

Abstract
Activating transcription factor 3 (ATF3) is a member of the ATF/cAMP-response element binding protein (CREB) family of transcription factors. In response to stress stimuli, ATF3 forms dimers to activate or repress gene expression. Further, ATF3 modulates the immune response, atherogenesis, cell cycle, apoptosis, and glucose homeostasis. Recent studies have shown that ATF3 may also be involved in pathogenesis of other diseases. However, more studies are needed to determine the role of ATF3 in metabolic regulation.
Keywords: Activating transcription factor 3 (ATF3), Immune, Oncogenesis, Glucose homeostasis, Liver injury
1. Introduction
The activating transcription factor (ATF) family of proteins is a group of transcription factors consisting of ATFs, cAMP response element binding protein (CREB), cAMP response element modulator (CREM), and related proteins.1 This protein family has at least ten members that all share a basic-region leucine zipper (bZip) DNA binding domain.2,3 ATF was first identified in 1987, when it was shown to bind to early adenovirus promoters (E2, E3, and E4) via a consensus binding sequence (TGACGTCA). In the same year, CREB was shown to bind to the somatostatin gene promoter through the same consensus binding site.4 Consequently, these two proteins are grouped together as the ATF/CREB family of proteins as they: i) bind to the same consensus binding sequence, ii) both contain bZip domains, and iii) form either homo- or heterodimers with other proteins in the ATF/CREB family.2,5
The ATF3/CREB family now includes ATF1, CREB, CREM, ATF2, ATF3, ATF4, ATF5, ATF6, ATF7, and B-ATF.6 The unique feature of these proteins is the basic DNA binding region and bZip domain, which is responsible for forming homo- or heterodimers with other ATF/CREB family members.5 While the ATF/CREB proteins have similar bZip domains, some members of this family share additional homology. For example, ATF1, CREB, and CREM are very similar in regions containing phosphorylation sites.7–9 Likewise, the first 100 N-terminal residues and last 13 C-terminal residues of ATF2 and ATFa are similar.10 Furthermore, apart from their sequence homology, proteins of the ATF/CREB family are induced in response to many different stimuli and can activate or repress transcription.2,5 For example, ATF1 and CREB induce transcription in response to cAMP and calcium influx, while ATF2 induces transcription in response to viral infection.5 Conversely, ATF3 represses transcription upon DNA binding,2 but activates transcription of target genes upon heterodimerization with Jun proteins.5 Therefore, depending on homo- or heterodimer status and promoter context, ATF3 can serve as either a transcriptional activator or repressor.
2. Structure and regulation of the ATF3 gene
The human ATF3 gene spans over 15 kilobases and has four exons termed A, B, C, and E. Exon A encodes the 5′-untranslated region, exon B encodes the N-terminal domain containing 80 amino acids, exon C encodes a further 36 amino acids, while exon E encodes the bZip domain and 3′-untranslated region.5 In addition, there is an alternatively spliced form of ATF3 termed ATF3ΔZip, which contains an additional exon, exon D. Exon D contains a termination codon.5 Thus, ATF3ΔZip is a truncated version of full-length ATF3, whereby it shares homology with full-length ATF3 protein until exon C, and does not contain the bZip domain encoded by exon E. There are limited data on relative expression levels of ATF3 versus ATF3ΔZip. In human umbilical vein endothelial cells (HUVEC) stimulated by anisomycin (a known ATF3 stimulus), protein expression of ATF3ΔZip was approximately 34% that of full-length ATF3 protein.5 To investigate the effect of both proteins on transcription of target genes, a promoter containing four consensus ATF3 binding sites was constructed. In transient transfection assays, ATF3 alone markedly repressed promoter activity, whereas ATF3ΔZip had no effect. Co-transfection of ATF3 with ATF3ΔZip significantly inhibited ATF3-mediated suppression of promoter activity,5 indicating that ATF3ΔZip can antagonize transcriptional activity of ATF3.
Analysis of the ATF3 gene promoter shows that it has binding sites for several transcription factors. Some of these are binding sites for ATF/CRE, activator protein 1 (AP1), and nuclear factor-κB (NF-κB), suggesting that ATF3 may be inducible by stress signals such as cAMP, UV radiation, calcium influx, and cytokines.11 Further, the ATF3 gene also has binding sites for Myc/Max and E2F, suggesting that ATF3 may also be involved in cell cycle regulation.11
Consistent with the presence of promoter binding sites for stress signals, ATF3 expression is induced in response to stress and plays a potential role in mediating stress responses.11,12 In animal studies, stress stimuli include ischemia, ischemia with reperfusion, axotomy, wounding, toxicity, and seizure. In cell culture experiments, stimuli include serum factors, cytokines, genotoxic agents, cell death-inducing agents, and the adenoviral protein, E1A.12 While there are limited data with regards the pathways involved in ATF3 induction, it is thought that the c-Jun NH(2)-terminal kinase/stress-activated protein kinase (JNK/SAPK) pathway mediates regulation of ATF3 via stress signals. In addition, p53 and interleukin (IL)-6 are required for induction of ATF3 under certain conditions.12 Studies from in vitro transfection and transcription assays suggest that ATF3 homodimers act as transcription inhibitors, while ATF3 heterodimers (with partners such as ATF2, c-Jun, Jun B, and Jun D) act as transcription activators.2,5,12
3. Role of ATF3 in immune function
Toll-like receptors (TLRs) are pathogenic sensors. Upon ligand activation, TLRs give rise to various intracellular signaling pathways that increase expression of inflammatory cytokines and chemokines to activate the adaptive immune system.13 In addition, TLRs have been shown to modulate tumor development.14
In bone marrow-derived macrophages (BMDM), activation of various TLRs increases protein expression of ATF3,15 suggesting that ATF3 plays a role in TLR signaling. Another study showed that ATF3 is specifically upregulated after infection with Streptococcus pneumonia via TLR2 and TLR4.16 Consistent with these observations, primary macrophages from Atf3−/− mice show increased expression of IL-6, IL-12, and tumor necrosis factor (TNF)-α,15 indicating that ATF3 is a negative regulator of immune responses. In a separate study, BMDM from Atf3−/− mice had significantly lower survival rates after stimulation with several TLR ligands.17 Apoptotic rates in response to lipopolysaccharide (LPS) signaling were also significantly higher in these macrophages. This latter study further showed that ATF3 acts downstream of JNK signaling after TLR activation.17 In a mouse model of ischemia/reperfusion (IR) injury, ATF3 ablation in LPS-stimulated BMDM decreased levels of nuclear factor erythroid-derived 2-related factor 2/heme oxygenase-1 (NRF2/HO-1) and phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT), resulting in enhanced TLR4/NF-κB activation.18 These results demonstrate a role of ATF3 in mediating NRF2/HO-1 signaling for regulation of TLR4-driven inflammatory responses in IR-stressed liver. Interestingly, NF-κB contains an ATF/CRE consensus sequence in its promoter. In addition, ATF3 may also interact with histone deacetylase 1 (HDAC1).19 Altogether, these latter findings offer further insight into the mechanism by which ATF3 inhibits transcription of NF-κB target genes. Systemic lupus erythematosus (SLE) is an auto-immune disease that increases the risk of cardiovascular disease via increased oxidized high-density lipoprotein (HDL).20 To mimic this model of SLE, macrophages were treated with either normal HDL or SLE HDL.21 Accordingly, SLE HDL activated NF-κB and increased inflammatory cytokine production. Further analysis revealed that NF-κB activation was due to failure of SLE HDL to induce ATF3 in macrophages.21 In a model of intestinal Crohn’s disease, ATF3 co-operates with NF-κB to downregulate cytokine production, further strengthening the role of ATF3 in amelioration of inflammation.22
Consistent with the anti-inflammatory action of ATF3, Atf3−/− mice show increased expression of pro-inflammatory cytokines (such as IL-6 and IL-12b) with LPS stimulation, as well as increased susceptibility to endotoxic shock-induced death compared with wild-type controls.23 However, in contrast, one study showed that increased susceptibility to bacterial and fungal infections in sepsis was due to IL-6 repression via ATF3 induction.24 In support of this observation, Atf3−/− mice were protected against bacterial and fungal infections, while Atf3−/−Il6−/− mice were susceptible to infection.24 Furthermore, Atf3−/− mice were protected from murine cytomegalovirus (MCMV) infection,25 with the underlying mechanism that ATF3 reduced expression of interferon (IFN)-γ, a potent regulator of natural killer cell activity.25 Despite these findings, Atf3−/− mice show increased levels of high mobility group box 1 (HMGB1), an important pro-inflammatory molecule that plays an crucial role in the end stages of sepsis.26 Adeno-associated virus (AAV)-mediated ATF3 gene transfer reduced LPS-induced mortality in Atf3−/− mice.26 In addition, Atf3−/− mice show increased TNFα expression in the spleen upon ligand activation of TLR9.19 Activated T lymphocytes isolated from Atf3−/− mice show increased levels of IL-4, IL-5, and IL-13.27 ATF3 was also shown to reduce transcription of all three genes by directly binding to their promoters after heterodimer formation with JunB, a member of the AP-1 family of proteins.27 Furthermore, there is increased expression of chemokine (C-C motif) ligand 1 (CCL1), CCL2, CCL4, CCL5, CCL7, CCL8, and CCL11 in Atf3−/− mouse lung,27 with a consensus ATF/CRE site detected in the promoter of CCL4. BMDM derived from Atf3−/− secrete lower CCL4 levels upon LPS treatment.27 In neutrophils, ATF3 reduced expression of chemokines, such as C-X-C motif ligand 1 (CXCL1), but promoted neutrophil chemotaxis.28 In primary human monocytes and macrophages, IFN-γ significantly induced ATF3 expression, which in turn bound to the promoter of matrix metalloproteinase 1 (MMP-1), reducing its transcription.29 IFN-β induced ATF3 expression, which binds to a distal regulatory sequence of IFN-β to reduce its transcription.30 Taken together, these observations indicate that ATF3 plays a vital role in modulating immune responses and maintaining normal host defense mechanisms (Fig.1A and 1B).
Fig.1. ATF3 is a negative regulator of the inflammatory response.
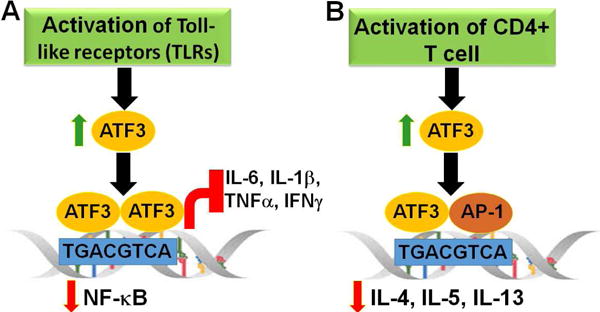
Protein expression of ATF3 is maintained at low levels in non-stimulated immune cells. There is rapid induction of ATF3 upon activation e.g., by cytokines, toll-like receptors (TLRs). ATF3 forms dimers and binds to its consensus binding sequence, TGACGTCA, on the promoter of nuclear factor-κB (NF-κB), resulting in repression of its expression, as well as downstream targets (A). In activated T-lymphocytes, ATF3 forms a heterodimer with JunB, an AP-1 family protein. This heterodimer then directly binds to the promoter of cytokines such as interleukin (IL)-4, IL-5, and IL-13 to inhibit their expression (B).
4. Role of ATF3 in mediating the anti-inflammatory effect of HDL
High-density lipoprotein has anti-atherogenic effects largely because of its ability to promote reverse cholesterol transport and protect against inflammatory responses. In contrast, the anti-inflammatory effect of HDL has not been well addressed. De Nardo et al.31 found that HDL induces ATF3 expression and protects against TLR-induced inflammation in a manner that is fully dependent on ATF3 in vitro and in vivo. In macrophages, ATF3 inhibits the gene encoding cholesterol 25-hydroxylase and prevents foam cell formation.32 As a result, Atf3 deletion in apolipoprotein e (Apoe)−/− mice promoted atherosclerosis progression.32 In addition, ATF3 inhibits tenascin C-induced foam cell formation by suppressing TLR4 in LPS-stimulated THP-1 macrophages.33 These data clearly show that ATF3 plays an atheroprotective role in macrophages.
5. Role of ATF3 in oncogenesis
One of the most striking roles of ATF3 has been in oncogenesis. ATF3 expression is elevated in a host of human cancers, most strikingly in breast cancer, prostate cancer, and Hodgkin’s lymphoma.19 Furthermore, ATF3 exhibits oncogenic properties in breast cancer cell lines, prostate cancer cells lines, and Hodgkin’s lymphoma cell lines, wherein it promotes cancer cell survival.34–36 While the exact mechanism remains unclear, in HeLa cells, ATF3 directly binds to the promoter for growth arrest and DNA damage-inducible protein (GADD153),37 a pro-apoptotic gene that induces cell cycle arrest. Therefore, ATF3 may promote oncogenesis in these cancers possibly by reducing GADD153 expression (Fig.2A).
Fig.2. ATF3 functions as an oncogene or tumor suppressor.
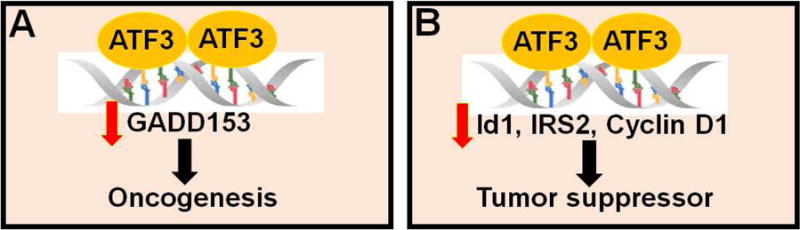
ATF3 may function as an oncogene by suppressing the pro-apoptotic gene, growth arrest and DNA damage-inducible protein (GADD153) (A), or a tumor suppressor by inhibiting the pro-survival genes, inhibitor of DNA binding 1 (Id1), insulin receptor substrate 2 (IRS2), and cyclin D1 (B).
Conversely, ATF3 is expressed at lower levels in human colorectal cancer cells,38 suggesting that it may also have a pro-apoptotic role. Indeed, the pro-apoptotic function of ATF3 may be related to transforming growth factor (TGF)-β signaling. TGFβ is a pro-apoptotic protein that stimulates ATF3 transcription via mothers against DPP homolog 3 (Smad3) activation. Subsequently, ATF3 and Smad3 form a complex that binds to the promoter and represses transcription of inhibitor of DNA binding 1 (Id1), a pro-survival gene. Thus, transcriptional repression of Id1 may be one of the mechanisms by which ATF3 promotes apoptosis19 (Fig.2B).
In transgenic mouse models, ATF3 over-expression in epithelial cells leads to increased incidence of squamous cell and mammary carcinomas, indicating that ATF3 plays an oncogenic role.19 In contrast, selective over-expression of pancreatic ATF3 leads to repression of insulin receptor substrate 2 (IRS2) transcription and increased apoptosis. IRS2 is implicated in diabetes and is a promoter of tumorigenesis and metastasis.39–40 Repression of IRS2 by ATF3 presents yet another mechanism by which ATF3 may act as an apoptopic promoter.19 Most importantly, ATF3 has been shown to bind to and repress activity of the cyclin D1 promoter, a protein that mediates G1 to S phase transition in the cell cycle19 (Fig.2B). Taken together, these data indicate that depending on the context and type of cancer, ATF3 may act as either an oncogenic or apoptotic mediator.
6. Role of ATF3 in maintenance of normal glucose homeostasis
ATF3 is induced in beta cells by many stimuli such as high glucose, fatty acids, reactive oxygen species (ROS), and endoplasmic reticulum (ER) stress.41 Functionally, ATF3 is pro-apoptotic in beta cells, mostly via repression of the pro-survival gene, IRS2.19 To study the role of ATF3 in diabetes, Zmuda et al. fed global Atf3−/− mice a high-fat diet for 12 weeks.41 They found that Atf3−/− mice had better glucose tolerance but unchanged insulin resistance. There were no significant differences in insulin-stimulated Akt-phosphorylation in fat pads and skeletal muscle or inflammation in adipose tissue. In addition, fasting and non-fasting serum insulin levels were significantly lower in Atf3−/− mice. However, there were no differences in beta cell mass, apoptosis, islet area or number.41 Further investigations revealed that ATF3 specifically binds to the insulin promoter in INS-1 beta cells and primary islets to increase its expression.41 Conversely, a study in pancreatic cell lines found that ATF3 increased glucagon transcription, but had no effect on insulin transcription.42 In support of this finding, Wang et al.43 discovered an alternatively spliced isoform of ATF3, named ATF3b, which binds to the ATF/CRE site on the proglucagon gene promoter and increases its transcription. While another study showed that cardiomyocyte-specific knockout of ATF3 led to reduced peripheral glucose tolerance in high-fat diet-fed conditions.44 Hartman et al. further characterized the role of ATF3 in stress-induced β-cell apoptosis.45 They found that ATF3 expression in pancreatic islet cells was induced under stress conditions and ameliorated in islets of Atf3−/− mice.45 Increased ATF3 expression was at least partly dependent on NF-κB and JNK pathways, both of which are stress-related pathways implicated in diabetes.45 In addition, ATF3 expression was highly induced in pancreatic islets from patients with hyperinsulinemia and diabetes.45 Moreover, Zmude et al.46 showed that islets from Atf3−/− mice display greater viability under in vitro stress conditions. In addition, ATF3 increased apoptotic and inflammatory signaling in islet cells. To investigate the role of pancreatic ATF3 in vivo, islets from Atf3−/−mice and wild-type mice were transplanted into C57BL/6J mice to develop a syngeneic transplantation mouse model. Here, it was found that islets from Atf3−/− mice improved glucose homeostasis and significantly lowered apoptosis and inflammation in syngeneic mice.46 This study suggests that pancreatic ATF3 may play an important role in modulating glucose homeostasis. Lee et al.47 generated pancreas and hypothalamus-specific Atf3−/− mice by crossing floxed Atf3 mice with Pdx1-Cre mice. These mice display better glucose tolerance (likely due to reduced food intake and increased energy expenditure) as plasma glucagon or insulin levels were unchanged.47 They further showed that ATF3 regulates agouti-related protein (Agrp) expression, and concluded that hypothalamic ATF3 is involved in regulating glucose and energy metabolism in mice via Agrp regulation.47
To investigate the specific role of the liver in maintaining glucose homeostasis, another group generated liver-specific Atf33 transgenic mice under control of the transthyretin (TTR) promoter.48,49 The first batch of “founder” mice (n = 24) showed no detectable levels of the Atf3 transgene but transmitted the transgene to their offspring, which caused perinatal lethality. Consequently, offspring were obtained by crossing founder mice with BALB/c non-transgenic mice. These F1 hybrids survived almost 4 days, with some surviving up to 7 days. Since the TTR promoter may also be active in the pancreas, some F1 hybrids expressed the Atf3 transgene in the pancreas. Further, some F1 hybrids displayed severe pancreatic defects, including missing islets. In F1 hybrids displaying mild defects, there was abnormal distribution of hormone producing cells, with insulin producing cells at the periphery instead of the center.48 These mice showed a reduction in the number of all four endocrine producing cells. The Atf3 transgene was only expressed in pancreatic ducts where mitosis of endocrine producing cells occurs. Investigation of the mitotic state of these cells revealed significantly lower mitosis in the ductal epithelium of Atf3 transgenic mice.48 In order to eliminate any contribution of pancreatic ATF3 to the phenotype, F1 hybrids expressing the Atf3 transgene in the pancreas were not included.48 In addition, TTR-Atf3 transgenic mice showed elevated serum aspartate transaminase, alanine aminotransferase, and bile acids, defects in conjugation and release of bilirubin, and reduced insulin, glucagon, serum glucose, and white or brown adipose mass.49 ATF3 binds to an ATF/CRE site on the gluconeogenic gene promoter, phosphoenoylpyruvate carboxylase (PEPCK), thus suppressing its expression.49 These observations indicate that hepatic ATF3 suppresses gluconeogenesis.
7. Role of ATF3 in maintenance of glucose homeostasis with chronic ethanol feeding
Chronic ethanol consumption leads to reduced gluconeogenesis in response to increased glucagon levels.50 This is due to increased NADH, which reduces conversion of lactate to pyruvate, a major substrate for gluconeogenesis. Chronic alcohol consumption also causes β-cell dysfunction in the islets of Langerhans of the pancreas.50 This is manifested by reduced insulin secretion from β-cells, along with reduced gene expression of glucokinase, glucose transporter (GLUT)-2, and insulin. Interestingly, ethanol consumption also increases ATF3 expression in the pancreas and liver, most likely as a stress signal.50 Targeted disruption of ATF3 using small interfering RNAs leads to reversal of alcohol-dependent β-cell dysfunction, as well as glucokinase and insulin upregulation.50,51 Down-regulation of the pancreatic and duodenal homeobox factor-1 (PDX-1) gene acts as an important contributor to type 2 diabetes. ATF3 binds to the PDX-1 promoter in β-cells and suppresses its transcription, thereby providing an additional mechanism by which ATF3 alters glucose homeostasis.52 Similarly, increased ATF3 after ethanol consumption leads to reduced binding of PDX-1 to the glucokinase promoter.50,52 Ethanol also reduced fasting blood glucose levels by reducing recruitment of CREB and its co-activator, CREB regulated transcription coactivator 2 (CRTC2), to gluconeogenic gene promoters and suppressing their expression.51 Recruitment of CREB and CRTC2 to gluconeogenic promoters is dependent on ATF3, as ATF3 silencing reversed these effects of ethanol in fasted mice and cultured hepatocytes.51 Altogether, these data suggest that ATF3 plays a critical role in the ethanol-mediated glucose-lowering effect.
8. Role of ATF3 in diabetes and liver injury
In the fasted state, energy metabolism shifts from glucose oxidation to lipid breakdown in adipose tissue, resulting in flux of free fatty acids from adipose tissue to the liver53. When blood glucose levels are low, circulating catecholamines turn on cAMP signaling and reduce glucose uptake into adipocytes by reducing expression and translocation of the glucose uptake gene, GLUT4. In addition, catecholamines also reduce adiponectin expression, which is a key mediator of insulin sensitivity.53 Meanwhile, the hepatic gluconeogenic program is upregulated to maintain blood glucose levels. CREB mediates the effect of catecholamines and other fasting hormones on gene expression. Once phosphorylated, CREB recruits histone acetylase, CREB-binding protein (CBP), and CRTC to gene promoters.53 In the liver, CREB upregulates gluconeogenesis, while in adipose tissue it activates adipogenesis in pre-adipocytes. CREB expression in white adipose tissue (WAT) is upregulated under obese conditions.53 One study used adipocyte-specific CREB knockdown mice to investigate the role of ATF3 in CREB-mediated repression of adiponectin and GLUT4. It was found that adipose-specific knockdown of CREB led to an increase in ATF3 expression in both obese and high-fat diet-fed conditions.53 They further showed that ATF3 blocks transcription of both adiponectin and GLUT4 by directly binding, and thus repressing promoter activities of both genes in adipocytes. Additionally, expression of both adiponectin and GLUT4 is induced in WAT of Atf3−/− mice under high fat diet-fed conditions.53 These data indicate that CREB promotes insulin resistance in WAT by increasing ATF3 expression, which in turn suppress adiponectin and GLUT4 expression.
Tsai et al.54 investigated the role of ryanodine receptor 3 (Ryr3) in regulating adiponectin synthesis and secretion in adipose tissue. Knockdown of Ryr3 in the 3T3-L1 cell line, as well as diabetes (db/db) mice, led to an increase in promoter activity, synthesis, and secretion of adiponectin.54 In addition, in vivo ATF3 silencing led to improved insulin sensitivity and reduced fasting blood glucose levels.54 Overexpression of ATF3 in the 3T3-L1 cell line blocked the effect of RyR3 silencing on adiponectin, indicating that this latter effect is dependent on ATF3.54 Kim et al.55 investigated the role of ATF3 in AMPK-mediated amelioration of β cell dysfunction in obese Zucker diabetic fatty (ZDF) rats. They showed that ATF3 was significantly induced in 19-week-old ZDF rats, along with increased phosphorylation of AMP-activated protein kinase (AMPK), impaired glucose tolerance, and increased β cell dysfunction in islet cells.55 These effects were reversed with chronic 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) treatment and antioxidants. Furthermore, it was found that while ATF3 does not directly affect AMPK phosphorylation, it diminishes the role of AMPK in improvement of ER stress-mediated β cell dysfunction, while ATF3 knockdown reverses this phenomenon.55 Additionally, ATF3 silencing improved glucose homeostasis and reduced production of ROS and apoptosis.55 Taken together, these data suggest that ATF3 may be a potential contributor to development of type 2 diabetes.
Diabetic patients have a higher chance of developing chronic liver disease and hepatocellular carcinoma.56 Treatment with streptozotocin (STZ) is a standard method to induce β-cell damage, and thus type 1 diabetes in animals.56 STZ-induced damage to β-cells is dependent on signal transducer and activator of transcription 1 (STAT1), a known pro-apoptotic gene, as this effect is completely abolished in Stat1−/− mice.56 IFN-γ-associated induction of STAT1 leads to increased hepatic injury and apoptosis.56 Because there are limited data on the mechanisms by which STAT1 signaling causes liver injury, one group hypothesized that STZ-induced STAT1/IFN-γ signaling caused liver injury via ATF3. Indeed, STZ treatment upregulates IFN-γ and STAT1 in diabetic liver injury, along with increased hepatic ATF3 expression.56 STZ-induced liver damage is dependent on STAT1 as Stat1−/− mice are resistant to STZ-induced hepatic damage.56 Moreover, STZ-induced increase in hepatic ATF3 expression was abolished in Ifnγ−/− mice but not Stat1−/−mice. Nonetheless, STAT1 is required for ATF3 translocation from the cytosol to the nucleus in hepatocytes.56 In addition, the C-terminal of ATF3 is required for IFN-γ stimulated increase in STAT1 expression and apoptosis. ATF3 stabilized STAT1 by preventing ubiquitination and proteasomal degradation of STAT1 in hepatocytes.56 These data indicate that STZ-mediated diabetic liver injury may result from increased ATF3 expression and subsequent STAT1 stabilization.
9. Role of ATF3 in hepatic steatosis
Hepatic steatosis results from dysregulated triglyceride synthesis, breakdown, and secretion. Glucocorticoid (GC) therapy is prescribed to patients that have undergone organ transplant to minimize the immune response, or even to patients in an inflammatory state due to a pathological condition.57 One of the mechanisms through which GC inhibits inflammation is to induce expansion of myeloid-derived suppressor cells (MDSC), which in turn suppress other immune cells.58 Expansion of MDSC is also observed in high-fat diet-fed conditions.57 In addition to inhibiting inflammation, GC therapy causes unwanted side effects, such as liver steatosis and hyperglycemia. To investigate the relationship between GC-induced hepatic steatosis and MDSC, gene expression analysis was performed using MDSC derived from the liver of GC-treated mice. It was found that ATF3 expression was significantly reduced in GC-treated MDSC.57 Interestingly, GC failed to induce hepatic steatosis and MDSC accumulation in the liver of Atf3−/− mice. When MDSC from GC-treated WT mice and Atf3−/−mice were transplanted into WT mice, both significantly increased lipid accumulation in recipient liver compared with vehicle-treated WT and Atf3−/− controls. It was also found that glucocorticoid receptors (GR) directly bind to and repress promoter activity of ATF3 in primary MDSC isolated from bone marrow, spleen, or liver.57 In Atf3−/− mice, the proportion of immature myeloid cells to mature myeloid cells was significantly lower,57 suggesting that ATF3 promotes MDSC development. Further investigations revealed significantly induced expression of S100A9 (which is involved in myeloid cell differentiation) in Atf3−/− mice. Therefore, S100A9 was identified as a unique downstream target of ATF3 in MDSC, and silencing S100A9 significantly alleviated GC-induced hepatic steatosis phenotype.57 These data indicate that upon GC activation, GR binds to the ATF3 promoter and represses its expression, resulting in induction of S10049 and subsequent expansion and differentiation of MDSC and hepatosteatosis.57
Under diabetic and obese conditions, the liver packages excess triglycerides and cholesterol with ApoB into very low-density lipoprotein (VLDL) particles for secretion into the bloodstream. Sortilin 1 (Sort1) has recently been shown to increase post-transcriptional modification of ApoB, possibly by sending it to the proteasome for degradation, and thus reducing hepatic VLDL secretion.59 To investigate the role of ER stress in regulation of Sort1 expression, mice were injected with either DMSO or tunicamycin, a known ER stress inducer. In addition to inducing other ER stress-related proteins, tunicamycin increased Atf3 expression and reduced Sort1 expression.59 Further investigations revealed that ATF3 binds to the Sort1 promoter and reduces its expression. Use of siAtf3 in obese (ob/ob) mice showed increased Sort1 expression and reduced triglyceride and ApoB secretion.59 These data indicate that ATF3 may regulate hepatic VLDL secretion via the SORT1-ApoB pathway in ob/ob mice.
10. Conclusions
In summary, ATF3 plays important roles in regulating the immune response, cancer development, and glucose homeostasis. While at present, the role of ATF3 in liver injury is not well characterized, and there is no definite consensus on whether ATF3 regulates hepatic triglyceride levels. Given that ATF3 has a profound effect on repressing inflammation, it is tempting to speculate that ATF3 plays a role in pathogenesis of fatty liver disease. Moreover, current studies suggest that ATF3 may act as an oncogene or tumor suppressor, depending on the tissue. ATF3 also appears to have differing effects on glucose homeostasis: ATF3 inhibits gluconeogenesis in the liver, but down-regulates expression of GLUT4 and adiponectin in adipose tissue. The role of pancreatic ATF3 in glucose homeostasis is controversial, yet hypothalamic ATF3 appears to regulate glucose homeostasis by modulating food intake and energy metabolism (Fig.3). The role of ATF3 in macrophage foam cell formation is well defined, and studies have suggested that macrophage ATF3 protects against atherosclerosis. To better understand the role of ATF3 in immune responses, cancer development, and metabolic regulation, it is essential to use tissue-specific gain- or loss-of-function approaches in the future.
Fig.3. Role of ATF3 in metabolic regulation.
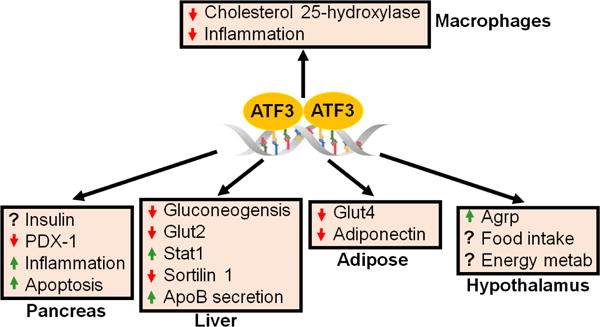
ATF3 appears to have distinct functions in different tissues. The role of ATF3 in the pancreas is controversial. In the liver, ATF3 inhibits gluconeogenesis and sortilin 1, and increases apolipoprotein B (ApoB) secretion. In adipose tissue, ATF3 may cause insulin resistance by inhibiting glucose transporter 4 (Glut4) and adiponectin expression. Loss of hypothalamic ATF3 inhibits agouti-related protein (Agrp) expression and food intake, and increases energy metabolism. In macrophages, ATF3 inhibits cholesterol 25-hydroxylase and inflammation, resulting in reduced foam cell formation.
Acknowledgments
This work was supported by USA National Institutes of Health (NIH) grants R01HL103227, R01DK102619, and R21AA024946 (to Y.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Edited by Peiling Zhu and Genshu Wang
Conflict of interest
The authors declare no conflicts of interest.
References
- 1.Hummler E, Cole TJ, Blendy JA, et al. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci U S A. 1994;91:5647–5651. doi: 10.1073/pnas.91.12.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen BP, Liang G, Whelan J, Hai T. ATF3 and ATF3 delta Zip. Transcriptional repression versus activation by alternatively spliced isoforms. J Biol Chem. 1994;269:15819–15826. [PubMed] [Google Scholar]
- 3.Hai T, Wolford CC, Chang YS. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Expr. 2010;15:1–11. doi: 10.3727/105221610x12819686555015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 5.Liang G, Wolfgang CD, Chen BP, Chen TH, Hai T. ATF3 gene. Genomic organization, promoter, and regulation. J Biol Chem. 1996;271:1695–1701. doi: 10.1074/jbc.271.3.1695. [DOI] [PubMed] [Google Scholar]
- 6.Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- 7.Foulkes NS, Borrelli E, Sassone-Corsi P. CREM gene: use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell. 1991;64:739–749. doi: 10.1016/0092-8674(91)90503-q. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez GA, Yamamoto KK, Fischer WH, et al. A cluster of phosphorylation sites on the cyclic AMP-regulated nuclear factor CREB predicted by its sequence. Nature. 1989;337:749–752. doi: 10.1038/337749a0. [DOI] [PubMed] [Google Scholar]
- 9.Hoeffler JP, Meyer TE, Yun Y, Jameson JL, Habener JF. Cyclic AMP-responsive DNA-binding protein: structure based on a cloned placental cDNA. Science. 1988;242:1430–1433. doi: 10.1126/science.2974179. [DOI] [PubMed] [Google Scholar]
- 10.Maekawa T, Sakura H, Kanei-Ishii C, et al. Leucine zipper structure of the protein CRE-BP1 binding to the cyclic AMP response element in brain. EMBO J. 1989;8:2023–2028. doi: 10.1002/j.1460-2075.1989.tb03610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hashimoto Y, Zhang C, Kawauchi J, et al. An alternatively spliced isoform of transcriptional repressor ATF3 and its induction by stress stimuli. Nucleic Acids Res. 2002;30:2398–2406. doi: 10.1093/nar/30.11.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. ATF3 and stress responses. Gene Expr. 1999;7:321–335. [PMC free article] [PubMed] [Google Scholar]
- 13.Akira S. TLR signaling. Curr Top Microbiol Immunol. 2006;311:1–16. doi: 10.1007/3-540-32636-7_1. [DOI] [PubMed] [Google Scholar]
- 14.Swann JB, Vesely MD, Silva A, et al. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A. 2008;105:652–656. doi: 10.1073/pnas.0708594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitmore MM, Iparraguirre A, Kubelka L, Weninger W, Hai T, Williams BR. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. 2007;179:3622–3630. doi: 10.4049/jimmunol.179.6.3622. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen CT, Kim EH, Luong TT, Pyo S, Rhee DK. TLR4 mediates pneumolysin-induced ATF3 expression through the JNK/p38 pathway in Streptococcus pneumoniae-infected RAW 264.7 cells. Mol Cells. 2015;38:58–64. doi: 10.14348/molcells.2015.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson MR, Xu D, Williams BR. Activating transcription factor 3 contributes to Toll-like receptor-mediated macrophage survival via repression of Bax and Bak. J Interferon Cytokine Res. 2013;33:682–693. doi: 10.1089/jir.2013.0007. [DOI] [PubMed] [Google Scholar]
- 18.Rao J, Qian X, Li G, et al. ATF3-mediated NRF2/HO-1 signaling regulates TLR4 innate immune responses in mouse liver ischemia/reperfusion injury. Am J Transplant. 2015;15:76–87. doi: 10.1111/ajt.12954. [DOI] [PubMed] [Google Scholar]
- 19.Thompson MR, Xu D, Williams BR. ATF3 transcription factor and its emerging roles in immunity and cancer. J Mol Med (Berl) 2009;87:1053–1060. doi: 10.1007/s00109-009-0520-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith CK, Vivekanandan-Giri A, Tang C, et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014;66:2532–2544. doi: 10.1002/art.38703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith CK, Seto NL, Vivekanandan-Giri A, et al. Lupus high-density lipoprotein induces proinflammatory responses in macrophages by binding lectin-like oxidised low-density lipoprotein receptor 1 and failing to promote activating transcription factor 3 activity. Ann Rheum Dis. 2017;76:602–611. doi: 10.1136/annrheumdis-2016-209683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng S, Abraham C. NF-kappaB1 inhibits NOD2-induced cytokine secretion through ATF3-dependent mechanisms. Mol Cell Biol. 2013;33:4857–4871. doi: 10.1128/MCB.00797-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilchrist M, Thorsson V, Li B, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 24.Hoetzenecker W, Echtenacher B, Guenova E, et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med. 2011;18:128–134. doi: 10.1038/nm.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenberger CM, Clark AE, Treuting PM, Johnson CD, Aderem A. ATF3 regulates MCMV infection in mice by modulating IFN-gamma expression in natural killer cells. Proc Natl Acad Sci U S A. 2008;105:2544–2549. doi: 10.1073/pnas.0712182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai PF, Cheng CF, Lin H, Tseng TL, Chen HH, Chen SH. ATF3 protects against LPS-induced inflammation in mice via inhibiting HMGB1 expression. Evid Based Complement Alternat Med. 2013;2013:716481. doi: 10.1155/2013/716481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khuu CH, Barrozo RM, Hai T, Weinstein SL. Activating transcription factor 3 (ATF3) represses the expression of CCL4 in murine macrophages. Mol Immunol. 2007;44:1598–1605. doi: 10.1016/j.molimm.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 28.Boespflug ND, Kumar S, McAlees JW, et al. ATF3 is a novel regulator of mouse neutrophil migration. Blood. 2014;123:2084–2093. doi: 10.1182/blood-2013-06-510909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho HH, Antoniv TT, Ji JD, Ivashkiv LB. Lipopolysaccharide-induced expression of matrix metalloproteinases in human monocytes is suppressed by IFN-gamma via superinduction of ATF-3 and suppression of AP-1. J Immunol. 2008;181:5089–5097. doi: 10.4049/jimmunol.181.7.5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labzin LI, Schmidt SV, Masters SL, et al. ATF3 is a key regulator of macrophage IFN responses. J Immunol. 2015;195:4446–4455. doi: 10.4049/jimmunol.1500204. [DOI] [PubMed] [Google Scholar]
- 31.De Nardo D, Labzin LI, Kono H, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol. 2014;15:152–160. doi: 10.1038/ni.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gold ES, Ramsey SA, Sartain MJ, et al. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J Exp Med. 2012;209:807–817. doi: 10.1084/jem.20111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo H, Wang J, Qiao C, Zhang X, Zhang W, Ma N. ATF3 inhibits Tenascin-C-induced foam cell formation in LPS-stimulated THP-1 macrophages by suppressing TLR-4. J Atheroscler Thromb. 2015;22:1214–1223. doi: 10.5551/jat.28415. [DOI] [PubMed] [Google Scholar]
- 34.Yin X, Dewille JW, Hai T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene. 2008;27:2118–2127. doi: 10.1038/sj.onc.1210861. [DOI] [PubMed] [Google Scholar]
- 35.Pelzer AE, Bektic J, Haag P, et al. The expression of transcription factor activating transcription factor 3 in the human prostate and its regulation by androgen in prostate cancer. J Urol. 2006;175:1517–1522. doi: 10.1016/S0022-5347(05)00651-8. [DOI] [PubMed] [Google Scholar]
- 36.Janz M, Hummel M, Truss M, et al. Classical Hodgkin lymphoma is characterized by high constitutive expression of activating transcription factor 3 (ATF3), which promotes viability of Hodgkin/Reed-Sternberg cells. Blood. 2006;107:2536–2539. doi: 10.1182/blood-2005-07-2694. [DOI] [PubMed] [Google Scholar]
- 37.Wolfgang CD, Chen BP, Martindale JL, Holbrook NJ, Hai T. gadd153/Chop10, a potential target gene of the transcriptional repressor ATF3. Mol Cell Biol. 1997;17:6700–6707. doi: 10.1128/mcb.17.11.6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bottone FG, Jr, Martinez JM, Collins JB, Afshari CA, Eling TE. Gene modulation by the cyclooxygenase inhibitor, sulindac sulfide, in human colorectal carcinoma cells: possible link to apoptosis. J Biol Chem. 2003;278:25790–25801. doi: 10.1074/jbc.M301002200. [DOI] [PubMed] [Google Scholar]
- 39.Wang A, Arantes S, Yan L, et al. The transcription factor ATF3 acts as an oncogene in mouse mammary tumorigenesis. BMC Cancer. 2008;8:268. doi: 10.1186/1471-2407-8-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 41.Zmuda EJ, Qi L, Zhu MX, Mirmira RG, Montminy MR, Hai T. The roles of ATF3, an adaptive-response gene, in high-fat-diet-induced diabetes and pancreatic beta-cell dysfunction. Mol Endocrinol. 2010;24:1423–1433. doi: 10.1210/me.2009-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YS, Kobayashi M, Kikuchi O, et al. ATF3 expression is induced by low glucose in pancreatic alpha and beta cells and regulates glucagon but not insulin gene transcription. Endocr J. 2014;61:85–90. doi: 10.1507/endocrj.ej13-0383. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Cao Y, Steiner DF. Regulation of proglucagon transcription by activated transcription factor (ATF) 3 and a novel isoform, ATF3b, through the cAMP-response element/ATF site of the proglucagon gene promoter. J Biol Chem. 2003;278:32899–32904. doi: 10.1074/jbc.M305456200. [DOI] [PubMed] [Google Scholar]
- 44.Kalfon R, Koren L, Aviram S, Schwartz O, Hai T, Aronheim A. ATF3 expression in cardiomyocytes preserves homeostasis in the heart and controls peripheral glucose tolerance. Cardiovasc Res. 2017;113:134–146. doi: 10.1093/cvr/cvw228. [DOI] [PubMed] [Google Scholar]
- 45.Hartman MG, Lu D, Kim ML, et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zmuda EJ, Viapiano M, Grey ST, Hadley G, Garcia-Ocaña A, Hai T. Deficiency of Atf3, an adaptive-response gene, protects islets and ameliorates inflammation in a syngeneic mouse transplantation model. Diabetologia. 2010;53:1438–1450. doi: 10.1007/s00125-010-1696-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee YS, Sasaki T, Kobayashi M, et al. Hypothalamic ATF3 is involved in regulating glucose and energy metabolism in mice. Diabetologia. 2013;56:1383–1393. doi: 10.1007/s00125-013-2879-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen-Jennings AE, Hartman MG, Kociba GJ, Hai T. The roles of ATF3 in glucose homeostasis. A transgenic mouse model with liver dysfunction and defects in endocrine pancreas. J Biol Chem. 2001;276:29507–29514. doi: 10.1074/jbc.M100986200. [DOI] [PubMed] [Google Scholar]
- 49.Allen-Jennings AE, Hartman MG, Kociba GJ, Hai T. The roles of ATF3 in liver dysfunction and the regulation of phosphoenolpyruvate carboxykinase gene expression. J Biol Chem. 2002;277:20020–20025. doi: 10.1074/jbc.M200727200. [DOI] [PubMed] [Google Scholar]
- 50.Kim JY, Song EH, Lee HJ, et al. Chronic ethanol consumption-induced pancreatic {beta}-cell dysfunction and apoptosis through glucokinase nitration and its down-regulation. J Biol Chem. 2010;285:37251–37262. doi: 10.1074/jbc.M110.142315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai WW, Matsumura S, Liu W, et al. ATF3 mediates inhibitory effects of ethanol on hepatic gluconeogenesis. Proc Natl Acad Sci U S A. 2015;112:2699–2704. doi: 10.1073/pnas.1424641112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jang MK, Park HJ, Jung MH. ATF3 represses PDX-1 expression in pancreatic beta-cells. Biochem Biophys Res Commun. 2011;412:385–390. doi: 10.1016/j.bbrc.2011.07.108. [DOI] [PubMed] [Google Scholar]
- 53.Qi L, Saberi M, Zmuda E, et al. Adipocyte CREB promotes insulin resistance in obesity. Cell Metab. 2009;9:277–286. doi: 10.1016/j.cmet.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsai SH, Chang EY, Chang YC, et al. Knockdown of RyR3 enhances adiponectin expression through an atf3-dependent pathway. Endocrinology. 2013;154:1117–1129. doi: 10.1210/en.2012-1515. [DOI] [PubMed] [Google Scholar]
- 55.Kim JY, Park KJ, Kim GH, et al. In vivo activating transcription factor 3 silencing ameliorates the AMPK compensatory effects for ER stress-mediated beta-cell dysfunction during the progression of type-2 diabetes. Cell Signal. 2013;25:2348–2361. doi: 10.1016/j.cellsig.2013.07.028. [DOI] [PubMed] [Google Scholar]
- 56.Kim JY, Lee SH, Song EH, et al. A critical role of STAT1 in streptozotocin-induced diabetic liver injury in mice: controlled by ATF3. Cell Signal. 2009;21:1758–1767. doi: 10.1016/j.cellsig.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu YF, Wei JY, Shi MH, Jiang H, Zhou J. Glucocorticoid induces hepatic steatosis by inhibiting activating transcription factor 3 (ATF3)/S100A9 protein signaling in granulocytic myeloid-derived suppressor cells. J Biol Chem. 2016;291:21771–21785. doi: 10.1074/jbc.M116.726364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li L, Zhang T, Diao W, et al. Role of myeloid-derived suppressor cells in glucocorticoid-mediated amelioration of FSGS. J Am Soc Nephrol. 2015;26:2183–2197. doi: 10.1681/ASN.2014050468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ai D, Baez JM, Jiang H, et al. Activation of ER stress and mTORC1 suppresses hepatic sortilin-1 levels in obese mice. J Clin Invest. 2012;122:1677–1687. doi: 10.1172/JCI61248. [DOI] [PMC free article] [PubMed] [Google Scholar]