

Abstract
A link between chromosome translocations and cancer has been recognized for several decades, as translocations sometimes generate fusion genes that deregulate normal cell growth. Fusion genes with oncogenic potential have now been described in many hematological and solid malignancies. Sequencing approaches have confirmed that numerous, non-clonal translocations are a typical feature of cancer cells, and have also revealed that many chromosome rearrangements are highly complex and contain sequence from multiple genomic sites. The factors and pathways that promote translocations are becoming clearer, with nonhomologous end-joining implicated as a major source of chromosome rearrangements.
Main text
Genomic instability, comprising sequence changes and chromosome rearrangements is considered to be the most prominent mechanism leading to the appearance of cancer1. Much research has therefore focused on the nature of the mutations that are present in cancer cells, and on the processes that lead to their appearance. While the importance of genetic changes in driving cancer has been appreciated for almost 100 years, recent technological advances have substantially increased our ability to study cancer-associated mutations. Furthermore, by studying DNA repair pathways that normally suppress the genomic instability that leads to mutation, we now understand better why mutations arise, and in several cases how we can manipulate cells to reduce the rate of mutation.
In this Review we focus on how translocations arise, with a particular emphasis on how [G]nonhomologous end-joining causes the appearance of many chromosome rearrangements, including the spectacularly complex chromosome translocations associated with [G]chromothrypsis2.
The nature of translocations
A translocation is an abnormal chromosome region containing rearranged genetic material, usually from two nonhomologous chromosomes (Fig 1 and Box 1). Translocations are not exclusive to cancer cells; screening of cells from developing embryos has revealed that a significant number of embryos of the order of 0.7 per 1000 live births) have cells that contain translocations3, 4. Although these de novo translocations are associated with developmental abnormalities in some cases, many balanced translocations do not cause noticeable pathology, suggesting that balanced translocations are well-tolerated in many instances. Untransformed primary mouse blood cells also contain a wide range of chromosomal translocations5–8. However, there is no question that translocations are particularly common in cancer cells. A large number of translocations have been catalogued9, 10 and are listed in databases such as the Database of Chromosome Rearrangements in Disease (dbCRID; http://dbcrid.biolead.org) and the Catalogue of Somatic Mutations in Cancer (COSMIC; http://www.sanger.ac.uk/genetics/CGP/cosmic/). Although the majority of pathological translocations that contain gene fusions that deregulate cell growth, such as BCR–ABL1, have historically been found in haematological malignancies, a growing number of such mutations have been found in solid tumors9. This is exemplified in prostate cancer, in which at least 40% of cases feature translocations between TMPRSS2 and a gene encoding the ETS transcription factor, ERG11.
Fig 1. Visualizing translocations.
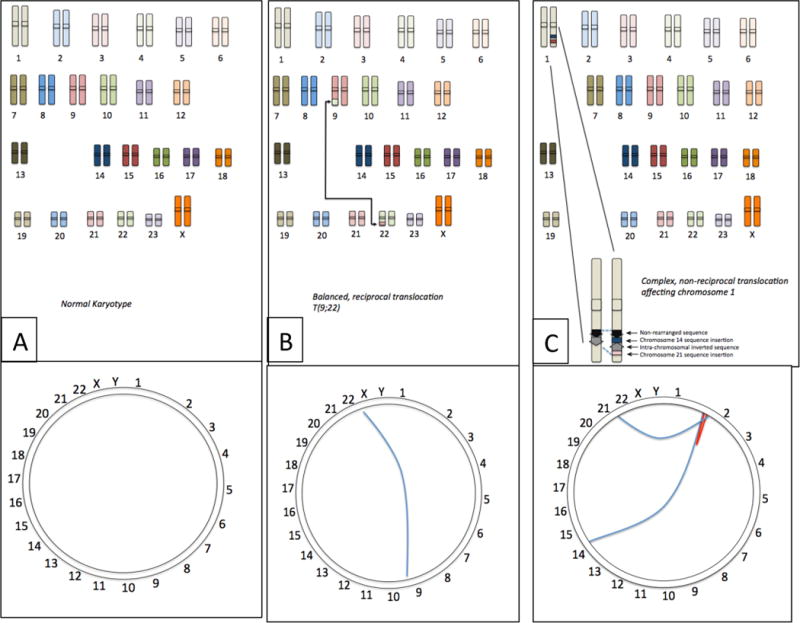
(A) The normal human chromosome set contains no rearrangements between chromosomes. The Circos plot18 shows this as a ring with the uninterrupted sequence of the chromosome running around the circumference. (B) Certain cancer cells contain balanced, reciprocal translocations, which join sequence from different chromosomes, such as the T(9;22) translocation from CLL, which exchanges sequence from chromosomes 9 and 22. Viewed as a Circos plot, this translocation can be visualized as a line connecting the breakpoints of the translocation on chromosomes 9 and 22. (C) Many translocations are more complex rearrangements involving multiple chromosomes. In this example, chromosome 1 contains a rearrangement involving translocated sequence from chromosomes 14 and 21, and an internal sequence inversion. Such complex translocations can be pictured using the Circos plot, where the blue lines indicate interchromosomal translocations and the red line shows the intra-chromosomal inversion.
At a Glance.
Cancer genome sequencing has demonstrated that translocations that fuse sequence from different chromosomes are typical features of cancer cells.
Translocations that create neomorphic fusion genes occur in both lymphoid malignancies and solid tumors.
A large number of translocations do not encode fusion genes and may not contribute to malignancy.
Translocations are frequently complex and involve sequence from multiple chromosomes, similar to ‘chromothrypsis’.
Many translocations arise as a consequence of ‘classical’ or ‘alternative’ pathways of non-homologous end-joining.
Mammalian cells have regulatory systems to bias DNA repair toward repair pathways that are less likely to contribute to translocation.
Frequency of DNA breakage is the metric that best predicts the likelihood of a particular genomic site being involved in a translocation.
Therapeutic intervention to reduce translocation frequency is a potential mechanism for reducing the risk of cancer.
The frequency of recurrent gene fusions varies depending on the specific type of cancer, but currently-known translocations are estimated to drive ~20% of cancer cases9. Next-generation sequencing of genomes and transcriptomes from primary human cancer cells is revealing new gene fusions that may be involved in driving tumorigenesis, including new examples found in colorectal carcinoma, breast cancer and acute lymphoblastic leukemia (ALL)12–14. Nonetheless, sequencing has shown that somatic mutations affecting the sequence of genes are significantly more common than chromosome rearrangements12–17.
Sequencing efforts have also revealed that cancer genomes do not typically contain a discrete number of coherent reciprocal translocations. Tumor cells more commonly contain a large number of complex translocations, featuring inter- and intra-chromosomal rearrangements (Fig 1). In most cases, it is not possible to draw definite conclusions about the mechanism or extent by which any one of these individual translocations contributed to the malignancy of the cancer cell. Furthermore, the frequency and type of translocations are not always shared among tumors of the same class. Sequence data from primary breast cancers showed between zero and 29 translocations per case13. Squamous cell lung cancer cells have a higher rate of translocations, with an average of 165 somatic rearrangements per cell, compared to 98 rearrangements per cell in non-small cell lung carcinoma and 90 rearrangements per genome in prostate tumors15–17. Rearrangements in cancer cells affect genic and non-genic DNA at approximately equal rates; however, a study in prostate cancer cells found an enrichment of rearrangements in transcribed regions, as measured by RNA Pol II ChIP15. A study in breast cancer cells also reported a somewhat elevated rate of rearrangements within the total area (including introns) of protein coding genes13. A minority of translocations form gene fusion events, but translocations may also contribute to tumorigenesis by interrupting the sequence of tumor suppressor genes, as observed for the tumor suppressor TTC28 in certain cases of colorectal carcinoma12. As is the case with mutations affecting gene sequence, many translocations seen in cancer cells are probably bystander mutations as opposed to being drivers of the disease. The number of translocations in tumor cells from a cohort of patients with lung adenocarcinoma was found not to correlate with clinical outcome17. Hence, tumors with a small number of translocations can be more aggressive and more difficult to treat than tumors with many translocations. Other types of mutations in addition to translocations clearly play an important role in driving growth and survival of cancer cells.
The frequency and complexity of cancer-associated translocations has required the development of new bioinformatic tools to analyse and display the huge volume of data that is being generated by cancer genome sequencing projects18 (Fig 1). A case of spectacular genomic rearrangement was revealed in a case of chronic myeloid leukemia (CML), with 42 intrachromosomal rearrangements affecting chromosome 4q19. Such highly complex, clustered translocations are referred to as ‘chromothrypsis’. These rearrangements can affect one or more chromosomes in a cell and are thought to be generated in a single catastrophic event. An initial estimate indicates that as many as 3% of all cancers exhibit such clustered rearrangements20. It is plausible that the same mechanisms cause chromothrypsis as cause the complex translocations seen in other cancer cells2. Complex translocations similar to chromothrypsis have been described, based on sequencing the genomes of cells from prostate cancer and ALL14, 21.
NHEJ as a source of genomic instability
Several pathways have been proposed to be involved in the formation of translocations22 (Fig 2). These pathways include non-homologous end-joining (NHEJ), breakage-fusion-bridge cycles (Box 2 and Fig 3), and replication-based mechanisms, such as [G]break-induced replication (BIR)23. Replication-based mechanisms are proposed to cause translocations by switching of the extending DNA strand from its template sequence to another homologous template during DNA replication, potentially resulting in non-homologous sequence being copied into the new DNA strand24, 25. However, in the absence of an appropriate inducible model, or genetic evidence for the requirement of specific factors in mediating replication-based translocations, it is challenging to quantify the contribution of such pathways to the overall frequency of translocations. Mechanisms based on homologous recombination may also cause translocations, such as [G]non-allelic homologous recombination (NAHR), which has been implicated in chromosome rearrangements that occur in the germline26.
Fig 2. Pathways to translocation.
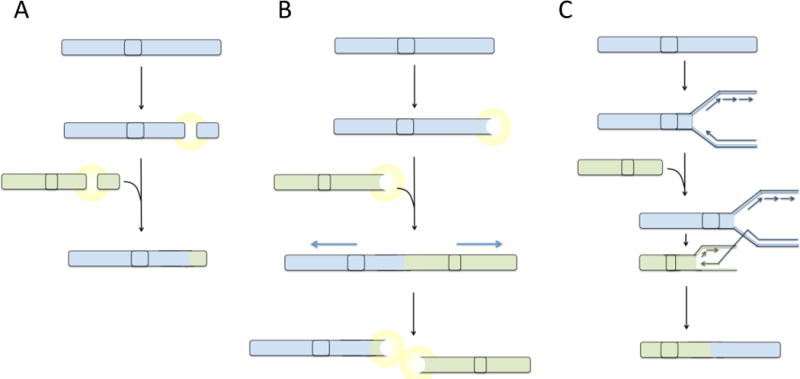
(A) Balanced reciprocal translocations are hypothesized to form as a consequence of fusion of two double-strand breaks that arise in the same cell. Following appearance of double-strand breaks, a signaling pathway is activated, which leads to ligation of the free DNA ends mediated by factors of the non-homologous end-joining pathway. (B) Telomere uncapping or attrition generates a DNA double-strand break response, potentially leading to fusion of telomeres generating end-to-end fusions. During anaphase, dicentric fusion chromosomes are pulled apart leading to the formation of translocations and double-strand breaks. Broken chromosomes act as substrates for additional rounds of fusion and breakage, generating increasingly complex translocations. (C) Hypothetically, translocations could arise by a replication-based mechanism by ‘switching’ of the DNA replication machinery to a site on a different chromosome with some degree of sequence homology to the original template. Extension of the replication fork at a site on a different chromosome would lead to a composite daughter strand being produced, containing sequence from two chromosomes. This composite chromosome would appear as a translocation. Highly complex translocations could be generated by multiple template switching events, generating an aberrant chromosome containing sequence from several different parts of the genome.
Box 1: Types of chromosomal translocations.
The development of techniques for visualizing and staining chromosomes using dyes such as quinacrine and Giemsa led to the identification of translocations starting in the 1950s, and significant disease-causing rearrangements are still being discovered today. A chromosome containing a translocation is termed a ‘derivative chromosome’, and the nature of the rearrangements affecting that chromosome are described by a systematic nomenclature. Robertsonian translocations are those in which the long arms of two acrocentric chromosomes are joined around a single centromeric region. ‘Reciprocal’ translocations describe exchange of genetic material between two chromosome arms. Such translocations can be classified as ‘balanced’ or ‘unbalanced’ depending on whether the translocation affects the copy number of any section of the genome, with a balanced translocation causing no change in overall copy number.
Many well-known pathological translocations fall into the class of apparently-balanced, reciprocal translocations between two non-homologous chromosomes. This group includes the Philadelphia chromosome, t(9;22) (translocation between chromosome 9 and 22); a translocation between chromosomes 11 and 22, t(11;22), seen in 85% of Ewing’s sarcoma; and a translocation of chromosomes 8 and 14, t(8;14), which is seen in 85% of cases of Burkitt’s lymphoma. Translocations such as these promote cancer by deregulating expression of key cellular transcription factors and signalling modulators to cause uncontrolled growth. In addition to the recurrent t(9;22) translocation in chronic myeloid leukaemia, which causes overexpression of ABL1 kinase, the t(11;22) translocation in Ewing’s sarcoma causes deregulated activity of Fli1, an ETS transcription factor, whereas t(8;14) in Burkitt’s lymphoma is a translocation that causes overexpression of the mitogenic MYC transcription factor.
Fig 3. Oncogene Amplification by Breakage Fusion Bridge Cycles.
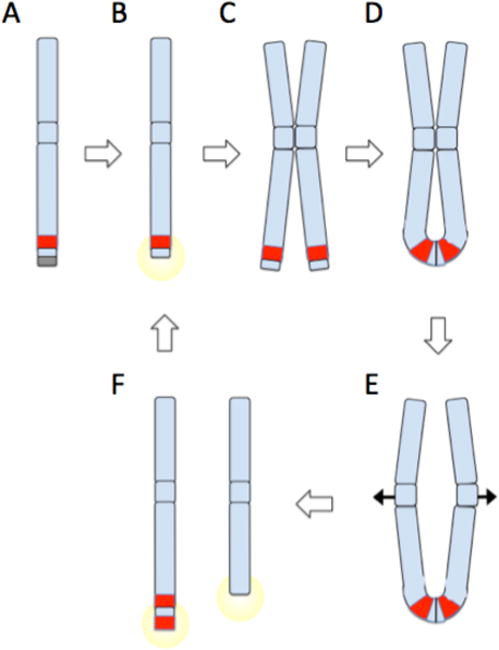
(A) Chromosomes are normally protected by telomeres (gray box). A sub-telomeric oncogene, shown in red, can become amplified by breakage fusion bridge cycles. (B) Telomere loss or double-strand breakage creates an unprotected DNA end, which triggers a DNA damage response. (C) Cancer cells with checkpoint defects will continue to grow despite DNA damage signaling, leading to duplication of the broken chromosome. (D) Ligation of broken chromatid ends produces an ‘anaphase bridge’, with a chromatin connection between the two sister chromatids. (E) As chromatids are drawn apart during anaphase, the anaphase bridge is subjected to increasing stress as centromeres are pulled to opposite poles of the dividing nucleus. Eventually, the anaphase bridge will shear, producing uneven derivative chromosomes as shown in (F). One derivative chromosome may capture sequence including a second copy of the oncogene from the broken sister chromatid. The broken chromosomes can act as substrates for further breakage fusion bridge cycles (B–F), potentially leading to dramatic amplification of oncogenes near telomeric sites. Oncogene amplification is a driver of malignant cell growth. If breakage fusion bridge cycles are combined with fusion of double strand breaks from other chromosomes, complex translocations can be built up featuring sequence from multiple chromosomes.
Translocations, in particular those translocations that generate gene fusions, are often assumed to form because of the joining of DNA double-strand breaks that arise at different sites on non-homologous chromosomes. In this case, the double-strand breaks are joined by an endogenous DNA repair pathway such as NHEJ (Fig 4). In mammalian cells, the best-characterized pathway for nonhomologous end-joining, which has come to be known as ‘classical’ NHEJ or C-NHEJ, initiates by binding of the Ku70 (also known as XRCC6)–Ku80 (also known as XRCC5) heterodimer to broken DNA ends27. DNA-PKcs (also known as PRKDC)–Artemis (also known as DCLRE1C) subsequently binds to the Ku70–K7u80–DNA complex and processes the DNA end through the nuclease activity of Artemis. Finally, a complex of XRCC4-like factor (XLF; also known as NEHJ1)–XRCC4–DNA ligase 4 (LIG4) joins the DNA ends (Fig 4). The importance of the NHEJ pathway in maintaining genomic stability is known from genetic studies in mice28–30 and from individuals with mutations in key NHEJ genes (Table 1).
Fig 4. Steps in classical and alternative end-joining.
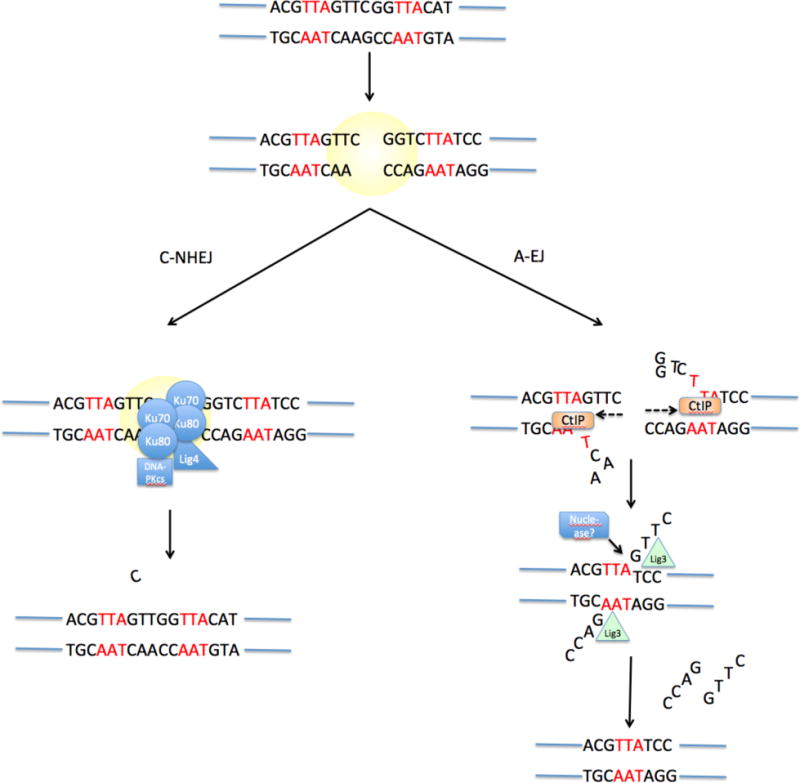
Upon appearance of a DNA double-strand break, two pathways can be active. Classical Non-Homologous End-Joining (C-NHEJ) involves binding of Ku70−Ku80 to the DNA break, followed by recruitment of DNA-PKcs and several other factors that mediate blunt-end ligation of the break by ligase 4 (LIG4). This process has no sequence requirements and may cause small-scale mutation such as the addition or deletion of a small number of nucleotides at the break junction. Alternative End-Joining (‘A-EJ’) involves exonucleolytic processing of the double-strand break to reveal stretches of potentially complementary sequence (microhomology, indicated in red) on either side of the break. This resection process may be mediated by the exonuclease CtIP. Following base-pairing at regions of microhomology, the ends are joined by an undetermined ligase enzyme (LIG).
Table 1.
Phenotypes of loss-of-function NHEJ mutations Bunting and Nusseznweig, 2013.
| NHEJ gene | Mouse knockout phenotype | Patient phenotype |
|---|---|---|
| XRCC6 (encoding Ku70) | Viable, SCID, small size, radiosensitivity, thymoma48, 49 | None known |
| XRCC5 (encoding Ku80) | Viable, SCID, small size, radiosensitivity, genomic instability and tumors especially with p53 deletion.45, 50–52 | None known |
| PRKDC (encoding DNA-PKcs) | Viable, SCID, some genomic instability, tumors with p5353–55 | Human hypomorph has SCID, radiosensitivity56 |
| DCLRE1C (encoding Artemis) | Viable, SCID, radiosensitivity, genomic instability57 | Null is SCID, radiosensitivity. Hypomorph is reduction in lymphocytes, instability, lymphoma58, 59 |
| NHEJ1 (encoding XLF) | Mild lymphocytopenia, radio-sensitivity60 | Cernunnos syndrome; Immunodeficiency, developmental delay, microcephaly, reduced growth, genomic instability61 |
| XRCC4 | Null is lethal with neuronal apoptosis; rescue with p53 is SCID, radiosensitivity, early B lymphoma, genomic instability47, 62 | None known |
| LIG4 | Knockout is lethal, neural apoptosis; rescue with p53 gives pro-B lymphoma, radiosensitivity; hypomorph is small, lymphopenic, reduced hematopoietic stem cell function.63, 64 | Lig4 syndrome; immunodeficiency, reduced growth, developmental issues, microcephaly, malignancy65, 66 |
DCLRE1C, DNA cross-link repair 1C; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; LIG4, DNA ligase 4; NHEJ, non-homologous end-joining; NHEJ1, NHEJ factor 1; PRKDC, protein kinase, DNA-activated, catalytic polypeptide; SCID, severe combined immunodeficiency; XLF, XRCC4-like factor; XRCC, X-ray repair cross-complementing protein.
Despite the appearance of chromosomal translocations in cells that lack NHEJ activity, several lines of evidence suggest that in certain cases, NHEJ contributes to the appearance of chromosomal translocations. Deficiency in RAD18 makes cells hypersensitive to camptothecin, an agent used in certain chemotherapy regimens on the basis of its ability to cause DNA double-strand breaks. However, this hypersensitivity is relieved by suppressing NHEJ31. Mutations in BRCA1, which is required for homologous recombination, or any of the Fanconi anaemia complementation group (FANC) genes, which excise DNA inter-strand crosslinks, predispose affected individuals to cancer owing to the requirement of these factors for normal DNA repair. However, several reports have concluded that ablation of NHEJ factors such as Ku70, Ku80 or LIG4 reduces genomic instability and the appearance of chromosome rearrangements in BRCA1- or FANC-deficient cells32–35. Chemical inhibition of DNA-PKcs has also been reported to reduce genomic instability in cell lines lacking BRCA1 and BRCA236. Collectively, these results suggest that when mammalian DNA repair pathways are defective, the NHEJ pathway can act to increase the amount of genomic instability and therefore accelerate the accumulation of mutations that contribute to cancer.
Genomic sequencing indicates that up to 50% of ovarian carcinoma cells have mutations affecting the homologous recombination pathway, making these cells particularly vulnerable to aberrant DNA repair by NHEJ37. Cells from breast cancer patients have a higher than expected frequency of mutations in FANCC, Bloom’s Syndrome gene (BLM; also known as RECQL3) and XRCC238–40. These genes are essential for error-free repair of DNA damage, hence cells with these mutations may over-use NHEJ for repair, leading to further accumulation of genetic abnormalities. Another form of evidence that NHEJ is important for chromosome translocations has come from study of the genetic requirements for the fusion of uncapped telomeres (Box 2). NHEJ is also considered to be the mechanism underlying the complex pattern of translocations and rearrangements seen in chromothrypsis19. NHEJ therefore plays an important role in shaping the genome of the cancer cell by contributing to error-prone pathways of DNA repair that lead to the appearance of mutation.
Classical versus alternative end-joining pathways
Early studies on the characteristics of end-joining activities in mammalian cells demonstrated the presence of two classes of products: those formed from the simple ligation of DNA ends, and those where small sections of shared sequence identity (microhomology) at the joined ends could be observed41, 42 (Fig 4). Yeast studies support the importance of Ku70–Ku80 for NHEJ, but additionally showed that in the absence of these factors, an alternative activity using microhomology can mediate joining with some deletion of DNA sequence around the break site43–45. Subsequent biochemical data and assays with end-joining substrates with different amounts of terminal homology showed that joining of ends with 6–8 bp of homology is not dependent on the Ku-mediated ‘classical’ pathway for NHEJ (C-NHEJ)46, 47, and instead requires a Ku-independent NHEJ pathway called alternative end joining (A-EJ), or microhomology-mediated end-joining (MMEJ). The existence of A-EJ accounts for translocations and chromosome rearrangements in cells lacking Ku70, Ku80 or LIG429. Notably, mice with targeted knockouts of Ku70-−Ku80, XRCC4 or LIG4, in combination with p53-deficiency, develop tumors with translocations featuring microhomology48, 49. Microhomology was also reported in 85% of translocations induced using a translocation reporter system in mouse embryonic stem (ES) cells50. A-EJ therefore appears to be capable of producing translocations, particularly when ‘classical’ NHEJ is deficient. In a system to measure the frequency of translocations between Igh and Myc in mouse B cells, deletion of Ku70 or LIG4 actually increased the rate of translocation, with A-EJ apparently providing the joining activity51. Translocations were also increased in a reporter system in mouse ES cells when XRCC4−XLF was inactivated52. These results suggest that NHEJ causes a relatively low rate of translocations, but in its absence, A-EJ becomes active and produces an increased number of chromosome rearrangements.
A-EJ is of particular interest because microhomology signatures have been reported at the breakpoints of chromosome rearrangements in primary human cancer cells53, 54. This raises the possibility that A-EJ, or some other microhomology-based mechanism is responsible for the formation of translocations. The amount of microhomology used in repair of a double stranded DNA break is the standard measure for distinguishing between C-NHEJ and A-EJ joining, but it is not clear how much microhomology is optimal for each pathway. Understanding the importance of A-EJ in the formation of translocations will require better characterization of the components of the pathway. The identification of factors that are required for A-EJ in mammalian cells has been aided by studies in yeast, which have suggested that factors such as Mre11, Rad50 and Sae2 are involved in A-EJ53. Studies using translocation reporter constructs in mouse embryonic stem cells have shown that the frequency of translocations between induced double-strand breaks on different chromosomes is reduced after knockdown of CtIP, an exonuclease that is considered the closest mammalian homolog of Sae255. Furthermore, the translocations that do occur show a reduced amount of microhomology at the breakpoints, supporting a role for CtIP in a pathway that produces translocations using microhomology.
A role for CtIP in A-EJ is plausible, according to a model in which limited exonculease resection of DNA double-strand breaks is necessary to uncover stretches of microhomology that can anneal and mediate joining (Fig 4). Intrachromosomal joining assays in mammalian ES cells and cell lines have likewise supported an involvement of Mre11 in Ku70–Ku80–XRCC4-independent end joining using microhomology56, 57. However, although modulation of end resection appears to be a key regulator of A-EJ, data from mouse B cells measuring induced intrachromosomal rearrangments showed no reduction of microhomology-mediated joining in cells after CtIP-knockdown or Mre11 inhibition58. The essential genetic makeup of A-EJ is therefore an ongoing question in the field, and multiple redundant processes may contribute to A-EJ. The DNA end-binding factor, Ku, has recently been suggested to have a key regulatory role in suppressing use of A-EJ, as depletion of human Ku86 was found to increase the use of A-EJ in cells lacking other C-NHEJ factors59
As A-EJ is active in the absence of LIG4 much interest has focused on which of the other two mammalian ligase enzymes, LIG1 or LIG3, is required for the joining of DNA ends in A-EJ. Depletion of either LIG1 or LIG3 reduces the use of microhomology-mediated end joining of cut plasmids in cell-free extracts60. Cells with a specific deficiency in nuclear LIG3 show a reduced frequency of translocations between targeted double-strand breaks on chromosomes 6 and 11, with the small number of remaining translocations showing reduced use of microhomology61. This supports a role of LIG3 for mediating A-EJ translocations in mammalian cells, and suggests that A-EJ is active even when all the factors of the ‘classical’ NHEJ pathway are present. This study further demonstrated that LIG1 can act as a backup ligase for LIG3 in A-EJ, because depletion of both LIG3 and LIG1 together reduces translocations to a lower rate than that seen in nuclear LIG3-deficienct cells. However, data from conditional deletion of XRCC1, the co-factor of LIG3, in B lymphocytes has produced conflicting data regarding the importance of LIG3 in A-EJ62. In B cells with deficiencies in C-NHEJ, deletion of XRCC1 or knockdown of LIG3 had no effect on translocations between MYC and the IgH locus (IGH). The relative importance of LIG3 and LIG1 in A-EJ is therefore still somewhat unclear.
Although A-EJ is an important pathway for formation of translocations, several lines of evidence suggest that conventional NHEJ still accounts for the majority of rearrangements. First, the measured amount of microhomology found in translocation reporter cell lines is quite low, with a mean of 1.36 bp50. Second, in two different inducible systems that generate experimental inter-chromosomal translocations, microhomology-mediated joining was observed in a minority of cases63, 64. Third, data from next generation sequencing projects using human cancer patients indicate a minor role for A-EJ. For example, one recent study used next-generation sequencing technology to characterize the breakpoints of 52 germline chromosomal rearrangements from human patients65. The majority of these rearrangements were thought to be balanced translocations. However, at the molecular level they almost invariably featured deletion of genetic sequence at the breakpoint junction. A significant number of the translocations were not formed by the simple joining of DNA breaks, but involved local fragmenting of the DNA with reassembly of inverted local sequence at the final translocation join. Of the 141 breakpoints identified through next generation sequencing, just 30.5% had regions of microhomology. In addition, chromosome rearrangements in prostate cancer cells usually do not contain microhomology sequences, and in breast cancer cells, microhomology is generally either absent or limited to 2bp or less13, 21. These findings suggest that microhomology-based mechanisms are responsible for a minority of de novo human translocations and conventional NHEJ is the primary pathway for formation of chromosome translocations.
Regulation of choice of HR and NHEJ in mammalian cells
C-NHEJ is the only double-strand break repair pathway that can join DNA ends with no homology at the repair site. Furthermore, C-NHEJ acts at blunt or minimally processed DNA ends, whereas some degree of resection of the double-strand break is required for HR, single strand annealing (SSA) and A-EJ. The regulation of double-strand break resection therefore acts as the key determinant in committing repair of a double-strand break to C-NHEJ or a homology-based pathway66, 67. One potential method for regulation of resection is kinetic: resection only proceeds after initial attempts at NHEJ of double-strand breaks have failed. This hypothesis has been supported by multiple lines of evidence using immunofluorescence to detect the accumulation of repair factors at break sites, plasmid rejoining assays and reporter constructs68–70. NHEJ seals DNA breaks with minor nucleotide deletions and additions at the breakpoint, and is capable of joining DNA breaks on different chromosomes. It is therefore surprising that cells use ‘quick and dirty’ repair by NHEJ instead of the slower, more accurate repair by HR. HR, which is template-based and much less error-prone might be expected to be the preferred pathway for faithful repair of double-strand breaks.
Resection of a DNA double-strand break is initiated by Mre11, as part of a complex with Rad50–Nbs1 (MRN) complex, or, with Xrs2 in yeast cells (known as the MRX complex). Resection becomes extensive following the action of CtIP. EXO1 and BLM–DNA2 have also been reported to generate single-stranded DNA overhangs at break sites in mammalian cells. Cyclin-dependent kinase (CDK) signalling regulates the activity of the resection apparatus, such that it is mainly active in the S and G2 phases of the cell cycle. Nonetheless, components of the NHEJ pathway remain active in S-phase and G2-phase cells and compete with HR for repair of double-strand breaks69, 71. Extensive resection can also occur in G1, at least in the absence of 53BP1 and H2AX72, 73. Thus, in addition to CDK activity, other levels of regulation must be present to ensure use of error-free HR versus mutagenic NHEJ.
One important regulatory mechanism for DNA end resection in mammalian cells is mediated bythe DNA damage response factor, p53 binding protein 1 (53BP1). 53bp1−/− cells are mildly sensitive to ionizing radiation compared to other cells with deficiencies in the NHEJ pathway, but 53bp1−/− B cells have a marked defect in their ability to mediate class switch recombination74, 75. Several lines of evidence suggest that 53BP1 may act to repress HR through blocking resection33, 34, 58, 73, 76–78. Whether 53BP1-mediated blocking of resection acts to achieve rapid repair of breaks at the expense of potential mutagenicity, or evolved to enable repair of induced double-strand breaks during the assembly of antigen receptor genes is not clear. Several recent reports have shown that 53BP1 inhibits resection of DNA double-strand breaks by recruiting the DNA damage response factor Rif179–83. Rif1 binds to sites within 53BP1 that are phosphorylated by the damage-response kinase, ATM. 53BP1–Rif1 represses the recruitment of BRCA1 to DNA damage sites in the G1 phase of the cell cycle, whereas BRCA1, in coordination with CtIP, prevents accumulation of Rif1 at break sites during S phase and G2 (Fig 5). In the absence of Rif1, cells accumulate genomic instability and a higher frequency of IGH–MYC translocations, but as seen previously with 53BP1, end-to-end fusions of deprotected telomeres are reduced in the absence of Rif1. These findings demonstrate the complex regulation of double strand break repair pathway choice in mammalian cells, and reinforce the idea that proper choice is essential to maintain genome integrity.
Fig 5. Regulation of DNA double-strand break repair pathways.
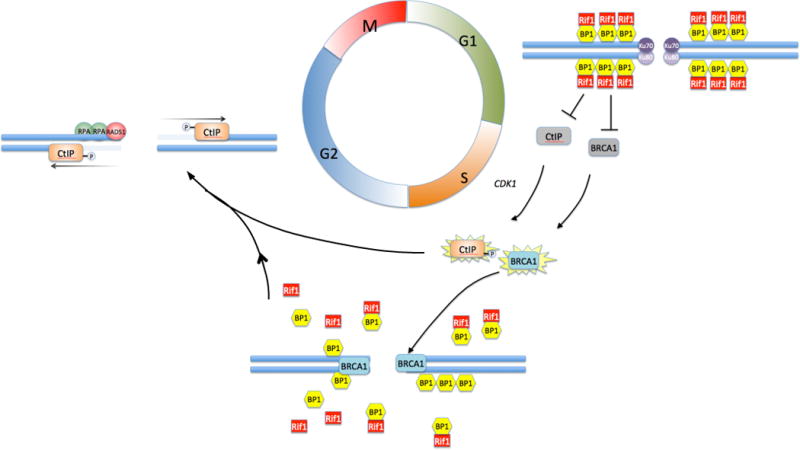
Non-homologous End-Joining is favored in G1, when the activities of BRCA1 and CtIP are repressed by a complex of 53BP1 and Rif1 that coats the chromatin in the vicinity of double-strand breaks. During the transition to S/G2, BRCA1 acquires the ability to bind at break sites despite the repressive effect of 53BP1 and Rif1. The mechanism for BRCA1 activation and recruitment is still unknown. Depletion of Rif1 and activation by cyclin dependent kinase 1 (CDK1)-mediated phosphorylation allows CtIP to become active at the break site, where it resects duplex DNA to form a 5′ single-strand overhang. This favors resection-dependent repair pathways, including A-EJ and HR. Commitment to HR is mediated by loading of Replication Protein A (RPA) and Rad51 at the break site.
DNA-PKcs, which associates with C-NHEJ factors in mammalian cells but is not present in yeast, is a candidate regulator of NHEJ in mammalian cells. As measured by reporter substrates, increased expression of DNA-PKcs represses HR, but this effect is not seen with mutant forms of DNA-PKcs that lack kinase activity84, 85. Further mutagenesis studies of DNA-PKcs revealed that autophosphorylation of T946, S1004 and T3950 inactivates NHEJ and promotes HR. DNA-PKcs autophosphorylation is therefore likely to be a critical mechanism for ensuring appropriate use of HR. This idea is supported by the observation that mice with targeted substitution of multiple DNA-PKcs autophosophorylation sites die at a very early age and are defective in HR86. Altogether, these findings are consistent with a model wherein NHEJ and HR factors are in active competition for repair of DNA double strand breaks. If NHEJ is not initially successful, displacement of NHEJ factors, perhaps by DSB resection, might enable error-free repair activities to dominate.
Factors favouring translocations
Experimentally-induced double-strand breaks on different chromosomes are known to significantly increase the rate of translocation between those chromosomes87. Evidence in favour of a DNA double-strand break intermediate in translocation has come from the study of translocations between the immunoglobulin constant region genes IGKC and IGLC1 and MYC in Burkitt’s lymphoma. First, double-strand breaks at the IgH locus are known to occur because of the action of the activation induced cytidine deaminase (AID) enzyme, which deaminates target cytidine residues leading to the appearance of staggered double-strand breaks88. In the absence of AID, translocations between these two genetic loci occur at vanishingly low frequency89, demonstrating the importance of double-strand breaks as a substrate for translocation. Experimental systems using site-specific, inducible DNA double-strand breaks have also shown that translocation between two sites is highly dependent on the frequency of double-strand breaks6, 7. Further evidence for a double-strand intermediate leading to translocation came from an analysis of translocation frequency in p53-knockout mice. One activity of the tumour suppressor gene TP53 is to promote apoptosis in cells with double-strand breaks, and deletion of p53 or upstream components of the DNA damage signalling pathway, such as the kinase ataxia telangiectasia mutated (ATM), increases the overall frequency of translocations90. This shows that providing an environment that is favourable to DNA double-strand breaks promotes translocation.
The appearance of recurrent translocations, such as IGH–MYC in Burkitt’s lymphoma, has posed the question of why certain translocations occur so commonly in specific malignancies. One possibility is that these recurrent translocations arise at no more common frequency than any other translocation, but are selected on the basis of their potential to drive survival and proliferation of the cancer cell. An additional, long-standing hypothesis is that recurrent translocations arise because the translocation partners are in particularly close proximity in the nuclei of cells from the affected tissue91, 92, 93. Chromosome Conformation Capture has been used to measure genomic interactions, and combining this technique with deep sequencing has recently enabled the measurement at base-pair resolution of how closely genomic loci interact. Using this approach, Hakim et al showed that IGH and MYC do not interact particularly closely in activated B cells5. In fact, even though IGH–MYC translocations are found in 85% of Burkitt’s lymphoma, 2,361 other genes interact with IGH more often than MYC. This suggests that nuclear proximity is not the key driver of recurrent, cancer-associated IGH–MYC translocations. Moreover, Rocha et al showed that there is a poor correlation between genes that physically interact with IGH and those that are AID targets94. The authors proposed that AID targets are situated within broader genomic domains that associate with IGH, but the current evidence seems to indicate that physical proximity within the nucleus is a minor determinant of translocation frequency between genes on different chromosomes.
Quantification of how frequently a double-strand break at a specific site forms translocations with other genomic loci has recently become feasible owing to the development of two similar techniques: High-Throughput Genome-Wide Translocation Sequencing (HTGTS), and Translocation-Capture Sequencing (TC-Seq)6, 7. These studies were conducted in B cells, and focused again on understanding what factors determine the likely translocation partners of double-strand breaks at IGH or MYC. In both studies, there was a strikingly high correlation between AID target sites and translocation frequency. This suggests that genes that are more often affected by double-strand breaks form translocations more readily. Transcriptional status is another factor influencing translocation frequency, as the majority of translocations were to coding sequences, and transcribed genes were more commonly subject to translocation than silent genes. Translocation partners for double-strand breaks are not strictly limited to closely-interacting chromosome domains, but when a large number of double-strand breaks are present, there is an increased frequency of inter-chromosomal translocation between partners with higher physical interaction95. All deep sequencing studies to date have shown that double-strand breaks on the same chromosome, particularly those lying nearby on the same chromosome, have the highest rate of joining, matching a previous study of the frequency of joining of breaks induced by the RAG1 and RAG2 recombinases96.
Taking these studies together, the primary predictor for whether genes take part in translocations is the frequency with which those genes undergo double-strand breakage. Hence, translocations between MYC and IGH are favoured in B cells because those regions are common sites for double-strand breaks in B cells. Active transcription also correlates with translocation, and up to 40% of translocations involve joining of a break to a sequence from the same chromosome. These intrachromosomal translocations are not seen recurrently in cancer cells. This suggests that many translocations do not contribute to tumorigenesis, and that oncogenic translocations that increase cell survival and proliferation are selected for during the evolution of a tumour.
As the frequency of double-strand breaks at a particular genomic site appears to be the key determinant of whether that site becomes involved in a translocation, it is obviously of major significance to understand the processes that produces double-strand breaks. DNA replication is a source of DNA double-strand breaks97. As the entire genome is replicated during cell division, any genomic site is a potential site for replication-associated double strand breaks. However, replication-associated DNA damage is not entirely random. Common Fragile Sites (CFSs) are regions of the genome that are prone to breakage during replication stress. Whereas CFSs are relatively stable in normal cells, cancer cells accumulate breaks and genomic aberrations, including translocations, at these sites98, 99. Breakage at CFSs in cancer cells appears to be a consequence of replication stress arising from accelerated, oncogene-mediated replication100–104. In addition to CFSs, a second class of genomic sites prone to double-strand breakage, and associated with translocations in human cancer, has recently been identified in murine B cells105. These ‘Early Replicating Fragile Sites’, or ERFSs, were identified using a chromatin immunoprecitipation (ChIP)–deep sequencing (Seq) approach to reveal sites that are preferentially bound by DNA damage response proteins after replication stress. ERFSs are distinct from CFSs because they are found around early replication origins, whereas CFSs replicate late in S phase. ERFSs also have a high-GC content, are commonly associated with repetitive DNA elements, and correlate with transcriptionally active genes in an open chromatin environment. Breakage at ERFSs is AID independent, hence these sites may also be present in other cell types. Some euchromatic regions are targets of both AID activity in G1 and ERFS fork collapse during S phase; however, whereas AID activity is limited to 1–2 kb of promoters, breakage at ERFS spans a much larger region ranging from 10–1000 kb105.
Chromatin and transcriptional status are likely to play a substantial role in determining the likelihood that a particular genomic site will be involved in a translocation. A correlation of translocations with transcriptional activity was noted from deep sequencing studies6, 7, and transcription has been reported to predispose genomic fragile sites to DNA breakage by causing increased collapse of DNA replication forks106. Although the existing data indicates that transcriptionally active regions are more prone to translocations, γH2AX, which signals DNA damage, accumulates more readily in euchromatic sites than in heterochromatin107, 108. Double-strand breaks also take longer to repair when located in heterochromatin109. Heterochromatin protein 1a (HP1α; also known as CBX5) paralogs are recruited to break sites110–112, and depletion of HP1α or the nucleosome assembly complex, chromatin assembly factor 1 (CAF1) inhibits repair by homologous recombination113.
Translational opportunities and perspectives
Sequencing-based approaches have enabled significant progress in recent years in understanding the nature and effect of chromosome translocations. We now have a much clearer idea of the frequency and complexity of translocations. Although translocations are almost invariably found when we study the genomic landscape of cancer cells, the importance of translocations to the onset of malignancy is still a matter of debate. In contrast to the situation with the characteristic clonal translocations identified in CML and Burkitt’s Lymphoma, many translocations do not appear to be primary drivers of cancer cell growth. We are still at an early stage in analysing the sequencing data that is pouring in, and making sense of how translocations influence cancer cell growth will be a major topic of research interest in the coming years. Another major challenge lies in understanding the cellular pathways that underpin the genomic complexity of cancer cells. What pathways are responsible for causing translocations? Is there a role for replication-based mechanisms in the formation of translocations, and what is the significance of microhomology at translocation junctions? Answering these questions will require us to build on our current understanding of the genes involved in translocation pathways, enabling us to test requirements for the appearance of translocations in vivo. Sequencing is demonstrating that many translocations are more complex than we had imagined before65. It will be interesting to see whether chromothrypsis2 arises by different pathways to translocations, or merely represents the most extreme end of a spectrum of chromosome rearrangements present in cancer cells.
Although we have made major progress in understanding nuclear phenomenon that influence the frequency of translocations, there are clearly outstanding issues to address relating to the impact of chromatin on genomic instability and translocation frequency. At the time of writing, there has been no published, genome-wide attempt to correlate chromatin status with translocation frequency. Such work would shed light on several interesting studies that have demonstrated how chromatin can affect the processing of DNA breaks. Future work is also likely to identify other factors in mammalian cells, which, as is the case with 53BP1, Rif1, Ku and DNA-PKcs are able to modulate the use of nonhomologous end-joining pathways. The activity of such factors could determine the frequency of translocations, by biasing repair of DNA breaks to error-prone end-joining pathways.
As translocations appear to be produced mainly by the C-NHEJ and A-EJ pathways, selective inhibition of end-joining pathways could potentially be used to prevent the appearance of cancer, or to block the appearance of further mutations that drive cancer growth and survival. Global inhibition of end-joining is unlikely to be a beneficial long-term treatment modality, based on observations in gene-targeted mice that correlate loss of end-joining activity with increased chromosome abnormalities and tumor incidence.
Nonetheless, cancer cells appear to make use of A-EJ pathways to join DNA double-strand breaks in aberrant ways that promote cancer growth114. Acquired resistance of BRCA2-deficient cells to poly (ADP ribose) polymerase (PARP) inhibition has been shown to occur by A-EJ-mediated internal deletions within the BRCA2 gene that restore its activity115, 116. These observations reinforce the importance of understanding the genetic requirements of A-EJ to enable specific targeting of this pathway.
Inhibitors of DNA ligases have been identified and shown to be toxic towards cancer cell lines and to synergize with methyl methanesulfonate treatment to increase cell killing117, 118. Inhibitors of DNA-PK have likewise shown promise, potentially based on their ability to bias DNA repair towards HR instead of more toxic pathways119. Although at the present time animal studies are lacking, in the future, agents that enable repair to be shifted from mutagenic pathways towards repair pathways that promote faithful DNA repair, as has been proposed for inhibition of 53BP1 in patients with BRCA1 deficiency34 could provide a new avenue for cancer treatment based on the prevention of mutation.
Box 2 Telomeres and translocations.
Telomeres normally protect the end of chromosomes, but incipient tumor cells are known to suffer from acutely-short telomeres120, 121. When telomeres become shortened or uncapped by loss of shelterin, the chromosome end is signalled as a double-strand break122. Normally, p53 signalling triggers apoptosis in response to this signal, but in telomerase-null mice in the absence of p53, end-to-end chromosome fusions are observed that correlate with a high frequency of epithelial cancer123. End-to-end chromosome fusions are thought to cause genomic instability because the cell will try to pull both centromeres from the fused chromosome into separate daughter cells, causing the fused chromosome to break, generating translocations and new DNA ends that form substrates for additional breakage-fusion-bridge cycles (Fig 3)124. Key intermediates in this process, i.e. dicentric chromosomes and anaphase bridges, have been observed in primary human tumors125. Cycles of BFB have been observed leading to amplification of genetic sequence near DNA break sites and complex translocation involving multiple chromosomes in B cells with combined deletion of DNA repair genes and p5348, 49. These complex translocations feature amplification of the c-myc oncogene, which is an essential driver of tumorigenesis in these cells. End-to-end fusion of uncapped telomeres is also dependent on the NHEJ factors, Ku70 and ligase 4 (LIG4)126, 127. Whereas loss of the shelterin component, TRF2, causes chromosome fusion by classical non homologous end joining (C-NHEJ) DNA repair pathways128, alternative end joining (A-EJ)-mediated chromosome fusions are observed in TRF2-deficient cells when Ku80 is absent. In mice, the shelterin proteins TRF1, TPP1, POT1a and POT1b combine with TRF2 to suppress A-EJ events78, 129. Although the significance of this effect in human cancer in not fully clear, mouse models of cancer arising from defective telomere function share many genomic features in common with human tumors130. Hence, both classical and alternative end-joining pathways are active in causing chromosome rearrangements arising from end-to-end fusions.
Glossary terms
- Non-homologous end-joining.
Joining of DNA double-strand breaks without extensive sequence homology by ligation of DNA ends
- Non-allelic homologous recombination (NAHR)
This process involves recombination between repetitive regions at different genomic sites and can lead to chromosome rearrangements as seen in genetic diseases such as Charcot-Marie-Tooth syndrome.
- Break-induced replication (BIR)
A modified homology-based repair pathway, where a broken DNA end is repaired by copying a large amount of sequence from an undamaged homologous partner, potentially leading to copying of the entire homologous sequence from the site of damage to the end of the chromosome23
- Chromothrypsis
Highly complex chromosome rearrangement event, characterized by extensive re-assortment of genetic fragments from one or more chromosomes2
Biographies
Samuel F Bunting
Dr Bunting’s discovery that the pathological effects of BRCA1 mutation can be rescued by deletion of 53BP1 was a stimulus for his current drive to discover mammalian regulators of DNA repair pathway choice. He is currently using mouse genetics to find targets for potential therapies to promote error-free DNA repair, and prevent the mutations that drive the growth of cancer cells.
Andre Nussenzweig
A physicist by training, Dr Nussenzweig’s past research in the field of DNA repair established the key role of Ku80 in mammalian nonhomologous end-joining and antigen receptor rearrangements. He subsequently demonstrated the vital role of H2AX phosphorylation in signalling DNA breaks in mammalian cells, and continues to study the intersection of DNA repair, chromatin biology and cellular aging.
Contributor Information
Samuel F Bunting, Department of Molecular Biology and Biochemistry, Rutgers, The State University of New Jersey.
Andre Nussenzweig, Laboratory of Genome Integrity, National Cancer Institute Center for Cancer Research, National Institutes of Health, Bethesda, MD.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Forment JV, Kaidi A, Jackson SP. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer. 2012;12:663–70. doi: 10.1038/nrc3352. [DOI] [PubMed] [Google Scholar]
- 3.Giardino D, et al. De novo balanced chromosome rearrangements in prenatal diagnosis. Prenat Diagn. 2009;29:257–65. doi: 10.1002/pd.2215. [DOI] [PubMed] [Google Scholar]
- 4.Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991;49:995–1013. [PMC free article] [PubMed] [Google Scholar]
- 5.Hakim O, et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484:69–74. doi: 10.1038/nature10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein IA, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiarle R, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–19. doi: 10.1016/j.cell.2011.07.049. These three papers used next-generation sequencing to systematically measure the frequency of transloctation between sites throughout the genome. Chiarle et al and Klein et al showed a strong proximity effect for joining of DNA breaks, with intra-chromosomal joining being preferred. Translocation is most closely linked to frequency of DNA breakage, but also correlates with transcription. Hakim et al extended these observations and showed that nuclear architecture is a relatively weak predictor of translocation frequency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato L, et al. Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes. Proc Natl Acad Sci U S A. 2012;109:2479–84. doi: 10.1073/pnas.1120791109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–45. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 10.Rabbitts TH. Commonality but diversity in cancer gene fusions. Cell. 2009;137:391–5. doi: 10.1016/j.cell.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 11.Tomlins SA, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. One of a series of reports from The Cancer Genome Atlas consortium, which has been sequencing cancer genomes in an effort to find the mutations that drive tumorigenesis. This study, focusing on colorectal carcinoma, revealed frequently mutated cancer genes and a pattern of recurrent and sporadic translocations and chromosome rearrangements. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephens PJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–10. doi: 10.1038/nature08645. This paper, one of several recent publications from Michael Stratton’s group exploring the genomic landscape of breast cancer, revealed new fusion genes formed by translocations in breast cancer cells, and also demosntrated the fequency and complexity of breast cancer translocations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imielinski M, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stephens PJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maher CA, Wilson RK. Chromothripsis and human disease: piecing together the shattering process. Cell. 2012;148:29–32. doi: 10.1016/j.cell.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger MF, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–6. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–64. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet. 2004;13(Spec No 1):R57–64. doi: 10.1093/hmg/ddh073. [DOI] [PubMed] [Google Scholar]
- 24.Llorente B, Smith CE, Symington LS. Break-induced replication: what is it and what is it for? Cell Cycle. 2008;7:859–64. doi: 10.4161/cc.7.7.5613. [DOI] [PubMed] [Google Scholar]
- 25.Ruiz JF, Gomez-Gonzalez B, Aguilera A. Chromosomal translocations caused by either pol32-dependent or pol32-independent triparental break-induced replication. Mol Cell Biol. 2009;29:5441–54. doi: 10.1128/MCB.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–5. doi: 10.1038/nature05723. This report provides evidence that in S cerevisiae, chromosome rearrangments can occur by a process inving Break Induced Repair with multiple rounds of strand invasion. [DOI] [PubMed] [Google Scholar]
- 27.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Difilippantonio MJ, et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–4. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferguson DO, et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci U S A. 2000;97:6630–3. doi: 10.1073/pnas.110152897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y, et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- 31.Saberi A, et al. RAD18 and poly(ADP-ribose) polymerase independently suppress the access of nonhomologous end joining to double-strand breaks and facilitate homologous recombination-mediated repair. Mol Cell Biol. 2007;27:2562–71. doi: 10.1128/MCB.01243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adamo A, et al. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;39:25–35. doi: 10.1016/j.molcel.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 33.Bunting SF, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell. 2012;46:125–35. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bunting SF, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. This paper demonstrated that a mammalian DNA damage response factor, 53BP1, represses the use of the error-free homologous recombination pathway for repair of DNA double-strand breaks and increases use of a mutational mechanism inving nonhomolgous end-joining that causes cancer in BRCA1-deficient mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pace P, et al. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 2010;329:219–23. doi: 10.1126/science.1192277. [DOI] [PubMed] [Google Scholar]
- 36.Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108:3406–11. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. Study from TCGA that suggested that up to 50% of ovarian carcinoma cases inve mutations that cause effect of inactivation of the homologous recombination pathway for DNA repair. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park DJ, et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am J Hum Genet. 2012;90:734–9. doi: 10.1016/j.ajhg.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson ER, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012;8:e1002894. doi: 10.1371/journal.pgen.1002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roth DB, Porter TN, Wilson JH. Mechanisms of nonhomologous recombination in mammalian cells. Mol Cell Biol. 1985;5:2599–607. doi: 10.1128/mcb.5.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth DB, Wilson JH. Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Mol Cell Biol. 1986;6:4295–304. doi: 10.1128/mcb.6.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res. 1996;24:4639–48. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boulton SJ, Jackson SP. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996;15:5093–103. [PMC free article] [PubMed] [Google Scholar]
- 45.Daley JM, Wilson TE. Rejoining of DNA double-strand breaks as a function of overhang length. Mol Cell Biol. 2005;25:896–906. doi: 10.1128/MCB.25.3.896-906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Difilippantonio MJ, et al. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med. 2002;196:469–80. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu C, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–21. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 48.Weinstock DM, Elliott B, Jasin M. A model of oncogenic rearrangements: differences between chromosomal translocation mechanisms and simple double-strand break repair. Blood. 2006;107:777–80. doi: 10.1182/blood-2005-06-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boboila C, et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med. 2010;207:417–27. doi: 10.1084/jem.20092449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–6. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai AG, et al. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–42. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. 2011;18:80–4. doi: 10.1038/nsmb.1940. Using a chromosome translocation reporter system, this report demonstrated that the putative exonuclease CtIP has a role in promoting chromosome translocations by an ‘alternative end-joining’ pathway, potentially by exposing microhomology at break points by end resection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rass E, et al. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–24. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 55.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–8. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bothmer A, et al. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J Exp Med. 2013;210:115–23. doi: 10.1084/jem.20121975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang L, et al. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008;36:3297–310. doi: 10.1093/nar/gkn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simsek D, et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011;7:e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boboila C, et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1) Proc Natl Acad Sci U S A. 2012;109:2473–8. doi: 10.1073/pnas.1121470109. Both of these papers (Simsek et al and Boboila et al) attempt to find the genetic requirements for the ‘Alternative End-Joining’ pathway, which seems to mediate a subset of chromosome rearrangments in cancer cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brunet E, et al. Chromosomal translocations induced at specified loci in human stem cells. Proc Natl Acad Sci U S A. 2009;106:10620–5. doi: 10.1073/pnas.0902076106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robbiani DF, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–38. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiang C, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. 2012;44:390–7. doi: 10.1038/ng.2202. S1. Another interesting report using next-generation sequencing, this study reported the remarkable observation that apparently balanced chromosome translocations frequently inve sequence from multiple chromosomes. This finding indicates that a model inving simple joining of double-strand breaks on different chromosomes may not account for the complexity of cancer-associated translocations. This report also showed that a minority of rearrangement breakpoints inved microhomology, suggesting that ‘classical’ NHEJ may cause the majority of translocations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 2010;17:11–6. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 65.Frank-Vaillant M, Marcand S. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell. 2002;10:1189–99. doi: 10.1016/s1097-2765(02)00705-0. [DOI] [PubMed] [Google Scholar]
- 66.Kim JS, et al. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol. 2005;170:341–7. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 2008;7:1765–71. doi: 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–42. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Difilippantonio S, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–33. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Helmink BA, et al. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature. 2011;469:245–9. doi: 10.1038/nature09585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manis JP, et al. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–7. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 72.Ward IM, et al. 53BP1 is required for class switch recombination. J Cell Biol. 2004;165:459–64. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bothmer A, et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell. 2011;42:319–29. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bouwman P, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–7. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buonomo SB, Wu Y, Ferguson D, de Lange T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J Cell Biol. 2009;187:385–98. doi: 10.1083/jcb.200902039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chapman JR, et al. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Virgilio M, et al. Rif1 Prevents Resection of DNA Breaks and Promotes Immunoglobulin Class Switching. Science. 2013 doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Escribano-Diaz C, et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 80.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 Regulates DSB Repair Using Rif1 to Control 5′ End Resection. Science. 2013 doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neal JA, et al. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol Cell Biol. 2011;31:1719–33. doi: 10.1128/MCB.01298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shrivastav M, et al. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair (Amst) 2009;8:920–9. doi: 10.1016/j.dnarep.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang S, et al. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J Cell Biol. 2011;193:295–305. doi: 10.1083/jcb.201009074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Richardson C, Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405:697–700. doi: 10.1038/35015097. This report describes an experimental system in ES cells to test factors that contribute to translocations. The authors report that the presence of DNA double-strand breaks on different chromosomes significantly increases the rate of translocation. [DOI] [PubMed] [Google Scholar]
- 85.Muramatsu M, et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 86.Ramiro AR, et al. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–8. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 87.Ramiro AR, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006;440:105–9. doi: 10.1038/nature04495. Landmark paper confirming the dependency of IgH:c-myc translocation on the ability of Activation-Induced Cytidine Deaminase to make DNA double-strand breaks, and demonstrating that mutations that decrease the cells’ ability to eliminate double-strand breaks cause an increase in translocation frequency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Misteli T. Higher-order genome organization in human disease. Cold Spring Harb Perspect Biol. 2010;2:a000794. doi: 10.1101/cshperspect.a000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Osborne CS, et al. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roix JJ, McQueen PG, Munson PJ, Parada LA, Misteli T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet. 2003;34:287–91. doi: 10.1038/ng1177. [DOI] [PubMed] [Google Scholar]
- 91.Rocha PP, et al. Close proximity to Igh is a contributing factor to AID-mediated translocations. Mol Cell. 2012;47:873–85. doi: 10.1016/j.molcel.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y, et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–21. doi: 10.1016/j.cell.2012.02.002. Combining high-throughput chromosome conformation capture and translocation sequencing, this group directly tested the hypothesis that nuclear architecture is a contributor to translocation frequency. They conclude that the relative proximity of chromosomes in the nucleus can affect the frequency of translocations between those chromosomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mahowald GK, et al. Aberrantly resolved RAG-mediated DNA breaks in Atm-deficient lymphocytes target chromosomal breakpoints in cis. Proc Natl Acad Sci U S A. 2009;106:18339–44. doi: 10.1073/pnas.0902545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 95.Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst) 2006;5:1126–35. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 96.Ozeri-Galai E, Bester AC, Kerem B. The complex basis underlying common fragile site instability in cancer. Trends Genet. 2012;28:295–302. doi: 10.1016/j.tig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 97.Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 98.Bester AC, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–46. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 100.Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 101.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 102.Barlow JH, et al. Identification of Early Replicating Fragile Sites that Contribute to Genome Instability. Cell. 2013;152:620–32. doi: 10.1016/j.cell.2013.01.006. This report identifies a new class of genomic sites, called ‘ERFS’, which are particularly frequently subject to DNA double-strand breaks following replication stress. Owing to their frequency of undergoing DNA breakage, ERFS represent potential hotspots for translocation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell. 2011;44:966–77. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 104.Cowell IG, et al. gammaH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS One. 2007;2:e1057. doi: 10.1371/journal.pone.0001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim JA, Kruhlak M, Dotiwala F, Nussenzweig A, Haber JE. Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J Cell Biol. 2007;178:209–18. doi: 10.1083/jcb.200612031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goodarzi AA, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 107.Ayoub N, Jeyasekharan AD, Venkitaraman AR. Mobilization and recruitment of HP1: a bimodal response to DNA breakage. Cell Cycle. 2009;8:2945–50. [PubMed] [Google Scholar]
- 108.Luijsterburg MS, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol. 2009;185:577–86. doi: 10.1083/jcb.200810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zarebski M, Wiernasz E, Dobrucki JW. Recruitment of heterochromatin protein 1 to DNA repair sites. Cytometry A. 2009;75:619–25. doi: 10.1002/cyto.a.20734. [DOI] [PubMed] [Google Scholar]
- 110.Baldeyron C, Soria G, Roche D, Cook AJ, Almouzni G. HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J Cell Biol. 2011;193:81–95. doi: 10.1083/jcb.201101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sallmyr A, Tomkinson AE, Rassool FV. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood. 2008;112:1413–23. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Edwards SL, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 113.Sakai W, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20. doi: 10.1038/nature06633. These two papers demonstrated the potentially dangerous effect of end-joining pathways in promoting intra-chromosomal rearrangments that allow recovery of BRCA2 function in cancer cells challenged with chemotherapy. An ‘Alternative End-Joining’ pathway is implicated in the development of chemoresistance in this case based on the presence of microhomology at the breakpoints of novel mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen X, et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68:3169–77. doi: 10.1158/0008-5472.CAN-07-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Srivastava M, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151:1474–87. doi: 10.1016/j.cell.2012.11.054. By developing specific inhibitors of a mammaliand DNA ligase enzyme, the authors of this report were able to demonstrate that targeting nonhomologous end-joining can have anti-cancer effects in combination therapy. This demonstrates the feasibility of using drugs that manipulate DNA repair pathways to achieve therapeutic goals. [DOI] [PubMed] [Google Scholar]
- 116.Davidson D, et al. Irinotecan and DNA-PKcs inhibitors synergize in killing of colon cancer cells. Invest New Drugs. 2012;30:1248–56. doi: 10.1007/s10637-010-9626-9. [DOI] [PubMed] [Google Scholar]
- 117.de Lange T, et al. Structure and variability of human chromosome ends. Mol Cell Biol. 1990;10:518–27. doi: 10.1128/mcb.10.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hastie ND, et al. Telomere reduction in human colorectal carcinoma and with ageing. Nature. 1990;346:866–8. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 119.d’Adda di Fagagna F, et al. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr Biol. 2001;11:1192–6. doi: 10.1016/s0960-9822(01)00328-1. [DOI] [PubMed] [Google Scholar]
- 120.Artandi SE, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–5. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 121.Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gisselsson D, et al. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci U S A. 2000;97:5357–62. doi: 10.1073/pnas.090013497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–8. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 124.Celli GB, Denchi EL, de Lange T. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat Cell Biol. 2006;8:885–90. doi: 10.1038/ncb1444. [DOI] [PubMed] [Google Scholar]
- 125.Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol. 2002;12:1635–44. doi: 10.1016/s0960-9822(02)01179-x. [DOI] [PubMed] [Google Scholar]
- 126.Rai R, et al. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010;29:2598–610. doi: 10.1038/emboj.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maser RS, et al. DNA-dependent protein kinase catalytic subunit is not required for dysfunctional telomere fusion and checkpoint response in the telomerase-deficient mouse. Mol Cell Biol. 2007;27:2253–65. doi: 10.1128/MCB.01354-06. [DOI] [PMC free article] [PubMed] [Google Scholar]