

Abstract
The successful development of high-affinity gapmer antisense oligonucleotide (ASO) therapeutics containing locked nucleic acid (LNA) or constrained ethyl (cEt) substitutions has been hampered by the risk of hepatotoxicity. Here, we present an in vitro approach using transfected mouse fibroblasts to predict the potential hepatic liabilities of LNA-modified ASOs (LNA-ASOs), validated by assessing 236 different LNA-ASOs with known hepatotoxic potential. This in vitro assay accurately reflects in vivo findings and relates hepatotoxicity to RNase H1 activity, off-target RNA downregulation, and LNA-ASO-binding affinity. We further demonstrate that the hybridization-dependent toxic potential of LNA-ASOs is also evident in different cell types from different species, which indicates probable translatability of the in vitro results to humans. Additionally, we show that the melting temperature (Tm) of LNA-ASOs maintained below a threshold level of about 55°C greatly diminished the hepatotoxic potential. In summary, we have established a sensitive in vitro screening approach for assessing the hybridization-dependent toxic potential of LNA-ASOs, enabling prioritization of candidate molecules in drug discovery and early development.
Keywords: high affinity, antisense oligonucleotides, LNA, hepatotoxicity, in vitro assay, apoptosis, off-target RNA cleavage, melting temperature
Introduction
For nearly 40 years, antisense oligonucleotides (ASOs) have been researched and refined to modify target gene expression. Recently, three ASOs with drug-like properties received approvals, and more than 100 ASOs are currently in clinical development.1 An ongoing challenge in ASO development is the optimization of the ASO design to ensure adequate patient safety. ASOs have known potential safety liabilities, recently reviewed by Chi et al.,2 which mainly involve (1) hybridization-independent binding of ASOs to cellular proteins, thereby interfering with their biological function and/or (2) hybridization-dependent downregulation of unintended target RNAs, thereby reducing the level of potentially essential proteins or regulatory RNAs. Following in vivo administration, ASOs mainly accumulate in the kidney and liver, where ASO-mediated toxicity is typically observed.3
Some 2′-sugar modifications, including locked nucleic acid (LNA) and constrained ethyl (cEt) substitutions, significantly increase the binding affinity of RNase H1-activating gapmer ASOs, i.e., ASOs containing a central deoxynucleotide region flanked on both ends by several modified ribonucleotides. Short (12–16 nt long) gapmer ASOs with LNA or cEt nucleotides in the flanks tend to exhibit higher potency than longer oligonucleotides built with lower-affinity chemistry.1 This shorter ASO design could enhance in vivo delivery and further mitigate some typical class effects of ASOs containing stability- and protein binding-increasing phosphorothioate (PS) backbone modifications.1, 2 Despite these advantages, ASOs with high-affinity modifications have been associated with a higher risk of inducing liver toxicity.4, 5, 6, 7, 8, 9, 10, 11, 12 Recent in vivo data suggest that the observed hepatotoxicity is hybridization dependent and requires RNase H1 activity.10, 12 Furthermore, the affinity of an ASO is considered to be associated with its hepatotoxic potential, likely because high-affinity ASOs can bind to and mediate cleavage of more unintended target RNAs than lower affinity ASOs.10, 11, 12, 13
Although our understanding of hybridization-dependent ASO toxicity is increasing, there are only a few reported in silico and/or in vitro approaches to predict this in vivo liability.6, 11, 12, 13, 14, 15 Although in silico methods can identify putative off-target sequences,11, 13 setting tolerable thresholds to minimize false positive off-target predictions remains difficult. Thus, in silico approaches can markedly reduce, but not eliminate, the risk of selecting oligonucleotides with hybridization-dependent toxicities.16 To assess this remaining risk, ASOs are routinely tested in animals (typically mice) or, as recently reported, primary mouse and human hepatocytes for their hepatotoxic potential.14
Here, we investigated whether the hepatotoxic potential of LNA-ASOs can also be assessed employing conventional tissue culture cells and methods. Applying lipotransfection for efficient nuclear delivery of 236 LNA-ASOs, we observed a clear association between induction of apoptosis in mouse 3T3 fibroblast cells in vitro and LNA-ASO hepatotoxicity determined in vivo in mice. Moreover, using the in vitro assay, we accurately reproduced recent mechanistic in vivo findings in relation to hybridization-dependent hepatotoxicity.10, 12 The cytotoxic properties of hepatotoxic LNA-ASOs also manifested in tumor-derived human tissue culture cell lines. These findings imply a general cytotoxic—rather than a cell-type-specific toxic—effect of certain high-affinity ASOs. Finally, our data suggest a relationship between the calculated melting temperature (Tm) of LNA-ASOs and their hepatotoxic potential.
Results
Apoptosis Induction in Mouse 3T3 Fibroblast Cells Differentiates Hepatotoxic from Well-Tolerated LNA-ASOs
Recently, Sewing et al.14 demonstrated that unassisted (gymnotic) delivery of LNA-ASOs into primary mouse and human hepatocytes can discriminate hepatotoxic from well-tolerated LNA-ASOs by various readouts, including apoptosis. Assuming hybridization- and RNase-H-1-dependent mechanisms underlying hepatotoxicity, we hypothesized that this oligonucleotide-mediated toxicity may be evident in other cell types following efficient entry of the oligonucleotides into the cell’s nucleus. Thus, we used Lipofectamine 2000 for nuclear delivery of LNA-ASOs into mouse 3T3 fibroblast cells and assessed apoptosis 24 hr later. As initial tool molecules, we selected six LNA-ASOs targeting mouse “Myeloid differentiation primary response 88” (Myd88) RNA (Table 1), of which three were known to be hepatotoxic and three were well-tolerated based on alanine aminotransferase (ALT) levels determined after 2 weeks of treatment with 5 × 15 mg/kg in mice.6 As evident from Figure 1, the hepatotoxic LNA-ASOs showed a concentration-dependent increase in caspase activity, whereas the well-tolerated LNA-ASOs did not markedly induce caspase activity at any tested concentration when compared to untreated cells (UTCs).
Table 1.
Structures and Key Data of Initial Set of Tool LNA-ASOs
| LNA-ASO Name | LNA-ASO Sequence | Target RNA | Target Species | ALT Increase Relative to Control | Hepatotoxic | Tm (°C) |
|---|---|---|---|---|---|---|
| LNA32 | EAAaggaaacacaEAT | Myd88 | mouse, rat | 1.09 | no | 44.5 |
| LNA33 | EAAatgctgaaacTAT | Myd88 | mouse | 1.02 | no | 44.0 |
| LNA39 | GTEagaaacaaccAEE | Myd88 | mouse, rat | 1.06 | no | 51.4 |
| LNA41 | EAEattccttgctETG | Myd88 | mouse, human | 44.57 | yes | 57.2 |
| LNA37 | GEEtcccagttccTTT | Myd88 | mouse, rat | 41.54 | yes | 64.3 |
| LNA43 | GATgcctcccaGTT | Myd88 | mouse, rat | 36.48 | yes | 55.9 |
Capital letters: LNA; small letters: DNA; E, 5-methylcytosin; all LNA-ASOs were fully phosphorothioated. The LNA-ASOs were designed to target Myd88, with some also targeting rat Myd88 or human MYD88, as indicated. ALT levels were determined 2 weeks after i.v. treatment of mice with 5 × 15 mg/kg.6 ALT levels are expressed as fold change relative to saline-treated animals. The theoretically calculated melting temperatures (Tms) are indicated.
Figure 1.

Apoptosis Induction In Vitro Reveals the Hepatotoxic Potential of LNA-ASOs
Mouse 3T3 fibroblast cells were transfected using Lipofectamine 2000 with well-tolerated and hepatotoxic LNA-ASOs, respectively. Caspase 3/7 activity was measured 24 hr later. Triplicate transfections were performed; the results are shown as a percentage change relative to UTCs. Data are mean ± SD.
To extend this observation and rule out any transfection or target-related toxicity, an additional 230 LNA-ASOs were tested in the in vitro assay at a fixed concentration of 100 nM. These 236 LNA-ASOs were designed to target 13 different genes and had all been evaluated in mice (n = 5) for their hepatotoxic potential.6 LNA-ASOs with a demonstrated lower hepatotoxic liability (i.e., mean ALT ≤5-fold change relative to saline-treated mice6) induced significantly lower caspase activities (p value = 5.4 × 10−12 by Wilcoxon rank sum test) than LNA-ASOs with a higher hepatotoxic liability (i.e., mean ALT >5-fold change relative to saline-treated mice6) (Figure 2).
Figure 2.
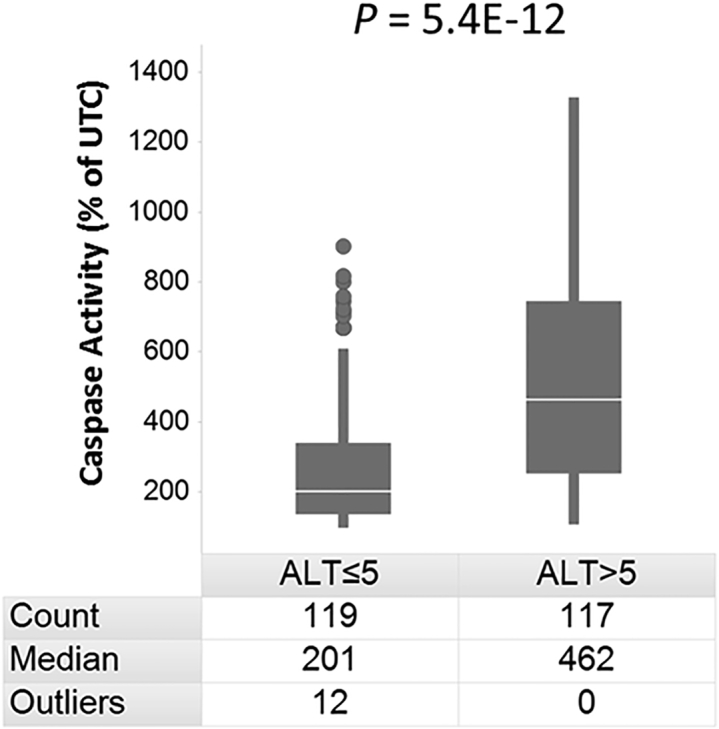
In Vitro Caspase 3/7 Induction Is Stronger for Hepatotoxic Than for Well-Tolerated LNA-ASOs
Mouse 3T3 fibroblast cells were transfected using Lipofectamine 2000 with 236 different LNA-ASOs at 100 nM. Caspase 3/7 activity was measured 24 hr later. Duplicate transfections were performed, and the values were calculated as average percentage change relative to UTCs. The LNA-ASOs were separated into two groups of lower and higher hepatotoxicity in vivo (i.e., mean ALT levels ≤5-fold or >5-fold, respectively, relative to saline-treated mice after intravenous (i.v.) treatment with 5 × 15 mg/kg LNA-ASOs6). Each group of LNA-ASOs is listed versus their respective caspase values determined in vitro in a boxplot diagram. For each group, the number (count) of included LNA-ASOs, the median caspase activities, and the number of outliers is shown. The level of significance as evaluated by Wilcoxon rank sum test is indicated above the boxplot.
Apoptosis Induction by Hepatotoxic LNA-ASOs In Vitro Is RNase H1 Dependent
Recently, it was demonstrated in vivo that oligonucleotide-mediated downregulation of RNase H1 expression in mouse liver suppressed hepatotoxicity of certain high-affinity ASOs.10, 12 To assess whether our in vitro assay also reproduces this finding, we pre-transfected mouse 3T3 fibroblast cells with a small interfering RNA (siRNA) against RNase H1 and a non-targeting control siRNA, respectively. Similar to the reported in vivo finding,10, 12 apoptosis was strongly suppressed when the hepatotoxic LNA41 or LNA37 were transfected into cells pre-transfected with a siRNA against RNase H1 but not with the control siRNA (Figure 3A). The siRNA-mediated downregulation of RNase H1 was confirmed by qPCR (Figure 3B).
Figure 3.

RNase H1 Knockdown Suppresses LNA-ASO Toxicity In Vitro
(A) Mouse 3T3 fibroblast cells were transfected with a siRNA against RNase H1 (light gray bars) and a control (ctr.) siRNA (dark gray bars) at 10 nM, respectively, using Lipofectamine 2000. 24 hr later, cells were transfected with the hepatotoxic LNA41 or LNA37 at 30 nM using Lipofectamine 2000. Caspase 3/7 activity was measured 24 hr later. (B) siRNA transfection was performed as in (A) and cells were harvested 48 hr later for assessing RNase H1 knockdown by qPCR. Results in (A) and (B) are shown as a percentage change relative to UTCs. Data (n = 3) are mean ± SD.
2′-OMe Modifications in the Gap Region of Hepatotoxic LNA-ASOs Suppress Toxicity In Vitro
Substitution of several unmodified DNA nucleotides in the gap region with 2′-sugar modifications of hepatotoxic LNA-ASOs has recently been shown to interfere with RNase H1 activity, resulting in reduced hepatotoxicity in mice compared to the parent ASOs.10, 12 To investigate whether this finding can also be observed in our in vitro system, we substituted DNA nucleotides in the gap region of hepatotoxic LNA41 and LNA37 with two (resulting in LNA41’ and LNA37’) and ten (resulting in LNA41” and LNA37”) 2′-O-methyl (2′-OMe)-modified ribonucleotides, respectively. Similar to reported in vivo findings, 2′-OMe gap modifications strongly suppressed the toxic potential of hepatotoxic LNA-ASOs also in vitro. As evident from Figure 4, caspase activity induced by LNA41 and LNA37 was potently suppressed when the DNA gap region of the ASOs was completely modified with ten 2′-OMe-modified ribonucleotides (i.e., LNA41” and LNA37”). Two 2′-OMe gap modifications in LNA37’ were sufficient to suppress caspase values to similar levels as the fully 2′-OMe gap-modified LNA37”. In contrast, the two 2′-OMe gap modifications in LNA41’ seemed to suppress caspase activity less potently but still considerably.
Figure 4.

2′-OMe Modifications in the Gap Region of Hepatotoxic LNA-ASOs Suppress Toxicity In Vitro
Left: schematic illustration of the parent LNA41 and LNA37 ASOs and their 2′-OMe-modified versions LNA41’ and LNA37’ (with two 2′-OMe-modifications in the gap region) and LNA41” and LNA37” (with ten 2′-OMe modifications in the gap region). Right: caspase activity measured 24 hr after transfection of mouse 3T3 fibroblast cells with the indicated LNA-ASOs at 30 nM. Results are shown as a percentage change relative to UTCs. Data (n = 3) are mean ± SD.
2′-OMe Modifications in the Gap Region of Hepatotoxic LNA-ASOs Suppress Downregulation of Their Intended Target Sequence
To assess the impact of 2′-OMe modifications in the gap region of LNA41 and LNA37 on their downregulatory potential, we determined Myd88 expression by qPCR 24 hr after transfection of the different LNA-ASOs into mouse 3T3 fibroblast cells (Figure 5). Complete modification of the gap region with ten 2′-OMe ribonucleotides in LNA41” and LNA37” potently abrogated Myd88 downregulation. Consistent with the above experiment (see Figure 4), two 2′-OMe gap modifications were sufficient to potently suppress LNA37’-mediated downregulation of Myd88 expression, whereas the downregulatory potential of LNA41’ was only slightly affected by the two 2′-OMe gap modifications.
Figure 5.

2′-OMe Modifications in the Gap Region of Hepatotoxic LNA-ASOs Suppress Downregulation of Their Intended Target
Myd88 expression was determined 24 hr after transfection of mouse 3T3 fibroblast cells with the indicated LNA-ASOs at 30 nM. Results are shown as a percentage change relative to UTCs. Data (n = 3) are mean ± SD.
Gene Expression Analysis Suggests an Association between Apoptosis Induction In Vitro and the Number of Genes De-regulated by a Hepatotoxic LNA-ASO
For further insight into the apoptosis-inducing mechanisms of hepatotoxic LNA-ASOs, we investigated gene expression changes following in vitro transfection of a hepatotoxic LNA-ASO (LNA41) and its less toxic 2′-OMe-gap-modified version (LNA41’), respectively, into 3T3 cells. Cells were harvested at 3 and 6 hr after transfection, respectively, to preferentially capture direct off-target effects as opposed to secondary downstream effects. Gene array analysis was performed considering only genes for analysis that were at least 2-fold de-regulated at the 6-hr time point. Using these settings, 293 genes were found to be significantly downregulated by the parent LNA41, whereas only 78 of these genes were also affected by the partial gap-modified LNA41’ (Figure 6A). For most of the affected genes, the downregulatory effect was already evident at 3 hr. In line with recent in vivo data,10, 11, 12 pathway analysis suggested that none of the downregulated off-target transcripts could unequivocally be associated with apoptosis (data not shown). In accordance with the Myd88 qPCR data (Figure 5), the partial gap modification affected the on-target (Myd88) downregulatory potential of LNA41’ only modestly (Figure 6A; red dashed ovals). LNA41 also significantly upregulated 60 genes at 6 hr, whereas gap-modified LNA41’ induced only seven of these genes (Figure 6B). As with the downregulated genes, this effect was already evident at 3 hr for most of the upregulated genes. Several genes upregulated by toxic LNA41 were genes associated with cellular stress and, in particular, with apoptosis, including Bcl2l11,17 Gadd45a,18 Trp53inp1,19 and Cdkn1a20 (Table S2).
Figure 6.

Effect of Hepatotoxic LNA41 and Its 2′-OMe-Gap-Modified Version LNA41’ on Gene Expression in Mouse Fibroblast Cells
Mouse 3T3 fibroblast cells were transfected with LNA41 and 2′-OMe-modified LNA41’, respectively, at 30 nM in triplicates, and were then harvested at 3 and 6 hr, respectively. Gene array analysis was performed and only genes were considered, which were significantly (p value ≤ 0.01) de-regulated by at least 2-fold (indicated by the dashed red lines; number of respective genes identified are in blue) from the untreated control (dashed black lines) at the 6-hr time point for LNA41. (A) shows the downregulatory trends of the identified genes between 3 and 6 hr after transfection. The red dashed oval and the arrow indicate the downregulation trends for the LNA41 target transcript Myd88; the remaining Myd88 transcript levels are indicated as fold change relative to control. (B) shows the upregulatory trends of the identified genes between 3 and 6 hr after transfection
Apoptosis Induction In Vitro and Hepatotoxicity Observed In Vivo Associate with the Binding Affinity of LNA-ASOs
Increasing evidence indicates that the binding affinity of an ASO is associated with its hepatotoxic potential.10, 11, 12 An ASO’s binding affinity can be characterized by the Tm, which is the temperature at which half of the target RNA-DNA oligonucleotide duplexes dissociate to their single stranded form under standard conditions. The Tm can be determined experimentally,21 or, as done in the present study, can be predicted using a nearest-neighbor algorithm with experimentally determined thermodynamic parameters.22 The three well-tolerated Myd88-targeting LNA-ASOs all had a calculated Tm < 52°C, whereas the toxic Myd88-targeting LNA-ASOs all had a Tm > 55°C (Table 1). To explore this potential association further, we calculated the theoretical Tm for the additional 230 LNA-ASOs used in the assessment above. When all 236 LNA-ASOs were grouped into well-tolerated (i.e., mean ALT levels ≤5-fold from saline-treated mice) and hepatotoxic LNA-ASOs (i.e., mean ALT levels >5-fold from saline-treated mice), the hepatotoxic LNA-ASOs had a significantly (p value = 7.9 × 10−5) higher Tm (median Tm = 57.1°C) than the well-tolerated LNA-ASOs (median Tm = 54.2°C) (Figure 7A). Next, the caspase values determined in vitro for the different LNA-ASOs were analyzed relative to the calculated Tms. The 236 LNA-ASOs were grouped into different Tm ranges and the respective caspase values for each range were listed in a boxplot diagram. Figure 7B shows a clear association between the different Tm ranges and the respective caspase values determined in vitro.
Figure 7.

Higher Affinity LNA-ASOs Tend to Be More Toxic In Vitro and In Vivo
(A) The 236 LNA-ASOs were separated into two groups with in vivo mean ALT levels ≤5-fold and >5-fold from saline-treated mice, respectively. For each group, the respective theoretical Tm values were calculated and summarized in a boxplot diagram. (B) The 236 LNA-ASOs were separated into four Tm ranges, as indicated. For each Tm range, the oligonucleotides’ respective caspase values were listed in a boxplot diagram. In addition, for each group in (A) and (B) the number (count) of included LNA-ASOs, the median Tm (for A) and caspase (for B) values, and the number of outliers are shown. The level of significance as evaluated by Wilcoxon rank sum test is indicated above the boxplots in (A).
Tm Calculations Aid the Design of LNA-ASOs with Low In Vitro Toxic Potential
To investigate whether the above findings can be used to design well-tolerated LNA-ASOs, we designed 10 LNA-ASOs with a Tm < 50°C. As a control, we also designed 10 LNA-ASOs with a Tm > 60°C (presented in Table S1). All ASOs targeted mouse glyceraldehyde 3-phosphate dehydrogenase (Gapdh) and were subjected to the 3T3 caspase assay. As evident from Figure 8, 80% of the LNA-ASOs with a Tm < 50°C displayed low caspase values (i.e., <200% of UTC), whereas only 20% of the LNA-ASOs with a Tm > 60°C were within this lower caspase range (p value = 0.005).
Figure 8.

Melting Temperature, Tm, Calculations Facilitate the Design of LNA-ASOs with Low Toxic Potential
Mouse 3T3 fibroblast cells were transfected using Lipofectamine 2000 with ten low-binding affinity (Tm < 50°C; dark gray bars) and ten high-binding affinity (Tm > 60°C; black bars) LNA-ASOs targeting mouse Gapdh, respectively, at 30 nM. Caspase 3/7 activity was measured 24 hr later. Results are shown as a percentage change relative to UTCs. Data (n = 3) are mean ± SD.
Modulating the Toxic Potential of an LNA-ASO by Tuning Its Binding Affinity
The data so far suggest that binding affinity is a major determinant for hybridization-dependent oligonucleotide toxicity. Therefore, we speculated that reducing the binding affinity of a toxic LNA-ASO should consequently reduce its toxicity. To test this, we used the toxic mGAPDH_LNA1189 from the previous experiment (see Figure 8 and Table S1). We changed the number and position of high-affinity LNA modifications in the wings of mGAPDH_LNA1189 while keeping the length and sequence constant. To maintain protection from nucleases, we substituted only the middle LNA modifications in the wings with DNA nucleotides. The different substitutions resulted in mGAPDH_LNA1189 versions with different calculated Tms. As displayed in Figure 9, in vitro toxicity of the parent mGAPDH_LNA1189 was gradually reduced by reducing its binding affinity.
Figure 9.

Effect of the Wing Design on Binding Affinity and LNA-SSO Toxicity
Left: schematic illustration of the parent mGAPDH_LNA1189 and its DNA-substituted wing variants with their respective calculated Tms. Right: caspase activity measured 24 hr after transfection of mouse 3T3 fibroblast cells with the indicated LNA-ASOs at 30 nM. Results are shown as a percentage change relative to UTCs. Data (n = 3) are mean ± SD.
Hepatotoxic LNA-ASOs Also Induce Apoptosis in Tumor-Derived Human Tissue Culture Cell Lines
Finally, we investigated whether the toxic effect of LNA-ASOs, which was initially demonstrated in vivo,6 and which could here also be demonstrated in vitro in mouse fibroblast cells, can also be observed in human cells. To address this, toxic LNA41 and LNA43 and well-tolerated LNA32 and LNA33, respectively, were transfected into different human cell lines, and caspase activity was assessed 24 hr later. The difference in toxicity among the LNA-ASOs observed in mouse fibroblast cells was recapitulated (compare with Figure 1) in tumor-derived human cell lines, including HeLa, HepG2, andA549 cells (Figure 10).
Figure 10.

Selected Hepatotoxic LNA-ASOs Also Exert Their Toxic Potential in Different Human Tumor Cells
A549, HeLa, and HepG2 cells were transfected using Lipofectamine 2000 with the well-tolerated LNA32 and LNA33 (light gray bars) and the hepatotoxic LNA41 and LNA43 (dark gray bars), respectively, at 30 nM. Caspase 3/7 activity was measured 24 hr later. Triplicate transfections were performed, and the results are shown as a percentage change relative to UTCs. Data are mean ± SD.
Discussion
Current research has greatly improved our understanding of the potential mechanisms of ASO-induced liver toxicity. Convincing in vivo evidence now suggests that RNase-H1-mediated cleavage of off-target RNAs represents an important mechanistic cause underlying ASO-mediated hepatotoxicity.10, 12 Until recently, in vivo testing of ASOs was the only way to assess the hepatotoxic potential of ASOs. Sewing et al.,14 however, has now demonstrated that the hepatotoxic potential of LNA-ASOs can be assessed in vitro employing unassisted (gymnotic) delivery of LNA-ASOs into primary mouse and human hepatocytes.
Here, we investigated whether we can use conventional tissue culture cells and transfection methods to also assess the hepatotoxic potential of LNA-ASOs. Remarkably, when 236 different LNA-ASOs (targeting 13 different gene transcripts) with known hepatotoxic potential were lipotransfected into mouse 3T3 fibroblast cells, a significantly higher apoptosis induction was measured for hepatotoxic LNA-ASOs compared to well-tolerated LNA-ASOs (Figure 2). Moreover, in accordance with recently published in vivo data,10, 11, 12, 13 our subsequent in vitro investigations further support the crucial role of RNase H1 activity (Figures 3 and 4), the relation to the number of downregulated genes (Figure 6A), and the impact of binding affinity (Tm) on LNA-ASO toxicity (Figures 7, 8, and 9). The concordance between the present in vitro data and the in vivo findings supports the in vitro screening approach for the assessment of the hepatotoxic potential of ASOs and suggests that the molecular mechanisms underlying LNA-ASO toxicity in vivo and in vitro are similar.
Previous data indicated that liver injury induced by certain LNA-ASOs in mice occurs through initiation of apoptosis in hepatocytes, and depending on the hepatotoxic potential of the LNA-ASO, can be detected as early as 24 hr after administration.9 Accordingly, our in vitro data show apoptosis induction by hepatotoxic LNA-ASOs 24 hr after assisted delivery. In particular, we found that a selected toxic LNA-ASO induced apoptosis-associated stress response genes, including Bcl2l11,17 Gadd45a,18 Trp53inp1,19 and Cdkn1a,20 already 3–6 hr after transfection (Table S2). The rapid induction of an apoptosis-related signal suggests that the initial trigger of oligonucleotide-induced toxicity may be more closely linked to the intrinsic RNA cleavage events than generally assumed.10, 11, 12 Supporting this notion are previous in vivo data demonstrating a rapid induction of stress response signals already 8 hr following administration of a hepatotoxic LNA-ASO into mice.5 In this study, a hepatotoxic LNA-ASO induced many gene and pathway changes associated with DNA replication, cell cycle, cell death, cell growth, and proliferation in the liver of mice. Preliminary phosphoproteome analyses performed in our lab demonstrated distinct stress-related phosphorylation signals already 1 hr after transfection of a hepatotoxic but not of a well-tolerated LNA-ASO in vitro (A.D., unpublished data). Together, these early stress signals suggest that the trigger of oligonucleotide-induced toxicity may be closely linked to intrinsic RNA cleavage events and not necessarily to downregulation of a specific transcript or set of transcripts. Therefore, we hypothesize that ASO-induced toxicity is primarily related to the amount of RNA cleavage (≈RNA damage), which may also explain the correlation observed in vivo10, 11 and the association demonstrated here (Figure 6A) between LNA-ASO toxicity and the number of downregulated (≈cleaved/damaged) transcripts. Moreover, this hypothesized mechanism may also explain why, so far, no biological pathways could be identified that are commonly downregulated by hepatotoxic ASOs.5, 10, 11
Recently, it has been demonstrated that RNA cleavage products generated by ASOs are processed by the RNA surveillance machinery.23 Eukaryotes have evolved RNA surveillance mechanisms to monitor RNA quality and rapidly respond to and eliminate faulty transcripts. This essential process prevents the accumulation of defective proteins and acts as a quality control filter to both uphold fidelity and fine-tune gene expression.24, 25 It is tempting to speculate that toxicity observed for some high-affinity LNA-SSOs might be related to the amount of ASO-generated RNA fragments sequestering or activating factors crucial for maintaining the quality of the RNA surveillance process. The gradual sequestering (or activation) of these factors may act as a measure of RNA cleavage: once a certain fraction of these factors is sequestered (or activated), apoptosis may be induced. However, whether this hypothesized mechanism explains how cells sense and respond to exaggerated oligonucleotide-mediated RNA cleavage must be further investigated. Obviously, a large amount of RNA cleavage events induced by some ASOs might also increase the probability of hitting essential genes so that the mechanism(s) of toxicity can be expected to be a combination of RNA damage and ASO-specific silencing of essential genes (potentially explaining the outliers found in our study).
Of particular note, apoptosis induction by RNA cleavage is also considered to be the underlying cause of cytotoxicity observed for certain ribonucleases.26 Remarkably, we found that selected hepatotoxic LNA-ASOs, similar to cytotoxic ribonucleases,26 exerted their toxic potential in cells derived from tumors of different origin(s), including HepG2, HeLa, and A549 cells (Figure 10). Although the precise mechanisms leading to apoptosis induction by cytotoxic RNases are not yet fully understood, it is possible that exaggerated off-target RNA cleavage mediated by certain high-affinity ASOs triggers similar mechanisms.
Our data suggest a general cytotoxic—rather than a cell-type-specific toxic—effect of ASOs with observed hepatotoxicity. This may be expected because the high number of downregulated off-targets by an ASO with high binding affinity is likely similar in different cell types and species. In agreement with previous in vivo studies,10, 12 our data suggest that the binding affinity of an LNA-ASO is a major determinant for hybridization-dependent toxicity (Figures 7, 8, and 9). Moreover, our data also suggest that LNA-ASOs with a calculated Tm < 55°C have a likely lower hepatotoxic potential than LNA-ASOs with a higher Tm (as suggested by the threshold of Tm ≈ 55°C, which seems to roughly separate hepatotoxic from non-hepatotoxic LNA-ASOs; Figure 7A). Such an affinity threshold above which toxicity seems to increase suggests that the cells can potentially tolerate RNA cleavage events to a certain degree before inducing apoptosis. The number of gross RNA cleavage events mediated by LNA-ASOs with a Tm < 55°C may fall more frequently into the tolerable range than the number induced by LNA-ASOs with a Tm ≥ 55°C.
One might reason that LNA-ASOs with a lower Tm are less potent than LNA-ASOs with a higher Tm. However, as recently demonstrated, too high a binding affinity of an ASO to its target can also reduce its potency, suggesting that an optimal affinity exists that is dependent on various target and cellular factors.26 We found that for gapmer LNA-ASOs, which are 16 nt long, a predicted Tm between 50°C and 55°C is within a range that roughly balances potency toward the intended target versus the hybridization-dependent toxic potential associated with the binding to unintended targets.
In silico methods can identify putative off-target sequences, making them an invaluable tool for designing oligonucleotide-based compounds. However, most of the predicted off-target sequences are not confirmed in vivo or in vitro.11, 13, 16 We here present an example illustrating the shortcoming of in silico off-target predictions, solely relying on identifying interactions based on sequence complementarity. In this example, four LNA-ASOs (the four different mGAPDH_LNA1189 wing design variants; Figure 9) had the same sequence but different numbers of LNA modifications in the wings, resulting in different calculated Tms. In terms of sequence complementarity, in silico approaches would predict the same number and identity of potential off-target sequences for these four LNA-ASOs and, hence, the same off-target-related toxic potential. However, our in vitro assessment of these oligonucleotides suggests that the various LNA wing design variants of mGAPDH_LNA1189 affect their common off-targets differently, causing differing grades of toxicity in vitro (Figure 9). A plausible explanation, which follows previous findings,13 is that the lower affinity mGAPDH_LNA1189 variants will stably bind to and cleave fewer predicted off-target sequences with partial mismatches than the higher affinity variants. Of interest, an implementation of our Tm < 55°C threshold into the design of these LNA-ASOs would have selected the well-tolerated versions while filtering out the more toxic versions. Finally, this example also supports using high-affinity modifications in the wings of ASOs because variations of their number and position in the wings allow fine-tuning of the binding affinity for optimally designed ASO molecules. However, a reduction of the Tm (by, e.g., reducing the LNA content in the wings) of a given potent, but toxic, LNA-ASO should not be considered as a general strategy to reduce the LNA-ASO’s toxic liability because the affinity reduction will likely also reduce its potency. We instead suggest designing LNA-ASOs with a “moderate” Tm (e.g., between 50°C and 55°C, as suggested by our data) and testing these oligonucleotides for efficacy.
Our data suggest that substitution of a few unmodified DNA nucleotides in the gap region with, e.g., 2′-OMe modifications, might also reduce the toxic potential of LNA-ASOs (Figure 4). Although 2′-OMe modifications increase the Tm of an ASO and potentially its potency when located in the wings, their positioning and content in the gap region can interfere with RNase H1 activity.27 Hence, 2′-OMe gap modifications can reduce the ASO’s on- and off-target cleavage potential, which likely also reduces the hybridization-dependent toxic potential observed in the present study (Figure 4). However, our data suggest that depending on the LNA-ASO sequence, slight 2′-OMe gap modifications could have a different impact on the on-target compared to the off-target sites (Figures 5 and 6A). Further investigation is required to understand how widely the gap modification approach can be used to reduce off-target effects while at the same time retaining activity on the intended target.
We acknowledge that ASOs may also cause non-hybridization-related toxicities and that the overall number and expression of potential off-targets for a given ASO and consequently the hybridization-related toxicities likely vary between cell types. This is reflected in the differing sensitivity of the three cell lines tested (Figure 10). To broadly explore the hybridization-dependent toxic potential of ASOs, we propose to include a set of different human cell types for assessment and propose to consider only those ASOs for further development, which will demonstrate the lowest apoptosis score in all cell types tested. In our hands, caspase 3/7 values <200% of the level found in untreated cells may indicate low, hybridization-related oligonucleotide toxicity (as suggested by Figure 2). Additional transcriptome and pathway analyses implemented in in vitro and in animal studies will help to prioritize LNA-ASOs for further development.11
Overall, our finding of a Tm < 55°C threshold level for LNA-ASO toxicity has the potential to improve the design of well-tolerated and efficacious molecules. Moreover, our in vitro screening approach represents a valuable addition to the set of in vitro assays that can identify and deselect ASOs with potential toxicity liabilities,11, 14, 15 further improving the discovery of safer ASO drugs.
Materials and Methods
Cell Culture
Mouse 3T3 cells were cultured in DMEM, A549 cells were cultured in Ham’s F12K medium, and HeLa and HepG2 cells were cultured in MEM. All media were supplemented with 10% (v/v) fetal bovine serum. Cells were cultured at 37°C and 5% CO2.
Oligonucleotides and Transfection
All LNA-ASOs and derivatives listed in detail in Figures 4 and 9 and Tables 1 and S1 were derived from Exiqon (Denmark). siRNAs, including control siRNA (#1027280) and mouse RNase H1 siRNA (#1027416), were purchased from QIAGEN. 1 day before transfection, cells were plated in 100 μL growth medium without antibiotics in a 96-well plate at a density that resulted in 60%–70% cell confluency at the time of transfection. Lipofectamine 2000 (Invitrogen) was used for transfections in 96-well plates. Oligonucleotides or siRNAs were diluted to the required concentration to a total volume of 25 μL in Opti-MEM (Invitrogen) and mixed with 25 μL transfection complex (0.25 μL Lipofectamine 2000 and 24.75 μL Opti-MEM). After 20 min incubation, 50 μL antibiotic-free medium was added to the solution and mixed. After removing the medium from the wells, 100 μL oligonucleotide:transfection agent solution was added to the cells and incubated for 24 hr (LNA-ASO transfections) or 48 hr (siRNA transfections), respectively. All transfections were performed in triplicates.
Apoptosis Assay
Caspase-3/7 activity was determined 24 hr after oligonucleotide transfection using the Caspase-Glo 3/7 Assay (Promega) according to the manufacturer’s instruction on a VICTOR3 plate reader (Perkin Elmer).
RNA Preparation and qPCR Experiments
mRNA purification from mouse 3T3 cells was performed using the RNeasy Mini Kit (#74104; QIAGEN), including an RNase-free DNase I treatment according to the manufacturer’s instructions. 50 ng RNA were used per sample, and a one-step RT-PCR was performed using the LightCycler Multiplex RNA Virus Master Kit (#06754155001; Roche) according to the manufacturer’s protocol for a LightCycler 96 (Roche Diagnostics). The primer probes for mouse Myd88, RNase H1, and GAPDH were Mm00440338_m1, Mm00488036_m1, and Mm99999915_g1 (all from Thermo Fisher Scientific), respectively. Analysis was done by the ΔΔCt threshold method to determine expression relative to GAPDH mRNA. Each analysis reaction was performed in triplicates, with three samples per condition.
RNA Preparation and Microarray Experiments
mRNA purification from mouse 3T3 cells was performed using the RNeasy 96 Kit (QIAGEN), including an RNase-free DNase I treatment, according to the manufacturer’s instructions. The mouse WG-6 v2 Expression BeadChip (Illumina) was processed in accordance with the manufacturer’s instructions. 750 ng total RNA were used for cRNA in vitro transcription and labeling with the Illumina TotalPrep RNA Amplification Kit (Life Technologies). Hybridization was carried out in accordance with the Illumina Hybridization System Manual, and data analysis was performed as described previously.28
Tm Calculations
A nearest-neighbor model was used to calculate theoretical Tm of the LNA-ASOs binding to RNA, as previously described.22 The nearest-neighbor model combines published thermodynamic parameters for LNA binding to DNA with parameters for DNA binding to RNA, and was validated on experimentally measured Tm for LNA-ASOs binding to RNA.22
Statistical Analysis
The data of experiments are expressed as the mean ± SD. Comparisons of data between two groups were analyzed with the Kolmogorov-Smirnov test and Wilcoxon rank sum.
Author Contributions
A.D. conceived the study, supervised the experiments, and wrote the manuscript; P.H.H. coordinated and analyzed the transfection of 236 LNA-ASOs into 3T3 cells and co-wrote the manuscript; Y.B. and C.B. performed the experiments; M.B. performed the data analysis; M.E., T.S., and F.S. revised and approved the manuscript.
Conflicts of Interest
All authors are employees of F. Hoffmann-La Roche. The funder provided support in the form of salaries for authors but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
The authors thank Marianne Rudolph Hanson and Jon Bodnar for critically reading the manuscript, and Ulrich Certa, Erich Koller, Kevin Brady, Adrian Roth, Annie Moisan, and Sabine Sewing for insightful discussions.
Footnotes
Supplemental Information includes two tables and can be found with this article online at https://doi.org/10.1016/j.omtn.2017.11.004.
Supplemental Information
References
- 1.Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chi X., Gatti P., Papoian T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today. 2017;22:823–833. doi: 10.1016/j.drudis.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Frazier K.S. Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol. 2015;43:78–89. doi: 10.1177/0192623314551840. [DOI] [PubMed] [Google Scholar]
- 4.Swayze E.E., Siwkowski A.M., Wancewicz E.V., Migawa M.T., Wyrzykiewicz T.K., Hung G., Monia B.P., Bennett C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007;35:687–700. doi: 10.1093/nar/gkl1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kakiuchi-Kiyota S., Koza-Taylor P.H., Mantena S.R., Nelms L.F., Enayetallah A.E., Hollingshead B.D., Burdick A.D., Reed L.A., Warneke J.A., Whiteley L.O. Comparison of hepatic transcription profiles of locked ribonucleic acid antisense oligonucleotides: evidence of distinct pathways contributing to non-target mediated toxicity in mice. Toxicol. Sci. 2014;138:234–248. doi: 10.1093/toxsci/kft278. [DOI] [PubMed] [Google Scholar]
- 6.Hagedorn P.H., Yakimov V., Ottosen S., Kammler S., Nielsen N.F., Høg A.M., Hedtjärn M., Meldgaard M., Møller M.R., Orum H. Hepatotoxic potential of therapeutic oligonucleotides can be predicted from their sequence and modification pattern. Nucleic Acid Ther. 2013;23:302–310. doi: 10.1089/nat.2013.0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seth P.P., Jazayeri A., Yu J., Allerson C.R., Bhat B., Swayze E.E. Structure activity relationships of α-L-LNA modified phosphorothioate gapmer antisense oligonucleotides in animals. Mol. Ther. Nucleic Acids. 2012;1:e47. doi: 10.1038/mtna.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stanton R., Sciabola S., Salatto C., Weng Y., Moshinsky D., Little J., Walters E., Kreeger J., DiMattia D., Chen T. Chemical modification study of antisense gapmers. Nucleic Acid Ther. 2012;22:344–359. doi: 10.1089/nat.2012.0366. [DOI] [PubMed] [Google Scholar]
- 9.Burdick A.D., Sciabola S., Mantena S.R., Hollingshead B.D., Stanton R., Warneke J.A., Zeng M., Martsen E., Medvedev A., Makarov S.S. Sequence motifs associated with hepatotoxicity of locked nucleic acid--modified antisense oligonucleotides. Nucleic Acids Res. 2014;42:4882–4891. doi: 10.1093/nar/gku142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burel S.A., Hart C.E., Cauntay P., Hsiao J., Machemer T., Katz M., Watt A., Bui H.H., Younis H., Sabripour M. Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acids Res. 2016;44:2093–2109. doi: 10.1093/nar/gkv1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamola P.J., Maratou K., Wilson P.A., Rush K., Mullaney T., McKevitt T., Evans P., Ridings J., Chowdhury P., Roulois A. Strategies for in vivo screening and mitigation of hepatotoxicity associated with antisense drugs. Mol. Ther. Nucleic Acids. 2017;8:383–394. doi: 10.1016/j.omtn.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasuya T., Hori S., Watanabe A., Nakajima M., Gahara Y., Rokushima M., Yanagimoto T., Kugimiya A. Ribonuclease H1-dependent hepatotoxicity caused by locked nucleic acid-modified gapmer antisense oligonucleotides. Sci. Rep. 2016;6:30377. doi: 10.1038/srep30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamola P.J., Kitson J.D., Turner G., Maratou K., Eriksson S., Panjwani A., Warnock L.C., Douillard Guilloux G.A., Moores K., Koppe E.L. In silico and in vitro evaluation of exonic and intronic off-target effects form a critical element of therapeutic ASO gapmer optimization. Nucleic Acids Res. 2015;43:8638–8650. doi: 10.1093/nar/gkv857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sewing S., Boess F., Moisan A., Bertinetti-Lapatki C., Minz T., Hedtjaern M., Tessier Y., Schuler F., Singer T., Roth A.B. Establishment of a predictive in vitro assay for assessment of the hepatotoxic potential of oligonucleotide drugs. PLoS One. 2016;11:e0159431. doi: 10.1371/journal.pone.0159431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moisan A., Gubler M., Zhang J.D., Tessier Y., Dumong Erichsen K., Sewing S., Gérard R., Avignon B., Huber S., Benmansour F. Inhibition of EGF uptake by nephrotoxic antisense drugs in vitro and implications for preclinical safety profiling. Mol. Ther. Nucleic Acids. 2017;6:89–105. doi: 10.1016/j.omtn.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagedorn P.H., Hansen B.R., Koch T., Lindow M. Managing the sequence-specificity of antisense oligonucleotides in drug discovery. Nucleic Acids Res. 2017;45:2262–2282. doi: 10.1093/nar/gkx056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Connor L., Strasser A., O’Reilly L.A., Hausmann G., Adams J.M., Cory S., Huang D.C. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takekawa M., Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 19.Okamura S., Arakawa H., Tanaka T., Nakanishi H., Ng C.C., Taya Y., Monden M., Nakamura Y. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol. Cell. 2001;8:85–94. doi: 10.1016/s1097-2765(01)00284-2. [DOI] [PubMed] [Google Scholar]
- 20.Gartel A.L., Tyner A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 21.You Y., Tataurov A.V., Owczarzy R. Measuring thermodynamic details of DNA hybridization using fluorescence. Biopolymers. 2011;95:472–486. doi: 10.1002/bip.21615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedersen L., Hagedorn P.H., Lindholm M.W., Lindow M. A kinetic model explains why shorter and less affine enzyme-recruiting oligonucleotides can be more potent. Mol. Ther. Nucleic Acids. 2014;3:e149. doi: 10.1038/mtna.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lima W.F., De Hoyos C.L., Liang X.H., Crooke S.T. RNA cleavage products generated by antisense oligonucleotides and siRNAs are processed by the RNA surveillance machinery. Nucleic Acids Res. 2016;44:3351–3363. doi: 10.1093/nar/gkw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houseley J., LaCava J., Tollervey D. RNA-quality control by the exosome. Nat. Rev. Mol. Cell Biol. 2006;7:529–539. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- 25.Lykke-Andersen J., Bennett E.J. Protecting the proteome: eukaryotic cotranslational quality control pathways. J. Cell Biol. 2014;204:467–476. doi: 10.1083/jcb.201311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang E.F., Ng T.B. Ribonucleases of different origins with a wide spectrum of medicinal applications. Biochim. Biophys. Acta. 2011;1815:65–74. doi: 10.1016/j.bbcan.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Monia B.P., Lesnik E.A., Gonzalez C., Lima W.F., McGee D., Guinosso C.J., Kawasaki A.M., Cook P.D., Freier S.M. Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene expression. J. Biol. Chem. 1993;268:14514–14522. [PubMed] [Google Scholar]
- 28.Zhang J.D., Berntenis N., Roth A., Ebeling M. Data mining reveals a network of early-response genes as a consensus signature of drug-induced in vitro and in vivo toxicity. Pharmacogenomics J. 2014;14:208–216. doi: 10.1038/tpj.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.