

Abstract
BACKGROUND
Aging is a highly complex biological process driven by multiple factors. Its progression can partially be influenced by nutritional interventions. Vitamin E is a lipid-soluble anti-oxidant that is investigated as nutritional supplement for its ability to prevent or delay the onset of specific aging pathologies, including neurodegenerative disorders.
PURPOSE
We aimed here to investigate the effect of vitamin E during aging progression in a well characterized mouse model for premature aging.
METHOD
Xpg−/− animals received diets with low (~2.5 mg/kg feed), medium (75 mg/kg feed) or high (375 mg/kg feed) vitamin E concentration and their phenotype was monitored during aging progression. Vitamin E content was analyzed in the feed, for stability reasons, and in mouse plasma, brain, and liver, for effectiveness of the treatment. Subsequent age-related changes were monitored for improvement by increased vitamin E or worsening by depletion in both liver and nervous system, organs sensitive to oxidative stress.
RESULTS
Mice supplemented with high levels of vitamin E showed a delayed onset of age-related body weight decline and appearance of tremors when compared to mice with a low dietary vitamin E intake. DNA damage resulting in liver abnormalities such as changes in polyploidy, was considerably prevented by elevated amounts of vitamin E. Additionally, immunohistochemical analyses revealed that high intake of vitamin E, when compared with low and medium levels of vitamin E in the diet, reduces the number of p53-positive cells throughout the brain, indicative of a lower number of cells dying due to DNA damage accumulated over time.
CONCLUSIONS
Our data underline a neuroprotective role of vitamin E in the premature aging animal model used in this study, likely via a reduction of oxidative stress, and implies the importance of improved nutrition to sustain health.
Keywords: Vitamin E, neurodegeneration, aging, DNA damage repair, anti-aging interventions
Introduction
Aging is a highly complex biological process whose progression is generally associated with time-dependent accumulation of cellular damage (1, 2). For instance, the integrity of DNA is continually challenged by exogenous and endogenous threats including reactive oxygen species (ROS), leading to time-dependent accumulation of DNA lesions, which interfere with vital DNA metabolism and may cause cellular dysfunction and cell death (3, 4). At high concentrations, ROS are detrimental and responsible for compromising normal cellular functioning (5). Young and healthy cells are equipped with complex anti-oxidant defense systems to prevent the high reactivity of ROS, and various DNA repair mechanisms to repair (oxidative) DNA lesions (4, 5).
One such DNA repair system is the nucleotide excision repair (NER) pathway that is primarily responsible for clearing helix-distorting DNA lesions (6). This pathway consists of two branches using a distinct manner of damage recognition. One scans the whole genome for lesions and is named global genome (GG) NER while the other is triggered by transcription stalled by DNA damage and therefore called transcription-coupled NER. In total, NER employs over 30 proteins, including the endonuclease XPG (also known as ERCC5). In both sub-branches of NER, XPG cuts the DNA 5–6 nucleotides downstream of the damage, as part of a dual incision mechanism for excision of the damage (6, 7). Additionally, XPG has also been implicated in other processes such as homologous recombination repair of double stranded DNA breaks and removal of small oxidative DNA lesions by base excision repair (8, 9). Mutations that affect activity or expression levels of the endonuclease XPG are rare and cause a spectrum of severe symptoms, including striking hypersensitivity to sun exposure, early cessation of growth, accelerated aging features (including progressive neurodegeneration and body weight decline) and reduced life expectancy (10) a constellation of features characteristic of the rare transcription-coupled NER condition Cockayne syndrome (CS). Some XPG mutations strongly predispose patients to cancer and pigmentation abnormalities in sun-exposed parts of the skin, typical of the rare (GG-) NER disorder xeroderma pigmentosum (XP), or a combination of XP/CS.
Recently, a new DNA repair-deficient mutant mouse (Xpg−/−) was generated to mimic the genetic defects in Cockayne syndrome-type XP-G patients. This model is characterized by shortened lifespan, of about 18 weeks, and accelerated onset of multiple progressive aging features such as prominent neurodegeneration, including loss of hearing, vision and motor performance, cognitive decline, early development of tremors, imbalance and paresis, as well as cachexia, age-related abnormalities in multiple tissues and organs including liver, kidney, skeleton, intestine, skin, retina, muscles, vascular system and heart and overall frailty, strikingly resembling Cockayne syndrome in man (11, 12) (and unpublished results).
Alterations in macronutrients can have a great impact on health and aging of mice and other organisms (13). Xpg−/− mice are very sensitive to nutritional changes and respond remarkably well to dietary interventions (14). Indeed, a reduction of feed intake by 30%, known as diet restriction, that in normally aging wild type mice would yield in about 30% lifespan extension, drastically extended the lifespan of these animal models by 80% and delayed the onset of many age-related neurological abnormalities, such as tremors, imbalance and paresis (14).
Vitamin E includes eight structurally-related lipid-soluble compounds with potent anti-oxidant properties consisting of four tocopherols and four tocotrienols: α (alpha), β (beta), γ (gamma) and δ (delta) (15). α-Tocopherol is the most abundant and bioavailable form of vitamin E in human and rodents (16, 17) and serves as a strong ROS scavenger that protects polyunsaturated fatty acids in the cellular membranes and in lipoproteins from peroxidation (18).
Since vitamin E is known as potent anti-oxidant, we hypothesize that supplementing Xpg−/− mice with vitamin E might reduce some of the aging features driven by (oxidative) DNA damage accumulation. Although a number of papers showed beneficial effects associated with vitamin E supplementation, few reports described inconsistent or negative results (19–21) and references within]. The difficulty in performing precise and uniform human studies resides in several important variables such as: age and health status of the individuals included in the study, nutritional status and diet, form of vitamin E administered, but also quantity and time of supplementation (19). Moreover, other important parameters to be considered are the genetic variations that may alter absorption, availability, metabolism, excretion and therefore net activity of these compounds. Working with animal models may reduce the variability associated with all these parameters and therefore in vivo studies represent excellent tools to unravel the function of specific nutrients during aging progression.
Materials and Methods
Mouse model
The generation, genotyping and characterization of Xpg−/− mice has been previously described (11). Xpg−/− mice were obtained by crossing Xpg+/− mice with a pure C57BL6J and pure FVB backgrounds to yield Xpg−/− pups with a F1 C57BL6J/FVB hybrid background. Typical unfavorable characteristics, like blindness in an FVB background or deafness in a C57BL6J background, do not occur in this hybrid background (11).
Housing conditions
Since Xpg−/− mice are smaller than their wildtype littermates, feed was administered within the cages and water bottles with long nozzles were used. Animals were maintained in a controlled environment (20–22°C, 12h light : 12h dark cycle) and were housed in individual ventilated cages under Specific Pathogen-Free conditions. All animals were bred on AIN93G synthetic pellets (Research Diet Services B.V., Wijk bij Duurstede, The Netherlands; gross energy content 4.9 kcal/g dry mass, digestible energy 3.97 kcal/g; with choline bitartrate replaced for choline chloride to avoid potential formation of bladder and kidney stones).
Study description
Immediately after birth, litters together with their mothers were distributed into three groups receiving three diets taking into account: day of birth, litter size, and genetic background of the mother. All diets were relabeled before entering the mouse facility and given in a blinded manner. After weaning, at 4 weeks of age, all mice were kept on the designated diet and were individually housed to accurately measure feed intake. The diets were: a) AIN93G synthetic pellets without the addition of any vitamin E (hereafter referred to as “low”, considering this diet still contains traces of vitamin E from the Casein protein source); b) AIN93G pellets containing medium levels (75 mg/kg) of vitamin E (all-rac-α-tocopheryl acetate; hereafter referred to as “medium”, which is normally present in this diet, and is slightly higher than the recommended dosage for mice); c) AIN93G pellets with addition of extra vitamin E to a final concentration of 375 mg/kg (hereafter referred to as “high”). Mice were analyzed at 8 and 12 weeks of age (Figure 1a).
Figure 1. Study design and vitamin E content in the three diets.
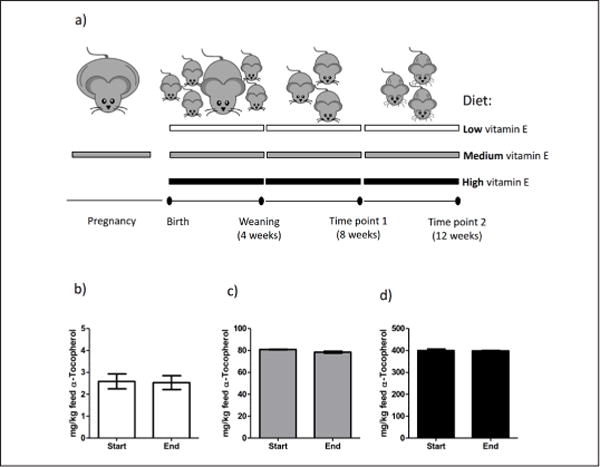
a) Schematic view of the study design. The three diets used in the study are indicated by the horizontal elongated rectangles: white (low vitamin E diet: traces), grey (medium vitamin E diet: 75 mg/kg), black (high vitamin E diet: 375 mg/kg). Lower black line represents timeline with black dots representing key events during the study. b-d) α-Tocopherol measurements in the three diets: low, medium, high respectively. “Start” and “End” indicate the beginning and the final stage of the supplementation study. Error bars represent standard error of the mean (s.e.m.).
Phenotype scoring: body weight and tremors
Mice were clinically examined on a daily basis by the animal caretakers. Moreover, they were weighed, visually inspected and phenotypically scored for age related symptoms in a blinded manner by two experienced research technicians. The onset of body weight loss was counted as the first week a decline in body weight was noted after their maximal body weight was reached. Whole body tremor was scored if mice showed trembling for a combined total of at least 10 seconds when put on a flat surface for 20 seconds.
Micronutrients measurements
Chemicals and reagents
Retinol (vitamin A), α-tocopherol (vitamin E), thiamin hydrochloride (vitamin B1), riboflavin (vitamin B2) and pyridoxal-5′-phosphate (vitamin B6) were purchased from Sigma-Aldrich. The corresponding deuterated internal standard, retinol-d5, thiamin-d3, riboflavine 13C4 15N2 and pyridoxal-d3 5-phosphate were purchased from medical isotopes and vitamin A-d5 acetate from Alsachim. Other chemicals used were analytical grade and obtained from either Sigma-Aldrich or Merck Millipore. Water used was passed through a Milli-Q water purification system (Millipore).
Instrumentation
Separation and quantification was performed on an Agilent 1290 ultra-high performance liquid chromatography (UHPLC) coupled with an API 4000 mass spectrophotometer (MS) from AB Sciex using multiple reaction monitoring (MRM) transitions. A turbo ion spray source operating in positive mode was used for water soluble vitamins determination and a photospray ionization source operating in positive mode was used for fat soluble vitamins determination.
For B-vitamin analysis, the analytical column was an Ascentis Express C8 from Supelco. Samples were eluted using a gradient from 100% of acidified water (with acetic acid) to 100% of acetonitrile, with help of heptafluorobutiric acid used as ion pairing reagent.
For vitamin A and vitamin E determination, the analytical column was a Halo C18 from Advanced material technologies. Samples were eluted using a gradient from 8% of acidified water (with formic acid) to 100% of an acidified methanol/acetonitrile solution.
Sample preparation
Water soluble vitamins in plasma, brain and liver: in order to remove the protein and extract the B-vitamins, an aliquot of plasma sample or a part of the tissues (brain or liver) was combined with a solution of trichloroacetic acid (50g/L) containing the internal standards. After centrifugation (and filtration for tissue samples) the supernatant was injected on the UHPLC-MS system.
Vitamin A and vitamin E in plasma: after addition of internal standards to an aliquot of plasma, proteins were removed with the addition of a mixture of tetrahydrofuran, acetonitrile and methanol. After filtration and centrifugation, the supernatant was injected on the UHPLC-MS system.
Vitamin A and vitamin E in liver: internal standards and vitamin C (as anti-oxidant) were added to the tissue. A saponification was conducted with a methanol/potassium hydroxide solution, followed by a liquid-liquid extraction with hexane and a concentration step, the resulting supernatant was injected on the UHPLC-MS system.
Vitamin A and vitamin E in brain: internal standards and water/ethanol solution were added to the tissue samples. After a liquid-liquid extraction with hexane and concentration step, the resulting supernatant was injected on the UHPLC-MS system.
Quantification
Identification and quantification were performed applying external calibrations (using dedicated internal standards). Moreover, quality control samples were prepared with the similar procedure as the unknown samples and analyzed at each run. These methods enable the measurement of water and fat soluble vitamins with good accuracy (85–115%), inter and intra-day precision (<15%) in plasma, liver and brain. In addition, lower limits of quantitation (LOQ) were determined for tocopherol (250 ng/mL in plasma and 5.00 ng/mg in tissue), retinol (25.0 ng/mL in plasma and 0.50 ng/mg in tissue), thiamine (5.00 ng/mL in plasma and 0.050 ng/mg in tissue), riboflavin (0.5 ng/mL in plasma and 0.020 ng/mg in tissue) and vitamin B6 (0.5 ng/mL in plasma and 0.020 ng/mg in tissue).
Vitamin E measurements in the feed
Vitamin E in feed was analyzed as reported in (22). Briefly after saponification with with ethanolic potassium hydroxide, the unsaponifiable residue of the sample is extracted with cyclohexane/diethyl ether. Quantification is carried out with normal phase HPLC and fluorescence detection using α-tocopherol as external standard. With this analytical method, the sum of free as well as esterified α-tocopherol (α-tocopheryl acetate) is determined.
Markers of oxidative stress
Redox state of thiol groups
Two independent research groups previously developed an assay for monitoring thiol redox states (23, 24). Both assays used chemical modifiers that can specifically bind covalently with, and thereby inactivate, free thiol groups. Reducing agents cannot remove these modifications. After subsequent reduction of oxidized thiol group, that were engaged in disulfide bonds, these could be labeled with a second compound, to monitor thiol-based redox changes using mass spectrometry or histology (23, 24). Both protocols were here combined into an immunoblotting-based assay: To quantify the SS/SH redox ratio ~20 mg of liver tissue was lysed and sonicated in 400 μl “reduced thiol labeling solution”, containing 50 mM Tris-Cl pH 7.5, 1% SDS, 1 mM EDTA, 10 mM N-Ethylmaleimide (NEM; Sigma-Aldrich), 20 μM Alexa 800 maleimide (Thermo Fisher Scientific), and 1 protease inhibitor cocktail tablet (Roche) per ml dH2O for 50x conc (25). After 5 minutes heating at 70°C and 30 minutes incubation at room temperature the labeling was quenched by adding fresh NEM to a final concentration of 100 mM. 100 μl homogenate was precipitated in 1 ml cold Acetone:MeOH:EtOH (2:1:1) and re-suspended in 50 mM Tris-Cl pH 7.5, 1% SDS, 10 mM TCEP (Sigma-Aldrich) by vigorously pipetting up and down. Samples were heated for 5 minutes at 70°C, incubated for 30 minutes at room temperature and precipitated again with 1 ml cold Acetone:MeOH:EtOH (2:1:1). Pellets were re-dissolved in 200 μl “oxidized thiol labeling solution”, containing 50 mM Tris-Cl pH 7.5, 1% SDS, 10 mM NEM, and 20 μM Alexa 680 maleimide (Thermo Fisher Scientific), to label the thiols groups previously engaged in disulfide bonds. After 30 minutes incubation at room temperature and another round of precipitation, samples were dissolved in SDS-sample buffer and run on PAGE. Gel was fixed in 1% Orto-phosphoric acid, 50% EtOH and scanned using the Odyssey (Li-Cor Biosciences, USA). The ratio between the signal intensity per lane was quantified using the instruments software.
Protein carbonylation
For determination of the protein carbonyl content ~20 mg of liver tissue was lysed in 400 μl SDS-sample buffer. 50 μg homogenate was transferred to a new tube, supplemented with SDS to a total concentration of 12%, and incubated for 30 minutes at room temperature with 2 volumes of 20 mM 2,4-Dinitrophenylhydrazine (DNPH; Sigma-Aldrich) in 10% Trifluoroacetic acid (TCA) or 2 volumes 10% TCA only as negative control. The reaction was neutralized with 1.7 starting volume of 2M Tris base containing 30% glycerol and the protein samples were transferred by dotblot to PVDF membrane (Millipore). Carbonylation was visualized with anti-DNPH (Sigma-Aldrich; D8406; 1:2,000) in 5% BSA in PBS-Tween 0.05% and rat anti-mouse IgE-HRP (SouthernBiotech; 1130–05; 1:5,000), using ECL+ (PerkinElmer) on the ImageQuant LAS4000 mini (GE Healthcare Life Sciences). Signal intensities were quantified in ImageQuant TL software and corrected for total protein by Ponceau S. All samples were analyzed at least in duplo.
Histological analysis of nuclear DNA content
Fresh frozen liver tissue was embedded in TissueTek and sliced in 10 μm thick cryosections. Sections were shortly dried, post-fixed for 10 minutes in 4% paraformaldehyde, and stained with hematoxylin. Images were generated using the NanoZoomer Digital slide scanner (Hamamatsu Photonics, Japan) and the nuclei size was quantified using NDP view software in a blinded manner. A similar number of images was quantified for each group.
Oil Red O staining
Tissues were snap-frozen in liquid nitrogen, embedded in TissueTek and sliced in 10 μm thick cryosections and mounted on Superfrost Plus slides. Oil red O solution was applied to liver sections for 5 min. Slides were washed twice in water, 15 min each wash, and counterstained with hematoxylin. Oil red O images were generated using the NanoZoomer Digital slide scanner (Hamamatsu Photonics, Japan) and the Oil Red O intensity was quantified using Fiji software.
TUNEL staining
To quantify apoptotic cells in the retina, eyes were fixed overnight in 10% phosphate-buffered formalin, paraffin-embedded, sectioned at 5 μm, and mounted on Superfrost Plus slides. Paraffin sections were employed for TdT-mediated dUTP Nick-End Labeling (TUNEL) assay using a commercial kit (Apoptag Plus Peroxidase in situ apoptosis detection kit, Millipore). Sections were deparaffinized and incubated as described by the manufacturer.
Histological examination of neurodegeneration
Mice were anaesthetized with pentobarbital and transcardially perfused with 4% paraformaldehyde. Brains were carefully dissected out, post-fixed for 1.5 h in 4% paraformaldehyde, cryoprotected, embedded in 12% gelatin, rapidly frozen, and sectioned at 40 μm using a freezing microtome or stored at −80°C until use. Brain tissues of mice from different dietary regimes were embedded together, in one gelatin block per time point, to avoid fluctuations in antibody affinity. To quantitatively assess protein regulation, all immunohistochemical procedures were performed simultaneously for each antibody.
The extent of astrogliosis was assessed using rabbit anti-GFAP (DAKO; Z0334; 1:15,000) as primary antibody with a biotinylated secondary antibody from Vector Laboratories (BA1000; 1:200). All sections were processed free floating using the ABC method (ABC, Vector Laboratories, USA) with diaminobenzidine (0.05%; Sigma-Aldrich) as the chromogen. Immunoperoxidase-stained sections were imaged using the NanoZoomer Digital slide scanner with the NDP view software (Hamamatsu Photonics, Japan). Mean intensities were quantified using Fiji software.
The extent of neuronal genomic stress was assessed using rabbit anti-p53 (Leica; NCL-p53-CM5p; 1:1,000) with Cy3-conjugated goat-anti-rabbit (Jackson ImmunoResearch; 111-165-144; 1:200) and DAPI for cell nuclei. Immunofluorescence sections were analyzed using a Zeiss LSM700 confocal microscope. P53 quantification was performed using Fiji software.
Statistics
Statistical differences were calculated using a one-way ANOVA including Bonferroni’s multiple comparison test or Dunnett t (2-sided) using IBM SPSS Statistics 21. For the vitamin E quantification in the feed (two groups), and the vitamin measurements in liver and brain (too low n) statistics was assessed with a t-test. Plasma vitamin levels were assessed with a one-way ANOVA and Bonferroni’s multiple comparison test. Onset of tremors and body weight decline were statistically analyzed with survival curve analysis using the product limit method of Kaplan and Meier with Log-rank (Mantel-Cox) test in GraphPad Prism. Significance is indicated in the tables and figures by (*) p values < 0.05; (**) p values <0.01; (***) p values <0.001.
Results
Concentration of vitamin E and other micronutrients
We first measured the concentration of α-Tocopherol in the three diets at the beginning and at the end of the study (Figure 1b–d). As expected, the measured values were in agreement with the theoretical concentrations (~2.5, 75, and 375 mg/kg feed respectively) and we did not observe any change in vitamin E stability over time (Figure 1b–d, compare “Start” with “End” stage). Secondly, we examined the concentration of vitamin E, and a few additional other micronutrients that potentially could affect brain development, in plasma, liver, and brain of the experimental animals at 8 and 12 weeks. Increasing values of vitamin E in the feed, corresponded in all three tissues to significant increasing values of vitamin E specifically (Table 1 and Table 2).
Table 1.
Vitamin E levels in Xpg−/− mice at 8 and 12 weeks
| Micronutrient (time point) | Diet | Plasma ± s.e.m. (nmol/L) | Liver ± s.e.m. (nmol/g) | Brain ± s.e.m. (nmol/g) |
|---|---|---|---|---|
| α-Tocopherol (8 weeks) | Low | 1878 ± 93 a=*** | 63 ± 14 a=* | 107 ± 12 a=** |
| Medium | 20214 ± 1214 b=** | 1772 ± 418 b=ns | 276 ± 9 b=ns | |
| High | 26478 ± 1140 c=*** | 5282 ± 1455 c=* | 344 ± 53 c=* | |
| α-Tocopherol (12 weeks) | Low | 1767 ± 100 a=*** | 46 ± 12 a=** | 114 ± 9 a=** |
| Medium | 17829 ± 901 b=*** | 931 ± 172 b=* | 348 ± 39 b=ns | |
| High | 25773 ± 972 c=*** | 3808 ± 423 c=** | 643 ± 111 c=* |
Amount of α-Tocopherol in the plasma (nmol/L, n=7), liver and brain (nmol/g, n=2 for all and n= 3 for high vitamin E diet at 12 weeks) of 8 and 12 weeks old Xpg−/− mice. Errors represent s.e.m. values. a = compares low with medium vitamin E group; b = medium vs high; c = high vs low; ns: not significant.
Table 2.
Micronutrient levels in Xpg−/− mice at 8 and 12 weeks
| Micronutrient (time point) | Diet | Plasma ± s.e.m. (nmol/L) | Liver ± s.e.m. (nmol/g) | Brain ± s.e.m. (nmol/g) |
|---|---|---|---|---|
| Retinol (8 weeks) | Low | 1107 ± 91 | 3090 ± 1263 | 2.3 ± 0.98 |
| Medium | 1124 ± 56 | 2779 ± 1134 | 2.2 ± 0.91 | |
| High | 1131 ± 73 | 3034 ± 1250 | 2.3 ± 0.98 | |
| Retinol (12 weeks) | Low | 1026 ± 84 | 4765 ± 1944 | 2.9 ± 1.26 |
| Medium | 1068 ± 38 | 4566 ± 1867 | 3.2 ± 1.36 | |
| High | 1072 ± 66 | 5055 ± 380 | 2.6 ± 0.52 | |
| Thiamin (8 weeks) | Low | 226 ± 11 | 19 ± 0.49 | 1.43 ± 0.04 |
| Medium | 229 ± 11 | 19 ± 0.94 | 1.28 ± 0.0 | |
| High | 252 ± 18 | 19 ± 1.84 | 1.13 ± 0.11 | |
| Thiamin (12 weeks) | Low | 226 ± 41 | 15 ± 0.38 | 1.02 ± 0.04 |
| Medium | 177 ± 7 | 15 ± 1.58 | 1.05 ± 0.04 | |
| High | 173 ± 15 | 15 ± 0.41 | 1.09 ± 0.07 | |
| Riboflavin (8 weeks) | Low | 172 ± 11 | 6.4 ± 0.23 | 1.35 ± 0.05 |
| Medium | 181 ± 13 | 6.1 ± 0.08 | 1.32 ± 0.03 | |
| High | 170 ± 13 | 6.6 ± 0.66 | 1.30 ± 0.03 | |
| Riboflavin (12 weeks) | Low | 154 ± 15 | 5.6 ± 0.10 | 1.09 ± 0.03 |
| Medium | 157 ± 5 | 5.6 ± 0.11 | 1.01 ± 0.03 | |
| High | 167 ± 11 | 6.1 ± 0.26 | 1.06 ± 0.08 | |
| Vitamin B6 (8 weeks) | Low | 129 ± 10 | 31 ± 1.6 | 2.9 ± 0.3 |
| Medium | 161 ± 18 | 34 ± 0.8 | 3.1 ± 0.3 | |
| High | 161 ± 18 | 33 ± 0.0 | 3.3 ± 0.08 | |
| Vitamin B6 (12 weeks) | Low | 275 ± 72 | 36 ± 0.4 | 3.6 ± 0.00 |
| Medium | 222 ± 44 | 39 ± 2.8 | 3.5 ± 0.08 | |
| High | 218 ± 20 | 39 ± 1.6 | 3.4 ± 0.12 |
Amount of retinol (vitamin A), thiamin (vitamin B1), riboflavin (vitamin B2) and pyridoxal 5′phosphate (vitamin B6) in the plasma (nmol/L), liver and brain (nmol/g) of 8 and 12 weeks old Xpg−/− mice. Values represent averages of n>2 biological replicates: plasma n=7, liver and brain n=2 and n=3 for high vitamin E diet at 12 weeks. Errors represent s.e.m. values.
Feed intake, body weight and onset of tremors
No clear differences were observed in average feed intake by the lack or increase of vitamin E (Figure 2a and b; females and males respectively), excluding indirect effects of dietary restriction for which Xpg−/− mice are very sensitive (14). Concomitantly, no obvious differences in average body weight of Xpg−/− mice fed with diets containing different dosages of vitamin E were measured (Figure 2c and d; females and males respectively). Only a minor difference in body weight at 4 weeks of age could be observed, likely due to the random distribution of litters at birth over the different diets (Figure 2c). Subsequently, both females and males gained in body weight until approximately 8 weeks of age. After reaching their maximal body weight they all gradually declined with age as consequence of the aging-associated deterioration, which includes cachexia, like in Cockayne syndrome patients. The onset of body weight decline was significantly delayed in a vitamin E dose-dependent manner (Figure 2e). The animals receiving the highest amount of vitamin E in their feed showed a median delay of 2 weeks in onset of body weight decline compared to the animals with the lowest amount of vitamin E given (Figure 2e; p=0.0268).
Figure 2. Vitamin E prevents age-related decline in body weight and onset of tremors in Xpg−/− mice.
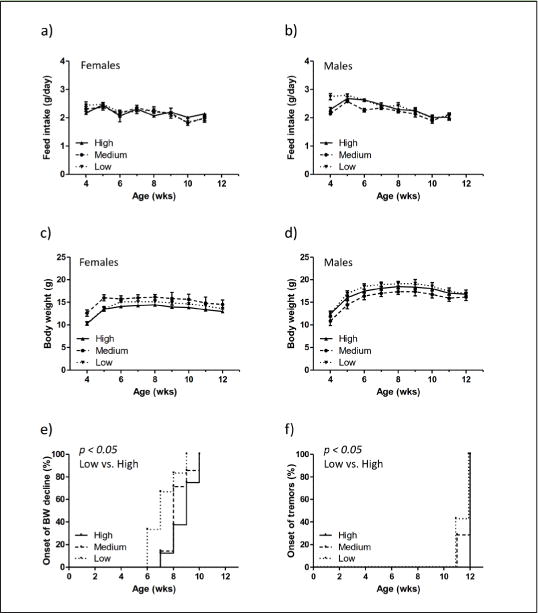
a–b) Feed intake expressed in grams (g)/day measured weekly from 4 to 12 weeks (wks) of age in Xpg−/− females (a, n=4) and Xpg−/− males (b, n=6). c–d) Body weight curves of Xpg−/− female (c, n=4) and male (d, n=6) mice measured weekly. For all graphs (a-d) the “n” indicates the number of animals (for each diet group) available until the first time point (8 weeks of age, Figure 1a). e) Onset of body weight (BW) decline measured in the Xpg−/− mice designated for time point 2 (12 weeks of age). Per animal the first week of BW decline was expressed in percentage (%) of animals presenting a reduced body weight at a given time (in weeks). n=6–8 animals per group *p=0.0268 (between low and high vitamin E using the Log-rank Mantel-Cox test). f) Onset of tremors (neurological abnormality) with age in the Xpg−/− mice fed with different amounts of vitamin E in their feed given as % of animals with tremors. n=7–8 animals per group *p=0.0455 (between low and high vitamin E using the Log-rank Mantel-Cox test). Importantly, animals were scored in a fully blind manner (see Material & Methods section). For all graphs in Figure 2, error bars indicate s.e.m.
Appearance of tremors, a neurodegenerative aging feature occasionally observed in old animals, was also monitored during the visual examination of the Xpg−/− mice. About 40% of the mice fed on a diet containing low vitamin E developed tremors at an age of 11 weeks, while no signs of tremors were present yet in the mice fed on high content of vitamin E (Figure 2f; p=0.0455). However, at 12 weeks of age all mice showed signs of tremors, similarly to what was previously observed for these mice (11). This minor observational difference prompted us to further investigate aging features in more detail.
Oxidative status
Since vitamin E is known as a potent anti-oxidant, we verified whether increasing amounts of vitamin E were associated with reduced oxidative stress levels, using two distinct assays. Preliminary evidence indicated that the overall intracellular redox state (SS/SH ratio) was significantly reduced in liver homogenates with medium and high concentrations of vitamin E in the feed (Figure 3a; p=0.000064 and p=000004 relatively compared to low vitamin E). This dose-dependent shift was, however, not observed for the specific detection of protein carbonyl groups (Figure 3b). Due to the limited amount of brain tissue available for direct measurements, we could not measure the oxidation status of brains and the analysis was therefore limited to only liver tissue.
Figure 3. High vitamin E reduces polyploidization in the liver of Xpg−/− mice.
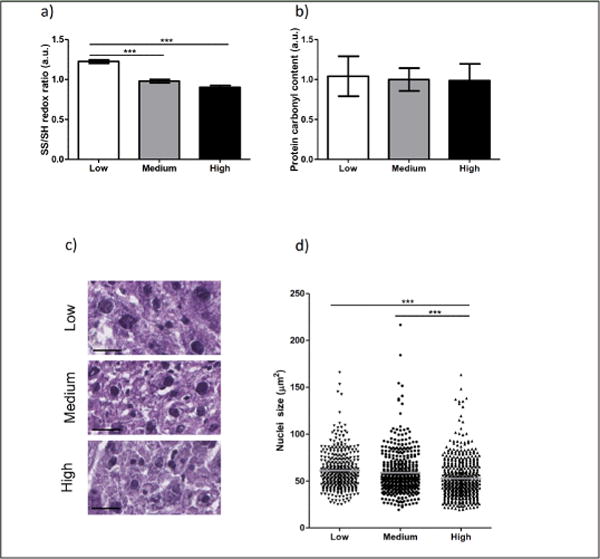
a) Immuno-blot analysis of the overall liver redox state. The ratio between oxidized (SS) and reduced (SH) thiol groups was determined for low (white bar) and high (black bar) vitamin E relative to the level of normal (gray bar) vitamin E. b) Dot-blot analysis of the protein carbonyl content in liver samples of low (white bar) and high (black bar) amounts of vitamin E relative to normal (gray bar) vitamin E. For a) and b) n>2 liver pieces of 2–3 animals/group. c–d) Representative images (c) and average nucleus size of hepatocytes (d, gray lines) in the liver of 12 weeks old Xpg−/− mice fed with low (triangle down), medium (circle), or high (triangle up) vitamin E in the diet. Equal numbers of images were quantified from n>3 liver slices of 2–3 animals/group. Each point corresponds to one nucleus. Scale bar = 25μm. Error bars represent s.e.m. Statistical significance is measured by one-way ANOVA (***p<0.001).
Cellular damage in liver
Considering the delayed body weight decline and onset of tremors associated with a diet containing high amount of vitamin E (Figure 2), specific markers of aging were studied more in detail. We first focused on the liver, an organ not only important for vitamin E metabolism, but also very sensitive to oxidative stress (26). With reduced levels of vitamin E, more polyploid hepatocyte nuclei were observed (Figure 3c). Quantification of similar areas of liver indeed yielded in an increased average nuclei size and thereby less total hepatocytes per section. In turn, supplementation with high vitamin E reduced the hepatocyte nuclei size, compared to both diets with low and medium vitamin E levels (Figure 3c and d; p=9.364E-8 and p=0.000043 respectively). We also quantified the percentage of cells containing an increased nuclear size double the normal value (set as bigger than 80μm2). This was reduced in a vitamin E dose dependent manner, 18.5% (low), 15.8% (medium), to 9.6% (high), concomitantly with an increased number of hepatocytes present in a similar area (356, 429, and 499 respectively). Additionally, fat accumulation in the liver did not result in any obvious changes by dietary alterations of vitamin E (Supplementary Figure).
Cellular damage in nervous system
Since Xpg−/− animals display numerous features of nervous system aging that primarily can be halted by nutritional changes like diet restriction (14), we next investigated the effects of vitamin E changes on retina and brain. TUNEL staining confirmed cell death in the outer nuclear layer of the retina at 12 weeks of age, but no improvement by vitamin E supplementation could be detected (Figure 4a and b). Changing vitamin E content in the diet also did not result in macroscopical changes in brain (data not shown), or immunohistological changes in the glial fibrillary acidic protein (GFAP) expression, a marker for astrocytosis (Figure 4c and d).
Figure 4. Vitamin E does not affect retina degeneration and astrogliosis in Xpg−/− mice.
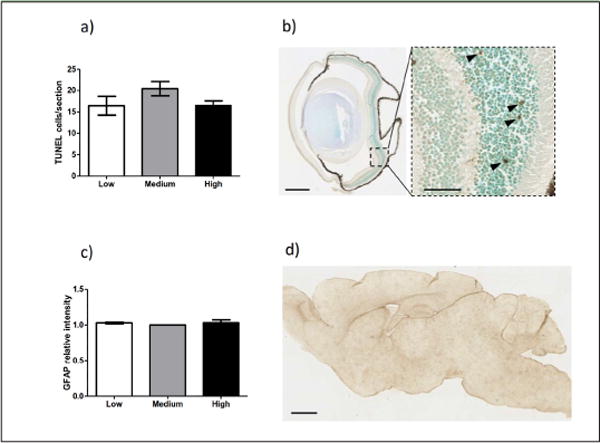
a) Quantification of TUNEL-positive cells in the outer nuclear layer of retinal sections of 12 weeks old Xpg−/− mice on low (white bar), medium (gray bar), and high (black bar) vitamin E (n=5 animals per group). b) Representative image of TUNEL staining in the eye of a Xpg−/−mouse on low vitamin E diet. Scale bar = 500μm. Dying cells (TUNEL +) are indicated by black arrowheads in the higher magnification on the right. Scale bar = 50μm. c) Quantification of the relative intensity of consecutive sagittal brain sections immunoperoxidase-stained for GFAP. Brains from animals on low (white bar), medium (gray bar), and high (black bar) vitamin E diets were analyzed. The amount of signal per slice for the normal vitamin E diet was set on 1 (n=3 animals per group). d) Representative picture of a sagittal brain section of a 12 weeks old Xpg−/− mouse on low vitamin E immunoperoxidase-stained for GFAP. Scale bar = 1mm. For all graphs in Figure 4, error bars indicate s.e.m.
As marker for genotoxic stress in the nervous system, we subsequently studied the expression of transcription factor p53. Immunohistochemical analysis showed a significant reduced percentage of p53-positive cells in the brain of animals fed with high vitamin E content (Figure 5a). The reduced cellular degeneration in the brain was already observed in 8 weeks old Xpg−/− mice (Figure 5b, p=0.025 low vs. high and p=0.023 medium vs. high) and persisted in older animals (Figure 5c, p=0.053 low vs. high and p=0.042 medium vs. high). Of note, no differences were observed when brains from animals fed on low vitamin E diet were compared to those fed on a diet containing medium vitamin E levels (Figure 5b and c).
Figure 5. Vitamin E reduces the number of p53 positive cells in the brains of Xpg−/− mice.
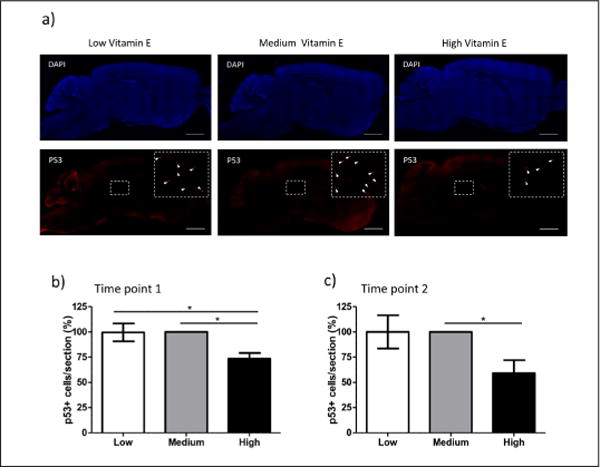
a) Representative images showing the genotoxic stress biomarker p53 in brain slices of Xpg−/− mice animals at 12 weeks of age fed on diets containing low, medium and high vitamin E. Upper panels show the DAPI staining (blue = cellular nuclei). Lower panels show the p53-positive cells (red staining). Enlargements show zoom areas with maximized contrast to visualize p53-positive cells (white arrows). Scale bar = 1mm. b–c) Quantification of the % of p53-positive cells in the brain of 8 weeks (b) and 12 weeks (c) old Xpg−/− mice fed on a diet with low, medium and high vitamin E content. For all analyses n=4. Error bars represent the s.e.m. Statistical significance is measured by one-way ANOVA (*p<0.05). The number of p53-positive cells in Xpg−/− mice fed with medium vitamin E diet was set as 100%.
Discussion
The results of this study show for the first time, that modifying the concentration of a single micronutrient (vitamin E) in the diet of the Xpg−/− premature aging murine model, was sufficient to ameliorate some of the cellular and phenotypic deficiencies. Since XPG is involved in multiple DNA repair systems, various types of DNA lesions will accumulate with age that differ in rates and outcome between distinct organs [27]. We therefore primarily investigated the effect of vitamin E on the consequences of DNA damage in liver and nervous system.
The liver plays an important role in maintaining body homeostasis via the processing of nutrients, hormones, and metabolic waste products and by metabolic (in) activation of toxic compounds. It undergoes substantial changes in structure, composition and function in old age, thereby affecting systemic aging and disease predisposition (28, 29). One of the age-related changes in liver induced upon DNA damage, is the ability of hepatocytes to undergo polyploidization (4n, 8n, 16n and higher) (30). The level of polypoidization thereby shows a correlation with accumulation of DNA damage (14, 31). Vitamin E protected against polyploidization in a dose-dependent manner (Figure 3c and d). A logical explanation would be that increasing the amount of vitamin E via dietary changes would, via its anti-oxidant properties, prevent ROS from accumulating and damaging the DNA. Since direct measurements of oxidative DNA lesions are very challenging and often variable we investigated some intracellular intermediates in between lipids and DNA. Although protein carbonylation, a footprint of ROS damage associated with many neurodegenerative diseases and progeria (32), was not reduced by the slight physiological increase in the high vitamin E diet, likely due to the relatively high levels of other vitamins and anti-oxidants as feed preservatives, overall redox levels, reflecting a more general antioxidant change, showed a shift towards a more reduced state upon increasing the amount of vitamin E (Figure 3a and b). This however does not rule out alternative protective functions of vitamin E (18).
In brain, p53 is a ubiquitously expressed transcription factor activated by multiple types of DNA damage and therefore often used as biomarker for neurodegeneration (11). P53 analysis showed that consumption of feed with high vitamin E content was sufficient to reduce the number of p53 positive cells throughout the brain suggesting a neuroprotective role for vitamin E (Figure 5). Of note, no difference was observed when a diet deprived in vitamin E was compared with the diet containing medium vitamin E levels. This is in agreement with the vitamin E measurements between liver and brain of mice on the low vitamin E diet. It also substantiates the importance of adequate coverage of the organism with vitamin E and that, under increased oxidative pressure, a high vitamin E diet is needed to reduce the degenerating changes. Without further optimization, the current dosage of vitamin E supplementation did not yield a subsequent beneficial effect in astrogliosis in the brain or cell death in the retina (Figure 4). However, the reduced liver polyploidization and lower activation of the DNA damage response marker p53, indicates that higher levels of vitamin E might prevent or reduce age-dependent DNA damage accumulation.
Vitamin E is a lipid-soluble compound whose antioxidant properties have been investigated during aging progression or in pathologies typical of old age (17, 20, 21). The difficulty in performing precise and uniform human studies resides in several important variables such as: age and health status of the individuals included in the study, nutritional status and diet, form of vitamin E administered, but also quantity and time of supplementation. Other important parameters to be considered are also the genetic variations that can alter the vitamin E metabolism in the tissues as it was demonstrated by Goncalves and collaborators (33). Therefore, to investigate whether the observed and above mentioned preventive effects were indeed associated with alterations in vitamin E, we examined whether the orally supplemented α-Tocopherol was concentrated in the targeted organs studied. We measured vitamin E concentration in plasma, liver and brain of all experimental animals and confirmed that the diet enriched in vitamin E increased the concentration of this micronutrient in the analyzed tissue, while the diet with low vitamin E corresponded to vitamin E deficiencies (Table 1). Of note, brain showed the smallest reduction in vitamin E in animals subjected to the low vitamin E diet. These data confirm that the brain expresses high levels of the alpha-tocopherol transfer protein resulting in an enrichment and consequentially higher levels of vitamin E in the brain [34]. Moreover, our data corroborate what previously reported by Ulatowski and collaborators (35), who reported that vitamin E plays an important role in protecting the central nervous system, and specifically the cerebellar cortex by oxidative status with consequential deficits in motor coordination and cognitive functions following vitamin E deficiencies. Additionally, our findings demonstrate that a relatively brief period of vitamin E shortage leads to almost complete depletion of vitamin E from the tissue of these animals, emphasizing the importance for an elevated vitamin E supplementation under at-risk conditions. Other studies show that, in normal rats under vitamin E-deficient diet, a longer delay was observed for vitamin E concentrations to change in tissues studied (36, 37). The reason for this apparent discrepancy may be that in those studies lower levels of vitamin E were used and that we are dealing with a mouse model of premature aging in which the phenotype is fast developing. This may lead to an increased rate of metabolic processes and differences in vitamin E accumulation. Moreover, and in agreement to our results also in those studies the vitamin E supplementation led to marked elevation of its concentrations in different organs [36]. In addition, we measured the concentration of other micronutrients whose metabolism could have been affected by vitamin E content. Vitamin A, vitamin B1, B2 and B6 did not change in plasma, liver and brain as a function of the administered diet (Table 2) and therefore confirmed that vitamin E was the only variable explaining the observed parameters.
In conclusion, the data suggest that a diet rich in vitamin E delays the onset of specific aging features in the Xpg−/− premature aging model. In particular, we showed that a slight physiological increase in vitamin E reduces the number of damaged cells in liver and brain possibly in part due to an overall reduction of the oxidative stress status in these tissues. Our findings are of great potential for the implementation of focused therapeutic protocols in patients and imply the importance of improved nutrition to sustain health.
Supplementary Material
{kind=link}
Acknowledgments
We thank Yvette van Loon, Lucien Rooth and the animal caretakers for general assistance with mouse experiments. Pier Giorgio Mastroberardino and Chiara Milanese are acknowledged for their support with the oxidative stress measurements. We acknowledge financial support of the National Institute of Health (NIH)/National Institute of Ageing (NIA) (PO1 AG017242), European Research Council Advanced Grant DamAge and Proof of Concept Grant Dementia to JHJH, the European commission ITN Address (GA-316390) and ITN Marriage (GA-316964), the KWO Dutch Cancer Society (5030), the Dutch CAA Foundation and the Royal Academy of Arts and Sciences of the Netherlands (academia professorship to JHJH). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author contributions: G.L.F., J.H.J.H., W.P.V., and M.H.M. designed the research and wrote the manuscript. N.V., S.B., R.M.C.B., S.E., E.C., C.G., N.S. and P.W. contributed to editing the manuscript. S.B., R.M.C.B., and W.P.V. performed and analyzed the mouse cohorts and phenotypical scoring. S.E., E.C., and C.G., performed the quantitative vitamin measurements. N.V., S.B., and W.P.V. quantified oxidative stress parameters. N.V. performed the analysis of liver polyploidization and fat accumulation. N.V. and R.M.C.B. assessed the degree of retina degeneration. G.L.F. and W.P.V. characterized neuropathological changes.
Conflict of interests: G.L.F, S.E., E.C., C.G., N.S., P.W., and M.H.M. are employees of DSM Nutritional Products. All other authors declare no conflict of interests.
Statement of ethical approval: This study was performed in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and was approved by the Dutch Ethical Committee (permit # 139-12-13), in full accordance with European legislation.
References
- 1.Lopez-Otin C, et al. The hallmarks of aging. Cell. 2013;153(6):1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vermeij WP, Hoeijmakers JH, Pothof J. Genome Integrity in Aging: Human Syndromes, Mouse Models, and Therapeutic Options. Annu Rev Pharmacol Toxicol. 2016;56:427–45. doi: 10.1146/annurev-pharmtox-010814-124316. [DOI] [PubMed] [Google Scholar]
- 3.von Zglinicki T, Burkle A, Kirkwood TB. Stress. DNA damage and ageing – an integrative approach. Exp Gerontol. 2001;36(7):1049–62. doi: 10.1016/s0531-5565(01)00111-5. [DOI] [PubMed] [Google Scholar]
- 4.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 5.Dan Dunn J, et al. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015;6:472–85. doi: 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marteijn JA, et al. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15(7):465–81. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- 7.Mu D, Hsu DS, Sancar A. Reaction mechanism of human DNA repair excision nuclease. J Biol Chem. 1996;271(14):8285–94. doi: 10.1074/jbc.271.14.8285. [DOI] [PubMed] [Google Scholar]
- 8.Klungland A, et al. Base excision repair of oxidative DNA damage activated by XPG protein. Mol Cell. 1999;3(1):33–42. doi: 10.1016/s1097-2765(00)80172-0. [DOI] [PubMed] [Google Scholar]
- 9.Trego KS, et al. Non-catalytic Roles for XPG with BRCA1 and BRCA2 in Homologous Recombination and Genome Stability. Mol Cell. 2016;61(4):535–46. doi: 10.1016/j.molcel.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zafeiriou DI, et al. Xeroderma pigmentosum group G with severe neurological involvement and features of Cockayne syndrome in infancy. Pediatr Res. 2001;49(3):407–12. doi: 10.1203/00006450-200103000-00016. [DOI] [PubMed] [Google Scholar]
- 11.Barnhoorn S, et al. Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genet. 2014;10(10):e1004686. doi: 10.1371/journal.pgen.1004686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harkema L, Youssef SA, de Bruin A. Pathology of Mouse Models of Accelerated Aging. Vet Pathol. 2016;53(2):366–89. doi: 10.1177/0300985815625169. [DOI] [PubMed] [Google Scholar]
- 13.Solon-Biet SM, et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014;19(3):418–30. doi: 10.1016/j.cmet.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vermeij WP, et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature. 2016;537(7620):427–431. doi: 10.1038/nature19329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamal-Eldin A, Appelqvist LA. The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids. 1996;31(7):671–701. doi: 10.1007/BF02522884. [DOI] [PubMed] [Google Scholar]
- 16.Rigotti A. Absorption. transport, and tissue delivery of vitamin. E Mol Aspects Med. 2007;28(5–6):423–36. doi: 10.1016/j.mam.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Brigelius-Flohe R, Traber MG. Vitamin E: function and metabolism. FASEB J. 1999;13(10):1145–55. [PubMed] [Google Scholar]
- 18.Mocchegiani E, et al. Vitamin E-gene interactions in aging and inflammatory age-related diseases: implications for treatment. A systematic review. Ageing Res Rev. 2014;14:81–101. doi: 10.1016/j.arr.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Fata La, Weber GP, Mohajeri MH. Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients. 2014;6(12):5453–72. doi: 10.3390/nu6125453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohajeri MH, Leuba G. Prevention of age-associated dementia. Brain Res Bull. 2009;80(4–5):315–25. doi: 10.1016/j.brainresbull.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 21.Mohajeri MH, Troesch B, Weber P. Inadequate supply of vitamins and DHA in the elderly: implications for brain aging and Alzheimer-type dementia. Nutrition. 2015;31(2):261–75. doi: 10.1016/j.nut.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 22.Holler U, et al. Microwave-assisted rapid determination of vitamins a and e in beverages. J Agric Food Chem. 2003;51(6):1539–42. doi: 10.1021/jf0260038. [DOI] [PubMed] [Google Scholar]
- 23.Vermeij WP, Alia A, Backendorf C. ROS quenching potential of the epidermal cornified cell envelope. J Invest Dermatol. 2011;131(7):1435–41. doi: 10.1038/jid.2010.433. [DOI] [PubMed] [Google Scholar]
- 24.Horowitz MP, et al. Single-cell redox imaging demonstrates a distinctive response of dopaminergic neurons to oxidative insults. Antioxid Redox Signal. 2011;15(4):855–71. doi: 10.1089/ars.2010.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vermeij WP, Backendorf C. Reactive oxygen species (ROS) protection via cysteine oxidation in the epidermal cornified cell envelope. Methods Mol Biol. 2014;1195:157–69. doi: 10.1007/7651_2013_51. [DOI] [PubMed] [Google Scholar]
- 26.Li S, et al. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int J Mol Sci. 2015;16(11):26087–124. doi: 10.3390/ijms161125942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vermeij WP, Hoeijmakers JH, Pothof J. Aging: not all DNA damage is equal. Curr Opin Genet Dev. 2014;26:124–30. doi: 10.1016/j.gde.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Le Couteur DG, et al. Old age and the hepatic sinusoid. Anat Rec (Hoboken) 2008;291(6):672–83. doi: 10.1002/ar.20661. [DOI] [PubMed] [Google Scholar]
- 29.Hilmer SN, et al. Age-related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology. 2005;42(6):1349–54. doi: 10.1002/hep.20937. [DOI] [PubMed] [Google Scholar]
- 30.Duncan AW, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467(7316):707–10. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White RR, et al. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat Commun. 2015;6:6790. doi: 10.1038/ncomms7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Drug Metab Rev. 1998;30(2):225–43. doi: 10.3109/03602539808996310. [DOI] [PubMed] [Google Scholar]
- 33.Goncalves A, et al. Cluster-determinant 36 (CD36) impacts on vitamin E postprandial response. Mol Nutr Food Res. 2014;58(12):2297–306. doi: 10.1002/mnfr.201400339. [DOI] [PubMed] [Google Scholar]
- 34.Gohil K, et al. Mice lacking alpha-tocopherol transfer protein gene have severe alpha-tocopherol deficiency in multiple regions of the central nervous system. Brain Res. 2008;1201:167–76. doi: 10.1016/j.brainres.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ulatowski L, et al. Vitamin E is essential for Purkinje neuron integrity. Neuroscience. 2014;260:120–9. doi: 10.1016/j.neuroscience.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clement M, Bourre JM. Graded dietary levels of RRR-gamma-tocopherol induce a marked increase in the concentrations of alpha- and gamma-tocopherol in nervous tissues, heart, liver and muscle of vitamin-E-deficient rats. Biochim Biophys Acta. 1997;1334(2–3):173–81. doi: 10.1016/s0304-4165(96)00090-6. [DOI] [PubMed] [Google Scholar]
- 37.Gaedicke S, et al. Dietary vitamin E, brain redox status and expression of Alzheimer’s disease-relevant genes in rats. Br J Nutr. 2009;102(3):398–406. doi: 10.1017/S000711450819122X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.