

Abstract
ONC201/TIC10 is a first-in-class small molecule inducer of TRAIL that causes early activation of the integrated stress response. Its promising safety profile and broad-spectrum efficacy in vitro have been confirmed in Phase I/II trials in several advanced malignancies. Binding and reporter assays have shown that ONC201 is a selective antagonist of the dopamine D2-like receptors, specifically, DRD2 and DRD3. We hypothesized that ONC201’s interaction with DRD2 plays a role in ONC201’s anticancer effects. Using cBioportal and quantitative reverse-transcription polymerase chain reaction analyses, we confirmed that DRD2 is expressed in different cancer cell types in a cell type–specific manner. On the other hand, DRD3 was generally not detectable. Overexpressing DRD2 in cells with low DRD2 levels increased ONC201-induced PARP cleavage, which was preceded and correlated with an increase in ONC201-induced CHOP mRNA expression. On the other hand, knocking out DRD2 using CRISPR/Cas9 in three cancer cell lines was not sufficient to abrogate ONC201’s anticancer effects. Although ONC201’s anticancer activity was not dependent on DRD2 expression in the cancer cell types tested, we assessed the cytotoxic potential of DRD2 blockade. Transient DRD2 knockdown in HCT116 cells activated the integrated stress response and reduced cell number. Pharmacological antagonism of DRD2 significantly reduced cell viability. Thus, we demonstrate in this study that disrupting dopamine receptor expression and activity can have cytotoxic effects that may at least be in part due to the activation of the integrated stress response. On the other hand, ONC201’s anticancer activity goes beyond its ability to antagonize DRD2, potentially due to ONC201’s ability to activate other pathways that are independent of DRD2. Nevertheless, blocking the dopamine D1-like receptor DRD5 via siRNA or the use of a pharmacological antagonist promoted ONC201-induced anticancer activity.
Introduction
Dopamine receptors respond to the neurotransmitter dopamine. These receptors are G-protein coupled receptors (GPCRs) and can be divided into two major groups: D1-like and D2-like. The D1-type receptors (DRD1 and DRD5) generally associate with the Gαs/olf subunit and, consequently, activate adenylyl cyclase. By contrast, D2-like receptors (DRD2, DRD3, and DRD4 receptors) usually couple with Gαi/o subunit and inhibit adenylyl cyclase activity [1]. Dopamine receptors have been studied mostly in the context of neurobiology. Their role in cancer remains unclear and appears to be highly tumor type specific. In a number of cancer types, D2-like receptor activation inhibits cancer cell proliferation [2] or induces apoptosis. However, in different contexts, D2-like receptor antagonism has been shown to have anticancer effects [3], [4], [5], [6]. The mechanism of this efficacy involves, at least in part, the activation of the cAMP/PKA pathway [3].
Computational methods have suggested that the first-in-class small molecule ONC201 may be a selective antagonist of the dopamine receptors of the D2-like class. In vitro experiments have confirmed that ONC201 is a direct competitive antagonist of dopamine receptors DRD2 and DRD3, with a Ki of 3 μM [7], [8]. Patients receiving ONC201 in Phase I/Phase II clinical trials have shown a two-fold induction of serum prolactin, a marker for DRD2 receptor engagement [9].
ONC201 (originally known as TIC10) is a promising first-in-class small molecule that we screened and identified by its ability to induce expression of the death ligand Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) in a p53-independent manner [10]. ONC201 treatment results in dual inactivation of Akt and ERK, Foxo3a activation, and TRAIL induction. More recently, we and others reported that ONC201 induces an early activation of the integrated stress response (ISR), as indicated by the consequent upregulation of ATF4 and one of ATF4’s transcriptional target, CHOP. ONC201’s ability to provoke the ISR is dependent on two eIF2α kinases, HRI and PKR [11]. In the study reported here, we explored the possibility that the putative binding of ONC201 with DRD2 and DRD3 may contribute to ISR activation and anticancer activity.
Materials and Methods
Cell Culture
The human tumor cell lines were acquired from the American Type Culture Collection or the Fox Chase Cancer Center Cell Culture Facility. Cell line authentication was performed using short tandem repeat methodology [12]. All cell lines used in this study were confirmed to be free of mycoplasma contamination. The human lung, colorectal, liver, and prostate cancer cell lines were cultured in RPMI or McCoy's 5A medium. Human breast, glioblastoma, and neuroblastoma cell lines were maintained in Dulbecco's minimum essential medium. The ONC201/TIC10-resistant derivative of RKO colorectal cancer cells was previously described [11]. Nontumorigenic cells were cultured in Eagle's minimum essential medium. All basal media were supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution.
Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
RNA was isolated using RNeasy kit (Qiagen) or Quick-RNA Miniprep kit (Zymo Research) according to manufacturers' instructions. RNA was quantitated using a Nanodrop spectrophotometer. cDNA was synthesized using a SuperScript II RT kit, while real-time PCR was performed using a Quantitect SYBR Green PCR mix. Relative amounts of target mRNA were quantitated using the 2ΔΔCt method using GAPDH as internal control. At least three technical replicates per biological replicate were analyzed.
Immunoblotting
Cultures were washed twice with PBS, and the cells were lysed into lysis buffer (50 mM HEPES, 100 mM NaCl, 10 mM EDTA, 0.5% NP40, 10% glycerol, supplemented with 0.0001% Tween 20, 0.1 mM PMSF, 0.1 mM NaVO4, 0.5 mM NaF, 5 μg/ml leupeptin, 0.1 mM DTT). The proteins were quantified with the Bio-Rad protein assay and loaded equally onto 4% to 12% NuPage SDS-polyacrylamide gels (Invitrogen). Proteins were electrophoretically transferred to PVDF membranes. After blocking with 5% BSA or 5% milk, membranes were incubated with primary antibody overnight. Subsequently, incubation with appropriate secondary antibodies labeled with horseradish peroxidase was performed. Signal was visualized using chemiluminescence detection. The primary antibodies used in this study were: antibodies against PKA substrate (cat. # 9621S), ATF4 (cat. # 11815S), CHOP (cat. # 2895S), phospho-ERK1/2 (cat. # 4377S), ERK (cat. # 9102S), and PARP (cat. # 9542S) (Cell Signaling); against Ran (cat. # 610341) (BD Biosciences); and against β-actin (Sigma). Secondary antibodies were acquired from Pierce (cat. # 31430 and 31,460).
Plasmid Overexpression
Plasmid DNA constructs were acquired from Addgene and amplified according to Addgene recommendations. Plasmid DNA was isolated using QIAprep Spin midiprep kit (Qiagen) according to manufacturer's instructions. A total of 0.5 to 1 μg DNA was transfected into cells using Lipofectamine 2000. Vehicle or ONC201 was added 24 hours later, when indicated. GFP-DRD2 expression was verified by GFP-fluorescence imaging with a Nikon Eclipse-Ti-U inverted microscope.
Knockout of DRD2 and DRD3 Expression Using CRISPR
pLCV2E (Addgene #78852) was generated by PCR amplification of the C-terminus of the ORF containing eSpCas9(1.1) (Addgene #71814). PCR primers contained EcoRV and BamHI restriction sites to facilitate cloning into plasmid pLCV2 (Addgene #52961). This fragment contains the three point mutations which enhance specificity of codon-optimized SpCas9 and maintains the ORF of the polypeptide encoding puromycin acetyltransferase. The plasmid was sequence verified between restriction sites.
Single-guide RNAs (sgRNAs) were chosen using sgRNA Designer. For DRD2, RefSeq NM_016574.3 was used. On-target scores >0.6 were chosen such that several high-scoring sgRNAs could be screened using the same amplicon in a Surveyor or TIDE assay. Off-target scores were largely neglected since the eSpCas9 enzyme has low off-target rates. All sgRNAs were targeted to sequences encoding the 7-transmembrane domain. All chosen sgRNAs had a native 5′ G nucleotide which enhances expression from a U6 promoter and avoids potential mismatches which may limit on-target efficiency. Guides were cloned into the BsmBI site of pLCV2E, and sequence was confirmed. Sequences for single-guide RNAs and amplicon primers are as follows:
Guide RNA’s for DRD2 deletion:
| Guide 1 | GATGGAGGAGTAGACCACGA |
|---|---|
| Guide 3 | GATCTTGATGTAGACCAGCA |
| Guide RNAs for DRD3 deletion | |
| Guide 4 | GCACGGGACAGAGCTCCTGT |
| Guide 5 | GTATTAAAGCCAAACAGAAG |
| Guide 6 | GAAGCCATCACTGTTACCTG |
| Primer sequences for sequence verification | |
| Hs DRD2 forward | GTAGACCTAGAAGGGCACGC |
| Hs DRD2 reverse | GTAGACCTAGAAGGGCACGC |
| Hs DRD3 forward | ACGAACTTCTTTCATCTGGTCT |
| Hs DRD3 reverse | AAGCTTTGCATTCTGTGGCA |
One 10-cm dish of nearly confluent HEK293T cells was transfected with 4 μg of psPAX2, 2 μg of CMV-VSVG, and 4 μg of pLCV2E-sgRNA plasmid using 30 μl of Lipofectamine 2000 in 5 ml RPMI. Six hours later, the medium was replaced with DMEM/10% FBS. Two days later, the conditioned medium was collected and centrifuged. The supernatant was used to infect HCT116, HT29, or SKNSH cells with 8 μg/ml Polybrene. After 2 days, cells were trypsinized, plated at 10% confluency, and then treated with 1 μg/ml puromycin. Cells were passaged in selective medium at least two times prior to collection for DNA isolation.
To verify knockout of DRD2 expression, DNA was isolated from pooled selected cells and untransfected cells and diluted to 100 ng/μl. PCR was performed using Accuprime PFX (Life Technologies) using touchdown PCR. The starting amplification temperature was 65°C and was decreased by 0.5°/cycle for 10 cycles. Amplification at 60°C was continued for 20 additional cycles. One microliter of PCR product was diluted and sequenced (GeneWiz). Unmodified DNA from wt cells was used as a negative control. .ab1 chromatograms were inputted into TIDE [13] (https://tide-calculator.nki.nl/) to determine indel rates.
Viability and Apoptosis Assays
Viability was assayed using the CellTiter-Glo (CTG) assay according to the manufacturer's instructions (Promega). A total of 1 × 104 cells were seeded in 96-well black plates and incubated for 24 hours prior to addition of agents. To assay viability by a different method, trypan blue dye exclusion assays were performed. The cells were cultured in 24-well plates, treated with vehicle or ONC201, harvested, and stained with trypan blue. Viable cells were counted using a Cellometer cell counter.
Sub-G1 analyses were performed to quantitate apoptosis. After treatment, floating and adherent cells were fixed in 95% ethanol and stained with propidium iodide in the presence of RNase A. Cell cycle profile and SubG1 analyses were performed using a Coulter-Beckman Elite Epics cytometer.
Long-term treatment effects were assessed by performing clonogenic assays. A total of 500 or 2500 cells/well were plated in 6-well plates. After 24-hour incubation, medium was replaced with that containing DMSO vehicle or ONC201. After 72 hours of treatment, fresh medium without agents was added. Cells were cultured for 7 to 10 days after ONC201 removal, with replenishment of medium after 3 to 4 days. Cells were washed twice with PBS, fixed, and stained with 0.25% crystal violet-methanol solution for 30 minutes. After washing stain off with water, plates were allowed to dry and colonies were counted manually.
Transient Knockdown Using siRNA
Cells were plated in medium with 10% FBS but without antibiotic and incubated overnight. siRNA (a pool of three target-specific siRNAs) (Santa Cruz Biotechnology) (40 nM final concentration) was transfected into cells using 9 μl Lipofectamine RNAiMax. After 24 hours, vehicle or ONC201 was added. In the case of double knockdowns, the final concentration of each siRNA was 20 nM.
Stable Knockdown Using shRNA
Five different lentiviral plasmid shRNA constructs for DRD2 (Origene) were amplified, and DNA was isolated using plasmid mini kit (Qiagen) according to manufacturer's instructions. Lentiviral particles were generated by transfecting HEK293T cells with shDRD2 DNA, packaging plasmids pCMV-VSV-G, and pCMV delta R8.2 in 2:1:1 ration. After 72 hours, lentiviral particles were collected, diluted with equal volume of media, and added to cultures of HCT116 cells. Stable transfectants were selected with 1 μg/ml puromycin. Knockdown efficiency was assessed by qRT-PCR analysis for DRD2 mRNA expression.
Statistics
Data are presented as means ± SEM. To assess the statistical significance of the differences, unpaired Student's t test with Holm-Sidak correction for multiple comparisons (maximum of three comparisons were made) was performed with P < .05 deemed as statistically significant. Measurements from three biological replicates per treatment group were compared. Unless otherwise noted in the figure legend, comparisons were made against the vehicle control.
Results
Expression and Activity of D2-Like Receptors in Colorectal Cancer Cells
Given that dopamine receptors are mostly studied in the context of neurobiology, we explored the expression pattern of the receptors across different cancer cell types. To begin to interrogate the impact of this molecular interaction on ONC201’s anticancer activity, we utilized the cBioPortal for Cancer Genomics resource to assess DRD2 mRNA expression across multiple cancer types. We found that DRD2 expression is highly variable across different cancers (Figure 1A). By contrast, expression of DRD3 was not detectable in most cancers tested (Figure S1) (http://www.cBioPortal.org) [14], [15]. We evaluated the expression of the different dopamine receptors in seven cancer cell lines, including the ONC201/TIC10-resistant derivative of RKO colorectal cancer cells, and two nontumorigenic lines (Table 1). The D1-like receptors (DRD1 and DRD5) were more highly expressed in the tumor cell lines than in the normal lines, as indicated by their lower delta Ct values. By contrast, the expression of the D2-like receptors was highly cell-type specific. A delta Ct value higher than 19.93 indicates that the receptor mRNA level is >10,000-fold lower than that of the housekeeping gene GAPDH. Similar to the results from using cBioportal, DRD3 expression was not appreciable in any of the nine cell types tested.
Figure 1.


D2-like receptors are expressed and functional in colorectal cancer cells. (A) Assessment of DRD2 mRNA expression in different cancer tissue samples using cBioportal. (B) Viability assessment after 72 hours of treatment with dopamine or sumanirole, as indicated in legend. Western blot analyses of PKA substrate phosphorylation in (C) HCT116 cells treated with 500 μM phosphodiesterase inhibitor IBMX and 10 μM adenylyl cyclase activator forskolin (F), or treated with 10 μM ONC201, in the presence or absence of 1 μM protein kinase A inhibitor peptide 6-22 (PKI); (D) Hep3B cells treated with different doses of ONC201 for 24 hours; (E) HCT116, RKO, and ONC201-resistant RKO cells (RKOr1) treated with 10 μM (HCT116) or 5 μM (RKOp and RKOr1) ONC201 for indicated times.
Table 1.
The Expression of the Different Dopamine Receptors Is Variable Across Different Cell Types (Delta Cts versus GAPDH Are Shown)
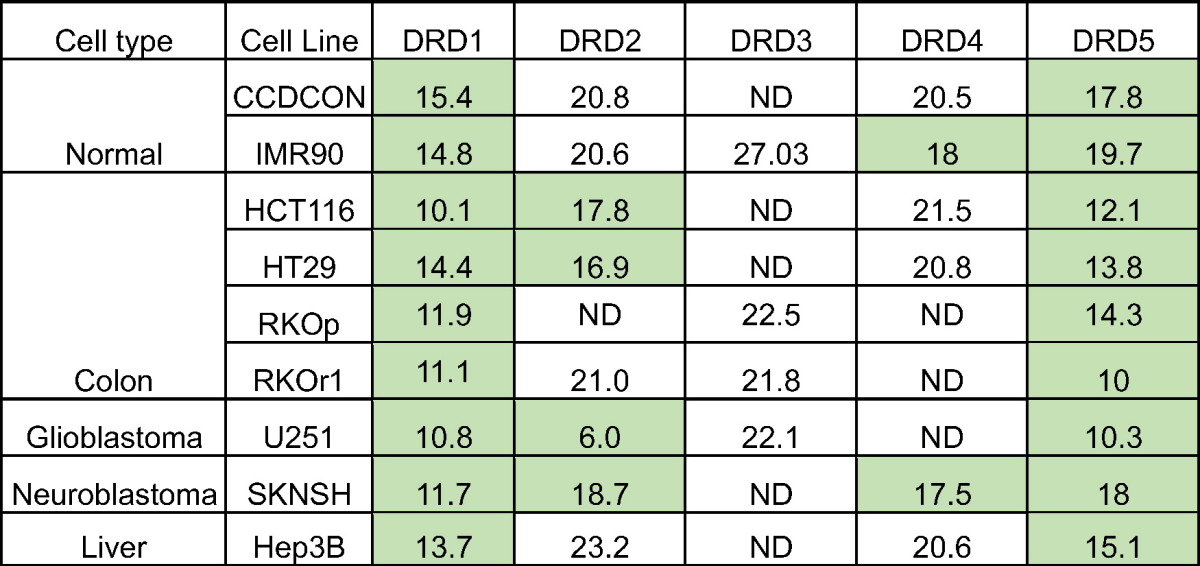
Delta Cts higher than 19.93 indicate that they are 10,000-fold less abundant than GAPDH. Data highlighted in green indicate appreciable receptor mRNA expression in indicated cell lines.
ND, not detected even if Ct value of GAPDH is ≤17.
Engagement of the D2-like receptors can promote cell proliferation [16], [17]. Thus, to confirm that the expressed dopamine receptors, as determined by qPCR, are functional in the cancer cell types to be used in the study, we assayed the impact of dopamine and the selective D2-like receptor agonist sumanirole on cell viability of HCT116 and RKO colorectal cancer cells. Both dopamine and sumanirole induced an increase in cell viability (Figure 1B).
The binding of ligands to dopamine receptors can perturb cyclic AMP (cAMP) levels as a result of alteration in adenylyl cyclase activity [1]. D2-like receptor antagonists can induce an increase in cAMP [18], [19] and activate cAMP-dependent protein kinase (PKA) [20], [21]. Thus, we assessed ONC201’s effects on PKA activity. As a positive control for activation of PKA, HCT116 cells were treated with forskolin, an adenylyl cyclase activator, in the presence of the phosphodiesterase inhibitor 3-isobutyl 1-methylxanthine (IBMX) (Figure 1C). ONC201 treatment induced PKA substrate phosphorylation with differential kinetics in cancer and normal cell lines (Figure 1, D-E).
Cell-Type Specific Role of DRD2 in ONC201's Anticancer Effect
To assess whether the putative binding of ONC201 with DRD2 contributes to ONC201-induced ISR activation and ONC201’s anticancer effects, we modulated the expression of DRD2 and performed assays for ATF4 and CHOP expression and PARP cleavage.
We overexpressed DRD2 in two cancer cell lines that did not express appreciable levels of DRD2 (Table 1)—the colorectal cancer cell line RKO and the hepatocellular carcinoma cell line Hep3B—to assess the impact of increased receptor levels on sensitivity to ONC201. Fluorescence imaging confirmed DRD2 expression, especially in the cell membrane (Figure 2A). Overexpressing the receptor increased the extent of PARP cleavage resulting from ONC201 treatment (Figure 2B). The increase in anticancer activity of ONC201 in DRD2-overexpressing RKO cells was correlated with an upregulation in CHOP mRNA expression (Figure 2C). Although DRD2 overexpression increased ONC201-induced PARP cleavage in wild-type RKO cells, DRD2 overexpression was not sufficient to increase PARP cleavage in ONC201/TIC10-resistant RKO cells (Figure S2, A-B).
Figure 2.


ONC201’s anticancer effect is not dependent on DRD2 in a subset of cancer cell types. (A) Fluorescence imaging to verify expression of GFP-DRD2 in the membrane of transfected cells. (B) Western blot analyses of PARP cleavage in RKO cells transfected with GFP-DRD2 construct for 24 hours and subsequently treated with 5 μM ONC201 for 72 hours. Blots are representative of two experiments. (C) qRT-PCR analyses of CHOP and DR5 mRNA expression in RKO cells transfected with GFP-DRD2 and subsequently treated with 5 μM ONC201 for 24 hours. (D) Cell count after 48-hour treatment with ONC201. (E) Viability assessment after 72 hours of treatment with ONC201 or L-741,626. ǂP < .05 versus viability of wild-type cells similarly treated. (F) qRT-PCR analyses of basal dopamine receptor expression of wild-type and DRD2 CRISPR/Cas9 knockout cells (transfected with guide 3 RNA). Data are means ± SE from at least two biological replicates.
We knocked out DRD2 protein expression via CRISPR-Cas9–mediated gene disruption. DNA sequencing confirmed a high percentage of indel mutations in cells that were selected after transfecting with guide RNA #3 (Figure S2C). Knocking out DRD2 in HCT116 and HT29 colorectal cancer cells did not significantly abrogate ONC201’s effects on cell number. In SKNSH neuroblastoma cells, the knockout of DRD2 increased the effect of ONC201 on cell count, albeit its effect was not statistically significant (P = .08 when wt SKNSH plus ON201 was compared to Guide 3 [G3] SKNSH plus ONC201) (Figure 2D). The D2-receptor antagonist L-741,626 decreases cell viability, like ONC201. Knocking out DRD2 partially abrogated the cytotoxic effect of L-741,626 but not of ONC201 (Figure 2E). Clonogenic assays confirmed that DRD2 knockout did not significantly affect ONC201’s anticancer activity in HCT116 and HT29 cells (Figure S2D).
We explored whether the absence of a significant impact of DRD2 knockout on ONC201-induced qRT-PCR analyses can be explained by a compensatory overexpression of other dopamine receptors. qRT-PCR analyses showed that there was no increase in expression of the other receptors. Instead, expression of DRD1 was also significantly reduced with DRD2 gene deletion in HCT116 cells (Figure 2F). DRD4 was not detected in both HCT116 and HT29 cells, and DRD2 and DRD3 mRNAs were also not detected in G3 HT29 cells.
We have previously shown that breast cancer cells respond to ONC201 [22]. Thus, similar CRISPR/Cas-9 deletion experiments were performed with two breast cancer cell lines, MDA-MB231 and SUM149PT. Moreover, given that ONC201 has been shown to bind to another D2-like receptor, DRD3 [8], we assessed the impact of DRD3 knockout on ONC201 anticancer effects. Similar to what we have observed with DRD2, knockout of DRD2 or DRD3 was not sufficient to abrogate ONC201’s cytotoxicity in the two breast cancer cell lines (Figure S2, E-F).
Consequences of Transient Knockdown of DRD2 in Colorectal Cancer Cells
Although ONC201’s anticancer activity cannot be explained entirely by ONC201’s interaction with DRD2, we explored the contribution and potential of DRD2 antagonism as a therapeutic strategy, particularly in colorectal cancer cells. As has been previously shown [11], ONC201 decreases cell count. Transiently reducing DRD2 expression levels using siRNA was also sufficient to cause a decrease in cell number. Nevertheless, ONC201 had a significantly greater effect on reducing cell count than DRD2 knockdown (Figure 3A). DRD2 knockdown was not sufficient to cause apoptosis as assessed by PARP cleavage. However, DRD2 knockdown exacerbated ONC201-induced PARP cleavage (Figure 3B). This increase in ONC201-induced apoptosis as a result of DRD2 knockdown was preceded by an increase in ATF4 protein expression at 12 hours and CHOP protein expression at 24 hours (Figure 3C).
Figure 3.


Transient knockdown of DRD2 in colorectal cancer cells can activate the ISR and reduce cell number. (A) Viable cell count, (B) Western blot analyses of PARP cleavage and CHOP expression, (C) Western blot analyses of ATF4 and CHOP protein expression in cells after DRD2 siRNA-mediated knockdown for 24 hours and subsequent treatment with 10 μM ONC201 for indicated times. *P < .05 versus vehicle-treated scramble siRNA- transfected; ǂP < .05 versus vehicle-treated DRD2 siRNA-transfected. (D) qRT-PCR analyses of DRD1 and DRD2 mRNA expression were performed to verify knockdown and monitor for potential compensatory overexpression of DRD1 receptor. Data are means ± SE from three biological replicates. ǂP < .05 versus viability of control shRNA cells similarly treated. (E) Western blot analyses for PKA substrate phosphorylation in stably transfected control and DRD2 shRNA cells treated with 10 μM ONC201 for 24 hours. (F) Cell proliferation rate assessment of control and DRD2 shRNA-transfected cells was performed by enumerating cell number after indicated times of cell culture. Data are means ± SE from three biological replicates. ǂP < .05 versus viability of control shRNA cells similarly treated.
As an alternative strategy to knock down DRD2 expression, we stably transfected HCT116 cells with DRD2 shRNA. Quantitative PCR results confirmed DRD2 knockdown in the transfectants. Moreover, one of the two clones (shRNA #43) showed a downregulation of DRD1 (Figure 3D). Similar to ONC201’s effects, knocking down DRD2 resulted in PKA activation (Figure 3E). In contrast to the effect of transient DRD2 knockdown on cell number (Figure 3A), the growth rate of HCT116 cells was not consistently downregulated when DRD2 expression levels were stably reduced (Figure 3F).
These results suggest that transient knockdown of DRD2 can reduce colorectal cancer cell number, at least in part, as a consequence of the activation of the integrated stress response.
Effects of Selective D2-Like Receptor Antagonists in Colorectal Cancer Cells
Given that transient knockdown of DRD2 decreased colorectal cancer cell number, we assessed the cytotoxic effects of other D2-selective antagonists. Although all the antagonists tested were specific for the D2-like receptor class, they had different preferential selectivity for DRD2 or DRD3. Remoxipride had similar micromolar affinities for the D2-like receptors as ONC201 (Table 2). Out of the D2-antagonists tested, L-741,626 and PG01037 also [23] significantly decreased cell viability. Their respective EC50s were in the micromolar range (Figure 4A). L-741,626 did not generally induce apoptosis (except in RKO cells) (Figure 4, B-C) or activate the ISR, at least under the conditions tested in this study (Figure 4D). PG01037 also did not induce PARP cleavage (Figure S3). ONC201 has been shown to be cancer selective [24]. By contrast, L-741,626 had significant cytotoxic activity against normal cells (Figure 4E).
Table 2.
Affinity of the Different D2-Like Receptor Antagonists Tested in This Study
|
Ki (nM) | |||||
|---|---|---|---|---|---|
| ONC201 | L-741, [33] | Risperidone | PG01037 [23], [34] | Remoxipride | |
| D2-like receptors | |||||
| DRD2 | 3000 | 2.4 | 0.3a | 93.3 | 30a |
| DRD3 | 3000 | 100 | 0.7 | 1600 | |
| DRD4 | 220 | 375 | 2800 | ||
| Other activity | High selectivity for serotonin receptor (0.4 nM) [35] | ||||
Radioligand independent [36].
Figure 4.

The anticancer effects of ONC201 are distinct from other selective DRD2 antagonists. (A) CTG assay images, (B) apoptosis measured by sub-G1 analyses, and Western blot analyses of (C) PARP cleavage and (D) ATF4 protein expression in different cancer cell types treated with different selective DRD2 antagonists (O: 10 μM ONC201 for HCT116, SKNSH, and U251, and 5 μM ONC201 for RKO; L: 25 μM L741626; R: 10 μM). (E) Nontumorigenic CCD-CON and IMR90 cells and HCT116 and SKNSH cancer cells treated with indicated doses of L-741,626 for 72 hours. *P < .05 versus vehicle treated; ǂP < .05 versus L-741,626 treated.
Effects of DRD5 Downregulation on Sensitivity to ONC201
DNA sequencing of ONC201-resistant RKO cells uncovered a missense mutation in dopamine receptor DRD5 [25]. Given that all of the cancer cell types tested in this study express DRD5 (Table 1), we transiently knocked down DRD5 and assessed whether ONC201-induced anticancer activity would be increased. Knocking down DRD5, but not the other D1-like receptor DRD1, increased ONC201-induced PARP cleavage (Figure 5A). Alternatively, we assessed whether treating cancer cells with a selective D1/D5 antagonist, SCH39166 hydrobromide [26], can increase ONC201’s effects on cell viability. The two colorectal cell lines HCT116 and RKO were relatively resistant to up to 50 μM SCH39166 (Figure S4). On the other hand, the neuroblastoma cell line SKNSH and the glioblastoma cell line U251 were sensitive to 50 μM SCH39166. Nevertheless, antagonizing D1/D5 with SCH39166 significantly increased susceptibility to ONC201 (Figure 5B).
Figure 5.

Downregulation of the D1-like receptor DRD5 increases sensitivity to ONC201. (A) Western blot analyses of PARP cleavage in cells after DRD1 or DRD5 siRNA-mediated knockdown for 24 hours and subsequent treatment with 10 μM ONC201 for 72 hours. (B) Viability assessment of indicated cell lines treated with vehicle, 5 μM ONC201, 50 μM SCH39166, or ONC201-SCH39166 combination for 72 hours.*P < .05 versus vehicle-treated cells; ǂP < .05 versus SCH39166-treated cells.
Discussion
The ability of ONC201 to bind to the D2-like receptor proteins, as has been observed in heterologous systems, provides an opportunity to explore the therapeutic potential of targeting these GPCR proteins in the context of cancer. There have been a number of papers that show the potential anticancer activity of D2-like receptor antagonists. In pancreatic cells [3] and glioblastoma [5], DRD2 knockdown is sufficient to significantly reduce cell viability. Single-agent treatment with D2-like receptor antagonists has been shown to be antitumorigenic against cervical and endometrial cancer, melanoma cells [27], [28], and breast cancer stem cells [4]. A limitation of these latter studies, however, is the lack of evidence that the effects of the antagonists were due to the D2-like receptors and not due to off-target effects. Moreover, a number of the antagonists that have been tested (e.g., haloperidol, thioridazine) have other targets aside from the D2-like receptors. By contrast, ONC201, L-741,626, PG01037, and remoxipride have not been shown to have other direct-binding targets aside from the D2-like receptors. Thus, these small molecules are appropriate tools to test the anticancer effects of D2-like receptor antagonism. We demonstrated in this study that most of the D2-receptor antagonists tested had cytotoxic effects. On the other hand, only ONC201 had significant proapoptotic effects. We explored the effect of DRD2 knockdown on the viability of colorectal cancers, in particular. We demonstrated that transient knockdown of DRD2 can reduce cell number and activate the integrated stress response. Nevertheless, the stable downregulation of DRD2 did not have significant effects on cell viability of colorectal cancer cells. Altogether, these results suggest that DRD2 antagonism by itself is insufficient for potent anticancer efficacy, at least against colorectal cancer cells.
In this study, modulating DRD2 expression influenced the expression of the proapoptotic protein CHOP but not the TRAIL receptor DR5. In contrast, ONC201 induces DR5 upregulation in an ATF4- and CHOP-dependent manner [11]. The lack of upregulation of DR5 may explain in part why DRD2 knockdown alone did not induce apoptosis. It is possible that ATF and/or CHOP need to cooperate with other transcription factors for DR5 to be expressed. This is plausible given that DR5 has been shown to be regulated not only by ATF4 and CHOP but also by p53, ATF3, Elk-1, Sp1, and NFkappaB [29]. Moreover, interactions on the DR5 promoter, between different DR5 transcriptional activators, such as ATF3 and p53 have been shown [30].
There has been increasing evidence that although D1-like and D2-like receptors mediate opposing pathways, heteromers of receptors from the two classes [31] activate distinct signaling cascades. Although ONC201’s anticancer effects do not rest entirely on DRD2, we have shown that blocking DRD5 can increase sensitivity to ONC201. It is possible that knocking down or pharmacologically blocking DRD5 affects its interaction with DRD2 and, consequently, DRD2’s ability to bind to ONC201.
We have shown in this study that downregulation of DRD2 whether through transient knockdown of the receptor or pharmacological antagonism can have cytotoxic effects towards cancer cells. Other GPCR antagonists, for instance, opioid receptor antagonists, have also been proposed to have anticancer activity [32]. On the other hand, we have shown that potent anticancer effects of the small molecule ONC201 are superior to other DRD2-selective antagonists. This may be attributed at least in part to ONC201’s ability to engage other pathways that are not directly related to DRD2 antagonism. Nevertheless, the demonstration that blocking DRD2 can have antiproliferative activity in colon cancer cells is helpful not only in understanding the effects of DRD2-targeted drugs but also in thinking of new ways of using these important neurobiological therapeutics.
Acknowledgments
Acknowledgements
Oncoceutics provided ONC201. This work was presented in part at the 2016 and 2017 American Association for Cancer Research meetings. This work was supported by grants from the National Institutes of Health (R01 CA173453) and the American Cancer Society to W. S. E.-D. W. S. E.-D. is an American Cancer Society Research Professor.
Footnotes
Disclosure of Potential Conflict of Interest: W. S. E.-D. is a co-founder and shareholder of Oncoceutics Inc. and is fully compliant with NIH and institutional disclosure guidelines. J. E. A. is an employee and shareholder of Oncoceutics Inc. No potential conflicts of interest were disclosed by the other authors.
Author's Contributions: Conception and design: C. L. B. K., W. S. E.-D. Development of methodology: C. L. B. K., P. A., J. E. A., W. S. E.-D. Acquisition of data: C. L. B. K., M. D. R., A. L., J. M. W., D. T. D. Analysis and interpretation of data: C. L. B. K., M. D. R., A. L., J. M. W., P. A., D. T. D., J. E. A., W. S. E.-D. Writing, review, and/or revision of the manuscript: C. L. B. K., M. D. R., A. L., J. M. W., P. A., D. T. D., J. E. A., W. S. E.-D. Administrative, technical, or material support: C. L. B. K., M. D. R., A. L., J. M. W., P. A., D. T. D., J. E. A., W. S. E.-D. Study supervision: W. S. E.-D.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neo.2017.10.002.
Appendix A. Supplementary data
Figure S1. Assessment of DRD3 mRNA expression in different cancer tissue samples using cBioportal.
Figure S2. (A) Fluorescence imaging to verify expression of GFP-DRD2 in the membrane of transfected RKOr1 cells. (B) Western blot analyses of PARP cleavage in cells transfected with GFP-DRD2 construct for 24 hours and subsequently treated with 5 μM ONC201 for 72 hours. (C) Indel rates in DRD2 CRISPR clones of colorectal cancer cell lines HCT116 and HT29, and a neuroblastoma cell line SKNSH. (D) Clonogenic assays on wt and DRD2 CRISPR clones treated with indicated concentrations of ONC201 for 72 hours and subsequently maintained in drug-free medium for 7 to 10 days. Dose-response curves representing cell viability, as assessed by CTG assays, of wild-type and (C) DRD2 and (D) DRD3 CRISPR clones after treatment with ONC201 for 72 hours.
Figure S3. Western blot analyses of PARP cleavage in cells treated with 25 μM PG01037 for 72 hours.
Figure S4. CTG assay images in indicated cells treated with SCH39166 hydrobromide for 72 hours.
References
- 1.Beaulieu JM, Espinoza S, Gainetdinov RR. Dopamine receptors — IUPHAR Review 13. Br J Pharmacol. 2015;172:1–23. doi: 10.1111/bph.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roy S, Lu K, Nayak MK, Bhuniya A, Ghosh T, Kundu S, Ghosh S, Baral R, Dagupta PS, Basu S. Activation of D2 dopamine receptors in CD133+ve cancer stem cells in non–small cell lung carcinoma inhibits proliferation, clonogenic ability, and invasiveness of these cells. J Biol Chem. 2017;292:435–445. doi: 10.1074/jbc.M116.748970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jandaghi P, Najafabadi HS, Bauer AS, Papadakis AI, Fassan M, Hall A, Monast A, von Knebel Doeberitz M, Neoptoloemos JP, Costello E. Expression of DRD2 is increased in human pancreatic ductal adenocarcinoma and inhibitors slow tumor growth in mice. Gastroenterology. 2016;151:1218–1231. doi: 10.1053/j.gastro.2016.08.040. [DOI] [PubMed] [Google Scholar]
- 4.Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell. 2012;149:1284–1297. doi: 10.1016/j.cell.2012.03.049. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Zhu S, Kozono D, Ng K, Futalan D, Shen Y, Akers JC, Steed T, KUshwaha D, Schlabach M. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget. 2014;5:882–893. doi: 10.18632/oncotarget.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basu S, Dasgupta PS. Role of dopamine in malignant tumor growth. Endocrine. 2000;12:237–241. doi: 10.1385/ENDO:12:3:237. [DOI] [PubMed] [Google Scholar]
- 7.Allen JE, Kline CL, Prabhu VV, Wagner J, Ishizawa J, Madhukar N, Lev A, Baumeister M, Zhou L, Lulla A. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016;7:74380–74392. doi: 10.18632/oncotarget.11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madhukar NS, Khade PK, Huang L, Gayvert K, Galletti G, Stogniew M, Allen JE, Giannakakou P, Elemento O. bioRxiv; 2017. A new big-data paradigm for target identification and drug discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein MN, Bertino JR, Kaufman HL, Mayer T, Moss R, Silk A, Chan N, Malhotra J, Rodriguez-Rodriguez L, Aisner J. First-in-human clinical trial of oral ONC201 in patients with refractory solid tumors. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen JE, Krigsfeld G, Mayes PA, Patel L, Dicker DT, Patel AS, Dolloff NG, Messaris E, Scata KA, Wang W. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction and potent antitumor effects. Sci Transl Med. 2013;5:1–13. doi: 10.1126/scitranslmed.3004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci Signal. 2016;9:ra18. doi: 10.1126/scisignal.aac4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ASN-0002-2011 AA. Authentication of human cell lines: standardization of STR profiling. 2011. [Google Scholar]
- 13.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jecobsen A, Bryne CJ, Heuer ML, Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Y, Kokkonen GC, Wang X, Neve KA, Roth GS. D2 dopamine receptors stimulate mitogenesis through pertussin toxin-sensitive G proteins and Ras-involved ERK and SAP/JNK pathways in rat C6-D2L glioma cells. J Neurochem. 1998;71:980–990. doi: 10.1046/j.1471-4159.1998.71030980.x. [DOI] [PubMed] [Google Scholar]
- 17.Taylor M, Grundt P, Griffin SA, Newman AH, Luedtke RR. Dopamine D3 receptor selective ligands with varying intrinsic efficacies at adenylyl cyclase inhibition and mitogenic signaling pathways. Synapse. 2010;64:251–266. doi: 10.1002/syn.20725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaneko M, Sato K, Horikoshi R, Yaginuma M, Yaginuma N, Shiragata M, Kumashiro H. Effect of haloperidol on cyclic AMP and inositol trisphosphate in rat striatum in vivo. Prostaglandins Leukot Essent Fatty Acids. 1992;46:53–57. doi: 10.1016/0952-3278(92)90059-r. [DOI] [PubMed] [Google Scholar]
- 19.Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci U S A. 2008;105:13656–13661. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dwivedi Y, Rizavi HS, Pandey GN. Differential effects of haloperidol and clozapine on [(3)H]cAMP binding, protein kinase A (PKA) activity, and mRNA and protein expression of selective regulatory and catalytic subunit isoforms of PKA in rat brain. J Pharmacol Exp Ther. 2002;301:197–209. doi: 10.1124/jpet.301.1.197. [DOI] [PubMed] [Google Scholar]
- 21.Sprenger JU, Nikolaev VO. Biophysical techniques for detection of cAMP and cGMP in living cells. Int J Mol Sci. 2013;14:8025–8046. doi: 10.3390/ijms14048025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ralff MD, Kline CLB, Kucukkase OC, Wagner J, Lim B, Dicker DT, Prabhu VV, Oster W, El-Deiry WS. ONC201 demonstrates antitumor effects in both triple-negative and non–triple-negative breast cancers through TRAIL-Dependent and TRAIL-independent mechanisms. Mol Cancer Ther. 2017;16:1290–1298. doi: 10.1158/1535-7163.MCT-17-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar R, Riddle L, Griffin SA, Grundt P, Newman AH, Luedtke RR. Evaluation of the D3 dopamine receptor selective antagonist PG01037 on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology. 2009;56:944–955. doi: 10.1016/j.neuropharm.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen JE, Crowder RN, El-Deiry WS. First-in-class small molecule ONC201 induces DR5 and cell death in tumor but not normal cells to provide a wide therapeutic index as an anti-cancer agent. PLoS One. 2015;10:e0143082. doi: 10.1371/journal.pone.0143082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madhukar N, Prabhu VV, Dardenne EE, Doherty F, VanEngelenburg A, Tarapore R, Garnett MJ, McDermott U, Benes C, Oster W. Abstract 2792: The small molecule imipridone ONC201 is active in tumor types with dysregulated DRD2 pathway. Cancer Res. 2017;77 [Google Scholar]
- 26.Tice MA, Hashemi T, Taylor LA, Duffy RA, McQuade RD. Characterization of the binding of SCH 39166 to the five cloned dopamine receptor subtypes. Pharmacol Biochem Behav. 1994;49:567–571. doi: 10.1016/0091-3057(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 27.Kang S, Dong SM, Kim BR, Park MS, Trink B, Byun HJ, Rho SB. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis. 2012;17:989–997. doi: 10.1007/s10495-012-0717-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gil-Ad I, Shtaif B, Levkovitz Y, Nordenberg J, Taler M, Korov I, Weizman A. Phenothiazines induce apoptosis in a B16 mouse melanoma cell line and attenuate in vivo melanoma tumor growth. Oncol Rep. 2006;15:107–112. [PubMed] [Google Scholar]
- 29.Edagawa M, Kawauchi J, Hirata M, Goshima H, Inoue M, Okamoto T, Murakami A, Maehara Y, Kitajima S. Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum (ER) stress–induced sensitization of p53-deficient human colon cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)–mediated apoptosis through up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib. J Biol Chem. 2014;289:21544–21561. doi: 10.1074/jbc.M114.558890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taketani K, Kawauchi J, Tanaka-Okamoto M, Ishizaki H, Tanaka Y, Sakai T, Miyoshi J, Maehara Y, Kitajima S. Key role of ATF3 in p53-dependent DR5 induction upon DNA damage of human colon cancer cells. Oncogene. 2012;31:2210–2221. doi: 10.1038/onc.2011.397. [DOI] [PubMed] [Google Scholar]
- 31.Blasiak E, Lukasiewicz S, Szafran-Pilch K, Dziedzicka-Wasylewska M. Genetic variants of dopamine D2 receptor impact heterodimerization with dopamine D1 receptor. Pharmacol Rep. 2017;69:235–241. doi: 10.1016/j.pharep.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Singleton PA, Moss J, Karp DD, Atkins JT, Janku F. The mu opioid receptor: a new target for cancer therapy? Cancer. 2015;121:2681–2688. doi: 10.1002/cncr.29460. [DOI] [PubMed] [Google Scholar]
- 33.Kulagowski JJ, Broughton HB, Curtis NR, Mawer IM, Ridgill MP, Baker R, Emms F, Freedman SB, Marwood R, Patel S. 3-((4-(4-Chlorophenyl)piperazin-1-yl)-methyl)-1H-pyrrolo-2,3-b-pyridine: an antagonist with high affinity and selectivity for the human dopamine D4 receptor. J Med Chem. 1996;39:1941–1942. doi: 10.1021/jm9600712. [DOI] [PubMed] [Google Scholar]
- 34.Grundt P, Carlson EE, Cao J, Bennett CJ, McElveen E, Taylor M, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR. Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J Med Chem. 2005;48:839–848. doi: 10.1021/jm049465g. [DOI] [PubMed] [Google Scholar]
- 35.Leysen JE, Janssen PM, Megens AA, Schotte A. Risperidone: a novel antipsychotic with balanced serotonin-dopamine antagonism, receptor occupancy profile, and pharmacologic activity. J Clin Psychiatry. 1994;(55 Suppl):5–12. [PubMed] [Google Scholar]
- 36.Seeman P, Corbett R, Van Tol HH. Atypical neuroleptics have low affinity for dopamine D2 receptors or are selective for D4 receptors. Neuropsychopharmacology. 1997;16:93–110. doi: 10.1016/S0893-133X(96)00187-X. [discussion 1-35] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Assessment of DRD3 mRNA expression in different cancer tissue samples using cBioportal.
Figure S2. (A) Fluorescence imaging to verify expression of GFP-DRD2 in the membrane of transfected RKOr1 cells. (B) Western blot analyses of PARP cleavage in cells transfected with GFP-DRD2 construct for 24 hours and subsequently treated with 5 μM ONC201 for 72 hours. (C) Indel rates in DRD2 CRISPR clones of colorectal cancer cell lines HCT116 and HT29, and a neuroblastoma cell line SKNSH. (D) Clonogenic assays on wt and DRD2 CRISPR clones treated with indicated concentrations of ONC201 for 72 hours and subsequently maintained in drug-free medium for 7 to 10 days. Dose-response curves representing cell viability, as assessed by CTG assays, of wild-type and (C) DRD2 and (D) DRD3 CRISPR clones after treatment with ONC201 for 72 hours.
Figure S3. Western blot analyses of PARP cleavage in cells treated with 25 μM PG01037 for 72 hours.
Figure S4. CTG assay images in indicated cells treated with SCH39166 hydrobromide for 72 hours.