

Abstract
The extracellular signal-regulated kinase (ERK)/MAPK (mitogen-activated protein kinase) cascade has been established as a potent regulator of gene transcription in long-term memory formation, but the precise mechanisms of this regulation are poorly understood. ERK does not directly affect many of its nuclear targets, but rather must act through intermediary kinases. In this study, we investigated the role of mitogen- and stress-activated protein kinase 1 (MSK1), a nuclear kinase downstream of ERK, in chromatin remodeling during hippocampus-dependent memory formation. Mice lacking MSK1 show impaired Pavlovian fear conditioning and spatial learning, as well as a deficiency in histone phosphorylation and acetylation in the hippocampus after fear training. In addition, hippocampal slices from MSK1 knock-out mice exhibit a deficiency in both histone phosphorylation and acetylation after activation of the ERK pathway in vitro. In vivo injections of a histone deacetylase inhibitor, sodium butyrate, fail to alleviate the fear conditioning deficit in MSK1 knock-out mice. Finally, MSK1 knock-out mice demonstrate a deficiency in cAMP response element-binding protein (CREB) phosphorylation after fear training, which persists after sodium butyrate injection. This suggests that CREB phosphorylation and histone acetylation represent parallel targets of MSK1 function. Our study identifies MSK1 as an important regulator of chromatin remodeling in long-term memory.
Keywords: epigenetics, memory, histones, MAPK, MSK1, transcription
Introduction
Long-term memory formation involves complex biochemical cascades leading to changes in gene expression, accomplished partly by epigenetic mechanisms that remodel chromatin (Tsankova et al., 2004). These include post-translational modifications of histones, which regulate the dynamic interplay between the native inhibitory state of chromatin and a transcriptionally active state (Jenuwein and Allis, 2001; Berger, 2002). Previous studies have focused on neuronal regulation of histones, particularly in memory and synaptic plasticity (Swank and Sweatt, 2001; Guan et al., 2002; Alarcon et al., 2004; Korzus et al., 2004; Levenson et al., 2004; Chwang et al., 2006; Kim et al., 2007). These studies suggest that chromatin remodeling contributes significantly to regulation of gene expression and neuronal function.
The regulation of histone phosphorylation is particularly intriguing, given the importance of the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) cascade in learning and memory. In mammalian memory tasks, activation of ERK in the hippocampus is necessary for consolidation of the memory (Atkins et al., 1998; Blum et al., 1999). Once activated, ERK phosphorylates many targets implicated in gene transcription, protein synthesis, and synaptic plasticity (Impey et al., 1998; Adams et al., 2000; Davis et al., 2000; Goldin and Segal, 2003). Previously, ERK was shown to regulate histone phosphorylation and acetylation in hippocampal CA1 neurons (Levenson et al., 2004; Chwang et al., 2006), although the precise downstream molecular mechanisms are as of yet unknown.
One important histone kinase that has been identified is mitogen- and stress-activated protein kinase-1 (MSK1), a nuclear kinase downstream of ERK/MAPK and p38/MAPK (Deak et al., 1998). MSK1 mediates the mitogen- and stress-induced phosphorylation of histone H3, cAMP response element-binding protein (CREB), ATF-1 (activating transcription factor 1), HMG-14 (high-mobility group protein-14), and other transcription factors in vitro (Deak et al., 1998; Thomson et al., 1999; Wiggin et al., 2002; Soloaga et al., 2003). Although MSK1 appears to affect multiple targets important for plasticity and memory, currently, its role in behavioral learning remains mostly unknown. Animals lacking MSK1 display an impairment of locomotor sensitization to cocaine, and have decreased phosphorylation of CREB and histone H3 in striatal neurons (Brami-Cherrier et al., 2005). Furthermore, MSK1 is the major CREB kinase activated during contextual fear training (Sindreu et al., 2007). However, it is not known whether MSK1 is required for memory formation and chromatin remodeling in the hippocampus.
In this study, we investigated the contribution of MSK1 to behavioral learning and the modulation of histones in the hippocampus. We found that the absence of MSK1 leads to deficits in multiple hippocampus-dependent tasks: long-term contextual fear conditioning, passive avoidance, and the Morris water maze. We also found that MSK1 knock-out animals have a selective deficiency in histone H3 phosphorylation and acetylation, both markers of transcriptional activation. Treatment of animals with a histone deacetylase (HDAC) inhibitor, sodium butyrate, leads to an enhancement of fear memory in wild-type mice, but not in MSK1 knock-out mice. Finally, we found that trained MSK1 knock-out mice have deficient CREB phosphorylation that persists after treatment with sodium butyrate. These results establish the importance of MSK1 in regulating hippocampal chromatin remodeling in long-term memory.
Materials and Methods
Animals.
MSK1 knock-out mice were kindly provided by Dr. J. Simon Arthur (University of Dundee, Dundee, Scotland) and bred at Baylor College of Medicine. MSK1 knock-out animals were backcrossed with wild-type C57BL/6 mice for at least eight generations. Genotyping of animals was performed on offspring of heterozygote breedings, as previously described (Wiggin et al., 2002), and littermate controls were used in all behavioral experiments. All animals were housed under a 12 h light/dark schedule and given access to food and water ad libitum. All experiments were performed in compliance with the Baylor College of Medicine Institutional Animal Care and Use Committee as well as national regulations and policies.
Fear conditioning.
All behavioral experiments were performed in the neurobehavior core at the Baylor College of Medicine Transgenic Mouse Facility, supported by the Baylor Mental Retardation and Developmental Disabilities Research Center. Before each experiment, animals were transported to the testing room and allowed to acclimate for at least 30 min. Animals were placed in the fear conditioning apparatus and allowed to explore for 2 min. Each animal then received two pairings of a white noise tone (30 s, 90 dB) immediately followed by an electric foot shock (2 s, 0.5 mA). The pairings of tone and shock were separated by 2 min. After the final shock, the animal was left in the chamber for an additional 20 s before being removed. Freezing behavior was measured during the training trial and at various time points after training (FreezeFrame Actimetrics, Wilmette, IL). For the contextual test, the animal was placed into the same chamber and freezing was measured for 5 min. For the cued test, the environment of the training chamber was altered (Plexiglas inserts, different odor). After the animal was placed into this novel context and allowed to explore for 3 min, the tone was played continuously for 3 min, and freezing behavior was recorded. For biochemistry experiments, the hippocampi were removed 1 h after fear conditioning training and processed for histone extraction or cytoplasmic/nuclear extraction.
Baseline behaviors.
A number of tests were performed to measure baseline behaviors. For the open field analysis, the animal was placed in the center of a large Plexiglas box (43 × 42 × 30 cm) and locomotor activity was measured for 30 min (Versamax; AccuScan, Columbus, OH). The ratio of distance spent in the center of the field to total distance traveled was calculated. For the rotarod task, the animal was placed onto an accelerating rotating rod (UGO Basile, Comerio, Italy) (4 rpm to 40 rpm over 5 min) and allowed to run until the animal either rotated completely around the rod or fell off. The latency time until the animal lost control was measured. For the hot plate test, the animal was placed onto a 52°C surface and the latency time until the animal lifted or licked its paws was recorded.
Shock thresholds were measured by placing the animal into a training chamber and administering successive shocks from 0 to 0.5 mA. The thresholds to flinching, jumping, and vocalization behaviors were recorded. For prepulse inhibition, the animal was placed into an enclosed startle tube, and the force that the animal exerted on the underlying support plate was measured in response to a loud noise (Startle; San Diego Instruments, San Diego, CA). Multiple trials were conducted with noises up to 120 dB and prepulses of 74, 78, and 82 dB immediately preceding the noise. The percent dimunition of the force in response to 120 dB was measured for the different prepulses.
assive avoidance.
Animals were placed into a training chamber consisting of a dark compartment and a lighted compartment. On the first day, animals were placed into the lighted side, and the latency for the animal to step over to the dark compartment was measured. Afterward, an electric foot shock (2 s, 0.5 mA) was delivered, and the animal was removed. On the second day, animals were placed into the lighted side, and the latency for the animal to step into the dark compartment was again measured, with a maximum time of 5 min.
Morris water maze.
After acclimating to the testing room for at least 30 min, animals were trained to find a hidden platform in one quadrant of an opaque pool (130 cm diameter) using visual–spatial cues. Animals were given two blocks of four training trials per day for a total of 4 d, or 8 blocks of trials. The order of start positions was varied randomly for each trial. The latency for the animal to find the platform was recorded, and the maximum time the animal was allowed to search for the platform was 60 s. After the final training trial, a probe trial was performed in which the platform was removed and the animal's swim pattern was tracked for 60 s (EthoVision; Noldus, Wageningen, The Netherlands). For the visible platform test, animals were trained to locate a submerged platform with a highly visible dark square object mounted above. Latency for the animal to find the platform was recorded.
Nissl stain.
Animals were killed by decapitation, and the brains were removed, fixed in Bouin's solution overnight, and embedded in paraffin wax. Ten micrometer sagittal sections were cut, mounted on slides, and processed for Nissl staining. Sections were rehydrated successively with xylene, 100% ethanol, 95% ethanol, 75% ethanol, water, and PBS. Sections were then stained with cresyl violet solution (0.5% cresyl violet, 60 mm sodium acetate, 0.34 m acetic acid) for 10 min, washed, and dehydrated again. Images were digitally captured using a Zeiss (Thornwood, NY) Stemi SV11 microscope and XCAP V2.2 for Windows (EPIX, Buffalo Grove, IL).
Hippocampal slice preparation.
Animals were killed by decapitation, and the brains were removed and immersed in ice-cold cutting solution (CS) [containing (in mm) 100 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 glucose, and 0.6 ascorbate]. The caudal portion of the brain containing hippocampus and entorhinal cortex was isolated, and 400 μm transverse slices were prepared using a vibratome (Leica, Bannockburn, IL) and stored in ice-cold CS. The hippocampus was isolated from cortical tissue in all slices, and all hippocampi were equilibrated in 50% CS and 50% artificial CSF (ACSF) [containing (in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 25 glucose] for 45 min at room temperature. Slices were removed and further incubated in 100% ACSF for 45 min at room temperature, then 100% ACSF for 1 h at 32°C. Drugs or vehicle were then added to the slices for 1 h before slices were removed for processing. All solutions were continuously bubbled with 95% O2/5% CO2.
Histone extraction.
Samples were homogenized for 12 strokes at 4000 rpm using Teflon grinders. All steps were performed on ice, and all centrifugations were done at 4°C. Homogenized samples were centrifuged at 7700 × g for 1 min. The supernatant was stored at −80°C as the cytoplasmic fraction. The pellet containing the nuclear fraction was resuspended in 1 ml of 0.4 N H2SO4 for 30 min to extract histones. Samples were centrifuged at 14,000 × g for 10 min. The supernatant was transferred to a fresh tube, and 250 μl of trichloroacetic acid with 4 mg/ml deoxycholic acid was added to precipitate proteins. The precipitate was incubated on ice for 30 min, then centrifuged at 14,000 × g for 30 min. The supernatant was discarded, and the protein pellet was washed with 1 ml of acidified acetone (0.1% HCl) and 1 ml of pure acetone for 5 min each, with centrifugation at 14,000 × g for 5 min after each wash. The final protein pellet was resuspended in 10 mm Tris pH 8 and stored at −80°C.
Cytoplasmic/nuclear extraction.
Samples were processed for cytoplasmic/nuclear extraction using the NE-PER nuclear and cytoplasmic extraction reagents (Pierce Biotechnology, Rockford, IL). All steps were performed on ice, and all centrifugations were done at 4°C. Samples were homogenized in PBS with protease inhibitors and centrifuged at 500 × g for 2–3 min. The supernatant was removed and the pellet resuspended in cytoplasmic extraction reagent (CER I) with protease inhibitors. After incubating on ice for 10 min, a second CER (CER II) was added. Samples were vortexed and centrifuged at 14,000 × g for 5 min. The supernatant was stored at −80°C as the cytoplasmic fraction. The remaining pellet was resuspended again in nuclear extraction reagent with protease inhibitors. After vortexing and incubating on ice every 10 min for a total of 40 min, the samples were centrifuged at 14,000 × g for 10 min. The supernatant was stored at −80°C as the nuclear fraction.
Western blotting.
Protein concentrations were measured using commercially available reagents (protein assay concentrate; Bio-Rad Laboratories, Hercules, CA). Four times sample buffer (final concentration: 6.25 mm Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 1.25% 2-mercaptoethanol, and 0.1% bromophenol blue) was added to each sample. 1 μg of protein from each sample was loaded and run on a 4% acrylamide stacking gel and 10% acrylamide resolving gel. Proteins were transferred to polyvinylidene difluoride membranes which were processed for immunoblotting. Membranes were first blocked in 5% milk in Tris-buffered saline with Tween (TTBS) (150 mm NaCl, 20 mm Tris-HCl pH 7.5, 0.05% Tween 20) for 45 min at room temperature, then incubated with primary antibody overnight at 4°C. Primary antibodies and dilutions used were anti-MSK1 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-MSK2 (1:500; Invitrogen, Calsbad, CA), anti-TATA-binding protein (TBP) (1:1000; Santa Cruz Biotechnology), anti-p44/42 MAPK (1:1000; Cell Signaling Technology, Danvers, MA), anti-phospho-p44/42 MAPK (1:1000; Cell Signaling Technology), anti-histone H3 (1:500; Millipore, Billerica, MA), anti-phospho-histone H3 (Ser10, 1:500; Millipore), anti-acetyl-histone H3 (Lys14, 1:1000; Millipore), anti-dimethyl-histone H3 (Lys9, 1:1000; Millipore), anti-acetyl-histone H4 (Lys5/8/12/16, 1:1000; Millipore, Billerica, MA), anti-histone H4 (1:1000; Millipore), anti-phospho-CREB (Ser133, 1:1000; Millipore), and anti-CREB (1:500; Cell Signaling Technology).
Subsequently, membranes were washed with TTBS and incubated with secondary antibody for 2.5 h at room temperature. Secondary antibodies used were horseradish peroxidase-conjugated goat anti-rabbit IgG heavy and light chain (Jackson ImmunoResearch, West Grove, PA) and horse anti-goat IgG heavy and light chain (Vector Laboratories, Burlingame, CA). Finally, membranes were washed with TTBS and 1× TBS, and then immunolabeled by chemiluminescence (GE Healthcare, Piscataway, NJ). Luminescence was recorded onto BioMax MR films (Kodak Scientific Imaging Systems, Rochester, NY). ImageJ (National Institutes of Health, Bethesda, MD) was used to quantify integrated densities of each band on film.
Statistical analysis.
Western blot immunodensities were analyzed using a one-way ANOVA followed by Tukey's multiple comparison test. Comparisons in freezing behavior, latencies, or platform crossings were analyzed using a one-way ANOVA followed by Tukey's multiple-comparison test. Significant differences were denoted by p < 0.05.
Results
MSK1 is highly expressed in the hippocampus and MSK1 knock-out mice display normal brain morphology
We first compared MSK1 knock-out and wild-type littermate mice to determine both MSK1 expression levels and overall brain morphology. We found that MSK1 is highly expressed in whole brain and hippocampus relative to other organs, whereas samples from MSK1 knock-out animals lack the immunoreactive band at 90 kDa on Western blots (Fig. 1A). Also, MSK2 expression in these tissues is unaffected, showing that this paralog is unlikely to mask biochemical and behavioral defects in MSK1 knock-out mice (Fig. 1A). Nissl stains of sagittal brain sections from wild-type and MSK1 knock-out animals showed no gross alterations in cortical morphology or hippocampal structure (Fig. 1B,C). This confirms previous results in which there were no obvious developmental brain alterations in these knock-out mice (Brami-Cherrier et al., 2005).
Figure 1.

MSK1 expression and brain morphology. A, MSK1 is highly expressed in the brain and hippocampus in wild-type mice, whereas MSK1 expression is absent in MSK1 knock-out mice. MSK2 expression is equivalent in wild-type and MSK1 knock-out mice. TBP was used as a loading control. B, Nissl stains of sagittal brain sections, showing no difference in brain morphology between wild-type and MSK1 knock-out mice. C, Close-up view of the hippocampus.
MSK1 knock-out mice have a deficit in long-term contextual fear memory
We hypothesized that MSK1, as a target of ERK signaling, would be important for proper associative fear conditioning. In our behavioral paradigm, we trained wild-type and MSK1 knock-out animals by pairing two sets of mild foot shocks with an associated context and an auditory cue. We found that 24 h after the training period, MSK1 knock-out mice exhibited significantly less freezing when reexposed to the context alone, whereas they exhibited less, but not significantly different, levels of freezing after reexposure to the cue in a different context (Fig. 2A). To assess whether the freezing deficit applies specifically to long-term memory, we tested short-term memory by measuring freezing behavior 1 h after training. We found that there were no significant differences in freezing between wild-type and MSK1 knock-out mice 1 h after training (Fig. 2B). This indicates that MSK1 knock-out animals are able to process the sensory stimulus of the foot shock and form an associative memory with both the context and the auditory cue, but have a deficit in 24 h memory.
Figure 2.

MSK1 knock-out mice have a contextual fear conditioning deficit. A, Quantification of freezing behavior in wild-type (n = 19) and MSK1 knock-out mice (n = 13) during training, and contextual and cued tests 24 h post-training. MSK1 knock-out mice displayed significantly less freezing than wild-type mice during the contextual test. B, Quantification of freezing behavior in wild-type (n = 10) and MSK1 knock-out mice (n = 9) during contextual and cued tests 1 h post-training. C, Quantification of freezing behavior in wild-type (n = 12) and MSK1 knock-out mice (n = 11) during contextual and cued tests 7 d post-training. Error bars indicate SEM. An asterisk denotes a significant difference (p < 0.05) as determined by Tukey's multiple-comparison test.
In addition, we tested remote memory by measuring freezing behavior 7 d after training. There were equivalent levels of freezing in wild-type and MSK1 knock-out mice when tested for contextual and cued fear memory after 7 d (Fig. 2C), indicating that the ability to form a remote memory is still intact. Thus, the contextual fear impairment appears to be selective for the 24 h time period.
MSK1 knock-out mice have normal baseline behaviors
As additional control experiments for fear conditioning, we used a battery of baseline behavioral tests to assess anxiety, sensory perception, and motor performance in wild-type and MSK1 knock-out mice. We found that there was no significant difference in anxiety levels in the open field analysis, as measured by the center-to-total distance ratio over a 30 min testing period (data not shown). We found that latencies on the accelerating rotarod task were similar between wild-types and MSK1 knock-outs and that performance improved similarly across eight trials (Fig. 3A), indicating no abnormal motor coordination or cerebellar function. There was no significant difference in nociception or general sensory perception on the hot plate test (data not shown), and shock thresholds to flinching, jumping, and vocalization behaviors were similar (Fig. 3B). Levels of prepulse inhibition were equivalent between wild-type and knock-out mice as a measurement of reflexes, hearing, and the acoustic startle response (Fig. 3C). Finally, swim speeds during the Morris water maze task were similar, and escape latencies in the visible platform test were equivalent (data not shown), indicating no differences in visual perception or motivation. These results show that MSK1 knock-out mice have a normal anxiety, sensory, nociceptive, and motor phenotype that cannot account for the observed difference in long-term contextual fear memory.
Figure 3.

Analysis of select baseline control behaviors in wild-type and MSK1 knock-out mice. A, Plot of latency times spent on rotarod device for wild-type (n = 12) and MSK1 knock-out mice (n = 8) over eight trials. B, Measurement of shock thresholds to flinching, jumping, and vocalization behaviors for wild-type (n = 13) and MSK1 knock-out mice (n = 11). C, Comparison of percent inhibition of acoustic startle response for wild-type (n = 18) and MSK1 knock-out mice (n = 12) with different prepulses. Error bars indicate SEM.
MSK1 knock-out mice have impaired passive avoidance and spatial learning
To further examine whether MSK1 is an important component of hippocampus-dependent memory, we tested wild-type and MSK1 knock-out mice in passive avoidance and the Morris water maze. In passive avoidance testing, animals learn to suppress their natural tendency to retreat from a lighted area to a darker area of the training chamber. We trained animals by placing them into an illuminated chamber and measured the latency with which the animals stepped over to the dark compartment, after which they received a mild foot shock. Twenty-four hours later, after representation into the lighted side, the latency for MSK1 knock-out animals to step over to the dark compartment was increased from the training day, but significantly shorter than for wild-type animals (Fig. 4A).
Figure 4.

MSK1 knock-out mice have impaired passive avoidance and spatial learning. A, Latency times for wild-type (n = 23) and MSK1 knock-out mice (n = 13) to step into the dark compartment of the training chamber in the passive avoidance task. MSK1 knock-out mice have significantly shorter latency time on day 2 after training. B, Plot of latency times to find the escape platform for wild-type (n = 14) and MSK1 knock-out mice (n = 18) over eight trial blocks in the Morris water maze. C, Time spent in each quadrant of the Morris water maze pool for wild-type (n = 14) and MSK1 knock-out mice (n = 18) during the probe trial. MSK1 knock-out mice spent significantly less time in the target quadrant than wild-type mice, lacking a spatially selective search strategy. D, Number of platform crossings in each quadrant for wild-type (n = 14) and MSK1 knock-out mice (n = 18) during the probe trial. MSK1 knock-out mice had significantly fewer platform crossings in the target quadrant than wild-type mice. Error bars indicate SEM. Asterisks denote a significant difference (p < 0.05) as determined by Tukey's multiple-comparison test.
Next, we tested spatial learning with wild-type and MSK1 knock-out animals in the Morris water maze task. In our training paradigm, animals underwent 2 blocks of 4 training trials a day, for a total of 4 d (eight blocks). Using spatial cues in the room, the animals were required to locate a hidden platform in one quadrant of the pool. Over the course of training, the latencies to find the escape platform decreased in a similar manner for both wild-type and MSK1 knock-out animals (Fig. 4B), showing that all animals were able to learn some strategy to find the hidden platform. However, during the probe trial in which the hidden platform was removed, the amount of time MSK1 knock-out animals spent searching in the target quadrant was significantly less than wild-types, and was not significantly different from time spent in other quadrants (Fig. 4C). Similarly, the number of platform crossings in the target quadrant was significantly less than for wild-types, but not significantly different when compared with other quadrants (Fig. 4D). Thus, MSK1 knock-out mice lack a spatially selective search strategy and were unable to locate the precise position of the hidden platform. Together, these results demonstrate that the loss of MSK1 affects several different forms of hippocampus-dependent memory, as measured by fear conditioning, passive avoidance, and the Morris water maze.
MSK1 regulates histone phosphorylation and acetylation after in vitro activation of ERK
We next asked whether the absence of MSK1 results in altered histone regulation in acute hippocampal slices in vitro after pharmacological activation of the ERK signaling pathway. Slices were assayed for histone modifications that signal for either activation or repression of transcription. Phosphorylation of histone H3 (Ser 10), acetylation of histone H3 (Lys 14), and acetylation of histone H4 (Lys 5/8/12/16) have been established as activator markers, whereas dimethylation of histone H3 (Lys 9) serves as a repressor marker (Berger, 2002). We first used forskolin (FSK, 50 μm), a drug that activates the cAMP/protein kinase A (PKA) pathway that leads to ERK activation. The phosphodiesterase inhibitor Ro20–1724 (100 μm) was added along with FSK to prevent breakdown of cAMP. When hippocampal slices were treated with FSK plus Ro20–1724 for 1 h, phosphorylation and acetylation of histone H3 were significantly increased in wild-type slices, but not in MSK1 knock-out slices. There were no significant changes in histone H3 dimethylation or histone H4 acetylation with FSK stimulation (Fig. 5A). A commonly accepted way to inhibit the ERK pathway is to inhibit its upstream kinase, ERK kinase (MEK). We found that preincubation of slices with U0126 (20 μm), a selective inhibitor of MEK, for 10 min followed by FSK blocked the increases in histone H3 phosphorylation and acetylation in wild-type slices, and had no significant effect in MSK1 knock-out slices (Fig. 5A). Levels of total histone H3 protein did not change with any treatment (Fig. 5A). This indicates that ERK activation through cAMP/PKA regulates selective changes in histone H3, and that these changes are mediated by MSK1.
Figure 5.

Regulation of histone modifications after in vitro stimulation of hippocampal slices. A, Quantification of immunoblot densities for phospho-histone H3 (P-H3), acetyl-histone H3 (Ac-H3), dimethyl-histone H3 (Di-Me-H3), and acetyl-histone H4 (Ac-H4). Treatment of wild-type hippocampal slices (n = 7) with FSK (50 μm) plus Ro20–1724 (100 μm) for 1 h significantly increased H3 phosphorylation and acetylation, whereas preincubation with U0126 (20 μm) for 10 min followed by FSK blocked these changes. Treatment of MSK1 knock-out hippocampal slices (n = 7) with FSK plus Ro20–1724 for 1 h did not result in any significant changes in histone modifications, and preincubation with U0126 for 10 min followed by FSK had no significant effect. Total histone H3 was unchanged. Representative immunoblots for P-H3, Ac-H3, and total H3 are shown for FSK treatments. B, Quantification of immunoblot densities for phospho-histone H3, acetyl-histone H3, dimethyl-histone H3, and acetyl-histone H4. Treatment of wild-type hippocampal slices (n = 8) with PDA (3 μm) for 1 h significantly increased H3 phosphorylation and acetylation, whereas preincubation with U0126 (20 μm) for 10 min followed by PDA blocked these changes. Treatment of MSK1 knock-out hippocampal slices (n = 8) with PDA for 1 h did not result in any significant changes in histone modifications, and preincubation with U0126 for 10 min followed by PDA had no significant effect. Total histone H3 was unchanged. Representative immunoblots for P-H3, Ac-H3, and total H3 are shown for PDA treatments. Drug-treated and vehicle-treated slices for each condition came from one individual animal. All drug-treated slices were compared with vehicle-treated (DMSO) controls and normalized to total protein loaded. Error bars indicate SEM. Asterisks denote significant differences (p < 0.05) as determined by Tukey's multiple-comparison test.
We also performed experiments using phorbol 12,13-diacetate (PDA; 3 μm), a drug that activates the protein kinase C (PKC) pathway leading to ERK activation. In a similar manner, treatment of acute hippocampal slices with PDA led to significant increases in histone H3 phosphorylation and acetylation in wild-type slices, but not in MSK1 knock-out slices. There were no significant changes in histone H3 dimethylation or histone H4 acetylation with PDA stimulation (Fig. 5B). Preincubation of slices with U0126 followed by PDA blocked the changes seen in histone H3 in wild-type slices, and had no significant effect in MSK1 knock-out slices (Fig. 5B). Levels of total histone H3 were unchanged (Fig. 5B). These data demonstrate that pharmacological activation of ERK through cAMP/PKA or PKC regulates selective changes in histone H3, and that MSK1 is a significant effector kinase downstream of ERK that mediates these changes.
MSK1 regulates histone phosphorylation and acetylation after fear training
To further support these findings, we asked whether the regulation of histone modifications in the hippocampus is altered in MSK1 knock-out mice with fear training. After our training paradigm (two pairs of mild foot shocks with an associated context and auditory cue), the hippocampi of wild-type and MSK1 knock-out animals were removed and assayed for histone modifications that serve as activator or repressor markers. We found that 1 h after training, phosphorylation and acetylation of histone H3 in the hippocampus were significantly increased in wild-type mice, although these modifications were not significantly changed in MSK1 knock-out mice (Fig. 6A). Dimethylation of H3 and acetylation of H4 were not significantly changed with training in either group of animals (Fig. 6A). Levels of total histone H3 were unchanged (Fig. 6A). Thus, the loss of MSK1 results in a selective deficiency in two markers of transcriptional activation. When we measured phosphorylation of ERK in the hippocampus 1 h after fear training, we found that there was a significant increase in ERK phosphorylation in both wild-type and MSK1 knock-out mice (Fig. 6B), showing that the activation of ERK signaling upstream of MSK1 is intact. There was no change in levels of total ERK (Fig. 6B). This indicates that the deficiency in histone phosphorylation and acetylation seen with MSK1 knock-out animals is not attributable to a loss of training-induced ERK activation.
Figure 6.
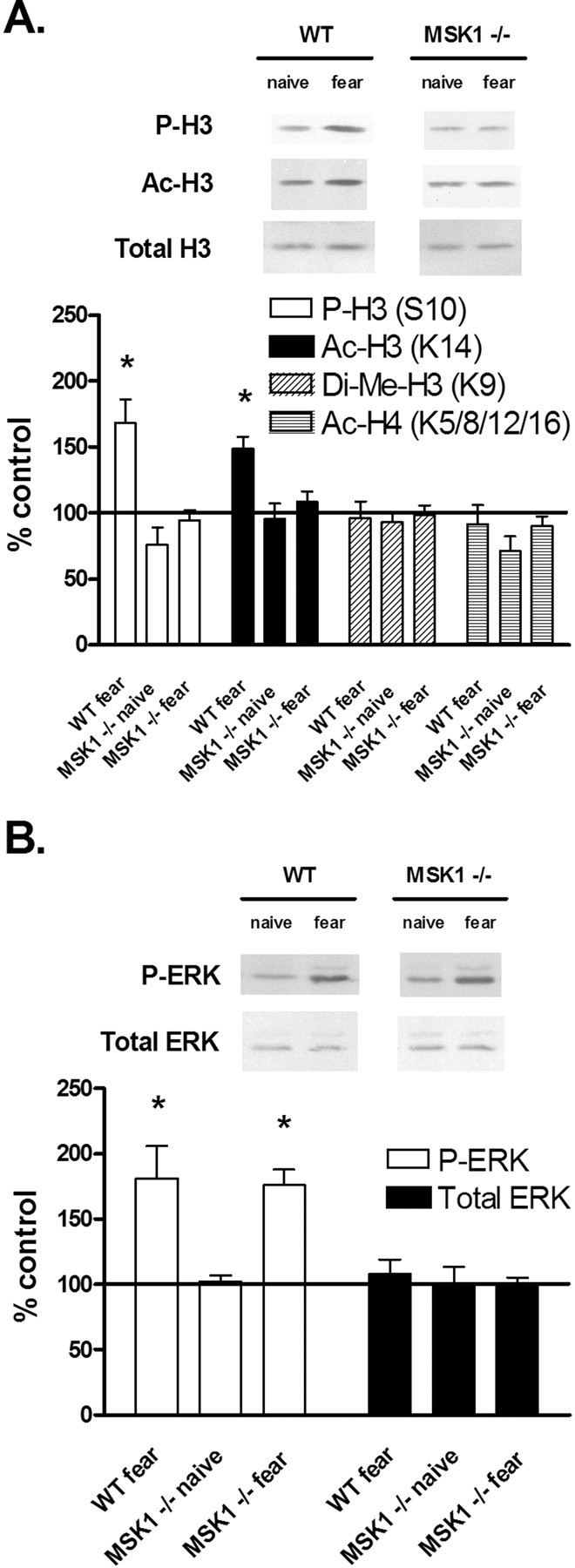
Regulation of histone modifications and ERK phosphorylation after fear conditioning training. A, Quantification of immunoblot densities for phospho-histone H3 (P-H3), acetyl-histone H3 (Ac-H3), dimethyl-histone H3 (Di-Me-H3), and acetyl-histone H4 (Ac-H4). Fear conditioning (2 pairs of tone and shock) in wild-type mice (n = 11) resulted in a significant increase in H3 phosphorylation and acetylation 1 h after training, whereas training in MSK1 knock-out mice (n = 11) did not result in significant changes in histone modifications. Total histone H3 was unchanged. Representative immunoblots for P-H3, Ac-H3, and total H3 are shown for fear-conditioned animals. B, Quantification of immunoblot densities for phospho-ERK (P-ERK) and total ERK. Fear conditioning (2 pairs of tone and shock) resulted in a significant increase in ERK phosphorylation 1 h after training for both wild-type mice (n = 11) and MSK1 knock-out mice (n = 11). Total ERK was unchanged. Representative immunoblots for P-ERK and total ERK are shown for fear-conditioned animals. Each sample came from one individual animal. All samples were compared with naive controls and normalized to total protein loaded. Error bars indicate SEM. Asterisks denote significant differences (p < 0.05) as determined by Tukey's multiple-comparison test.
An inhibitor of histone deacetylase fails to compensate for the fear Conditioning deficit in MSK1 knock-out mice
Thus far, we have observed hippocampus-dependent memory deficits as well as a biochemical deficit in histone phosphorylation and acetylation in MSK1 knock-out mice. We next hypothesized that these deficits might be alleviated by administration of a HDAC inhibitor, sodium butyrate. Previous studies have established that inhibition of HDAC activity can help promote transcriptional activation, induce growth arrest in cell proliferation models of cancer, and ameliorate the effects of depression and neurodegenerative diseases such as Huntington's (Ferrante et al., 2003; Schroeder et al., 2007). Interestingly, sodium butyrate injection in wild-type animals was found to enhance the formation of long-term memory (Korzus et al., 2004; Levenson et al., 2004), and inhibition of HDAC activity also enhanced long-term potentiation in vitro (Levenson et al., 2004). Therefore, we investigated whether the administration of sodium butyrate in MSK1 knock-out mice leads to increased histone acetylation in the hippocampus, and whether the long-term fear memory deficit could be ameliorated in these animals.
We performed high-dose intraperitoneal injections of sodium butyrate (NaB; 1.2 g/kg) in wild-type and MSK1 knock-out mice, and measured histone acetylation in the hippocampus after 1 h. We found that in both wild-type and MSK1 knock-out animals, there was a significant increase in histone H3 acetylation compared with saline-injected controls, confirming the efficacy of the treatment (Fig. 7A). There was no change in levels of total histone H3 (Fig. 7A). We then performed injections of NaB 1 h before fear training (two pairs of shocks with context and auditory cue) in wild-type and MSK1 knock-out mice. We found that NaB had no significant effect on freezing behavior during training for either group of animals (Fig. 7B). However, during the contextual test 24 h after training, freezing was increased in NaB-injected wild-type mice compared with saline-injected controls, whereas freezing in MSK1 knock-outs was unchanged (Fig. 7C). Similarly, during the cued test 24 h after training, injection with NaB enhanced freezing behavior in wild-type mice compared with injection with saline, whereas there was no change in freezing behavior in MSK1 knock-out mice (Fig. 7D). Interestingly, the NaB injection experiments revealed statistically significant deficits in both contextual and cued fear conditioning in MSK1 knock-out animals compared with wild-types (compare Figs. 2A, 7C,D). When measured 1 h after training, all animals displayed equivalent levels of freezing whether injected with NaB or saline, indicating that NaB has no effect on short-term memory (data not shown).
Figure 7.

NaB enhances fear memory in wild-type mice, but not in MSK1 knock-out mice. A, Representative immunoblots for Ac-H3 and total H3 are shown 1 h after NaB treatment compared with saline. Histone H3 acetylation increased in wild-type and MSK1 knock-out animals with NaB compared with saline-injected controls. Each sample came from one individual animal. B, Quantification of freezing behavior in wild-type (n = 12) and MSK1 knock-out mice (n = 18) during training. NaB had no significant effect on freezing in either group compared with saline. C, Quantification of freezing behavior in wild-type (n = 12) and MSK1 knock-out mice (n = 18) during the contextual test 24 h post-training. With saline injection, MSK1 knock-out mice displayed significantly less freezing than wild-type mice. However, with NaB injection, freezing in wild-type animals was enhanced, whereas freezing in MSK1 knock-out animals was unchanged. D, Quantification of freezing behavior in wild-type (n = 12) and MSK1 knock-out mice (n = 18) during the cued test 24 h post-training. E, Quantification of freezing behavior in wild-type (n = 8) and MSK1 knock-out mice (n = 11) during the contextual test 7 d post-training. F, Quantification of freezing behavior in wild-type (n = 8) and MSK1 knock-out mice (n = 11) during the cued test 7 d post-training. Error bars indicate SEM. Asterisks denote significant differences (p < 0.05) as determined by Tukey's multiple-comparison test.
The effects of NaB on remote fear memory were examined by performing injections of NaB or saline before training, and measuring freezing behavior 7 d later. For the contextual test, we found that NaB injection led to enhanced freezing in wild-type mice after 7 d, whereas there was no change in freezing in MSK1 knock-out mice (Fig. 7E). Likewise, for the cued test, NaB injection resulted in greater freezing in wild-type mice after 7 d, whereas there was no change in freezing in MSK1 knock-out mice compared with saline-injected controls (Fig. 7F).
These experiments establish several findings. First, inhibition of HDAC activity in wild-type animals in vivo enhanced long-term contextual and cued fear memory, as expected from previous results (Korzus et al., 2004; Levenson et al., 2004). Second, inhibition of HDAC activity in MSK1 knock-out animals in vivo did not significantly change long-term contextual and cued fear memory. This suggests that histone acetylation is not the only critical target of MSK1, as the impairment in hippocampus-dependent memory formation in MSK1 mutants cannot be alleviated by HDAC inhibition.
MSK1 knock-out mice have impaired CREB phosphorylation after fear training and HDAC inhibition
MSK1 has been identified previously as a major CREB kinase in the hippocampus (Sindreu et al., 2007). Given that the efficacy of HDAC inhibition depends on normal activity of CREB and CREB-binding protein (CBP) (Vecsey et al., 2007), a possible reason why fear memory was not rescued in MSK1 mutants is because CREB activation was still impaired. In a final set of experiments, we investigated the activation of CREB signaling in MSK1 knock-out mice. We asked whether the administration of sodium butyrate can affect CREB phosphorylation levels, and whether the regulation of CREB phosphorylation is altered after fear training and HDAC inhibition.
We trained wild-type and MSK1 knock-out mice (two pairs of cue and shock) and assayed for CREB phosphorylation in the hippocampus using nuclear extracts. We found that 1 h after training, CREB phosphorylation was significantly increased in wild-type mice, although it was not significantly changed in MSK1 knock-out mice (Fig. 8A). Levels of total CREB were unchanged (Fig. 8A). This is consistent with previous results in which fear training induces the activation of both MSK1 and CREB in hippocampal neurons (Sindreu et al., 2007). We then performed intraperitoneal injections of sodium butyrate (1.2 g/kg) in wild-type and MSK1 knock-out mice, and measured CREB phosphorylation in the hippocampus after 1 h. We found that there was no change in CREB phosphorylation in either wild-type or MSK1 knock-out animals with NaB injection compared with saline-injected controls (Fig. 8B). There was no change in levels of total CREB (Fig. 8B).
Figure 8.

Regulation of CREB phosphorylation after fear conditioning training and HDAC inhibition. A, Quantification of immunoblot densities for phospho-CREB (P-CREB) and total CREB. Fear conditioning (2 pairs of tone and shock) in wild-type mice (n = 6) resulted in a significant increase in CREB phosphorylation 1 h after training, whereas training in MSK1 knock-out mice (n = 6) did not result in significant changes in CREB phosphorylation. Total CREB was unchanged. Representative immunoblots for P-CREB and total CREB are shown for fear-conditioned animals. B, Representative immunoblots for phospho-CREB and total CREB are shown 1 h after NaB treatment compared with saline. There was no change in CREB phosphorylation in either wild-type or MSK1 knock-out animals with NaB compared with saline-injected controls. C, Quantification of immunoblot densities for phospho-CREB and total CREB. In wild-type mice (n = 6), fear conditioning resulted in increased CREB phosphorylation in both saline- and NaB-treated animals. In MSK1 knock-out mice (n = 6), training did not result in significant changes in CREB phosphorylation in both saline- and NaB-treated animals. Thus, the deficit in CREB phosphorylation persists in MSK1 knock-out mice even after administration of NaB. Representative immunoblots for P-CREB and total CREB are shown for each treatment. Each sample came from one individual animal. All samples were compared with naive controls and normalized to total protein loaded. Error bars indicate SEM. Asterisks denote significant differences (p < 0.05) as determined by Tukey's multiple-comparison test.
Finally, we performed injections of NaB 1 h before fear training in wild-type and MSK1 knock-out mice. We found that CREB phosphorylation was significantly increased in wild-type animals in both saline- and NaB-treated groups (Fig. 8C). However, there was no significant change in CREB phosphorylation in MSK1 knock-out animals in both saline- and NaB-treated groups (Fig. 8C). Levels of total CREB were unchanged (Fig. 8C). These data demonstrate that the absence of MSK1 leads to a deficiency in CREB phosphorylation with fear training, and this deficiency persists even after administration of the HDAC inhibitor sodium butyrate.
Discussion
This study shows that MSK1 knock-out mice have a deficiency in histone regulation and hippocampus-dependent memory formation, and inhibition of HDAC activity in vivo fails to ameliorate the fear memory deficit in these animals. Furthermore, trained MSK1 knock-out mice have an impairment in CREB phosphorylation that persists after HDAC inhibition. These results establish MSK1 as a key regulator of chromatin structure in hippocampal neurons, and a critical effector kinase downstream of ERK. This suggests a model in which MSK1, as a final component of the pathway, is a juncture point at which the activity of cell surface receptors (NMDA, AMPA, various neurotransmitters), PKC, cAMP/PKA, MEK, and ERK is coupled with chromatin remodeling in the nucleus (Fig. 9). This model is consistent with recent studies in which the coactivation of PKA, ERK/MAPK, MSK1, and CREB was found to depend on Ca2+-stimulated adenylyl cyclase activity (Sindreu et al., 2007). Thus, it appears that MSK1 plays a complex role in regulating transcription: MSK1 mediates the readout of ERK signaling, and targets chromatin as well as CREB and other transcription factors.
Figure 9.

Model of MSK1 in chromatin regulation in memory formation. The activity of cell surface receptors is coupled with the activation of PKC, PKA, MEK, ERK, and finally MSK1. MSK1 regulates a diverse set of targets in the nucleus, including histones (phosphorylation and acetylation), transcription factors (e.g., CREB), and HATs, leading to upregulation of gene transcription. GLU, Glutamate; DA, dopamine; 5HT, 5-hydroxy-tryptophan; NMDA-R, NMDA receptor; AMPA-R, AMPA receptor; NT-R, neurotransmitter receptor; AdC, adenylyl cyclase.
Our behavioral assays demonstrate that MSK1 contributes to several forms of hippocampus-dependent memory. We found that MSK1 knock-out animals have a deficiency in contextual fear conditioning, but not cued fear conditioning, and display deficits in passive avoidance and spatial learning. Furthermore, the contextual fear deficit is present at 24 h, but not at 1 h or 7 d. These results indicate that there is a dissociation between the contextual and cued variants of the test with respect to MSK1, and there is a specific time window over which the fear impairment is manifested. Although fear responses to both contextual and cued stimuli require the integrity of the amygdala, contextual conditioning is more sensitive to hippocampal function (Phillips and LeDoux, 1992; Kim et al., 1993). This strongly suggests that MSK1 plays a more prominent role in hippocampus-dependent rather than amygdala-dependent forms of learning. In addition, the temporal dissociation of contextual fear in MSK1 knock-outs suggests that MSK1 functions critically in the 24 h time period during which gene transcription is known to occur in the hippocampus (Levenson et al., 2004). All of these data implicate the hippocampus as an essential site for MSK1 function in the mammalian brain.
We found that MSK1 mutants lack regulation of histone H3 phosphorylation and acetylation, and the loss is not attributable to a loss of ERK activation upstream. These results support the hypothesis that histone phosphorylation is catalyzed by MSK1 in hippocampal neurons. The identity of the histone H3 kinase has been debated at some length, with MSK1 and ribosomal S6-kinase-2 (RSK2) being the primary candidates. It has been reported that RSK2 phosphorylates histone H3 from analysis of Coffin–Lowry cells (Sassone-Corsi et al., 1999), although in vitro assays with purified RSK2 showed that the kinase failed to phosphorylate histone H3 (Strelkov and Davie, 2002). Later studies from mouse embryonic fibroblasts demonstrated that MSK1 is indeed the kinase that phosphorylates histone H3 (Soloaga et al., 2003). However, these and the present study suggest a more complex picture of kinase regulation. For example, how does MSK1 achieve specificity in targeting a group of histone proteins? It is possible that MSK1 phosphorylates histones indiscriminately as part of a general metaplastic signal, or binds to specific target sequences or nuclear factors to affect transcription of a unique subset of genes. Also, what is the role of RSK2, Aurora, and other kinases? These may certainly phosphorylate histones at different residues or at different locations within the genome, in keeping with a “histone code” hypothesis of memory formation (Wood et al., 2006).
However, the regulation of histone acetylation is accomplished by the presence of histone acetyltransferases (HATs), complexes that directly catalyze acetylation (Fig. 9). Several families of HATs have been identified, including Gcn5, a transcriptional coactivator, and p300/CBP (Brownell et al., 1996; Ogryzko et al., 1996; Reifsnyder et al., 1996). Histone acetylation has generally been associated with upregulation of gene transcription: several coactivator complexes have intrinsic histone acetyltransferase activity, for example mediated by Gcn5 and CBP. We found that MSK1 regulates histone acetylation as a downstream effector of the ERK pathway. MSK1 may thereby target a subset of these HATs, achieving regulatory control of histone acetylation and thereby priming chromatin into a transcriptionally active state. In particular, it will be interesting to determine whether MSK1 regulates CBP, because it is the binding partner of CREB. MSK1 might coordinately regulate both CREB and CBP, or, alternately, CBP might be regulated independently of MSK1. The most parsimonious model is the first scenario, but the second model cannot be excluded at present based on available data.
HDAC inhibitors function in general to upregulate transcription by inducing histone hyperacetylation (Van Lint et al., 1996). Sodium butyrate has been shown to significantly upregulate histone acetylation in the brain, and has been investigated as a possible treatment for disorders such as depression and Huntington's disease (Ferrante et al., 2003; Schroeder et al., 2007). Our experiments show that a single sodium butyrate injection robustly enhanced long-term and remote fear memory in wild-type mice, but had no significant effect in MSK1 knock-out mice. Furthermore, sodium butyrate injection had no significant effect on CREB phosphorylation in MSK1 knock-out mice. We interpret this as evidence that MSK1 has multiple parallel targets that contribute to proper consolidation of long-term memory, including both CREB and histone acetylation through HATs. These targets likely affect the transcription of a wide variety of genes that regulate memory formation and plasticity. Our findings are consistent with previous studies demonstrating that the effects of HDAC inhibition are mediated by the CREB:CBP transcriptional complex (Vecsey et al., 2007). One caveat to this interpretation is that sodium butyrate, although a broad spectrum HDAC inhibitor, may not target the relevant HDAC isoform necessary to reverse the effects of loss of MSK1. Furthermore, the histone type whose acetylation contributes to memory enhancement after HDAC inhibition may be different from histone H3. Regardless, selective amelioration of defects in histone acetylation is unable to alleviate the memory defect in our fear paradigm. This supports a combinatorial requirement for histone modifications as well as the necessity of intact CREB signaling in memory formation.
Our findings support the general hypothesis that there is an epigenetic “histone code” for memory formation (Wood et al., 2006). Multiple post-translational histone modifications may be integrated together, combinatorially driving gene expression patterns by remodeling the structure of chromatin or by recruiting signaling complexes (Jenuwein and Allis, 2001). Thus, there is a mapping of multiple histone alteration states onto subsets of genes that are transcribed as a result of those changes. We have shown that the ERK/MAPK cascade regulates chromatin structure in the hippocampus, and MSK1 is a key mediator of the effects of ERK activation. Interestingly, histone H3 phosphorylation at Ser 10 and acetylation at Lys 14, both transcriptional activation markers, are significantly increased with fear training; however, histone H4 acetylation at Lys 5/8/12/16, also an indicator for activation, is unchanged. At a minimum, this shows that patterns of histone regulation are specific for the type of modification as well as the residue on which it occurs. In addition, overall patterns of histone modifications do not necessarily reflect those at the single-gene level. However, we hypothesize based on our findings that MSK1 regulates histone phosphorylation and acetylation as part of a “histone code” that is interpreted as a pattern of gene transcription specific to hippocampus-dependent memory.
Our results also support the idea that chromatin acts as a signal integration platform (Schreiber and Bernstein, 2002). Each of the core histones contains N-terminal tails on which the majority of histone modifications take place (Peterson and Laniel, 2004). Each modification may be targeted by separate, independent signaling pathways that converge after reaching the nucleus, and chromatin itself may serve as a scaffold on which disparate signals are integrated. We have shown that phosphorylation and acetylation of histone H3 are targeted by ERK and MSK1 in hippocampal neurons; however, studies in cultured embryonic fibroblasts have also implicated the p38 MAPK and SAPK2 pathways in targeting histone phosphorylation and acetylation (Thomson et al., 1999). Both mitogen-activated and stress-activated pathways may target histones independently, and their effects on transcriptional output may be additive, redundant, or even antagonistic. additional work will be needed to determine precisely what other signaling pathways may be involved, and how their relative contributions in histone regulation are integrated to produce a coordinated transcriptional output.
In summary, we demonstrated in this study that MSK1 regulates hippocampal chromatin remodeling in memory formation. MSK1 knock-out mice display deficits in long-term fear conditioning, passive avoidance, and spatial learning, and show abnormal regulation of histones both in vivo and in hippocampal slices in vitro. The fear deficit is not alleviated by treatment with the histone deacetylase inhibitor sodium butyrate. Furthermore, MSK1 knock-out mice exhibit a deficit in CREB activation after fear training and sodium butyrate treatment. We conclude that MSK1 plays a significant role in the hippocampus as a nuclear kinase downstream of ERK signaling.
Footnotes
This work was supported by grants from the National Institutes of Health (MH57014, NS37444, NS13546), National Alliance for Research on Schizophrenia and Depression, the American Health Assistance Foundation, and the Evelyn F. McKnight Brain Research Foundation (J.D.S.). W.B.C. was supported in part by grants from the Baylor College of Medicine Medical Scientist Training Program, the C. Thomas Caskey Scholarship Fund, and the Chinese American Doctors Association of Houston. We acknowledge Farah D. Lubin, Susan Campbell, Felecia Hester, Agnieszka E. Mlodnicka, Sadia Waheed, Se Y. Kim, and David Shim for advice and technical support.
References
- Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem. 2000;75:2277–2287. doi: 10.1046/j.1471-4159.2000.0752277.x. [DOI] [PubMed] [Google Scholar]
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- Blum S, Moore AN, Adams F, Dash PK. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Herve D, Darragh J, Corvol JC, Pages C, Arthur SJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- Chwang WB, O'Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin M, Segal M. Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur J Neurosci. 2003;17:2529–2539. doi: 10.1046/j.1460-9568.2003.02694.x. [DOI] [PubMed] [Google Scholar]
- Guan Z, Guistetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Rison RA, Fanselow MS. Effects of amygdala, hippocampus, and periaqueductal gray lesions on short- and long-term contextual fear. Behav Neurosci. 1993;107:1093–1098. doi: 10.1037//0735-7044.107.6.1093. [DOI] [PubMed] [Google Scholar]
- Kim SY, Levenson JM, Korsmeyer S, Sweatt JD, Schumacher A. Developmental regulation of Eed complex composition governs a switch in global histone modification in brain. J Biol Chem. 2007;282:9962–9972. doi: 10.1074/jbc.M608722200. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14:R546–551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Reifsnyder C, Lowell J, Clarke A, Pillus L. Yeast SAS silencing genes and human genes associated with AML and HIV-1 Tat interactions are homologous with acetyltransferases. Nat Genet. 1996;14:42–49. doi: 10.1038/ng0996-42. [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285:886–891. doi: 10.1126/science.285.5429.886. [DOI] [PubMed] [Google Scholar]
- Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Sindreu CB, Scheiner ZS, Storm DR. Ca2+-stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron. 2007;53:79–89. doi: 10.1016/j.neuron.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelkov IS, Davie JR. Ser-10 phosphorylation of histone H3 and immediate early gene expression in oncogene-transformed mouse fibroblasts. Cancer Res. 2002;62:75–78. [PubMed] [Google Scholar]
- Swank MW, Sweatt JD. Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J Neurosci. 2001;21:3383–3391. doi: 10.1523/JNEUROSCI.21-10-03383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Hawk JD, Abel T. Combinatorial chromatin modifications and memory storage: a code for memory? Learn Mem. 2006;13:241–244. doi: 10.1101/lm.278206. [DOI] [PMC free article] [PubMed] [Google Scholar]