

Abstract
Low-density lipoprotein (LDL), one of the four major groups of lipoproteins for lipid transport in vivo, is emerging as an attractive carrier for the targeted delivery of theranostic agents. In contrast to the synthetic systems, LDL particles are intrinsically biocompatible and biodegradable, together with reduced immunogenicity and natural capabilities to target cancerous cells and to escape from the recognition and elimination by the reticuloendothelial system. Enticed by these attributes, a number of strategies have been developed for reconstituting LDL particles, including conjugation to the apolipoprotein, insertion into the phospholipid layer, and loading into the core. Here we present a tutorial review on the development of reconstituted LDL (rLDL) particles for theranostic applications. We start with a brief introduction to LDL and LDL receptor, as well as the advantages of using rLDL particles as a natural and versatile platform for the targeted delivery of theranostic agents. After a discussion of commonly used strategies for the reconstitution of LDL, we highlight the applications of rLDL particles in the staging of disease progression, treatment of lesioned tissues, and delivery of photosensitizers for photodynamic cancer therapy. We finish this review with a perspective on the remaining challenges and future directions.
TOC entry
This tutorial review introduces the concept of reconstituting low-density lipoprotein for the targeted delivery of different types of theranostic agents.
1. Introduction
Over the past several decades, researchers have been searching for an ideal carrier to selectively deliver theranostic agents to the diseased sites with maximal efficacy and minimal side effects.1–3 To this end, a large number of nanotechnology-inspired formulations that feature both the unique properties of nanomaterials and the theranostic capabilities of payloads have been developed. Excitingly, some of them have been successfully moved into clinical translation. For example, the US Food and Drug Administration (FDA) has approved the clinical use of liposomes and polymer-based nanoparticles for the delivery of various cancer therapeutics in vivo.4 Despite the successful applications of these formulations, the delivery systems still face major limitations and barriers, including short plasma half-life, variable encapsulation efficiency, uncontrolled drug release, poor tumor-targeting capability, and restricted diffusion into solid tumors.3,5 As such, there is a critical need to come up with more effective systems for targeted delivery and theranostic applications. In meeting this need, biological building blocks and/or biological design/fabrication principles have been actively explored to create new delivery systems. In particular, those materials directly copied or borrowed from nature, including lipoproteins, viruses, exosomes, and cell membranes, have attracted the most attention and is emerging as a biomedical frontier research.
Among various bio-inspired systems, lipoproteins exhibit a number of attractive features for drug delivery and theranostic applications.5,6 Lipoproteins are endogenous particles assembled from different combinations of lipids and apolipoproteins to offer distinctive sizes, morphologies, and biological functions (Table 1). Typically, each particle contains a hydrophobic core (composed of cholesteryl esters and triglycerides) that is enclosed by a monolayer of phospholipids and one or multiple lipid-binding proteins known as apolipoproteins.7 These particles circulate in the blood plasma and are responsible for lipid transport from the tissue of origin to the sites where they will be utilized or stored as a fuel. In general, four major types of lipoproteins can be separated from blood by ultracentrifugation based on their differences in density, including chylomicron (50–200 nm), very-low-density lipoprotein (VLDL, 28–70 nm), low-density lipoprotein (LDL, 20–25 nm), and high-density lipoprotein (HDL, 8–11 nm).7,8 Among them, LDL and HDL particles are the most relevant for use as delivery carriers owing to their compact sizes, typically, below 30 nm in diameter. The small sizes allow them to readily diffuse into the interfibrillar openings in a solid tumor.5 As such, a variety of reconstitution strategies have been developed to greatly enhance their capability as a class of superior carriers for the targeted delivery of theranostic agents.9
Table 1.
Physical properties and compositions of major lipoproteins in human plasma (data taken from ref. 7)
| Chylomicron | VLDL | LDL | HDL | |
|---|---|---|---|---|
| Diameter (nm) | 50–200 | 28–70 | 20–25 | 8–11 |
| Density (g/mL) | <1.006 | 0.95–1.006 | 1.006–1.063 | 1.063–1.210 |
| Composition (wt. %) | ||||
| Protein | 2 | 10 | 23 | 55 |
| Phospholipid | 9 | 18 | 20 | 24 |
| Free cholesterol | 1 | 7 | 8 | 2 |
| Cholesterol ester | 3 | 12 | 37 | 15 |
| Triglyceride | 85 | 50 | 10 | 4 |
| Apolipoprotein | A-IV | – | – | A-I, II, IV |
| B-48 | B-100 | B-100 | – | |
| C-II, III | C-I, II, III | – | C-I, II, III | |
| – | – | – | D | |
| E | E | – | E | |
| Biological function | Transport dietary lipids from the intestine to other tissues | Transport triglycerides from the liver to muscles and adipose tissues | Transport cholesterol to extrahepatic tissues | Reversely transport cholesterol from extrahepatic tissues to the liver |
As nanoscale carriers, lipoproteins have many unbeatable advantages, including the absolute biocompatibility and biodegradability, as well as reduced immunogenicity and the ability to escape the recognition and elimination by the reticuloendothelial system.10,11 Given that LDL is the only native lipoprotein that simultaneously possesses small particle size, high loading capacity, and natural tumor-targeting capability, here we will restrict our discussion to the use of reconstituted LDL (rLDL) particles for the targeted delivery of theranostic agents. Specifically, we start with a brief introduction to the molecular composition of LDL and the upregulation of the LDL receptor in neoplastic tissues. We then illustrate the advantages of using rLDL particles as a nature-inspired, nanoscale carrier for drug delivery, followed by a discussion of strategies commonly used for LDL reconstitution. Afterwards, we use a number of examples to highlight the theranostic powers of rLDL particles towards various types of diseases. Finally, we conclude this review by discussing several challenges and future directions.
2. LDL as an all-natural carrier
2.1. LDL and the LDL receptor
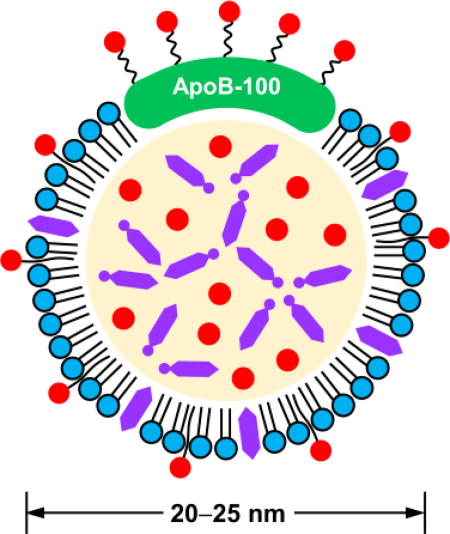
An LDL particle (20–25 nm in diameter) contains an amphiphilic phospholipid monolayer and a hydrophobic core that is rich in cholesteryl esters (Fig. 1).7 Each LDL particle consists of one apolipoprotein molecule of B-100 (ApoB-100), which covers half of the particle surface via the complex amphipathic α-helix protein–lipid interaction to stabilize this remarkable nanostructure.12 During systemic circulation, ApoB-100 can be specifically recognized by the LDL receptor on cell membrane, ultimately fulfilling the transport of cholesterol in vivo.
Fig. 1.

Two-dimensional illustration showing the composition and structure of an LDL particle.
The LDL receptor is a single-chain transmembrane glycoprotein that can be found in many different tissues and organs.13 The uptake of cholesterol-containing particles by cells is typically achieved through a process known as receptor-mediated endocytosis. In this process, the LDL receptor specifically recognizes ApoB-100 that is embedded in the phospholipid monolayer of an LDL particle. Upon the association of an LDL particle with the ligand-binding domain of the receptor, a clathrin-coated pit in the cell membrane will be formed. The coated pit then pinches off from the membrane, generating a vesicle that is subsequently transformed to an acidic endosome. The low pH in the endosome triggers the dislocation of the LDL particle from the receptor. Afterwards, the receptor is recycled back to the cell surface while the LDL particle is further delivered to a lysosome, in which it will be degraded into free cholesterol, fatty acids, and amino acids for utilization by the cell.
Cholesterol, an essential component of cell membrane, can be obtained by either de novo synthesis or cellular uptake of circulating LDL.11 Aberrant regulation of cholesterol level has been correlated with multiple types of tumors as cancerous cells have increased demand towards lipids in order to synthesize new membrane for rapid proliferation. In contrast to normal tissues, it is well established that the LDL receptor is upregulated in many malignant tissues, including colon cancer, prostate cancer, adrenal cancer, gynecological cancer, gynecological cancer, lung cancer, brain cancer, and melanoma.11,14 Such an aberrant distribution pattern forms the basis to employ LDL as a naturally selective carrier for the tumor-targeted delivery of theranostic agents. However, the LDL receptor is also abundant in two major organs, liver and adrenals, in order to facilitate their normal uptake of LDL.10 To reduce the side effects arising from the drug-loaded LDL particles, the activity of LDL receptor in these organs needs to be downregulated, preferably without affecting its function in the tumor tissues. To this end, a number of specific regulators have been identified, including bile salts (sodium taurolaurate and hydrocortisone sodium succinate), saturated fats, cholesterol with hydrogenated coconut oil, modified LDL (acetylated LDL, oxidized LDL, and glucosylated LDL), and Angiotensin-II inhibitors.10 Besides, redirecting the rLDL particles to alternative receptors by grafting new targeting ligands to ApoB-100 offers another means for reducing and thus minimizing the binding of LDL towards normal tissues.15,16
2.2. Advantages of using LDL for targeted delivery
As a naturally occurring material, LDL has many unique advantages over synthetic drug delivery systems.5,11 Firstly, LDL is inherently biocompatible and biodegradable. Upon cellular uptake, LDL is eventually degraded into recyclable biological units, including cholesterol, fatty acids, and amino acids, ensuring complete biodegradability. Secondly, LDL can escape from the surveillance of mononuclear phagocytic systems and is thus non-immunogenic. Thirdly, LDL particles have an ideal size for achieving long circulation. The LDL particles are typically smaller than 30 nm, protecting the particles from rapid renal clearance and/or recognition by the reticuloendothelial system and thus warranting prolonged circulation time (2–4 days). Moreover, such a small size also enables their diffusion into the interfibrillar openings in a solid tumor. Fourthly, LDL particles have high loading capacity and can be processed with multiple loading strategies. Each LDL particle has a relatively large hydrophobic core to accommodate a large quantity of hydrophobic therapeutics. The core-shell structure is also well-suited for direct drug loading. In addition, both the apolipoprotein and the phospholipid shell can be utilized for the loading of various theranostic agents.17 Lastly, LDL has a natural capability towards tumor targeting. As mentioned earlier, the LDL receptor is significantly upregulated in many neoplastic tissues, giving LDL the selective affinity towards tumor tissues. A detailed comparison of the LDL-based drug delivery system with other types of drug delivery carriers can be found in Table 2.
Table 2.
Comparison of LDL-based drug delivery system with other types of drug delivery carriers
| Drug delivery systems | Pros | Cons |
|---|---|---|
| LDL |
|
|
| Liposome |
|
|
| Polymeric nanoparticle |
|
|
2.3. Strategies for the reconstitution of LDL
Three major strategies have been explored for the reconstitution of LDL, namely, conjugation to the apolipoprotein (Fig. 2A), insertion into the phospholipid monolayer (Fig. 2B), and loading into the hydrophobic core (Fig. 2C). A comparison of the pros and cons of these three strategies can be found in Table 3.
Fig. 2.

Two-dimensional illustrations showing three major strategies for the reconstitution of LDL: (A) conjugation to the apolipoprotein, (B) insertion into the phospholipid monolayer, and (C) loading into the hydrophobic core.
Table 3.
The pros and cons of the three major strategies for the reconstitution of LDL
| Reconstitution strategy | Pros | Cons |
|---|---|---|
| Conjugation to the apolipoprotein |
|
|
| Insertion into the phospholipid monolayer |
|
|
| Loading into the hydrophobic core |
|
|
Conjugation to the apolipoprotein
This strategy refers to the covalent coupling of payload to the apolipoprotein via specific amino acid residuals, such as lysine, arginine, tyrosine, and cysteine.17 In view of the relatively small loading capacity, this method is primarily employed for attaching a contrast agent to the LDL particles to monitor their biodistribution in vivo.14,18 It has also been explored to reroute the targeting capability of LDL by introducing a new targeting ligand. Owing to the involvement of covalent bonding, this method can typically achieve stable labeling. However, any covalent modification to the crucial amino acid residuals will lead to irreversible inactivation of ApoB-100.19 To prevent the apolipoprotein from possible dysfunction, the head group of the phospholipid can serve as an alternative site for covalent conjugation.17
Insertion into the phospholipid monolayer
This strategy involves the non-covalent insertion of payload into the phospholipid monolayer by relying on the relatively weak interactions such as van der Waals forces.9 In general, the payload should have an amphiphilic structure to favor its insertion, ideally with the hydrophobic tail embedding in the phospholipid shell and the hydrophilic head extending into the surrounding aqueous environment.20 In contrast to the covalent coupling to the apolipoprotein, this method is much easier to implement. Nevertheless, an additional synthetic step is often required to give the payload molecules amphiphilicity. Moreover, the non-covalently bounded payloads are more prone to dissociation from the LDL particle and transfer into the membrane of neighboring cells because such a translocation process is favored by thermodynamics.19
Loading into the hydrophobic core
This strategy, firstly demonstrated by Krieger and coworkers, involves the replacement of endogenous lipids in the core with exogenous lipophilic compounds.21 The endogenous lipids are typically depleted by extraction with a non-polar organic solvent such as benzene, toluene, or heptane. The hydrophobic core is then reconstructed with either a drug/cholesteryl ester mixture or a drug-cholesteryl conjugate.22,23 It is estimated that each native LDL particle can carry a maximum of 1200–1300 cholesterol esters and 250–300 triglycerides in the core.9 As such, this method is particularly well-suited for the encapsulation of a large quantity of payloads. Because most theranostic agents are lipophilic in nature and thus have poor water-solubility, this loading strategy can substantially increase the bioavailability of drugs and thus enhance the treatment efficacy. Similar to polymeric nanoparticles, the release of drugs from the drug-loaded rLDL typically exhibits a sustained profile due to the restricted diffusion through the matrix in the hydrophobic core and the phospholipid monolayer. Nevertheless, the release kinetics is highly dependent on the physiochemical properties of drugs (e.g., hydrophobicity) and their interactions with the matrix in the core. In general, strong hydrophobicity indicates a slow release behavior, and vice versa, which can be readily interpreted using the “like dissolves like” rule.
Comparison of the three strategies for reconstitution
Since the different strategies result in payloads associated with LDL in different configurations and/or with different binding strengths, it is necessary to develop different methods for triggering their release. To this end, Zheng and coworkers reconstituted LDL particles with fluorescent dyes using the three different strategies and then directly compared their capabilities for controlled release and drug delivery (Fig. 3).22 Specifically, they conjugated fluorescein (FITC) to the apolipoprotein, inserted 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI) into the lipid monolayer, and loaded dioleyl fluorescein (Fluo-BOA) into the core. Since most drugs must escape from endolysosomes in order to reach the cytosol and then exert the therapeutic effect, they compared the capabilities of these rLDL for the cytosolic delivery of the fluorescent dyes with the assistance of disulfonated aluminium phthalocyanine (AlPcS2a), a photochemical internalization agent with an affinity towards the endolysosomal membrane. Specifically, they co-incubated rLDL and AlPcS2a with LDL receptor-overexpressed cells. Upon irradiation with a laser, the produced reactive oxygen species (ROS) efficiently destructed endolysosomes, leading to the release of the entrapped payloads. At 8 h post incubation, both the dye-labeled LDL particles and AlPcS2a were internalized into the endolysosomes of A549 cells for all three groups. After laser irradiation, AlPcS2a was evenly distributed in the cytoplasm and its fluorescence intensity was significantly enhanced owing to the decreased aggregation. For the apolipoprotein-conjugated FITC, irradiation of AlPcS2a resulted in a 2.4-fold signal enhancement at 5 min post laser irradiation (Fig. 3A). The dye release originated from the partially hydrolyzed ApoB-100 that was conjugated with FITC. For the surface-inserted DiI, a more pronounced fluorescence enhancement was found, with a 9.8-fold increase at 5 min post irradiation (Fig. 3B). Such an efficient cytosolic release was attributed to the transfer of DiI from LDL to the endolysosomal membrane that was subsequently disrupted upon laser irradiation. The quenched DiI became emissive after its release into the cytosol, leading to the recovery of fluorescence. For the core-loaded Fluo-BOA, however, laser irradiation did not result in any significant change to the fluorescence intensity as the hydrophobic dye preferred to stay in the core of LDL (Fig. 3C). These results indicated that laser irradiation induced efficient cytosolic release for payloads that were either conjugated to the apolipoprotein or inserted into the phospholipid monolayer, whereas the release of core-loaded payload could not be triggered using the same strategy. For the latter case, other triggering mechanisms, such as dissolution or melting of the core, could be explored.
Fig. 3.

Comparison of the three major strategies in cytosolic delivery of fluorescent payloads with the assistance of a photochemical internalization agent AlPcS2a: (A) conjugation of FITC to the apolipoprotein, (B) insertion of DiI into the phospholipid monolayer, and (C) loading of Fluo-BOA into the hydrophobic core. The left images show the cellular localization of rLDL and AlPcS2a in A549 cells before and at 5 min post laser irradiation, whereas the right plots display the changes in average fluorescence intensity before, immediate after, and at 2 min and 5 min post laser irradiation. Modified with permission from ref. 22, copyright 2011 Royal Society of Chemistry.
LDL-mimetic particles (LMPs)
Directly prepared from triglycerides, cholesterol esters, cholesterol, phospholipids, and ApoB-100 or short peptides containing the LDL receptor-binding domain, LMPs are emerging as promising carriers for the encapsulation of lipophilic/amphiphilic theranostic agents.8 Since native LDL cannot be obtained in large quantities and the isolated LDL is potentially compromised by pathogen infection,11,24 LMPs offer an alternative platform to overcome these limitations. Compared to the established methods for the reconstitution of LDL particles, the fabrication of LMPs is relatively more complex as multiple components are needed to maximally mimic the structure of native LDL.25 If the LMPs are not equipped with the LDL receptor-binding moiety, specific ligands are required to decorate the resultant nanoparticles for targeted drug delivery.26 Similar to core reconstitution, this method allows for the incorporation of a large quantity of payloads. However, the resultant LMPs typically have a particle size greater than 100 nm, resulting in increased recognition by the reticuloendothelial system and thereby shortened blood circulation time.
3. Reconstituted LDL for contrast enhancement and therapy
In view of the unique advantages and diverse loading strategies, LDL has been reconstituted with a number of contrast agents and therapeutics to explore its applications in staging disease progression and treating lesioned tissues.9,11,19 As an extension, photosensitizers have also been loaded into LDL particles to facilitate light-guided, site-specific photodynamic therapy (PDT).27 Some of these examples can be found in Table 4.
Table 4.
Different types of theranostic agents that have been used for the reconstitution of LDL
| Category | Examples | Ref. |
|---|---|---|
| Contrast agents | ||
| Radiotracer | 125I, 99mTc, 68Ga, and 111In | 14, 18, 19, 21, and 30 |
| MRI | Gd(III) | 31 |
| Fluorescent dyes | DiR and tris[(porphinato)zinc(II)] | 16 and 32 |
| X-ray | Au nanocrystals and poly-iodinated triglyceride | 11 and 33 |
| Therapeutic agents | ||
| Anticancer drug | DOX and 5-fluorouracil | 34, 35, and 36 |
| Antisense nucleotide | Phosphorothioate oligodeoxynucleotides | 24 |
| Neuroprotective nutrient | DHA | 38 and 39 |
| Anti-atherosclerotic drug | Dexamethasone palmitate | 40 and 41 |
| PDT agents | ||
| Conventional photosensitizer | Porfimer sodium | 29, 45, 46, and 47 |
| Optimized photosensitizer | Silicone phthalocyanine analogue, silicon naphthalocyanine, and bacteriochlorin e6 bisoleate |
3.1 Delivery of contrast agents for the diagnosis of diseases
Many types of contrast agents have been used for labeling LDL. In particular, radio-labeling can be traced back to more than 30 years ago. Among various radioactive isotopes, technetium-99m (99mTc) and iodine-125 (125I) are commonly used for monitoring the biodistribution of LDL particles in vivo. To this end, Lees and coworkers demonstrated the primary accumulation of 99mTc-labeled LDL in the liver, adrenals, and kidneys of healthy rabbits, where the half-life circulation time could be as long as 20 h.18 Versluis and coworkers observed the preferential accumulation of 125I-labeled LDL in the tumor tissues of B16 melanoma-bearing mice, which accounted for the highest uptake, followed by the liver, spleen and adrenals.14 Despite the large uptake in these normal organs, the LDL receptor-mediated, tumor-specific drug delivery is still achievable because the activity of the LDL receptor in these normal organs can be selectively downregulated through methods without affecting the uptake of LDL in cancerous cells (see Section 2.1).
In general, it is challenging to directly conjugate a radioactive isotope to the apolipoprotein. As an alternative, chelators capable of complexing with multiple types of radioactive isotopes were developed. To this end, diethylenetriaminepentaacetic acid (DTPA) has been employed for the chelation of radionuclides (e.g., gallium-68 (68Ga) and indium-111 (111In)). In early studies, the DTPA was often attached to LDL through conjugation to the apolipoprotein, resulting in the inactivation of ApoB-100. To overcome this drawback, DTPA was chemically modified with two hydrophobic stearyl tails (DTPA-SA) to favor non-covalent insertion into the phospholipid monolayer.20 However, when applied to in vivo studies, the radio-labeled structures were troubled by the leakage of radionuclide and/or rapid cellular secretion of radionuclide after lysosomal degradation. To address these issues, Xiao and coworkers synthesized 1,3-dihydroxypropan-2-one 1,3-diiopanoate (DPIP), an 125I-labeled hexa-iodinated glyceride analogue, for core reconstitution.28 Owing to the presence of an ethyl group adjacent to the ester group, the resultant rLDL exhibited resistance to lysosomal degradation. It was demonstrated that the uptake of LDL in cervical cancerous cells was 50-fold as great as in normal gynecological tissue, suggesting its potential for cancer diagnosis.
In terms of quantification of the biodistribution of radiolabeled LDL, each labeling approach has its unique advantages and disadvantages. For direct or indirect conjugation of radionuclides to the ApoB moiety, a variety of methods can be employed. However, the labeling efficiency tends to be limited as over-labeling typically leads to the inactivation of ApoB. In this respect, membrane insertion provides an excellent solution to this issue despite the number of available radionuclides is limited. Incorporation of radionuclides in the core can overcome the limitations of the above two approaches, but a specially designed chemical structure is needed to facilitate efficient loading of radionuclides. Nevertheless, all radio-labeled LDL particles are inevitably subjected to lysosomal degradation, leading to the secretion of radioactive metabolites from the cells. Such a process complicates the interpretation of biodistribution results as the amount of radioactivity in the samples does not reflect the actual uptake of LDL particles. In this respect, radioactive tyramine cellobiose (e.g., 125I-tyramine cellobiose) and DPIP have been developed for direct labeling and core incorporation, respectively, as both of them are either retained in cells after lysosomal processing or resistant to lysosomal degradation after cellular uptake.
The use of LDL for delivering magnetic resonance imaging (MRI) contrast agents has also been explored. In one study, Corbin and coworkers inserted the bifunctional chelator DTPA-SA into the phospholipid monolayer of LDL and then achieved the chelation of Gd(III) (Fig. 4A).29 They further studied the biodistribution of the Gd(III)-labeled LDL in HepG2 tumor-bearing mice. As shown in Fig. 4B, the liver exhibited enhancement in signal at 5 h post administration, whereas significant signal enhancement in the tumor was observed by 24 h and retained until 36 h. It is noteworthy that the contrast in the entire tumor was significantly enhanced, which possibly benefited from the facile diffusion of Gd(III)-labeled LDL in the solid tumor. In contrast with the commercial Gd(III)-DTPA-diamide chelate, the Gd(III)-labeled LDL particles showed almost 45-fold contrast enhancement in the tumor at 24 h post administration, indicating its potential for cancer diagnosis.
Fig. 4.

Diagnosis of diseases with various types of rLDL particles. (A, B) Gd(III)-labeled LDL for MRI visualization of tumor tissues. (A) Schematic illustration of a LDL particle whose surface has been inserted with Gd(III)-DTPA-SA. (B) T1-weighted axial spin-echo images in a HepG2 tumor-bearing nude mouse, where the arrowheads point to the liver and the arrows indicate the tumor. Modified with permission from ref. 29, copyright 2006 Elsevier. (C–E) Rerouting LDL by conjugation with folate (FA). (C) Schematic illustration of a DiR-labeled, FA-conjugated LDL particle (DiR-LDL-FA). (D) Fluorescence images of mice bearing KB (FA receptor-positive) and HT1080 (FA receptor-negative) dual tumors post intravenous injection of the DiR-LDL-FA particles. (E) Fluorescence signals of tumor extracts from FA inhibition assay in vivo, where free FA in 30-fold excess was used to block the endocytic pathway of DiR-LDL-FA. Modified with permission from ref. 16, copyright 2007 American Chemical Society. (F–H) Gold nanoparticle-labeled LDL (Au-LDL) for CT imaging of tumor-bearing mice. (F) Schematic illustration showing the structure of an Au-LDL particle. (G) TEM images showing the LDL particles before and after labeling with Au nanoparticles. (H) Spectral CT images of B16-F10 tumor-bearing mice injected with Au-LDL, where the accumulation of Au in the tumor (left image) and liver (right image) is highlighted in yellow. Modified with permission from ref. 12, copyright 2013 American Chemical Society.
Fluorescent probes have long been used for optical imaging and tracking of LDL particles. Ideally, their emission peaks should be tuned to the near-infrared (NIR) region (650–950 nm) to minimize the scattering from tissues and the absorption by hemoglobin and water to achieve deep penetration in vivo. To this end, Wu and coworkers synthesized a series of NIR fluorophores based on tris[(porphinato)zinc(II)] and incorporated them into the core of LDL for the imaging of B16 melanoma cells.30 In addition to the LDL receptor-mediated uptake, LDL can also be rerouted to alternative receptors by modifying ApoB-100 with other tumor-homing ligands. Zheng and coworkers demonstrated this concept by conjugating the lysine residue of ApoB-100 with folate (FA).15 As anticipated, the resultant FA-LDL switched their targeting capability from LDL receptor-positive HepG2 cells to FA receptor-positive KB cells. In a follow-up study, they investigated the tumor-targeting capability of 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide (DiR)-labeled, FA-conjugated LDL (DiR-LDL-FA) in nude mice bearing both FA receptor-positive KB tumor and FA receptor-negative HT1080 tumor (Fig. 4C).16 They also monitored in vivo biodistribution of the DiR-LDL-FA in dual tumor-bearing mice (Fig. 4D). Immediately after intravenous injection, fluorescence signals were detected over the entire body of the animal. At 24 h post injection, most of the fluorescent dyes were cleared from blood circulation. Interestingly, the preferential accumulation of DiR-LDL-FA in the KB tumor over HT1080 tumor became more obvious. Saturation of the FA receptor with excess free FA further confirmed the FA receptor-mediated uptake (Fig. 4E).
In recent years, exogenous X-ray contrast agents have been introduced into LDL to facilitate tumor-targeted imaging by computed tomography (CT). Hill and coworkers used 2-oleoylglycerol 1,3-bis(iodopanoate), a poly-iodinated triglyceride, to reconstitute the core of LDL.31 Incubation of the resultant rLDL with HepG2 cells led to the enhancement in imaging contrast as compared to the control group in which excess LDL was used to block the endocytic pathway. In extending the labeling category of exogenous substances, Allijn and coworkers developed a micelle-translocation method to label LDL with a variety of contrast agents, including Au nanoparticles and lipophilic or amphiphilic fluorophores (Fig. 4F).12 Specifically, the Au nanoparticles (2–3 nm in diameter) were coated with a layer of phospholipids and/or fluorescently labeled phospholipids to form micelles. After mixing with native LDL, sonication was applied to initiate the membrane fusion between the phospholipid-coated Au nanoparticles and LDL. TEM imaging confirmed the successful loading of the Au nanoparticles into the core of LDL (Fig. 4G). Spectral CT images demonstrated the significant accumulation of the Au-labeled LDL (Au-LDL) inside the tumor and liver of a B16-F10 tumor-bearing mouse at 24 h post injection (Fig. 4H). Further analysis revealed that most of the Au-LDL particles were internalized by the tumor-associated macrophages (TAMs) with minor uptake in the endothelial and cancerous cells. It is worth noting that the TAMs are recently recognized as an important therapeutic target due to their negative correlation with patient survival rates. As such, this method can be used to quantitatively monitor the response of TAMs to cancer therapy.
3.2 Incorporation of therapeutic drugs for the targeted treatment of diseases
Considering the natural targeting capability of LDL towards malignancies, the loading of therapeutic drugs (in particular, anticancer drugs) into LDL has been extensively explored. In comparison to free drugs, the use of drug-loaded LDL greatly improves the accumulation of drugs at the diseased site, holding great potential in the use of relatively small amount of therapeutics to achieve significant clinical outcomes. For example, Chu and coworkers demonstrated the enhanced uptake of doxorubicin (DOX)-loaded LDL relative to free DOX in HepG2 cells.32 Despite DOX-loaded LDL exhibited slightly improved anti-proliferative effect in tumor-bearing mice relative to free DOX, a remarkably reduced cardiotoxicity was found for the group treated with DOX-loaded LDL, indicating its improved safety as a tumor-targeting carrier. Radwan and coworkers reported the use of cholesterol-conjugated 5-fluorouracil-loaded LDL for improved treatment of cancer in a mouse model.33 To extend the spectrum of payloads being delivered, Bijsterbosch and coworkers conjugated cholesterol to phosphorothioate oligodeoxynucleotides and achieved intracellular delivery of oligodeoxynucleotides for antisense gene therapy.23 In a recent study, LDL was also used as a negatively charged species to interact with the positively charged chitosan via electrostatic assembly for DOX encapsulation. In contrast to free DOX, the resultant particles exhibited enhanced uptake by cancerous cells.34 To enable oral drug delivery, Zhou and coworkers recently developed a pH- and heat-dependent process to produce nanogels from a mixture of drug-loaded LDL and pectin (a natural polysaccharide), where the polysaccharide can serve as a shield to protect LDL from enzymatic degradation in the stomach and promote controlled drug release in the small intestine.35 The resultant nanogels had a spherical shape and uniform particle size of about 58 nm. Further studies demonstrated that the nanogels exhibited excellent stability and sustained release under simulated gastrointestinal conditions, paving the way for using rLDL in oral delivery of drugs and nutrients.
Apart from the conventional anticancer drugs, docosahexaenoic acid (DHA), a natural omega-3 polyunsaturated fatty acid, has also been used for cancer treatment due to its pronounced antitumor activity. To this end, Reynolds and coworkers incorporated DHA into LDL and further evaluated its cytotoxicity in both healthy (TIB-73) and malignant (TIB-75) murine liver cells.36 At 60 µM of DHA-loaded LDL, TIB-75 cells were completely killed whereas TIB-73 cells remained viable. They precluded that the selective cytotoxicity was caused by the enhanced peroxidation of DHA and subsequent production of ROS in cancerous cells. In addition to cancer therapy, DHA was also supplemented as a neuroprotective nutrient to maintain normal brain function, and the lack of DHA in the brain could be correlated with a variety of neurodegenerative diseases. Mulik and coworkers recently achieved the localized delivery of DHA-LDL to rat brains with the assistance of focused ultrasound (FUS, see Fig. 5A).37 Upon intravenous injection of microbubbles, a selected brain region in a normal rat was exposed to a pulsed ultrasound using an FUS transducer to transiently open the blood-brain barrier (BBB). Using Evans blue as a marker, they confirmed the FUS-mediated BBB opening at the targeted region (Fig. 5B). The permeability of DiR-labeled LDL (DiR-LDL) into BBB was further examined. As shown in Fig. 5C, the strong fluorescence of DiR was found in the targeted cortical region when FUS was imposed. In contrast, the fluorescence signal was barely detected for the group without FUS exposure. Quantitative analysis of DiR fluorescence in the rat brain indicated that FUS exposure resulted in 5–6-fold greater DiR intensity in the targeted region than in the non-targeted hemisphere, and >60-fold higher than the case without FUS exposure (Fig. 5D). As shown in Fig. 5E, with the assistance of FUS, the DiR-LDL not only passed through BBB but also entered the cells in the brain, which was confirmed by the punctate fluorescence within the cytoplasm of neurons adjacent to the blood vessels. For the DHA-loaded LDL particles delivered into the brain, FUS exposure led to 2-fold increase of DHA and 3-fold increase of DHA-derived metabolites in the targeted region relative to the non-targeted region. Histological evaluation revealed that no evident injury or hemorrhage was found in the FUS-treated brain region. This study provides a generic strategy for the targeted delivery of rLDL to the brain, holding great potential in the treatment of acute brain injury or brain cancer.
Fig. 5.

Delivery of DHA-loaded LDL to rat brains with the assistance of focused ultrasound (FUS). (A) Schematic illustration showing the strategy of localized opening of BBB in a rat model. (B) A brain slice harvested from an animal exposed to FUS showing the localized leakage of Evans blue into the corresponding brain region. (C) Whole brains (top) and cross-sectional views of the brains (bottom) injected with DiR-labeled LDL (DiR-LDL) in the presence and absence of FUS, respectively. (D) Quantitative measurement of DiR fluorescence in the brain treated with the DiR-LDL plus FUS and the DiR-LDL, respectively (** and *** indicate p < 0.005 and 0.001, respectively). (E) Fluorescence image from coronal cryosection of the brain. Representative areas of interest were expanded from the FUS-exposed region (left) and contralateral hemisphere (right), where DiR and cell nuclei are shown in red and blue, respectively. Images were captured at 20× magnification. Modified with permission from ref. 37, copyright 2016 Elsevier.
Excess oxidized LDL in the aortic vasculature typically results in the recruitment of macrophages. Upon uptake of the oxidized LDL particles, the recruited macrophages are then transformed into foam cells to aggravate the formation of atherosclerotic plaques. The progression of atherosclerosis, however, can be intervened by inhibiting the formation of foam cells with anti-inflammatory drugs. In comparison to native LDL particles, those being oxidized have a higher affinity towards atherosclerotic plaques, thereby providing a possibility for the targeted delivery of therapeutic drugs. Tauchi and coworkers demonstrated that the oxidized LDL reconstituted with a prodrug (dexamethasone palmitate, DP) had a remarkable inhibitory effect on the formation of foam cells.38 They also evaluated the effect of the DP-loaded LDL (DP-LDL) on the accumulation of cholesterol esters in the aorta of atherogenic mice.39 In contrast to free dexamethasone, injection of the DP-LDL into the atherogenic mice resulted in 100-fold less accumulation of cholesteryl esters in the aorta. Nevertheless, these results were based on short-term observation. Since LDL carries pro-atherogenic molecules (e.g., ApoB-100) that can aggravate disease progression, the use of LDL for the treatment of atherosclerotic plaques should be highly regulated.
As a substitute to LDL, LMPs have also been developed for the delivery of diverse therapeutic drugs. In general, the materials used for LMP fabrication originate from exogenous lipids and/or short peptides containing the LDL receptor-binding domain. The drawbacks derived from these subtle variations tend to attenuate their clinical applications, including the complicated purification steps, uncertain immune responses, and improperly functioned apolipoprotein. The use of all-natural materials is thus preferred to overcome these limitations. Recently, Wang and coworkers developed a bio-inspired disassembly-reassembly strategy for constructing LMPs (Fig. 6A).40 Specifically, the components of native lipoproteins in human plasma were isolated and purified using an organic solvent/isoelectric precipitation method. Together with the payload, the lipid-containing organic phase (in ethanol) was added dropwise into an apolipoprotein-containing aqueous phase at a weight ratio identical to that of native lipoproteins, leading to the formation of drug-loaded LMPs. Since human plasma sample was directly used for reconstitution, a mixture of lipoprotein-mimetic nanoparticles was obtained, including HDL-like (with ApoA), LDL-like (with ApoB-100), and multiple lipoprotein-anchored (with both ApoA and ApoB-100) nanoparticles. These nanoparticles entered cells via multi-targeting pathways, in which scavenger receptor class B type I (SR-BI) mediated the direct transmembrane delivery of HDL-like nanoparticles, LDL receptor facilitated the internalization of LDL-like nanoparticles, and SR-BI and/or other lipoprotein receptors initiated the uptake of multiple lipoprotein-anchored nanoparticles. These LMPs exhibited efficient intracellular delivery of paclitaxel (PTX) and enhanced antitumor efficacy towards HepG2 cells compared to free PTX, PTX-loaded liposomes, and PTX-loaded HDL-mimetic nanoparticles (rHDL). In vivo analysis in HepG2-bearing nude mice at 24 h post injection suggested that the DiR-loaded LMPs primarily accumulated in the tumor and liver, while the liposomes and rHDL exhibited rapid blood clearance and significant hepatic uptake, respectively (Fig. 6, B and C). Antitumor evaluation indicated that the tumor inhibitory effect of the PTX-loaded LMPs was as high as 70.51%, which was greater than those of free PTX, PTX-loaded liposomes, and PTX-loaded rHDL. In addition to cancer therapy, Meng and coworkers reported the use of LMPs for the targeted delivery of curcumin (a drug with effects on the reduction of neurotoxic Aβ aggregates and ameliorate cognitive deficits) to a rat model with Alzheimer’s disease, demonstrating their potential for brain-targeted drug delivery.41 Apart from ApoB-100, apolipoprotein E3 (ApoE3) has been used as an alternative component for the fabrication of LMPs due to its high affnity toward the LDL receptor.42,43 Since the LDL receptor is overexpressed both in BBB and glioblastoma cells, these LMPs were able to pass through BBB via ApoE3-enabled transcytosis and target glioblastoma cells via receptor-mediated uptake, holding great potential for the treatment of glioblastoma in vivo.
Fig. 6.

Fabrication of LMPs using a bio-inspired disassembly-reassembly strategy. (A) Schematic illustration of the disassembly-reassembly process. Although HDL was used as an example to demonstrate the reconstitution procedure, this strategy should be applicable to the reconstitution of LDL, as well as the mixture of different types of lipoproteins in human plasma. (B) Biodistribution of DiR-loaded liposomes (DiR-Lipos), DiR-loaded HDL-mimetic nanoparticles (DiR-rHDL), and DiR-loaded LMPs fabricated from a mixture of different types of lipoproteins (DiR-Mixed) in HepG2 tumor-bearing nude mice at 24 h post injection. The fluorescence images and X-ray images were overlaid, where the red circles outline the accumulation of fluorescent dyes in the tumor tissues. (C) Quantification of fluorescence intensity in ex vivo tumors at 24 h post injection (*** indicates p < 0.001). Modified with permission from ref. 40, copyright 2016 Elsevier.
3.3 Incorporation of photosensitizers for the targeted photodynamic therapy
PDT has been recognized as a promising and non-invasive approach to cancer therapy.44 In a typical procedure, the photosensitizers are systemically injected for blood circulation, followed by light irradiation at the diseased site to produce the cytotoxic ROS, which lead to the irreversible destruction of lesioned tissues. In contrast to direct administration of therapeutics, PDT greatly reduces the side effects of drugs on normal organs. Among various photosensitizers, porphyrin and its derivatives or synthetic counterparts are widely used owing to their improved adsorption in the NIR region and prolonged retention in the malignant lesions. Because most photosensitizers are hydrophobic or amphiphilic in nature, they cannot be directly administered in vivo. To improve the water-solubility and tumor-targeting capability of these photosensitizers for systemic circulation, suitable carriers need to be employed, and endogenous LDL is an attractive candidate. Early studies were mainly focused on the non-covalent mixing of photosensitizers with LDL or covalent grafting of photosensitizers to ApoB-100 for targeted cancer therapy. However, the photosensitizers developed in these early studies had relatively short absorption wavelengths, greatly limiting their clinical application.27 To red-shift the absorption peak and improve the loading capacity of photosensitizers in LDL, Zheng and coworkers developed a silicone phthalocyanine analogue SiPcBOA (tetra-t-butyl silicon phthalocyanine bisoleate, λAbs = 684 nm) for core reconstitution. In this case, the loading capacity could be as high as 3000–3500 molecules of SiPcBOA per LDL particle.45 In vitro PDT analysis showed that the SiPcBOA-LDL imposed a pronounced cytotoxicity towards HepG2 cells under light irradiation, whereas free SiPcBOA only induced limited cell damage under the same dosage.
To achieve the optimal NIR wavelength for deep tissue penetration, the same group developed a naphthalocyanine-based photosensitizer SiNcBOA (tetra-t-butyl silicon naphthalocyanine bisoleate) that had a maximum absorption at 810 nm, together with an extinction coefficient of 3.7×105 M−1.46 In vivo experiments suggested the targeted accumulation of SiNcBOA-LDL in the HepG2-bearing mice, with a tumor to normal muscle ratio of 8:1 at 2 h post administration. Subsequently, they synthesized bacteriochlorin e6 bisoleate (Bchl-BOA, λAbs = 748 nm) and evaluated its PDT efficacy in vivo (Fig. 7A).47 At 3 h post intravenous injection of Bchl-BOA-loaded LDL, the fluorescence intensity ratio between the harvested tumor and muscle tissue could reach 30:1. PDT was then performed using a 750-nm diode laser for tumor ablation at energy doses of 125–175 J cm−2. As shown in Fig. 7B, PDT treatment at a dose of 150 J cm−2 completely ablated xenograft tumor in the HepG2-bearing nude mouse at day 60 post injection. The survival curves at different energy doses confirmed the suppressed tumor growth and improved survival rates after PDT treatment (Fig. 7C).
Fig. 7.

Bchl-BOA-loaded LDL for PDT treatment in vivo. (A) Schematic illustration showing the structure of a Bchl-BOA-loaded LDL particle. (B) HepG2 tumor-bearing mice pre- and post PDT treatment: i) nude mouse bearing a tumor on its left flank, ii) 24 h post treatment (150 J cm−2, 2 µmol kg−1 Bchl-BOA-LDL) with remarkable burn/edema, iii) scab formation at day 9 post treatment, and iv) complete tumor ablation (arrowed) at day 60 post treatment. (C) Survival curves of the PDT-treated and control mice, where untreated mice, mice irradiated at the light dose of 175 J cm−2 only, and mice treated with Bchl-BOA-LDL alone were used as the controls (* indicates p < 0.0001). Modified with permission from ref. 47, copyright 2011 Future Medicine.
In addition to cancer therapy, the Zheng group also developed a method for cytosolic delivery of payloads using PDT-assisted endolysosomal membrane disruption as discussed in Section 2.3 (see Fig. 3).22 This study demonstrated that insertion into the phospholipid monolayer was the most efficient way to trigger quick cytosolic release of the payloads, offering a promising strategy to bypass the undesired endolysosomal entrapment. Using the same photochemical internalization approach, they further achieved enhanced gene silencing by rLDL particles whose surface was loaded with cholesterol-conjugated siRNAs (small interfering RNAs).48 Although these studies provide an efficient way to achieve the cytosolic delivery of payloads, both the concentration of photosensitizer and the irradiation dose still need to be optimized to ensure that the cell viability is minimally affected.
4. Concluding remarks and perspectives
Enabled by the innovations in chemistry, biochemistry, and materials science, the past few decades have witnessed a tremendous progress in using LDL as a natural carrier for the targeted delivery of theranostic agents. In particular, the delivered payloads have been expanded from conventional contrast agents and anticancer drugs to nucleic acids and photodynamic agents. The imaging modalities have been extended to cover optical and radioactive imaging, MRI, and CT. The treated diseases have also been broadened from cancer and atherosclerosis to the recently demonstrated brain-related lesions. It is envisioned that LDL and its derivatives will play pivotal roles in defeating a broader spectrum of diseases by integration with other advanced theranostic modalities. All these achievements have benefited enormously from the collaboration between researchers from different disciplines. We believe that with the continued development of cutting-edge techniques, more diseases and disorders in human beings can be addressed with the assistance of rLDL and related nanoparticles.
As a platform material, LDL is advantageous in providing large loading capacity, intrinsic biocompatibility, diverse surface chemistry, decreased immunogenic response, and natural tumor-targeting capability. Despite the attractiveness of LDL, it has been relatively slow in advancing rLDL towards clinical translation, largely because of the following issues. Firstly, the supply of LDL is limited. Since most LDL is isolated from blood, it is difficult to obtain this biomaterial in very large quantities. Besides, the size and composition of LDL show variations between batches. Secondly, there is a concern regarding the storage stability as LDL particles tend to aggregate upon long-term storage. Meanwhile, the pathogen contamination associated with LDL isolation poses a potential safety issue. Thirdly, the matrix for core reconstitution has potential adverse impacts. Most core-loading methods employ either cholesteryl esters or cholesterol-drug conjugates. However, extensive delivery of these compounds elevates the total cholesterol level in the blood, which has a strong correlation with the formation of atherosclerotic plaques. Fourthly, the delivery efficiency is limited by tumor heterogeneity. Although many malignancies are reported to have upregulated LDL receptor, some cancerous cells are able to develop the receptor with lost activity or even deficiency in the receptor.5 In either case, rLDL will become less effective in treating these cancers. Lastly, LDL still shows undesired accumulation in normal tissues. Since liver and adrenal are the two major organs rich in the LDL receptor, the drug-loaded LDL administered via intravenous injection is inevitably captured by and retained in these organs, thereby imposing off-target toxicity.
Despite these challenges, LDL still holds great potential in the treatment of lesioned tissues. Looking ahead, we envision that the following aspects should be explored to further advance the LDL-based delivery system. First of all, more suitable core-loading matrices need to be developed for the replacement of the commonly used cholesteryl esters. In this regard, we recently reported the use of a eutectic mixture of lauric acid and stearic acid (with a well-defined melting point at 39 °C) as the core matrix for reconstituting LDL (Fig. 8 A and B).49 Because lauric acid and stearic acid have a favorable effect on the increase of HDL (the “good” blood cholesterol) and the decrease of LDL (the “bad” blood cholesterol), respectively, they are considered safe materials for clinical applications. The rLDL could enable the metabolism-triggered drug release while preventing the payloads from endolysosomal degradation. When loaded with drugs, we also achieved the selective delivery of DOX into the LDL receptor-overexpressed cancerous cells (Fig. 8, C–F). In a follow-up study, we further demonstrated the fabrication of phase-change nanoparticles based on the same eutectic mixture and achieved NIR-triggered intracellular drug release.50 It is worth pointing out that in both studies, the use of a mixture of fatty acids was able to substantially increase the drug loading capacity because of the decreased crystallinity for the matrix upon solidification.
Fig. 8.

Reconstitution of LDL with fatty acids for the targeted delivery of drugs into cancerous cells. (A) Schematic illustration showing an LDL particle whose core has been reconstituted with a drug-loaded mixture of fatty acids. (B) DSC curves of lauric acid, stearic acid, and a eutectic mixture of lauric acid and stearic acid at a weight ratio of 4:1. (C, D) Comparison of the delivery of DOX-LDL into A549 and NIH-3T3 cells. Scale bars: 50 µm. (E) Cytotoxicity of DOX-LDL towards A549 and NIH-3T3 cells. (F) Cytotoxicity of DOX-LDL towards A549 and NIH-3T3 cells in the absence and presence of native LDL in 20-fold excess to confirm the LDL receptor-mediated uptake. Modified with permission from ref. 49, copyright 2017 Wiley-VCH.
Additionally, more systematic in vivo evaluation should be performed to ensure the safe use of rLDL in human. Although a number of studies have demonstrated the usefulness of rLDL in theranostics, few of them undertake long-term observation of rLDL behavior in vivo, such as how they interact with major tissues and/or organs and how the body will respond when rLDL particles are administered over a long period of time. To facilitate the clinical translation of rLDL, these questions need to be answered through systematic, long-term investigations. We believe that the answers can broaden our understanding of how rLDL exactly works, which, in turn, will aid us to fully explore the great potential offered by LDL. It is anticipated that the results from these studies will inspire more people to expand the applications of LDL in biomedicine.
Key learning points.
Conceptual introduction to the naturally-occurring nanocarrier LDL and its receptor;
Understanding the unique advantages of rLDL over synthetic drug delivery systems;
Major strategies for the reconstitution of LDL and fabrication of LDL-mimetic particles;
State-of-the-art in applying rLDL particles to various theranostic applications;
Challenges and future directions in moving rLDL particles towards clinical translation.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (R01 CA138527) and startup funds from the Georgia Institute of Technology.
Biographies

Chunlei Zhu
Chunlei Zhu received his B.S. in chemistry from Jilin University in 2008 and Ph.D. in organic chemistry from the Institute of Chemistry, Chinese Academy of Sciences in 2013 under the supervision of Professor Shu Wang. He joined the Xia group as a postdoctoral fellow from 2014 to 2017. His research interests include the development of phase-change nanoparticles for drug delivery and novel inverse opal scaffolds for tissue engineering.

Younan Xia
Younan Xia studied at the University of Science and Technology of China (B.S., 1987) and University of Pennsylvania (M.S., 1993), and received his Ph.D. from Harvard University in 1996 (with George M. Whitesides). He started as an assistant professor of chemistry at the University of Washington (Seattle) in 1997 and joined the department of biomedical engineering at Washington University in St. Louis in 2007 as the James M. McKelvey Professor for Advanced Materials. Since 2012, he holds the position of Brock Family Chair and GRA Eminent Scholar in Nanomedicine at the Georgia Institute of Technology.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Angew. Chem., Int. Ed. 2014;53:12320–12364. doi: 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- 2.Tibbitt MW, Dahlman JE, Langer R. J. Am. Chem. Soc. 2016;138:704–717. doi: 10.1021/jacs.5b09974. [DOI] [PubMed] [Google Scholar]
- 3.Petros RA, DeSimone JM. Nat. Rev. Drug Discovery. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 4.Anselmo AC, Mitragotri S. Bioeng. Transl. Med. 2016;1:10–29. doi: 10.1002/btm2.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabnis N, Lacko AG. Ther. Deliv. 2012;3:599–608. doi: 10.4155/tde.12.41. [DOI] [PubMed] [Google Scholar]
- 6.Thaxton CS, Rink JS, Naha PC, Cormode DP. Adv. Drug Delivery Rev. 2016;106:116–131. doi: 10.1016/j.addr.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson DL, Cox MM. In: Lehninger Principles of Biochemistry. 5. Freeman WH, editor. Company; New York: 2008. [Google Scholar]
- 8.Huang H, Cruz W, Chen J, Zheng G. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2015;7:298–314. doi: 10.1002/wnan.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng KK, Lovell JF, Zheng G. Acc. Chem. Res. 2011;44:1105–1113. doi: 10.1021/ar200017e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain A, Jain K, Kesharwani P, Jain NK. J. Nanopart. Res. 2013;15:1888. [Google Scholar]
- 11.Harisa GI, Alanazi FK. Saudi Pharm. J. 2014;22:504–515. doi: 10.1016/j.jsps.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allijn IE, Leong W, Tang J, Gianella A, Mieszawska AJ, Fay F, Ma G, Russell S, Callo CB, Gordon RE, Korkmaz E, Post JA, Zhao Y, Gerritsen HC, Thran A, Proksa R, Daerr H, Storm G, Fuster V, Fisher EA, Fayad ZA, Mulder WJM, Cormode DP. ACS Nano. 2013;7:9761–9770. doi: 10.1021/nn403258w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu B, Olenyuk Z, Okamoto CT, Hamm-Alvarez SF. Adv. Drug Delivery Rev. 2013;65:121–138. doi: 10.1016/j.addr.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Versluis AJ, van Geel PJ, Oppelaar H, van Berkel TJ, Bijsterbosch MK. Br. J. Cancer. 1996;74:525–532. doi: 10.1038/bjc.1996.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng G, Chen J, Li H, Glickson JD. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17757–17762. doi: 10.1073/pnas.0508677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, Corbin IR, Li H, Cao WG, Glickson JD, Zheng G. J. Am. Chem. Soc. 2007;129:5798–5799. doi: 10.1021/ja069336k. [DOI] [PubMed] [Google Scholar]
- 17.Hermanson G. Bioconjugate Techniques. Academic Press; San Diego: 1996. [Google Scholar]
- 18.Lees RS, Garabedian HD, Lees AM, Schumacher DJ, Miller A, Isaacsohn JL, Derksen A, Strauss HW. J. Nucl. Med. 1985;26:1056–1062. [PubMed] [Google Scholar]
- 19.Glickson JD, Lund-Katz S, Zhou R, Choi H, Chen IW, Li H, Corbin I, Popov AV, Cao W, Song L, Qi C, Marotta D, Nelson DS, Chen J, Chance B, Zheng G. Mol. Imaging. 2008;7:101–110. [PubMed] [Google Scholar]
- 20.Jasanada F, Urizzi P, Souchard J-P, Le Gaillard F, Favre G, Nepveu F. Bioconjugate Chem. 1996;7:72–81. doi: 10.1021/bc950073l. [DOI] [PubMed] [Google Scholar]
- 21.Krieger M, Smith LC, Anderson RG, Goldstein JL, Kao YJ, Pownall HJ, Gotto AM, Brown MS. J. Supramol. Str. 1979;10:467–478. doi: 10.1002/jss.400100409. [DOI] [PubMed] [Google Scholar]
- 22.Jin H, Lovell JF, Chen J, Ng K, Cao W, Ding L, Zhang Z, Zheng G. Photochem. Photobiol. Sci. 2011;10:810–816. doi: 10.1039/c0pp00350f. [DOI] [PubMed] [Google Scholar]
- 23.Bijsterbosch MK, Manoharan M, Dorland R, Waarlo IH, Biessen EA, van Berkel TJ. Biochem. Pharmacol. 2001;62:627–633. doi: 10.1016/s0006-2952(01)00705-5. [DOI] [PubMed] [Google Scholar]
- 24.Nikanjarn M, Gibbs AR, Hunt A, Budinger TF, Forte TM. J. Control. Release. 2007;124:163–171. doi: 10.1016/j.jconrel.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Su H-T, Li X, Liang D-S, Qi X-R. Oncotarget. 2016;7:51535–51552. doi: 10.18632/oncotarget.10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JY, Kim JH, Bae KH, Oh MH, Kim Y, Kim JS, Park TG, Park K, Lee JH, Nam YS. Small. 2015;11:222–231. doi: 10.1002/smll.201303277. [DOI] [PubMed] [Google Scholar]
- 27.Jin H, Chen J, Lovell JF, Zhang Z, Zheng G. Isr. J. Chem. 2012;52:715–727. [Google Scholar]
- 28.Xiao W, Wang L, Ryan JM, Pater A, Liu H. Radiat. Res. 1999;152:250–256. [PubMed] [Google Scholar]
- 29.Corbin IR, Li H, Chen J, Lund-Katz S, Zhou R, Glickson JD, Zheng G. Neoplasia. 2006;8:488–498. doi: 10.1593/neo.05835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu SP, Lee I, Ghoroghchian PP, Frail PR, Zheng G, Glickson JD, Therien MJ. Bioconjugate Chem. 2005;16:542–550. doi: 10.1021/bc0497416. [DOI] [PubMed] [Google Scholar]
- 31.Hill ML, Corbin IR, Levitin RB, Cao W, Mainprize JG, Yaffe MJ, Zheng G. Acad. Radiol. 2010;17:1359–1365. doi: 10.1016/j.acra.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Chu AC, Tsang SY, Lo EH, Fung KP. Life Sci. 2001;70:591–601. doi: 10.1016/s0024-3205(01)01441-2. [DOI] [PubMed] [Google Scholar]
- 33.Radwan AA, Alanazi FK. Molecules. 2014;19:13177–13187. doi: 10.3390/molecules190913177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian J, Xu S, Deng H, Song X, Li X, Chen J, Cao F, Li B. Int. J. Pharm. 2017;517:25–34. doi: 10.1016/j.ijpharm.2016.11.030. [DOI] [PubMed] [Google Scholar]
- 35.Zhou M, Wang T, Hu Q, Luo Y. Food Hydrocolloids. 2016;57:20–29. [Google Scholar]
- 36.Reynolds L, Mulik RS, Wen X, Dilip A, Corbin IR. Nanomedicine. 2014;9:2123–2141. doi: 10.2217/nnm.13.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mulik RS, Bing C, Ladouceur-Wodzak M, Munaweera I, Chopra R, Corbin IR. Biomaterials. 2016;83:257–268. doi: 10.1016/j.biomaterials.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tauchi Y, Zushida I, Yokota M, Chono S, Sato J, Ito K, Morimoto K. Biol. Pharm. Bull. 2000;23:466. doi: 10.1248/bpb.23.466. [DOI] [PubMed] [Google Scholar]
- 39.Tauchi Y, Zushida I, Chono S, Sato J, Ito K, Morimoto K. Biol. Pharm. Bull. 2001;24:925–929. doi: 10.1248/bpb.24.925. [DOI] [PubMed] [Google Scholar]
- 40.Wang R, Gu X, Zhou J, Shen L, Yin L, Hua P, Ding Y. J. Control. Release. 2016;235:134–146. doi: 10.1016/j.jconrel.2016.05.055. [DOI] [PubMed] [Google Scholar]
- 41.Meng F, Asghar S, Gao S, Su Z, Song J, Huo M, Meng W, Ping Q, Xiao Y. Colloids Surf. B: Biointerfaces. 2015;134:88–97. doi: 10.1016/j.colsurfb.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 42.Huang J-L, Jiang G, Song Q-X, Gu X, Hu M, Wang X-L, Song H-H, Chen L-P, Lin Y-Y, Jiang D, Chen J, Feng J-F, Qiu Y-M, Jiang J-Y, Jiang X-G, Chen H-Z, Gao X-L. Nat. Commun. 2017;8:15144. doi: 10.1038/ncomms15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajora MA, Ding L, Valic M, Jiang W, Overchuk M, Chen J, Zheng G. Chem. Sci. 2017;8:5371–5384. doi: 10.1039/c7sc00732a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J. CA Cancer J. Clin. 2011;61:250–281. doi: 10.3322/caac.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Marotta DE, Kim S, Busch TM, Wileyto EP, Zheng G. J. Biomed. Opt. 2005;10:41203. doi: 10.1117/1.2011429. [DOI] [PubMed] [Google Scholar]
- 46.Song L, Li H, Sunar U, Chen J, Corbin I, Yodh AG, Zheng G. Int. J. Nanomed. 2007;2:767–774. [PMC free article] [PubMed] [Google Scholar]
- 47.Marotta DE, Cao W, Wileyto EP, Li H, Corbin I, Rickter E, Glickson JD, Chance B, Zheng G, Busch TM. Nanomedicine. 2011;6:475–487. doi: 10.2217/nnm.11.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin H, Lovell JF, Chen J, Lin Q, Ding L, Ng KK, Pandey RK, Manoharan M, Zhang Z, Zheng G. Bioconjugate Chem. 2012;23:33–41. doi: 10.1021/bc200233n. [DOI] [PubMed] [Google Scholar]
- 49.Zhu C, Pradhan P, Huo D, Xue J, Shen S, Roy K, Xia Y. Angew. Chem., Int. Ed. 2017;56:10399–10402. doi: 10.1002/anie.201704674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu C, Huo D, Chen Q, Xue J, Shen S, Xia Y. Adv. Mater. 2017 doi: 10.1002/adma.201703702. [DOI] [PMC free article] [PubMed] [Google Scholar]