycobiota Group for helpful suggestions related to the manuscript. This work was funded by the US National Institutes of Health (grants DK098310 and AI123819 to I.D.I.), Kenneth Rainin Foundation (Innovator and Breakthrough awards to I.D.I), Swiss National Science Foundation (fellowship P2ZHP3_164850 to I.L.) and support from the Jill Roberts Institute for Research in IBD. The authors apologize to many contributors whose work could not be cited due to space limitations.
Glossary
- Mycorrhiza
a symbiotic relationship between a mycorrhizal fungus and the root of a plant.
- Symbiosis
an ecological relationship between different species that persistently live in close contact. Symbiosis include relationships such as mutualism, parasitism and commensalism.
- Dysbiosis
a generalizing term initially coined by Ilya Metchnikoff indicating a disruption of a microbial community resulting in a shift from its composition at steady state. Dysbiosis can be caused by multiple factors and can be either a cause or a consequence of disease. The term is undergoing revision in light of the recent advances of microbiome science.
- Internal transcribed spacer (ITS)
a sequence in the fungal genome positioned between 18S and 5.8S (ITS1) or 5.8S and 28S (ITS2) fungal rDNA widely used in mycobiome next generation sequencing. Because of their variability, the ITS regions are often sufficiently different to classify fungal genera and species.
- Onychomycosis
a fungal infection of the fingernails or toenails.
- Chromoblastomycosis
a chronic, localized, infection of the skin and subcutaneous tissue caused by pigmented fungi containing sclerotic bodies.
- Secukinumab
a human blocking monoclonal antibody that binds to interleukin-17A.
- Inflammatory Bowel Disease
a relapsing and remitting condition of complex etiology characterized by inflammation of the lower digestive tract with possible extraintestinal manifestations. The most common types are Crohn’s Disease and Ulcerative Colitis.
- Phaeohyphomycosis
a heterogeneous group of fungal infections characterized by the presence of pigmented fungal cells.
- Deep-seated dermatophytosis
a fungal infection of deep keratinized tissue (including skin, hair, and claws).
- Alpha diversity
a biodiversity measure of the mean species diversity within an individual environmental habitat.
- Operational taxonomic units (OTUs)
clusters of marker gene sequences (e.g 16S or ITS) based upon sequence similarity used for taxonomy-independent community analysis.
- Commensalism
a relationship between two species where one organism benefits from the other without affecting it.
- Richness
the number of different species represented in an ecological community.
Footnotes
CFI
The authors declare no competing interests.
References
- 1.Peay KG, Kennedy PG, Talbot JM. Dimensions of biodiversity in the Earth mycobiome. Nature reviews Microbiology. 2016;14:434–447. doi: 10.1038/nrmicro.2016.59. [DOI] [PubMed] [Google Scholar]
- 2.Remy W, Taylor TN, Hass H, Kerp H. Four hundred-million-year-old vesicular arbuscular mycorrhizae. Proc Natl Acad Sci U S A. 1994;91:11841–11843. doi: 10.1073/pnas.91.25.11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wainright PO, Hinkle G, Sogin ML, Stickel SK. Monophyletic origins of the metazoa: an evolutionary link with fungi. Science. 1993;260:340–342. doi: 10.1126/science.8469985. [DOI] [PubMed] [Google Scholar]
- 4.Casadevall A. Fungi and the rise of mammals. PLoS Pathog. 2012;8:e1002808. doi: 10.1371/journal.ppat.1002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown GD, Gordon S. Fungal beta-glucans and mammalian immunity. Immunity. 2003;19:311–315. doi: 10.1016/s1074-7613(03)00233-4. [DOI] [PubMed] [Google Scholar]
- 6.Fisher MC, et al. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown GD, et al. Hidden killers: human fungal infections. Sci Transl Med. 2012;4:165rv113. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 8.Lanternier F, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med. 2013;369:1704–1714. doi: 10.1056/NEJMoa1208487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Findley K, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498:367–370. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brune A. Symbiotic digestion of lignocellulose in termite guts. Nature reviews Microbiology. 2014;12:168–180. doi: 10.1038/nrmicro3182. [DOI] [PubMed] [Google Scholar]
- 11.Solomon KV, et al. Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes. Science. 2016;351:1192–1195. doi: 10.1126/science.aad1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belkaid Y, Harrison OJ. Homeostatic Immunity and the Microbiota. Immunity. 2017;46:562–576. doi: 10.1016/j.immuni.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dollive S, et al. Fungi of the murine gut: episodic variation and proliferation during antibiotic treatment. PLoS One. 2013;8:e71806. doi: 10.1371/journal.pone.0071806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann C, et al. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One. 2013;8:e66019. doi: 10.1371/journal.pone.0066019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.David LA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iliev ID, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokol H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2016;0:1–10. doi: 10.1136/gutjnl-2015-310746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metchnikoff E. The prolongation of life: optimistic studies. New York & London: G.P. Putnam’s Sons; 1908. pp. 161–183. [Google Scholar]
- 19.Hsiao EY, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsland BJ, Gollwitzer ES. Host-microorganism interactions in lung diseases. Nat Rev Immunol. 2014;14:827–835. doi: 10.1038/nri3769. [DOI] [PubMed] [Google Scholar]
- 21.Lewis JD, et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell host & microbe. 2015;18:489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liguori G, et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. Journal of Crohn’s & colitis. 2016;10:296–305. doi: 10.1093/ecco-jcc/jjv209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoarau G, et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. mBio. 2016;7:e01250–01216. doi: 10.1128/mBio.01250-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheeler ML, et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell host & microbe. 2016;19:865–873. doi: 10.1016/j.chom.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghannoum MA, et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010;6:e1000713. doi: 10.1371/journal.ppat.1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bittinger K, et al. Improved characterization of medically relevant fungi in the human respiratory tract using next-generation sequencing. Genome Biol. 2014;15:487. doi: 10.1186/s13059-014-0487-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindahl BD, et al. Fungal community analysis by high-throughput sequencing of amplified markers--a user’s guide. The New phytologist. 2013;199:288–299. doi: 10.1111/nph.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang J, Iliev ID, Brown J, Underhill DM, Funari VA. Mycobiome: Approaches to analysis of intestinal fungi. J Immunol Methods. 2015;421:112–121. doi: 10.1016/j.jim.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol. 2014;14:405–416. doi: 10.1038/nri3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motooka D, et al. Fungal ITS1 Deep-Sequencing Strategies to Reconstruct the Composition of a 26-Species Community and Evaluation of the Gut Mycobiota of Healthy Japanese Individuals. Frontiers in microbiology. 2017;8:238. doi: 10.3389/fmicb.2017.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krasner RI, Young G, Yudkofsky PL. Interactions of oral strains of Candida albicans and lactobacilli. Journal of bacteriology. 1956;72:525–529. doi: 10.1128/jb.72.4.525-529.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukherjee PK, et al. Oral mycobiome analysis of HIV-infected patients: identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog. 2014;10:e1003996. doi: 10.1371/journal.ppat.1003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hallen-Adams HE, Suhr MJ. Fungi in the healthy human gastrointestinal tract. Virulence. 2016:1–7. doi: 10.1080/21505594.2016.1247140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suhr MJ, Banjara N, Hallen-Adams HE. Sequence-based methods for detecting and evaluating the human gut mycobiome. Letters in applied microbiology. 2016;62:209–215. doi: 10.1111/lam.12539. [DOI] [PubMed] [Google Scholar]
- 35.Cutler JE, Deepe GS, Jr, Klein BS. Advances in combating fungal diseases: vaccines on the threshold. Nature reviews Microbiology. 2007;5:13–28. doi: 10.1038/nrmicro1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luan C, et al. Dysbiosis of fungal microbiota in the intestinal mucosa of patients with colorectal adenomas. Sci Rep. 2015;5:7980. doi: 10.1038/srep07980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Q, et al. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn’s disease. Journal of clinical gastroenterology. 2014;48:513–523. doi: 10.1097/MCG.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirsner JB. Historical origins of current IBD concepts. World journal of gastroenterology. 2001;7:175–184. doi: 10.3748/wjg.v7.i2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53:1–4. doi: 10.1136/gut.53.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ott SJ, et al. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand J Gastroenterol. 2008;43:831–841. doi: 10.1080/00365520801935434. [DOI] [PubMed] [Google Scholar]
- 41.Qiu X, et al. Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis. Sci Rep. 2015;5:10416. doi: 10.1038/srep10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chehoud C, et al. Fungal Signature in the Gut Microbiota of Pediatric Patients With Inflammatory Bowel Disease. Inflamm Bowel Dis. 2015;21:1948–1956. doi: 10.1097/MIB.0000000000000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Israeli E, et al. Anti-Saccharomyces cerevisiae and antineutrophil cytoplasmic antibodies as predictors of inflammatory bowel disease. Gut. 2005;54:1232–1236. doi: 10.1136/gut.2004.060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Standaert-Vitse A, et al. Candida albicans colonization and ASCA in familial Crohn’s disease. Am J Gastroenterol. 2009;104:1745–1753. doi: 10.1038/ajg.2009.225. [DOI] [PubMed] [Google Scholar]
- 45.Schaffer T, et al. Anti-Saccharomyces cerevisiae mannan antibodies (ASCA) of Crohn’s patients crossreact with mannan from other yeast strains, and murine ASCA IgM can be experimentally induced with Candida albicans. Inflamm Bowel Dis. 2007;13:1339–1346. doi: 10.1002/ibd.20228. [DOI] [PubMed] [Google Scholar]
- 46.Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The Central Nervous System and the Gut Microbiome. Cell. 2016;167:915–932. doi: 10.1016/j.cell.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strati F, et al. Altered gut microbiota in Rett syndrome. Microbiome. 2016;4:41. doi: 10.1186/s40168-016-0185-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strati F, et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome. 2017;5:24. doi: 10.1186/s40168-017-0242-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charlson ES, et al. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. American journal of respiratory and critical care medicine. 2012;186:536–545. doi: 10.1164/rccm.201204-0693OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delhaes L, et al. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community--implications for therapeutic management. PLoS One. 2012;7:e36313. doi: 10.1371/journal.pone.0036313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SH, et al. Global Analysis of the Fungal Microbiome in Cystic Fibrosis Patients Reveals Loss of Function of the Transcriptional Repressor Nrg1 as a Mechanism of Pathogen Adaptation. PLoS Pathog. 2015;11:e1005308. doi: 10.1371/journal.ppat.1005308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Willger SD, et al. Characterization and quantification of the fungal microbiome in serial samples from individuals with cystic fibrosis. Microbiome. 2014;2:40. doi: 10.1186/2049-2618-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Woerden HC, et al. Differences in fungi present in induced sputum samples from asthma patients and non-atopic controls: a community based case control study. BMC infectious diseases. 2013;13:69. doi: 10.1186/1471-2334-13-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noverr MC, Noggle RM, Toews GB, Huffnagle GB. Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect Immun. 2004;72:4996–5003. doi: 10.1128/IAI.72.9.4996-5003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim YG, et al. Gut Dysbiosis Promotes M2 Macrophage Polarization and Allergic Airway Inflammation via Fungi-Induced PGE2. Cell host & microbe. 2014;15:95–102. doi: 10.1016/j.chom.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farr A, et al. Effect of asymptomatic vaginal colonization with Candida albicans on pregnancy outcome. Acta obstetricia et gynecologica Scandinavica. 2015;94:989–996. doi: 10.1111/aogs.12697. [DOI] [PubMed] [Google Scholar]
- 57.Drell T, et al. Characterization of the vaginal micro- and mycobiome in asymptomatic reproductive-age Estonian women. PLoS One. 2013;8:e54379. doi: 10.1371/journal.pone.0054379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Merenstein D, et al. Colonization by Candida species of the oral and vaginal mucosa in HIV-infected and noninfected women. AIDS research and human retroviruses. 2013;29:30–34. doi: 10.1089/aid.2012.0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo R, et al. Increased diversity of fungal flora in the vagina of patients with recurrent vaginal candidiasis and allergic rhinitis. Microb Ecol. 2012;64:918–927. doi: 10.1007/s00248-012-0084-0. [DOI] [PubMed] [Google Scholar]
- 60.Oh J, et al. Temporal Stability of the Human Skin Microbiome. Cell. 2016;165:854–866. doi: 10.1016/j.cell.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oh J, et al. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013;23:2103–2114. doi: 10.1101/gr.159467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smeekens SP, et al. Skin microbiome imbalance in patients with STAT1/STAT3 defects impairs innate host defense responses. Journal of innate immunity. 2014;6:253–262. doi: 10.1159/000351912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalan L, et al. Redefining the Chronic-Wound Microbiome: Fungal Communities Are Prevalent, Dynamic, and Associated with Delayed Healing. mBio. 2016;7 doi: 10.1128/mBio.01058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strati F, et al. Age and Gender Affect the Composition of Fungal Population of the Human Gastrointestinal Tract. Frontiers in microbiology. 2016;7:1227. doi: 10.3389/fmicb.2016.01227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jo JH, et al. Diverse Human Skin Fungal Communities in Children Converge in Adulthood. The Journal of investigative dermatology. 2016;136:2356–2363. doi: 10.1016/j.jid.2016.05.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Conti HR, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hise AG, et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell host & microbe. 2009;5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Luca A, et al. IL-22 and IDO1 affect immunity and tolerance to murine and human vaginal candidiasis. PLoS Pathog. 2013;9:e1003486. doi: 10.1371/journal.ppat.1003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kashem SW, et al. Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity. 2015;42:356–366. doi: 10.1016/j.immuni.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferwerda B, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. 2009;361:1760–1767. doi: 10.1056/NEJMoa0901053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Glocker EO, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gessner MA, et al. Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect Immun. 2012;80:410–417. doi: 10.1128/IAI.05939-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Puel A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010;207:291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jhingran A, et al. Compartment-specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog. 2015;11:e1004589. doi: 10.1371/journal.ppat.1004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rivera A, et al. Dectin-1 diversifies Aspergillus fumigatus-specific T cell responses by inhibiting T helper type 1 CD4 T cell differentiation. J Exp Med. 2011;208:369–381. doi: 10.1084/jem.20100906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trautwein-Weidner K, et al. Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis. PLoS Pathog. 2015;11:e1005164. doi: 10.1371/journal.ppat.1005164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lionakis MS, et al. CX3CR1-dependent renal macrophage survival promotes Candida control and host survival. The Journal of clinical investigation. 2013;123:5035–5051. doi: 10.1172/JCI71307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Conti HR, et al. IL-17 Receptor Signaling in Oral Epithelial Cells Is Critical for Protection against Oropharyngeal Candidiasis. Cell host & microbe. 2016;20:606–617. doi: 10.1016/j.chom.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14:667–685. doi: 10.1038/nri3738. [DOI] [PubMed] [Google Scholar]
- 80.Netea MG, Joosten LA, van der Meer JW, Kullberg BJ, van de Veerdonk FL. Immune defence against Candida fungal infections. Nat Rev Immunol. 2015;15:630–642. doi: 10.1038/nri3897. [DOI] [PubMed] [Google Scholar]
- 81.Plato A, Hardison SE, Brown GD. Pattern recognition receptors in antifungal immunity. Seminars in immunopathology. 2015;37:97–106. doi: 10.1007/s00281-014-0462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taylor PR, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nature immunology. 2007;8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marakalala MJ, et al. Differential adaptation of Candida albicans in vivo modulates immune recognition by dectin-1. PLoS Pathog. 2013;9:e1003315. doi: 10.1371/journal.ppat.1003315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang C, et al. Inhibition of Dectin-1 Signaling Ameliorates Colitis by Inducing Lactobacillus-Mediated Regulatory T Cell Expansion in the Intestine. Cell host & microbe. 2015;18:183–197. doi: 10.1016/j.chom.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 85.Moyes DL, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016;532:64–68. doi: 10.1038/nature17625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pierce JV, Kumamoto CA. Variation in Candida albicans EFG1 expression enables host-dependent changes in colonizing fungal populations. mBio. 2012;3:e00117–00112. doi: 10.1128/mBio.00117-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Vries HS, et al. Genetic association analysis of the functional c.714T>G polymorphism and mucosal expression of dectin-1 in inflammatory bowel disease. PLoS One. 2009;4:e7818. doi: 10.1371/journal.pone.0007818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Plantinga TS, et al. Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin Infect Dis. 2009;49:724–732. doi: 10.1086/604714. [DOI] [PubMed] [Google Scholar]
- 89.van der Velden WJ, et al. Role of the mycobiome in human acute graft-versus-host disease. Biol Blood Marrow Transplant. 2013;19:329–332. doi: 10.1016/j.bbmt.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 90.Saijo S, et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity. 2010;32:681–691. doi: 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 91.Wuthrich M, et al. Fonsecaea pedrosoi-induced Th17-cell differentiation in mice is fostered by Dectin-2 and suppressed by Mincle recognition. European journal of immunology. 2015;45:2542–2552. doi: 10.1002/eji.201545591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ishikawa T, et al. Identification of distinct ligands for the C-type lectin receptors Mincle and Dectin-2 in the pathogenic fungus Malassezia. Cell host & microbe. 2013;13:477–488. doi: 10.1016/j.chom.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 93.Barrett NA, Maekawa A, Rahman OM, Austen KF, Kanaoka Y. Dectin-2 recognition of house dust mite triggers cysteinyl leukotriene generation by dendritic cells. J Immunol. 2009;182:1119–1128. doi: 10.4049/jimmunol.182.2.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakamura Y, et al. Dectin-2 deficiency promotes Th2 response and mucin production in the lungs after pulmonary infection with Cryptococcus neoformans. Infect Immun. 2015;83:671–681. doi: 10.1128/IAI.02835-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hu XP, et al. Dectin-2 polymorphism associated with pulmonary cryptococcosis in HIV-uninfected Chinese patients. Medical mycology. 2015;53:810–816. doi: 10.1093/mmy/myv043. [DOI] [PubMed] [Google Scholar]
- 96.Zhao XQ, et al. C-type lectin receptor dectin-3 mediates trehalose 6,6′-dimycolate (TDM)-induced Mincle expression through CARD9/Bcl10/MALT1-dependent nuclear factor (NF)-kappaB activation. The Journal of biological chemistry. 2014;289:30052–30062. doi: 10.1074/jbc.M114.588574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhu LL, et al. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity. 2013;39:324–334. doi: 10.1016/j.immuni.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 98.Wang T, et al. Dectin-3 Deficiency Promotes Colitis Development due to Impaired Antifungal Innate Immune Responses in the Gut. PLoS Pathog. 2016;12:e1005662. doi: 10.1371/journal.ppat.1005662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.LeibundGut-Landmann S, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nature immunology. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 100.Bishu S, et al. The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect Immun. 2014;82:1173–1180. doi: 10.1128/IAI.01335-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Drummond RA, et al. CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System. PLoS Pathog. 2015;11:e1005293. doi: 10.1371/journal.ppat.1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang X, et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. The Journal of allergy and clinical immunology. 2014;133:905–908e903. doi: 10.1016/j.jaci.2013.09.033. [DOI] [PubMed] [Google Scholar]
- 103.Lanternier F, et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species-induced meningoencephalitis, colitis, or both. The Journal of allergy and clinical immunology. 2015;135:1558–1568e1552. doi: 10.1016/j.jaci.2014.12.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.International HapMap C et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sokol H, et al. Card9 mediates intestinal epithelial cell restitution, T-helper 17 responses, and control of bacterial infection in mice. Gastroenterology. 2013;145:591–601e593. doi: 10.1053/j.gastro.2013.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lamas B, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nature medicine. 2016;22:598–605. doi: 10.1038/nm.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Drummond RA, Lionakis MS. Mechanistic Insights into the Role of C-Type Lectin Receptor/CARD9 Signaling in Human Antifungal Immunity. Frontiers in cellular and infection microbiology. 2016;6:39. doi: 10.3389/fcimb.2016.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Whibley N, et al. Delinking CARD9 and IL-17: CARD9 Protects against Candida tropicalis Infection through a TNF-alpha-Dependent, IL-17-Independent Mechanism. J Immunol. 2015;195:3781–3792. doi: 10.4049/jimmunol.1500870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Conti HR, Gaffen SL. IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida albicans. J Immunol. 2015;195:780–788. doi: 10.4049/jimmunol.1500909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hueber W, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Borghi M, et al. Pathogenic NLRP3 Inflammasome Activity during Candida Infection Is Negatively Regulated by IL-22 via Activation of NLRC4 and IL-1Ra. Cell host & microbe. 2015;18:198–209. doi: 10.1016/j.chom.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 114.Tomalka J, et al. A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathog. 2011;7:e1002379. doi: 10.1371/journal.ppat.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lev-Sagie A, et al. Polymorphism in a gene coding for the inflammasome component NALP3 and recurrent vulvovaginal candidiasis in women with vulvar vestibulitis syndrome. Am J Obstet Gynecol. 2009;200:303 e301–306. doi: 10.1016/j.ajog.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 116.Jaeger M, et al. Association of a variable number tandem repeat in the NLRP3 gene in women with susceptibility to RVVC. Eur J Clin Microbiol Infect Dis. 2016;35:797–801. doi: 10.1007/s10096-016-2600-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhao X, et al. JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nature medicine. 2017 doi: 10.1038/nm.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wirnsberger G, et al. Inhibition of CBLB protects from lethal Candida albicans sepsis. Nature medicine. 2016;22:915–923. doi: 10.1038/nm.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xiao Y, et al. Targeting CBLB as a potential therapeutic approach for disseminated candidiasis. Nature medicine. 2016;22:906–914. doi: 10.1038/nm.4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS computational biology. 2012;8:e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wolfson SA. Black hairy tongue associated with penicillin therapy. Journal of the American Medical Association. 1949;140:1206–1208. doi: 10.1001/jama.1949.02900500014003. [DOI] [PubMed] [Google Scholar]
- 122.Boris S, Barbes C. Role played by lactobacilli in controlling the population of vaginal pathogens. Microbes Infect. 2000;2:543–546. doi: 10.1016/s1286-4579(00)00313-0. [DOI] [PubMed] [Google Scholar]
- 123.Noverr MC, Huffnagle GB. Regulation of Candida albicans morphogenesis by fatty acid metabolites. Infect Immun. 2004;72:6206–6210. doi: 10.1128/IAI.72.11.6206-6210.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lynch AS, Robertson GT. Bacterial and fungal biofilm infections. Annual review of medicine. 2008;59:415–428. doi: 10.1146/annurev.med.59.110106.132000. [DOI] [PubMed] [Google Scholar]
- 125.Pande K, Chen C, Noble SM. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nature genetics. 2013;45:1088–1091. doi: 10.1038/ng.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fan D, et al. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nature medicine. 2015;21:808–814. doi: 10.1038/nm.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zelante T, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–385. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 128.de Bekker C, et al. Species-specific ant brain manipulation by a specialized fungal parasite. BMC evolutionary biology. 2014;14:166. doi: 10.1186/s12862-014-0166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lin CH, et al. Identification of a major epitope by anti-interferon-gamma autoantibodies in patients with mycobacterial disease. Nature medicine. 2016;22:994–1001. doi: 10.1038/nm.4158. [DOI] [PubMed] [Google Scholar]
- 130.Sterkel AK, et al. Fungal Mimicry of a Mammalian Aminopeptidase Disables Innate Immunity and Promotes Pathogenicity. Cell host & microbe. 2016;19:361–374. doi: 10.1016/j.chom.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Poulsen M, Boomsma JJ. Mutualistic fungi control crop diversity in fungus-growing ants. Science. 2005;307:741–744. doi: 10.1126/science.1106688. [DOI] [PubMed] [Google Scholar]
- 132.Kenrad E, Nelson CW. Infectious Disease Epidemiology Theory and Practice. Jones & Bartlett Learning. 2014 [Google Scholar]
- 133.Lane N. The unseen world: reflections on Leeuwenhoek (1677) ‘Concerning little animals’. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2015;370 doi: 10.1098/rstb.2014.0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pasteur L, Illo J. Pasteur and rabies: an interview of 1882. Medical history. 1996;40:373–377. doi: 10.1017/s0025727300061354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Henle J. On miasmata and contagie. Johns Hopkins Press; Baltimore: 1938. [Google Scholar]
- 136.Koch R. An Address on Bacteriological Research. British medical journal. 1890;2:380–383. doi: 10.1136/bmj.2.1546.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Evans AS. Causation and disease: the Henle-Koch postulates revisited. The Yale journal of biology and medicine. 1976;49:175–195. [PMC free article] [PubMed] [Google Scholar]
- 138.Fredricks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clin Microbiol Rev. 1996;9:18–33. doi: 10.1128/cmr.9.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Byrd AL, Segre JA. Infectious disease. Adapting Koch’s postulates. Science. 2016;351:224–226. doi: 10.1126/science.aad6753. [DOI] [PubMed] [Google Scholar]
- 140.Olesen SW, Alm EJ. Dysbiosis is not an answer. Nature microbiology. 2016;1:16228. doi: 10.1038/nmicrobiol.2016.228. [DOI] [PubMed] [Google Scholar]
- 141.Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014;16:1024–1033. doi: 10.1111/cmi.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]