

ABSTRACT
The membrane source for autophagosome biogenesis is an unsolved mystery in the study of autophagy. ATG16L1 forms a complex with ATG12–ATG5 (the ATG16L1 complex). The ATG16L1 complex is recruited to autophagic membranes to convert MAP1LC3B-I to MAP1LC3B-II. The ATG16L1 complex dissociates from the phagophore before autophagosome membrane closure. Thus, ATG16L1 can be used as an early event marker for the study of autophagosome biogenesis. We found that among 3 proteins in the ATG16L1 complex, only ATG16L1 formed puncta-like structures when transiently overexpressed. ATG16L1+ puncta formed by transient expression could represent autophagic membrane structures. We thoroughly characterized the transiently expressed ATG16L1 in several mammalian cell lines. We found that transient expression of ATG16L1 not only inhibited autophagosome biogenesis, but also aberrantly targeted RAB11-positive recycling endosomes, resulting in recycling endosome aggregates. We conclude that transient expression of ATG16L1 is not a physiological model for the study of autophagy. Caution is warranted when reviewing findings derived from a transient expression model of ATG16L1.
KEYWORDS: ATG16L1, ATG5, ATG12, autophagosome biogenesis, autophagy, RAB11, recycling endosome
Introduction
Autophagosome biogenesis is initiated at not well-defined intracellular membranes of the cell. Autophagy-specific protein complexes are recruited to the membrane to drive the creation of a double membrane-bound autophagosome.1,2 Evidence suggests that the phagophore membrane (the precursor to the autophagosome) could be generated from existing intracellular membranes, including the endoplasmic reticulum (ER) membrane,3-5 mitochondrial membrane,6 connection sites of the mitochondrial and ER membranes,7 and the ER-Golgi intermediate compartment (ERGIC) membrane.8,9 Recycling endosomes (REs) and the plasma membrane may also contribute membrane to autophagosome biogenesis.10-13
Several autophagy-specific protein complexes control autophagosome biogenesis. Hierarchical analysis has revealed that the most proximal complex in mammalian cells is the ULK1-RB1CC1-ATG13-ATG101 complex (hereafter referred to as the ULK1 complex) and that its function is governed by MTORC1 activity. The downstream PIK3C3 complex regulates the ATG12-TG5-ATG16L1 complex (referred to as the ATG16L1 complex). The ATG16L1 complex serves as an E3 ligase for MAP1LC3B-I/LC3B-I to conjugate with phosphatidylethanolamine (to generate LC3B-II).1,2,14-16 As the ATG16L1 complex specifies the membrane where LC3B-I is converted to LC3B-II to start autophagosome biogenesis,17-19 studies to identify the adaptor protein that recruits the ATG16L1 complex to the autophagic membrane20-25 and to identify the origin of the autophagic membrane using ATG16L1 as an early autophagosome biogenesis marker have been emerging.10-12,26
A previous study demonstrated that correct stoichiometry of ATG16L1 complex components (ATG16L1 and ATG12–ATG5) is critical for autophagosome biogenesis.17 Our observations indicate that transiently expressed ATG16L1, but not ATG5 and ATG12, forms puncta-like structures in the absence of autophagic stimulation. To determine whether transiently expressed ATG16L1 can be used as a marker for the study of autophagosome biogenesis, we thoroughly defined the outcome of transient expression of ATG16L1 in several mammalian cell lines. We found that transient expression of human ATG16L1 inhibits autophagosome biogenesis. In addition, transiently expressed ATG16L1 forms abnormal puncta-like structures, which proved to be RAB11+ REs.
Results
Characterization of endogenous ATG16L1 trafficking under autophagic stimulation
To better understand the function of transiently expressed ATG16L1, we first examined endogenous ATG16L1 trafficking in the process of autophagy. It has been demonstrated that ATG16L1 targets the autophagic membrane in complex with the ATG12–ATG5 conjugate under autophagic stimulation.1,16,17,19,27,28 With Atg5 gene deletion, the ATG12–ATG5 conjugate is not generated (Fig. 1A). ATG7 is the E1-like enzyme in the conjugation reaction of ATG5 with ATG12.29 Deletion of Atg7 abolished ATG12–ATG5 conjugate formation (Fig. 1A). The absence of the ATG16L1 complex led to a deficiency of LC3B lipidation, as demonstrated by the lack of LC3B-II in these cell models (Fig. 1A).
Figure 1.
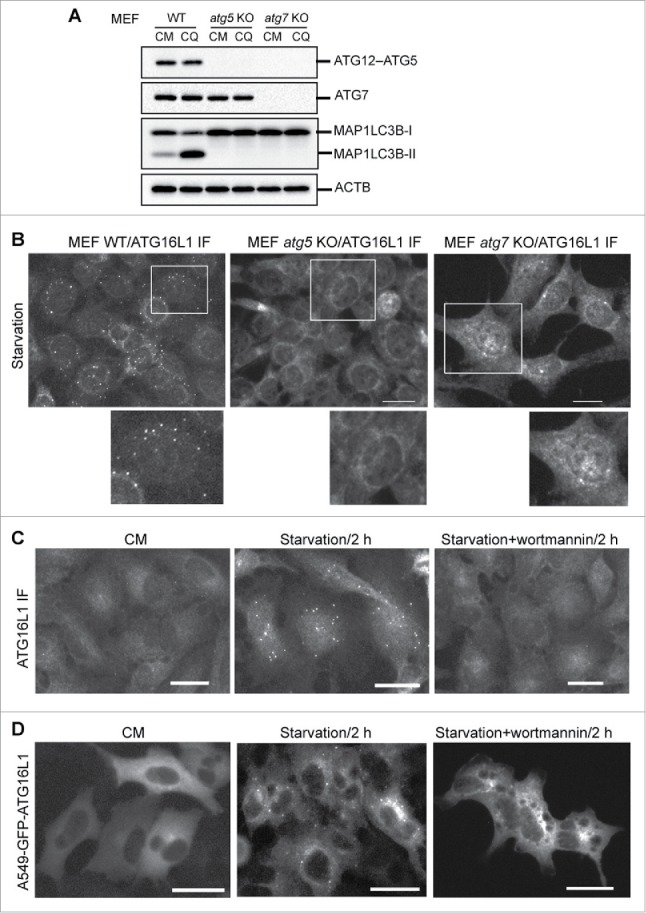
Phagophore targeting by endogenous ATG16L1 requires the ATG12–ATG5 conjugate and PtdIns3K activity. (A) Wild-type (WT), atg5 KO, or atg7 KO mouse embryonic fibroblasts (MEFs) were cultured in complete medium (CM) with or without the lysosome inhibitor chloroquine (CQ, 50 µM) for 2 h. Cells were then lysed for immunoblot assays using the antibodies as indicated. (B) WT, atg5 KO, or atg7 KO MEFs were starved in EBSS for 2 h. Cells were then fixed and probed with ATG16L1 antibody. (C) A549 cells were cultured in CM or starved in EBSS with or without the PtdIns3K inhibitor wortmannin (200 nM) for 2 h. Cells were then fixed and probed with ATG16L1 antibody. (D) A549 cells stably expressing GFP-ATG16L1 were cultured in CM or starved in EBSS with/without wortmannin (200 nM) for 2 h. Images were recorded by a fluorescence microscope. IF, immunofluorescence staining. Scale bar: 25 micron. Of note, images and immunoblots represent at least 3 experiments in all figures presented in this study.
The role of the ATG12–ATG5 conjugate in targeting the ATG16L1 complex to the autophagic membrane was scrutinized by verifying the capacity of ATG16L1 to form puncta in Atg5 and Atg7 knockout (KO) mouse embryonic fibroblasts (MEFs). As shown in Figure 1B, nutrient deprivation was unable to stimulate ATG16L1 puncta formation in both MEF atg5 KO and MEF atg7 KO cells compare with wild-type MEFs. These results demonstrate that the ATG12–ATG5 conjugate is obligatory for ATG16L1 to target the autophagic membrane. ATG16L1 alone (in the absence of the ATG12–ATG5 conjugate) under autophagic stimulation does not target the autophagic membrane and fails to function as an E3-like enzyme for LC3B-I to LC3B-II conversion.
We further established an A549 cell stably expressing green fluorescent protein (GFP)-tagged human ATG16L1. Parental A549 cells had few ATG16L1 puncta in nutrient-rich medium (complete medium, CM); whereas a significant number of ATG16L1 puncta were induced by metabolic stress (starvation), and ATG16L1 puncta formation was blocked by a class III phosphatidylinositol 3-kinase (PtdIns3K) inhibitor (Fig. 1C). Further, a similar observation was confirmed in A549 cells stably expressing GFP-ATG16L1. A number of GFP-ATG16L1 puncta were induced by nutrient deprivation, which can be blocked by a PtdIns3K inhibitor (Fig. 1D). These results demonstrate that stably expressed GFP-ATG16L1 appears to have a similar response to autophagic stimulation as endogenous ATG16L1.
Transiently expressed ATG16L1 does not reflect the functionality of endogenous ATG16L1
Next, we asked whether the functional trafficking of endogenous ATG16L1 was similar to that of transiently expressed ATG16L1, ATG12, and ATG5. Plasmids expressing human ATG16L1, ATG5, or ATG12 with an N-terminal GFP tag (500 ng per 6-well plate) were transfected into HeLa cells using Lipofectamine 2000 for 24 h. Cells were then maintained in either nutrient-rich medium (CM) or starvation media (Earle's balanced salt solution; EBSS) for 2 h. In the absence of autophagic stimulation, transiently expressed GFP-ATG16L1 formed puncta-like structures (Fig. 2A, upper right panel). Distinctly, PtdIns3K inhibition did not block GFP-ATG16L1 puncta formation (Fig. 2A, lower right panel). In contrast, transiently expressed GFP-ATG5 and GFP-ATG12 did not form puncta, nor did starvation induce puncta formation of GFP-ATG5 and GFP-ATG12 (Fig. 2A, left panels and middle panels, respectively).
Figure 2.
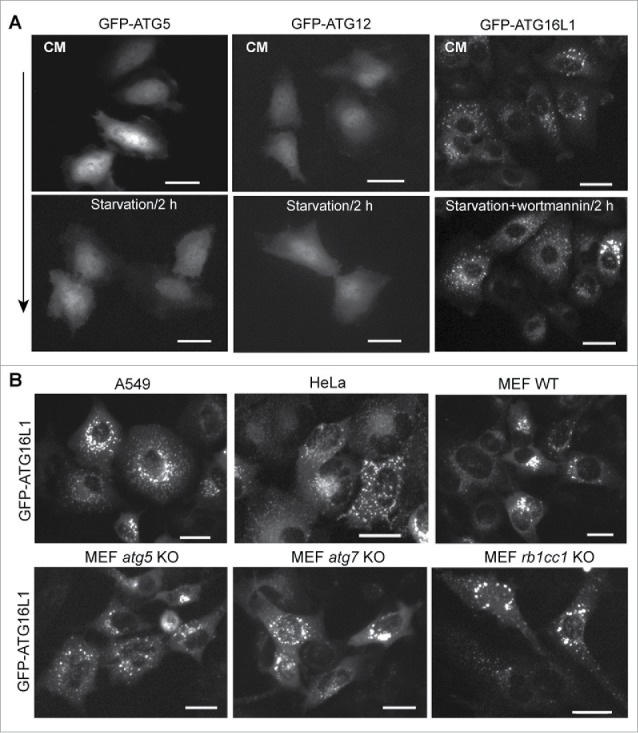
Puncta formation of transiently expressed ATG16L1 is independent of PtdIns3K activity and the ATG12–ATG5 conjugate. (A) HeLa cells were cultured in a 6-well plate at 2 × 105 cells per well for 16 h. Cells were then transfected with 500 ng of GFP-ATG5, GFP-ATG12, or GFP-ATG16L1 expression plasmid per 6-well plate for 20 h, respectively. Cells were starved in EBSS with/without wortmannin (200 nM) for 2 h. (B) A549 cells, HeLa cells, MEF WT, MEF atg5 KO, MEF atg7 KO, or MEF rb1cc1 KO cells were transduced with adenoviral vector expressing GFP-ATG16L1 at 2 MOI for 20 h, respectively. Scale bar: 25 micron.
These results demonstrate that transiently expressed proteins in the ATG16L1 complex do not reflect their endogenous counterparts. Endogenous ATG16L1 or stably expressed GFP-ATG16L1 can be induced to form puncta (autophagic membrane targeting) by autophagic stimulation, which can be blocked by a PtdIns3K inhibitor (Fig. 1C). In contradistinction to that physiological response, transiently expressed ATG16L1 forms puncta-like structures independent of autophagic stimulation and independent of PtdIns3K inhibition.
A further observation was made in Atg gene-deficient MEFs. Puncta-like structures formed with no autophagic stimulation following transient expression of GFP-ATG16L1 in all tested cell lines, even in the absence of ATG5 and ATG7. GFP-ATG16L1 generated puncta-like structures with equal efficiency in Atg5- or Atg7-deficient cell models (Fig. 2B, lower panels). Recent studies demonstrated that RB1CC1, a component of the ULK1 complex, interacts with ATG16L1 and recruits the ATG16L1 complex to the autophagic membrane.22,24 Transiently expressed GFP-ATG16L1 in Rb1cc1-deficient MEFs similarly formed puncta-like structures with equal efficiency when compare with all other cell models (Fig. 2B, lower panel). These data collectively indicate that transiently expressed ATG16L1 either forms protein aggregates or aberrantly targets intracellular membrane structures.
Puncta-like structures generated by transiently expressed ATG16L1 are not autophagosomal structures
We asked whether the ATG16L1+ structures formed by transient expression functioned as autophagosomal structures. First, we examined whether these transiently expressed ATG16L1 puncta were positive for endogenous ATG12–ATG5. Of note, overexpressed ATG16L1 formed puncta structures in nutrient-rich medium and starvation had no effect on this formation. As shown in Figure 3A, transiently expressed mCherry-ATG16L1 puncta were negative for ATG12. ATG12 antibody used for this assay was validated based on its positive puncta colocalization with GFP-LC3B under starvation conditions (Fig. S1A). In comparison, stably expressed GFP-ATG16L1 puncta induced by starvation were positive for ATG12 (Fig. 3B). The ULK1 complex is the most proximal autophagy-specific protein complex.2 ULK1 is recruited to the autophagic membrane under autophagic stimulation.30 We examined the localization of ULK1 with GFP-ATG16L1 puncta. ULK1 formed puncta with nutrient deprivation and these ULK1 puncta colocalized with GFP-LC3B (Fig. S1B) indicating that the ULK1 complex is indeed recruited to the autophagic membrane. We could not detect ULK1 within the GFP-ATG16L1+ puncta-like structures (Fig. 3C). In sharp contrast, stably expressed GFP-ATG16L1 puncta induced by starvation were positive for ULK1 (Fig. 3D).
Figure 3.
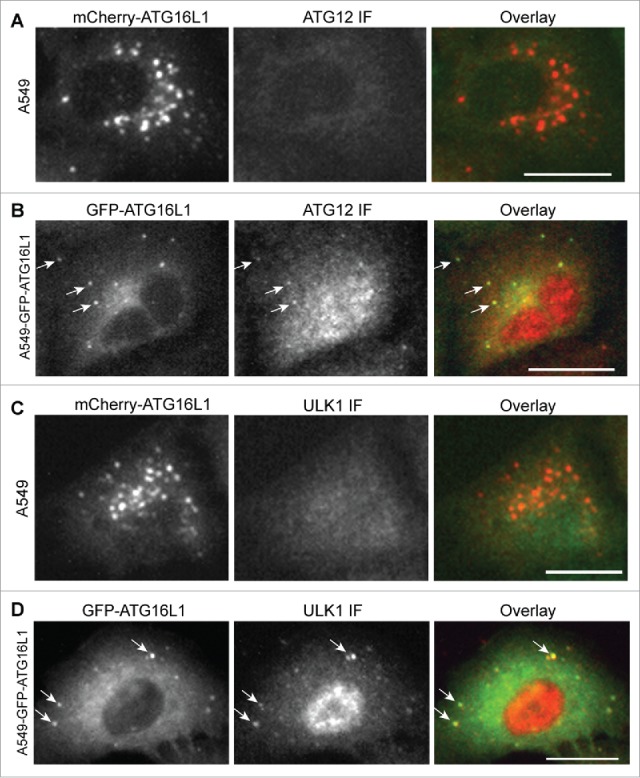
Transiently expressed ATG16L1+ puncta were negative for the ATG12–ATG5 conjugate and ULK1. (A) A549 cells were transduced with Ad-mCherry-ATG16L1 vector (MOI 2) for 16 h. Cells were then starved with EBSS for 2 h before they were fixed and probed with ATG12 antibody. (B) A549 cells stably expressing GFP-ATG16L1 were starved with EBSS for 2 h before they were fixed and probed with ATG12 antibody. (C) A549 cells were transduced with Ad-mCherry-ATG16L1 vector (MOI 2) for 16 h. Cells were then starved with EBSS for 2 h before they were fixed and probed with ULK1 antibody. (D) A549 cells stably expressing GFP-ATG16L1 were starved with EBSS for 2 h before they were fixed and probed with ATG12 antibody. Arrows point to starvation-induced GFP-ATG16L1 puncta positive for ATG12 or ULK1. Scale bar: 25 micron.
The interaction of ATG5 with ATG16L1 at its N-terminal domain has been well documented.19 Thus, it is not surprising to see colocalization of transiently expressed ATG16L1 with transiently expressed ATG5 in ATG16L1-containing puncta (Fig. S1C). In contrast, transiently expressed ATG12 did not demonstrate any colocalization with mCherry-ATG16L1+ structures (Fig. S1D).
An important question is whether these observations of ATG16L1 puncta formation could be caused by GFP or mCherry tag. To exclude this possibility, we generated a nontagged ATG16L1 expressing vector. As shown in Figure S1E (left), transiently expressed nontagged ATG16L1 formed puncta based on ATG16L1 antibody staining. GFP-ATG5 formed no puncta and were distributed throughout the cells (Fig. 1A and Fig. S1E, middle). When expression of GFP-ATG5 and nontagged ATG16L1 was co-transient, GFP-ATG5 interacted with nontagged ATG16L1 and was located in punctate structures (Fig. S1E, right). By using this model, we examined again the colocalization of nontagged ATG16L1 puncta with ATG12 and ULK1. The GFP-ATG5 and nontagged ATG16L1 puncta were negative for ULK1 and ATG12 (Fig. S2A).
Together, the data indicate that the transiently expressed GFP-ATG16L1+ structures lack ATG12–ATG5 and ULK1. They do not represent authentic autophagic structures.
Transient expression of ATG16L1 inhibits autophagosome biogenesis
To determine the effect of overexpressed ATG16L1 on autophagosome biogenesis, we first examined the autophagy flux in HeLa cells stably expressing GFP-LC3B. As shown in Figure 4A, HeLa-GFP-LC3B cells showed few GFP-LC3B puncta in nutrient-rich medium (CM). Extensive GFP-LC3B puncta were induced by EBSS for 3 h (starvation). EBSS starvation plus the lysosome inhibitor bafilomycin A1 (Baf A1) increased the GFP-LC3B puncta accumulation at perinuclear regions of the cells (starvation + Baf A1). In comparison, we examined HeLa-GFP-LC3B cells that transiently overexpressed mCherry-ATG16L1 for 16 h. Cells were then starved with EBSS for 3 h. A few GFP-LC3B puncta were observed in mCherry-ATG16L1-expressing cells (Fig. 4B, upper panel). As illustrated in the enlarged window, mCherry-ATG16L1 puncta were negative for GFP-LC3B. Furthermore, starvation + Baf A1-treated cells demonstrated an extensive number of GFP-LC3B puncta in cells lacking mCherry-ATG16L1 (Fig. 4B, lower panel, cells indicated with arrows). In sharp contrast, mCherry-ATG16L1-expressing cells (cell #1, 2, and 3) had few GFP-LC3B puncta (Fig. 4B, lower panel). One question is whether the inhibitory effect on autophagosome biogenesis may be due to adenoviral vector-mediated ATG16L1 gene expression. As shown in Figure S3, transient expression of ATG16L1 via Lipofectamine 2000-mediated plasmid transfection abolished GFP-LC3B puncta formation. These results demonstrate that transient expression of ATG16L1 inhibits autophagosome biogenesis.
Figure 4.
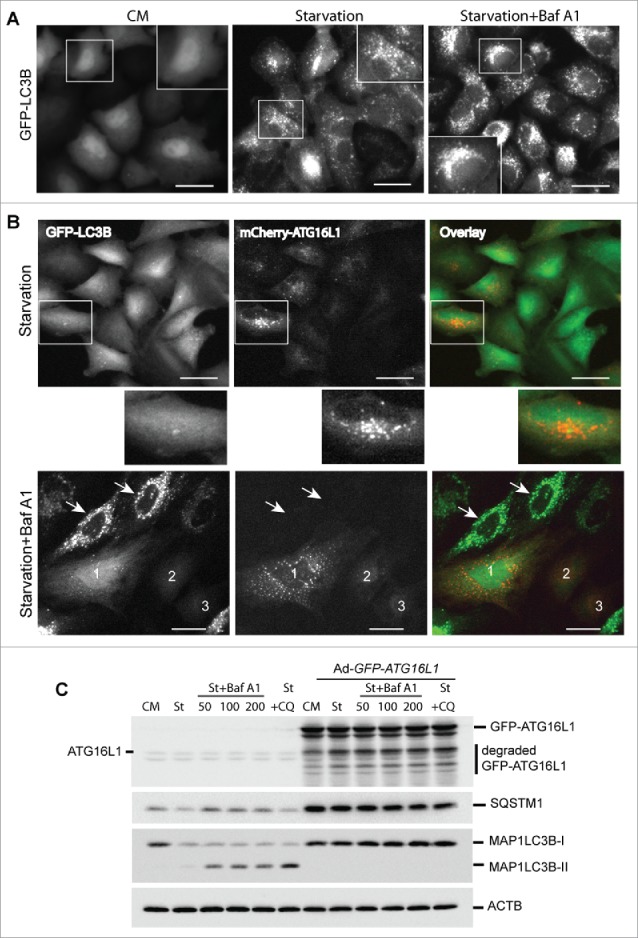
Transient expression of ATG16L1 inhibits autophagosome biogenesis. (A) HeLa cells stably expressing GFP-LC3B were cultured in complete medium (CM), starved in EBSS for 3 h (starvation), or starved in EBSS plus Baf A1 (100 nM) for 3 h (starvation + Baf A1). Boxed areas are enlarged to show the detailed GFP-LC3B puncta. Scale bar: 25 micron. (B) HeLa cells stably expressing GFP-LC3B were infected with Ad-mCherry-ATG16L1 (MOI 2) for 16 h. Cells were then starved in EBSS for 3 h (upper panel) or starved in EBSS plus baf A1 (100 nM) for 3 h (lower panel). Boxed area is enlarged to show the GFP-LC3B and mCherry-ATG16L1 puncta. Arrows point to cells with no mCherry-ATG16L1 expression. Cells with mCherry-ATG16L1 expression are labeled 1, 2, and 3. Scale bar: 25 micron. (C) HeLa cells were infected with Ad-GFP-ATG16L1 (MOI 2) for 16 h; cells were starved in EBSS, EBSS plus baf A1 (from left 50 nM, 100 nM and 200 nM, respectively), or chloroquine (100 µM). Cells were lysed and analyzed with immunoblot using ATG16L1, SQSTM1, or LC3 antibodies as indicated. St, starvation; Baf A1, bafilomycin A1; CQ, chloroquine.
We further examined this phenomenon using a biochemical assay. HeLa cells were transduced with a control adenoviral vector or with Ad-GFP-ATG16L1 for 16 h. As shown in Figure 4C, there was no appreciable autophagic response (LC3B-II level) with either starvation or starvation plus lysosome inhibitor treatment (bafilomycin A1 or chloroquine) in HeLa cells following transient overexpression of GFP-ATG16L1. A significant degree of SQSTM1 protein accumulation was observed, indicating a global inhibition of autophagy with ATG16L1 overexpression. In contrast, HeLa cells demonstrated normal autophagic flux exhibited by accumulation of LC3B-II with starvation and lysosome inhibitor treatments (Fig. 4C).
Although mCherry-ATG16L1 puncta were negative for GFP-LC3B, we observed a limited number of GFP-LC3B puncta in mCherry-ATG16L1-expressing cells. A previous study regarded the GFP-LC3B puncta as autophagosomal structures in ATG16L1-overexpressing cells.10 As SQSTM1 can bind LC3B31 in the context of SQSTM1 accumulation in ATG16L1-overexpressing cells, the likelihood of LC3B+ puncta in the setting of transient expression of ATG16L1 more likely represents protein aggregates, not autophagosome or autolysosome structures. To examine this hypothesis, we constructed vectors expressing GFP-tagged wild-type LC3B and the LC3BG120A mutant. LC3BG120A is a mutant LC3B, in which glycine 120 is replaced with an alanine. LC3BG120A cannot be cleaved by ATG4B to expose glycine 120 and thus cannot be lipidated and recruited to the autophagic membrane.32 Thus, whether GFP-LC3B is incorporated within the autophagic membrane or into protein aggregates can be determined utilizing these tools. Wild-type GFP-LC3B responded normally to nutrient deprivation. Inhibition of autolysosome-dependent degradation by Baf A1 led to accumulation of distinct GFP-LC3B+ puncta (Fig. 5A, right). In contrast, the GFP-LC3BG120A mutant did not respond to starvation or Baf A1, as demonstrated by the lack of GFP-LC3BG120A-positive puncta accumulation (Fig. 5B, right). Co-transient expression of mCherry-ATG16L1 and GFP-LC3BG120A led to GFP-LC3BG120A puncta (Fig. 5C). Some GFP- LC3BG120A+ puncta were close to or associated with mCherry-ATG16L1+ structures, but did not colocalize with each other. To confirm SQSTM1 protein aggregate formation upon transient expression of ATG16L1, A549 cells expressing GFP-ATG16L1 were probed with SQSTM1 antibody, and an accumulation of large SQSTM1 protein aggregates was clearly observed, with only a few of these SQSTM1 aggregates being associated with GFP-ATG16L1+ structures (Fig. S2B). In comparison, stably expressed GFP-ATG16L1 puncta induced by starvation were mostly colocalized with SQSTM1 (Fig. S2C). Of note, a previous study demonstrated that SQSTM1 is recruited to the autophagosome formation site under starvation conditions, which is distinct from SQSTM1 being degraded by autophagy.33 This result furthermore supports the idea that the transiently expressed ATG16L1 puncta are not indicative of autophagic membranes.
Figure 5.
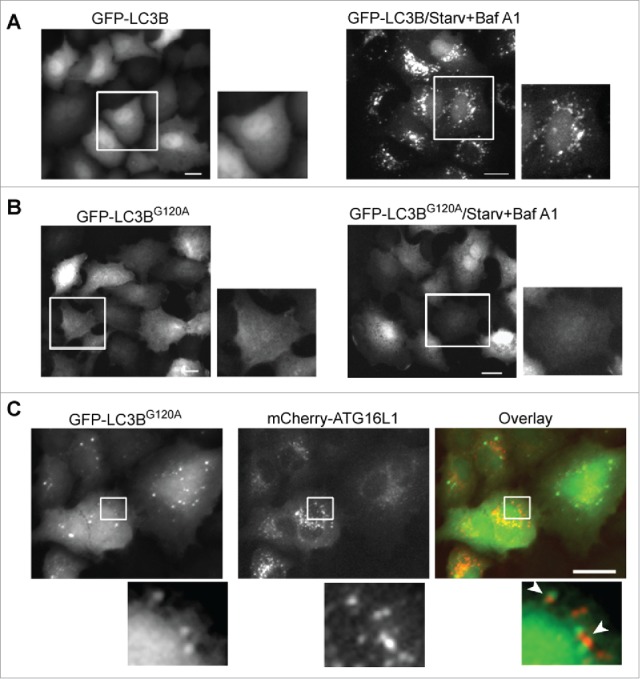
GFP-LC3B puncta represent protein aggregates in cells overexpressing ATG16L1. (A) A549 cells were infected with Ad-GFP-LC3B vector (MOI 2) for 16 h (left). Cells were then starved in EBSS with bafilomycin A1 (100 nM) for 2 h (right). (B) A549 cells were infected with Ad-GFP-LC3BG120A vector (MOI 2) for 16 h (left). Cells were then starved in EBSS with bafilomycin A1 (100 nM) for 2 h (right). (C) A549 cells were co-infected with Ad-mCherry-ATG16L1 and Ad-GFP-LC3BG120A at MOI 2 for 16 h. Boxed areas are enlarged to show the association between GFP-LC3BG120A puncta and mCherry-ATG16L1 puncta. Scale bar: 25 micron.
Collectively, these data demonstrate that GFP-LC3B+ puncta in the context of transient expression of ATG16L1 likely represent protein aggregates.
Transiently expressed ATG16L1 aberrantly targets RAB11+ recycling endosomes
A typical expression of a protein can form aggregates and be observed as puncta-like structures, as superfluous expression of SQSTM1 demonstrates.31 Alternatively, an overexpressed protein could aberrantly target intracellular membranes. We examined the membrane system of the cell to identify the membrane that is aberrantly targeted by the transiently expressed ATG16L1. GFP-ATG16L1+ puncta-like structures did not colocalize with the cis-Golgi membrane (Fig. 6A), the trans-Golgi membrane (Fig. 6B), the ERGIC membrane (Fig. 6C), the lysosome membrane (Fig. 6D), or the late endosome membrane (Fig. 6E).
Figure 6.
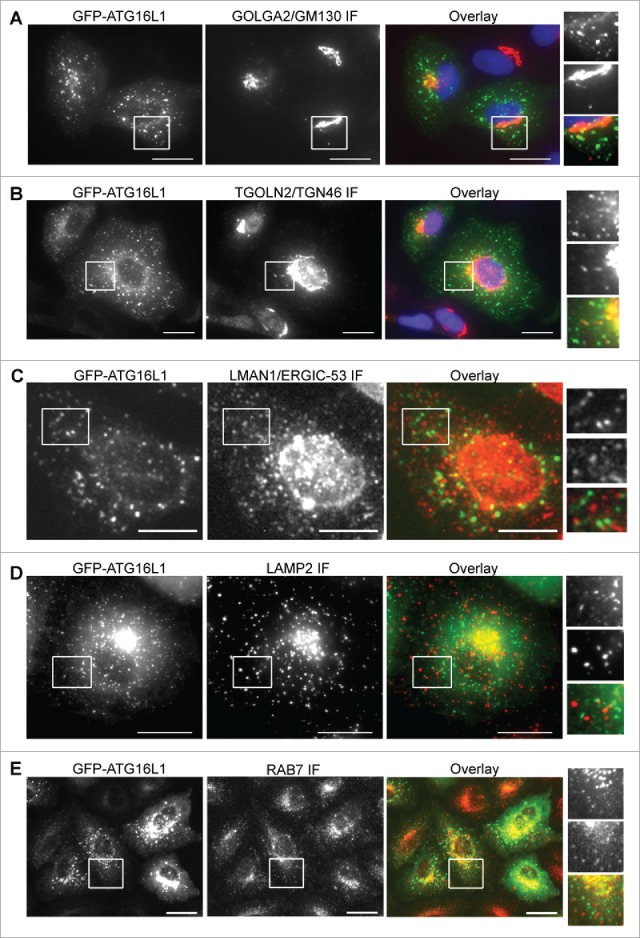
Identification of the aberrant targeted membrane by transiently expressed ATG16L1. A549 cells were infected with Ad-GFP-ATG16L1 (MOI 2) for 16 h. Cells were then fixed and probed with a cis-Golgi marker GOLGA2/GM130 (A), trans-Golgi marker TGOLN2/TGN46 (B), ER Golgi intermediate compartment marker (LMAN1/ERGIC-53) (C), lysosome marker LAMP2 (D), or late endosome marker RAB7 (E). Boxed areas are enlarged to show the association between GFP-ATG16L1+ puncta and corresponding subcellular compartments. Scale bar: 25 micron.
To examine whether the ATG16L1+ puncta colocalize with the early endocytic compartments, labeled TRF (transferrin) was loaded onto the A549 cells overexpressing GFP-ATG16L1. TRF colocalized with preformed GFP-ATG16L1+ puncta (Fig. 7A). Additionally, GFP-ATG16L1+ structures were stained positive for TFRC (transferrin receptor) (Fig. 7B). These findings suggest that overexpressed ATG16L1 may aberrantly target the early endocytic compartments.
Figure 7.

Transiently expressed ATG16L1 aberrantly targets RAB11+ recycling endosomes. (A) A549 cells were infected with Ad-GFP-ATG16L1 (MOI 2) for 16 h. Cells were then loaded with Alexa Fluor 555-conjugated TRF for 30 min. (B) A549 cells were infected with Ad-GFP-ATG16L1 (MOI 2) for 16 h. Cells were then fixed and probed with TFRC antibody. (C) A549 cells were infected with Ad-GFP-ATG16L1 (MOI 2). Cells were fixed and probed with RAB11 antibody. (D) A549 cells stably expressing GFP-ATG16L1 were starved in EBSS for 2 h. Cells were then fixed and probed with RAB11 antibody. Boxed area is enlarged to show the colocalization. Quantified colocalizations are labeled as indicated. Scale bar: 25 micron.
TFRC enters the endocytic membrane system as early endocytic vesicles and proceeds to early sorting endosomes with passage to fast recycling and slow REs.34 Colocalization of endocytic compartment-specific RAB proteins with ATG16L1+ puncta were examined to differentiate the specific early endocytic compartment atypically targeted by transiently expressed ATG16L1. RAB5+ endocytic vesicles and RAB5+ early endosomes were not identified in the colocalization with ATG16L1+ structures (Fig. S4A). Staining for the early endosome marker EEA1 further confirmed these findings (Fig. S4B). Similar to RAB5+ early endosomes, RAB4+ fast REs did not colocalize with ATG16L1+ structures (Fig. S4, C). These data, in addition to the colocalization of TRF with ATG16L1+ puncta-like structures, collectively imply that exogenous ATG16L1 may uncharacteristically target slow REs. Indeed, GFP-ATG16L1+ structures fully colocalized with RAB11+ REs (Fig. 7C). This finding was confirmed in wild-type MEFs (data not shown), MEF atg5 KO (data not shown), MEF atg7 KO (Fig. S4D), and HeLa cells (Fig. S4E). In contradistinction and importantly, stably expressed GFP-ATG16L1 puncta did not colocalize with RAB11+ REs (Fig. 7D). We furthermore confirmed that endogenous ATG16L1+ or WIPI2+ phagophores were not colocalized with TFRC antibody-labeled REs or RAB11+ REs or TRF-labeled REs (Fig. 8 and Fig. S5C). We validated the WIPI2 antibody for immunofluorescence staining. As shown in Figure S5A, a few WIPI2+ phagophores was observed in nutrient-rich medium (CM). In contrast, a number of WIPI2+ phagophores formed after starvation, which were blocked by wortmannin treatment. 100% of ATG16L1+ phagophores (by counting 30 cells) were colocalized with WIPI2+ puncta (Fig. S5B). Approximately 75 ± 5% of WIPI2+ phagophores were positive for ATG16L1 staining, which supports the notion that WIPI2 on the autophagic membrane may recruit the ATG16L1 complex onto the phagophore.23 This finding again accentuates the difference observed between endogenous ATG16L1 and exogenously overexpressed ATG16L1.
Figure 8.
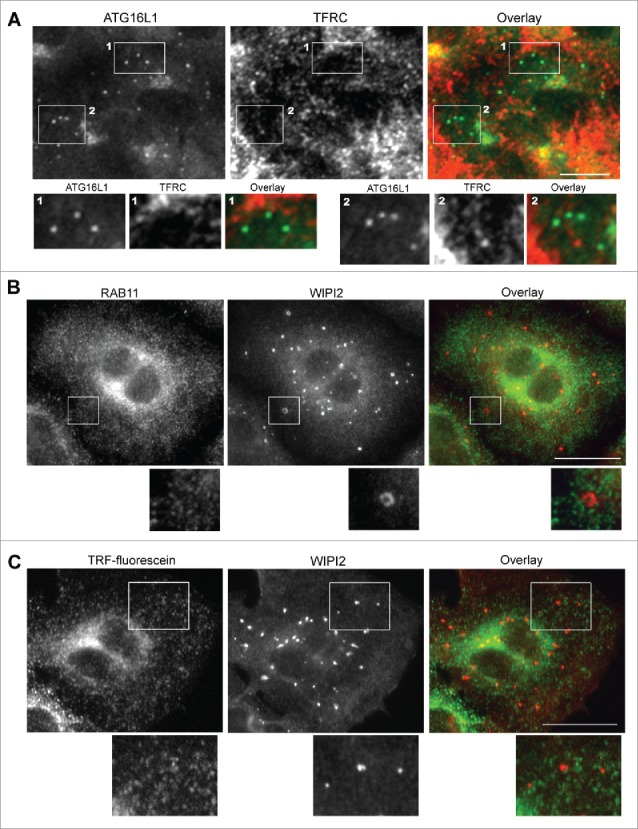
Endogenous ATG16L1+ or WIPI2+ phagophores do not colocalize with recycling endosomes. (A) A549 cells were starved with EBSS for 2 h. Cells were then fixed and co-probed with ATG16L1 antibody and TFRC antibody. Alexa Fluor 488-conjugated goat anti-rabbit IgG- and Alexa Fluor 647-conjugated donkey anti-mouse IgG secondary antibodies were used to detect ATG16L1 and TFRC+ RE, respectively. Boxed area 1 and 2 are enlarged to show the colocalization between ATG16+ puncta and TFRC-labeled recycling endosomes. (B) A549 cells were starved with EBSS for 2 h. Cells were then fixed and co-probed with RAB11 antibody and WIPI2 antibody. Alexa Fluor 488-conjugated goat anti-rabbit IgG and Alexa Fluor 647-conjugated donkey anti-mouse IgG secondary antibodies were used to detect RAB11+ REs and WIPI2+ phagophores, respectively. (C) A549 cells were starved with EBSS plus fluorescein-conjugated human TRF for 2 h. Cells were then fixed and probed with WIPI2 antibody. Alexa Fluor 647-conjugated donkey anti-mouse IgG secondary antibody was used to detect WIPI2+ phagophores. Boxed area is enlarged to show the details. Scale bar: 25 micron.
Transiently expressed ATG16L1 causes RAB11+ RE aggregates
Translocation of the ATG16L1 complex from the cytosol to the autophagic membrane is regulated by autophagy signaling and proceeds in a dynamic fashion during the autophagy process. Dissociation from the completed autophagosome is an essential process for recycling of the ATG16L1 complex. Yet, transient expression of ATG16L1 was observed to limit its capacity to dissociate from RAB11+ RE membranes and, in studying these epiphenomena, we were struck by the reality that REs were never observed to innately form such large structures in any cell model tested (data not shown). As shown in Figure 9A, overexpressed mCherry-ATG16L1 formed smaller puncta at an early time point (8 h, arrow). At 16 h post expression, these mCherry-ATG16L1 puncta had clustered to generate large puncta at the perinuclear regions of the cells (arrowhead). A single larger punctum was evident at 24 h (arrowheads). We postulated that the ATG16L1+ RAB11+ REs were able to receive more REs by homotypic tethering. To test this hypothesis, large GFP-ATG16L1+ structures were generated with overexpression of GFP-ATG16L1, followed by loading the cells with Alexa Fluor 555-labeled TRF (TRF-Alexa555). In time, the newly generated TRF-Alexa555+ vesicles advanced to tether with the smaller GFP-ATG16L1+ structures (Fig. 9B, arrows). Ring-like TRF-Alexa555+ REs were distributed peripherally around large pre-existing GFP-ATG16L1+ structures (Fig. 9B, arrowhead). These observations suggest that once formed, ATG16L1+ RAB11+ REs become a membrane center to receive newly generated REs and ATG16L1+ RAB11+ REs to form a larger membrane aggregate by homotypic tethering but not fusion, because an evident vesicle lumen was never observed.
Figure 9.
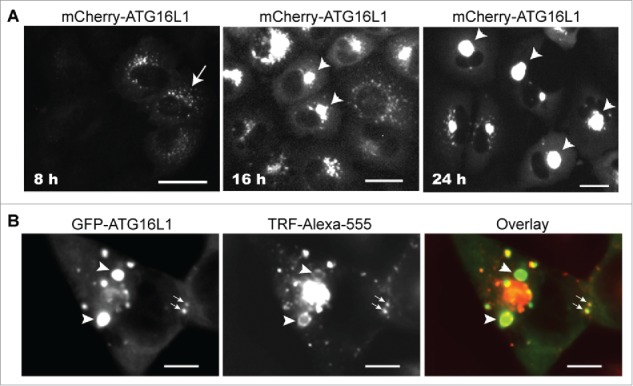
Transiently expressed ATG16L1 causes RAB11+ recycling endosome aggregates. (A) A549 cells were infected with Ad-GFP-ATG16L1 (MOI 4). Images were recorded at 8, 16, and 24 h post infection. Arrow points to puncta. Arrowheads point to GFP-ATG16L1+ aggregates. Scale bar: 25 micron. (B) A549 cells were infected with Ad-GFP-ATG16L1 (MOI 4) for 24 h to form larger GFP-ATG16L1+ structures (arrowhead). Cells were then loaded with Alexa Fluor 555-conjugated TRF for 30 min. Scale bar: 25 micron.
Discussion
One methodology to define membrane contribution to autophagosome biogenesis is to follow autophagy-specific protein trafficking under autophagic stimulation. Such strategies have led to the seminal findings of the contributing membrane, including the ER and mitochondrial membranes,4,6,7 to autophagosome biogenesis.
The functional ATG16L1 complex may or may not form in the correct stoichiometric manner when ATG16L1 is overexpressed. It would appear there is a mutual dependence between ATG16L1 and ATG12–ATG5 for accurate targeting of the complex, without which, autophagic membrane targeting is lost. For example, endogenous ATG16L1 does not target autophagic membrane in the absence of the ATG12–ATG5 conjugate (Fig. 1B) and the ATG12–ATG5 conjugate does not target the autophagic membrane without ATG16L1. 35
We consider that inhibition of autophagosome biogenesis by overexpressed ATG16L1 is a separate event from aberrantly targeting RAB11+ REs by overexpressed ATG16L1. Transiently expressed ATG16L1 is able to inhibit LC3B conversion when it has not yet reached the expression level to form punctate structures (Fig. 4). We also observed that the overexpressed coiled-coil domain of ATG16L1 was able to inhibit autophagosome biogenesis (data not shown). Our observations agree with those from the Fujita et al. study.17 In that study, Fujita et al. found inhibition of autophagy by overexpressed full-length mouse ATG16L1 or coiled-coil domain. They further found that overexpressed ATG16L1 did not interfere with any pre-existing ATG16L1 complexes, meaning that transiently expressed ATG16L1 does not replace endogenous ATG16L1 from an existing ATG16L1 complex. Overexpressed ATG16L1 lacks ATG12–ATG5 conjugate (Fig. 3 and Fig. S2A). This is easy to understand because there is limited endogenous ATG12–ATG5 conjugate available for overexpressed ATG16L1 to form a functional complex. The underlying mechanism of the inhibitory effect of overexpressed ATG16L1 on autophagosome biogenesis is unknown. We hypothesize that transiently overexpressed ATG16L1 forms a defective ATG16L1 complex due to the limited amount of endogenous ATG12–ATG5; moreover, excess ATG16L1 may titrate out an unknown factor that controls endogenous ATG16L1 complex autophagic membrane targeting, which results in the inability of pre-existing endogenous ATG16L1 complex to target the phagophore membrane for LC3B-I to LC3B-II conversion.
As for aberrant targeting of RAB11+ REs by overexpressed ATG16L1, we observed that the coiled-coil domain coordinating with the N-terminal fragment of overexpressed ATG16L1 caused RAB11+ RE targeting (data not shown). Deletion of the coiled-coil domain or N-terminal fragment (amino acids 1–79) of human ATG16L1 abolished the aberrant membrane targeting (data not shown). We hypothesize the following: 1) Complex formation of ATG12–ATG5 with ATG16L1 may regulate ATG16L1 complex conformation for the ATG16L1 that remains in the cytosol. The cytosolic ATG16L1 complex will be recruited onto the phagophore membrane by interaction with adaptor proteins on the phagophore under autophagic stimulation; the coiled-coil domain, which is responsible for forming a dimer of ATG16L1,36 may play a critical role in aberrant targeting via abnormal complex formation when overexpressed. 2) Alternatively, an unknown factor controls the ATG16L1 complex to correctly target to the phagophore membrane. When ATG16L1 is transiently overexpressed, the limited control factor loses that regulatory effect, which leads to ATG16L1 itself aberrantly targeting membrane under no autophagic stimulation. These hypotheses await future investigation.
Although the study of autophagy has made dramatic progress, the fundamental mystery of the origin of the autophagic membrane and how it grows to form an autophagosome is still under debate. Thus far, it has been suggested in mammalian cells that the membranes of the ER,3-5 mitochondria,6 Golgi,20 ER-Golgi intermediate compartment,9 RE,13 and plasma membrane,10 as well as other unlisted membranes, contribute to autophagosome formation. As we can see, on the one hand data suggest that multiple membranes from various sources contribute to autophagosome biogenesis. On the other hand, this is confusing (e.g., how is this coordinated, what SNARE proteins are involved?). We consider that the lack of standardized approaches and utilization of not well-validated tools could be the reason for these discrepancies. The ability to draw any conclusions regarding ATG16L1 intermolecular interactions and membrane targeting or trafficking as it is being overexpressed becomes severely limited. Conclusions derived from studies utilizing such methodologies should be reserved.
Materials and methods
Chemicals and antibodies
Wortmannin (Cell Signaling Technology, 9951); bafilomycin A1 (LC Laboratories, B1080); chloroquine diphosphate salt (Sigma-Aldrich, C6628); TGOLN2/TGN46 antibody (Sigma-Aldrich, T7576); Alexa Fluor 555-conjugated TRF (ThermoFisher Scientific, T35352); fluorescein-conjugated human TRF (ThermoFisher Scientific, T2871). RAB11 antibody (ThermoFisher Scientific, 71–5300); TFRC antibody (ThermoFisher Scientific, 236–15375); EEA1 antibody (BD Biosciences, 610457); GOLGA2/GM130 antibody (BD Biosciences, 610822); ATG16L1 antibody (Cell Signaling Technology, 8089); WIPI2 antibody (Bio-Rad, MCA5780GA); MAP1LC3B antibody (Cell Signaling Technology, 3868); ATG7 antibody (Cell Signaling Technology, 8558); anti-human ATG12 (Cell Signaling Technology, 2010) and anti-mouse ATG12 (Cell Signaling Technology, 2011); RAB7 antibody (Cell Signaling Technology, 9367); SQSTM1 antibody (Santa Cruz Biotechnology, sc-28359); ULK1 antibody (H-240; Santa Cruz Biotechnology, 33182); LAMP2 antibody (Santa Cruz Biotechnology, sc-18822); LMAN1/ERGIC-53 antibody (C-6; Santa Cruz Biotechnology, sc-365158); anti ACTB/β-actin (Santa Cruz Biotechnology, sc-47778); anti-rabbit IgG (Alexa Fluor 488 or 555 conjugate) (Cell Signaling Technology, 4412; 4413); anti-mouse IgG (Alexa Fluor 488 or 555 conjugate) (Cell Signaling Technology, 4408; 4409); donkey anti-mouse or anti-rabbit IgG (Alexa Fluor 647 conjugate; Jackson ImmunoResearch Laboratories, 715–605–150; 711–175–152). All restriction enzymes for gene expression vector constructions were obtained from New England Biolabs, Inc.
Cell culture and generation of stable cell lines
Cell culture media, DMEMs, were purchased from LONZA (12–614F); fetal bovine serum (ThermoFisher Scientific, SH30072.03); L-glutamine (LONZA, 17–605E); EBSS (ThermoFisher Scientific, SH30029.02). A549 (ATCC, CCL−185), HEK293 cell (ATCC, CRL-1573), HeLa cells (ATCC, CCL−2), MEF WT and MEF atg5 KO cells (kindly provided by Dr. Noboru Mizushima, The University of Tokyo), MEF atg7 KO cells (kindly provided by Dr. Masaaki Komatsu, Tokyo Metropolitan Institute of Medical Sciences), and MEF rb1cc1 KO cells (kindly provided by Dr. Jun-Lin Guan, University of Cincinnati college of Medicine) were grown in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 100 U/ml penicillin/streptomycin in a 5% CO2 incubator at 37°C. For nutrient starvation, cells were cultured in EBSS. Stable lines were made by infection of cells with a retroviral vector expressing a GFP fusion with the human MAP1LC3B gene (GFP-LC3B) and selected in growth medium supplemented with 800 µg/ml of G418 (ThermoFisher Scientific, BP7635). A549 cell stable expression of GFP-ATG16L1 was established by transfection with expression plasmid and selection with G418.
Expression vector construction
GFP or mCherry open reading frame was inserted into a type 5-based adenoviral shuttle vector.37 open reading frames of human ATG16L1 (NP_110430.5), human ATG5 (NP_001273035.1), human ATG12 (NP_004698.3), human RAB5A (NP_004153.2), and human RAB4A (NP_004569.2), were amplified using cDNA from HeLa cells by PCR and inserted at the C terminus of GFP or mCherry. pEGFP-C1 (Takara Biomedical Technology, 632470) was used to construct the GFP-ATG16L1 expression vector for establishing A549 cell line stable expression of GFP-ATG16L1. A retroviral vector (Takara Biomedical Technology, 631503) expressing GFP-LC3B was used to generate stable expression cell lines of A549 and HeLa cells, respectively. All inserted genes were sequenced. Adenoviral vectors were generated and purified as described.37
Exogenous expression of genes and in vitro cell treatment
Cells were infected with adenoviral vector expression genes (see details in figure legend). Alternatively, cells were transfected with expression plasmids using Lipofectamine 2000 (ThermoFisher Scientific, 11668) to express the indicated genes (details in the text and figure legends).
To label REs, cells were washed and incubated in EBSS for 30 min at 37°C before adding complete medium with Alexa Fluor 555-conjugated TRF (20 µg/ml). For colocalization study of TRF-labeled RE with ATG16L1 or WIPI2, fluorescein-conjugated human TRF (20 µg/ml) was added into EBSS-treated cells for 2 h. Cells were then washed and fixed in 4% paraformaldehyde in 1 × phosphate-buffered saline (PBS; LONZA, 17–516F) for 20 min before immunostaining or imaging using the EVOS FL Cell Imaging System (ThermoFisher Scientific, AMF4300).
Florescence microscopy and immunofluorescent staining
Cells were cultured in nutrient-rich medium or EBSS +/- chloroquine (CQ), bafilomycin A1, or wortmannin for the indicated times, and then were fixed in 4% paraformaldehyde in 1 × PBS for 15 min. Cells were washed 3 times for 10 min each with 1 × PBS and GFP-ATG16L1, mCherry-ATG16L1, or GFP-LC3B puncta and were recorded using fluorescence microscopy. For immunofluorescence staining, cells were grown on coverslips for the indicated times. Cells were then fixed with 4% paraformaldehyde in 1 × PBS for 15 min. After washing 3 times with 1 × PBS, cells were permeabilized using 0.1% Triton X-100 (Sigma-Aldrich, X100) in 1 × PBS, then blocked with 5% BSA (Sigma-Aldrich, A9418), and primary antibody and secondary antibody (Alexa Fluor 488-, Alexa Fluor 555- or Alexa Fluor 647-labeled) were sequentially applied. Stained cells were examined and recorded using the EVOS FL Cell Imaging System equipped with a Sony ICX285AQ color CCD (2/3´´, 1,360 × 1,024, 1.4 megapixels). 20 × (NA/0.4) or 40 × (NA/0.75) objective lens were used to observe and record fluorescent cell images. Images were saved as 16-bit color TIFF. For image figure presentation, we opened the TIFF file in Adobe Photoshop CSS Extended version 12.3 × 32. Images were brightness/contrast-adjusted. Green channel image, Red channel image and overlay image were separated as shown in the figures.
Immunoblot assays
Cells were rinsed with ice-cold 1 × PBS buffer, scraped, and collected by centrifugation at 4°C. Cells were then lysed in cell lysis buffer (Cell Signaling Technology, 9803) with protease inhibitor cocktails (Cell Signaling Technology, 5871). Lysates were centrifuged at 15,000 g for 15 min at 4°C, and supernatant fractions were collected. 10–20 µg of cell lysates were separated using SDS-PAGE gels and transferred to Immun-Blot PVDF membrane (Bio-Rad, 1620177). The membranes were blocked with 5% skim milk in 0.1% Tween 20 in 1 × PBS buffer and then incubated with primary antibodies. Blots were probed with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (ThermoFisher Scientific, 31430; 31460). Bands were visualized using Pierce ECL Western Blotting Substrate (ThermoFisher Scientific, 32106).
Supplementary Material
Abbreviations
- ATG5
autophagy-related 5
- ATG12
autophagy-related 12
- ATG16L1
autophagy-related 16-like 1 (S. cerevisiae)
- Baf A1
bafilomycin A1
- CM
complete medium
- CQ
chloroquine
- EBSS
Earle's balanced salt solution
- GFP
green fluorescent protein
- MAP1LC3B
microtubule-associated protein 1 light chain 3 β
- MOI
multiplicity of infection
- MTORC1
mechanistic target of rapamycin (serine/threonine kinase) complex 1
- PIK3C3
phosphoinositide-3-kinase, class 3
- PtdIns3K
class III phosphatidylinositol 3-kinase
- RAB11
RAB11, member RAS oncogene family
- RB1CC1
RB1-inducible coiled-coil 1
- RE
recycling endosome
- SQSTM1
sequestosome 1
- ULK1
unc-51 like kinase 1
- WIPI2 WD
repeat domain, phosphoinositide interacting 2
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. Noboru Mizushima (The University of Tokyo) for the MEF WT and MEF atg5 KO cells, Dr. Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science) for the MEF atg7 KO cells and Dr. Jun-Lin Guan (University of Cincinnati) for the MEF rb1cc1 KO cells.
Funding
This work was supported by the National Institutes of Health/National Cancer Institute Grant 3P30CA047904–21S5.
References
- [1].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6:764-76; PMID:20639694; http://dx.doi.org/ 10.4161/auto.6.6.12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Koyama-Honda I, Itakura E, Fujiwara TK, Mizushima N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 2013; 9:1491-9; PMID:23884233; http://dx.doi.org/ 10.4161/auto.25529 [DOI] [PubMed] [Google Scholar]
- [3].Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685-701; PMID:18725538; http://dx.doi.org/ 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 2009; 11:1433-7; PMID:19898463; http://dx.doi.org/ 10.1038/ncb1991 [DOI] [PubMed] [Google Scholar]
- [5].Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009; 5:1180-5; PMID:19855179; http://dx.doi.org/ 10.4161/auto.5.8.10274 [DOI] [PubMed] [Google Scholar]
- [6].Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656-67; PMID:20478256; http://dx.doi.org/ 10.1016/j.cell.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al.. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495:389-93; PMID:23455425; http://dx.doi.org/ 10.1038/nature11910 [DOI] [PubMed] [Google Scholar]
- [8].Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2013; 2:e00947; PMID:23930225; http://dx.doi.org/ 10.7554/eLife.00947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ge L, Zhang M, Schekman R. Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. Elife 2014; 3:e04135; PMID:25432021; http://dx.doi.org/ 10.7554/eLife.04135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12:747-57; PMID:20639872; http://dx.doi.org/ 10.1038/ncb2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell 2011; 146:303-17; PMID:21784250; http://dx.doi.org/ 10.1016/j.cell.2011.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013; 154:1285-99; PMID:24034251; http://dx.doi.org/ 10.1016/j.cell.2013.08.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Longatti A, Lamb CA, Razi M, Yoshimura S, Barr FA, Tooze SA. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol 2012; 197:659-75; PMID:22613832; http://dx.doi.org/ 10.1083/jcb.201111079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al.. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/ 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132-9; PMID:20056399; http://dx.doi.org/ 10.1016/j.ceb.2009.12.004 [DOI] [PubMed] [Google Scholar]
- [16].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- [17].Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092-100; PMID:18321988; http://dx.doi.org/ 10.1091/mbc.E07-12-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657-68; PMID:11266458; http://dx.doi.org/ 10.1083/jcb.152.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003; 116:1679-88; PMID:12665549; http://dx.doi.org/ 10.1242/jcs.00381 [DOI] [PubMed] [Google Scholar]
- [20].Itoh T, Fujita N, Kanno E, Yamamoto A, Yoshimori T, Fukuda M. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol Biol Cell 2008; 19:2916-25; PMID:18448665; http://dx.doi.org/ 10.1091/mbc.E07-12-1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Boada-Romero E, Letek M, Fleischer A, Pallauf K, Ramón-Barros C, Pimentel-Muiños FX. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. EMBO J 2013; 32:566-82; PMID:23376921; http://dx.doi.org/ 10.1038/emboj.2013.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, Virgin HW, Mizushima N. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep 2013; 14:284-91; PMID:23392225; http://dx.doi.org/ 10.1038/embor.2013.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell 2014; 55:238-52; PMID:24954904; http://dx.doi.org/ 10.1016/j.molcel.2014.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gammoh N, Florey O, Overholtzer M, Jiang X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat Struct Mol Biol 2013; 20:144-9; PMID:23262492; http://dx.doi.org/ 10.1038/nsmb.2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S, et al.. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 2013; 203:115-28; PMID:24100292; http://dx.doi.org/ 10.1083/jcb.201304188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Moreau K, Puri C, Rubinsztein DC. Methods to analyze SNARE-dependent vesicular fusion events that regulate autophagosome biogenesis. Methods 2015; 75:19-24; PMID:25461811; http://dx.doi.org/ 10.1016/j.ymeth.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mizushima N, Yoshimori T, Ohsumi Y. Mouse Apg10 as an Apg12-conjugating enzyme: analysis by the conjugation-mediated yeast two-hybrid method. FEBS Lett 2002; 532:450-4; PMID:12482611; http://dx.doi.org/ 10.1016/S0014-5793(02)03739-0 [DOI] [PubMed] [Google Scholar]
- [28].Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science 2008; 321:117-20; PMID:18599786; http://dx.doi.org/ 10.1126/science.1154822 [DOI] [PubMed] [Google Scholar]
- [29].Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature 1998; 395:395-8; PMID:9759731; http://dx.doi.org/ 10.1038/26506 [DOI] [PubMed] [Google Scholar]
- [30].Wirth M, Joachim J, Tooze SA. Autophagosome formation–the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol 2013; 23:301-9; PMID:23727157; http://dx.doi.org/ 10.1016/j.semcancer.2013.05.007 [DOI] [PubMed] [Google Scholar]
- [31].Gao W, Chen Z, Wang W, Stang MT. E1-like activating enzyme Atg7 is preferentially sequestered into p62 aggregates via its interaction with LC3-I. PloS one 2013; 8:e73229; PMID:24023838; http://dx.doi.org/ 10.1371/journal.pone.0073229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang W, Chen Z, Billiar TR, Stang MT, Gao W. The carboxyl-terminal amino acids render pro-human LC3B migration similar to lipidated LC3B in SDS-PAGE. PloS One 2013; 8:e74222; PMID:24040206; http://dx.doi.org/ 10.1371/journal.pone.0074222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 2011; 192:17-27; PMID:21220506; http://dx.doi.org/ 10.1083/jcb.201009067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sheff DR, Daro EA, Hull M, Mellman I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J Cell Biol 1999; 145:123-39; PMID:10189373; http://dx.doi.org/ 10.1083/jcb.145.1.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al.. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; http://dx.doi.org/ 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- [36].Fujita N, Saitoh T, Kageyama S, Akira S, Noda T, Yoshimori T. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J Biol Chem 2009; 284:32602-9; PMID:19783656; http://dx.doi.org/ 10.1074/jbc.M109.037671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Construction of adenovirus vectors through Cre-lox recombination. J Virol 1997; 71:1842-9; PMID:9032314 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.