

Abstract
In vitro models of the neuromuscular junction (NMJ) are emerging as a valuable tool to study synaptogenesis, synaptic maintenance, and pathogenesis of neurodegenerative diseases. Many models have previously been developed using a variety of cell sources for skeletal muscle and motoneurons. These models can advanced by integrating beneficial features of the native developmental milieu of the NMJ. We created a functional in vitro model of NMJ by bioreactor cultivation of transdifferentiated myocytes and stem cell-derived motoneurons, in the presence of electrical stimulation. In conjunction with a coculture medium, electrical stimulation resulted in improved maturation and function of motoneurons and myocytes, as evidenced by mature cellular structures, increased expression of neuronal and muscular genes, clusterization of acetylcholine receptors (AChRs) in the vicinity of motoneurons, and the response to glutamate stimulation. To validate the model and demonstrate its utility for pharmacological testing, we documented the potency of drugs that affect key pathways during NMJ signal transduction: (i) acetylcholine (ACh) synthesis, (ii) ACh vesicular storage, (iii) ACh synaptic release, (iv) AChR activation, and (v) ACh inactivation in the synaptic cleft. The model properly responded to the drugs in a concentration-dependent manner. We thus propose that this in vitro model of NMJ could be used as a platform in pharmacological screening and controlled studies of neuromuscular diseases.
Introduction
Neuromuscular junction (NMJ) is the synaptic connection between motoneurons and skeletal muscle. During development, NMJ forms in a series of elaborate signal interactions between axon terminals of motoneurons and plasma membrane of muscle, requiring intricate communication of cytokines, growth factors and cellular structures1, 2. Neuromuscular transmission is triggered when an action potential (AP) propagates to an axon terminal, causing influx of calcium ions through voltage-gated calcium channels and the release of neurotransmitter acetylcholine (ACh) from synaptic vesicles. ACh diffuses though synaptic cleft and activates nicotinic acetylcholine receptors (AChRs) on the plasma membrane of skeletal muscle, leading to AP and eventually to muscle contraction.
NMJ is widely used as a model to investigate synaptogenesis and pathophysiological development of neurodegenerative diseases3-6 because of its relatively large size and accessibility compared to central synapses7, 8. Many research groups have created in vitro coculture systems using combinations of cell lines, primary cells, and stem cell-derived neural and muscle cells. In particular, NMJ models were established in a serum-free culture of rat primary cells9, coculture of human stem cell-derived motoneurons and rat skeletal muscle10, low-density coculture of mouse stem cell-derived motoneurons and muscle cells11, and coculture of motoneurons and myotubes generated from the same line of human induced pluripotent stem cells12. However, the current models have not incorporated the environmental factors intrinsic to the native physiological niche of NMJ such as electrical stimulation. Also, the utility of the in vitro models of NMJ for investigating the pharmacological potency of drugs affecting synaptic communication has not been demonstrated.
We hypothesized that the provision of biomimetic environment guiding the organized development of motoneurons and myocytes would improve cell maturation and function during NMJ formation. To test this hypothesis, we integrated electrical stimulation into the co-culture of motor neuron and skeletal muscle cells. Using drug-induced contraction frequency of the coculture to quantify the drug potency, we demonstrated that the proposed NMJ model has utility in studying the pharmacological potency of the drugs that affect mechanisms involved in neuromuscular transmission. This simple, yet effective, coculture system can be used for pharmacological screening and to enhance the knowledge of pathogenesis of neurodegenerative diseases.
Materials and methods
Motoneuron derivation
Motoneurons used in the study were differentiated from an ES cell line (HBG3) derived from a transgenic mouse line (mHB9-Gfp1b), in which enhanced green fluorescent protein (eGFP) is expressed under the control of the mouse HB9 promoter. ES cell-derived motoneurons were obtained using methods described previously13. Briefly, HBG3 ES cells were grown on MEFs in ES cell medium (detailed compositions of each medium are provided in Supplemental Experimental Procedures). After 2 days, ES cell colonies were partially dissociated and cultured in DFK5 medium. Medium was replaced after 2 days and supplemented with 1 μM retinoic acid (Sigma), 300 nM Sonic hedgehog (Shh, R&D Systems), and 1 μM hedgehog agonist (R&D Systems). EBs were cultured for 4–5 days before being dissociated and sorted using magnetic-activated cell sorting by cluster of differentiation 2 (CD2), which was engineered to express under the HB9 promoter. Subsequently, dissociated cells were seeded at a density of 750 cells/mm2 in 24-well plates double-coated with poly-L-ornithine (Sigma) and laminin (Sigma) and cultured in CM coculture medium.
Magnetic-activated cell sorting
Upon dissociation and wash with L15 medium consisting of L15 solution (Sigma), 50 μM EDTA, 4% BSA (Sigma), 25 mM glucose, 2% Horse Serum (Sigma), 40 μg/mL DNase, 500 μg/ML Insulin (Sigma), 0.01M Putrecine (Sigma), 10 mg/mL Conalbumin (Sigma) and 30 μM Sodium Selenite (Sigma), 15 × 106 dissociated cells were incubated for 20 minutes at 4 °C in 80 μL of L15 medium supplemented with 10 μL of anti-rat CD2 antibody (Invitrogen). After washing again with L15 medium, cells were then incubated with anti-mouse magnetic microbeads (Miltenyi Biotec) for 20 minutes at 4 °C. Finally, cells were passed through a magnetic column to separate CD2-GFP-positive cells. Post-sorting, the cells were collected in motor neuron medium and cultured in well plates.
Direct reprogrammed skeletal myocytes from MEFs
Directly reprogrammed myocytes from MEFs used in the experiment were obtained using previously described methods14. Briefly, MEFs were initially cultured in high-glucose DMEM supplemented with 10% fetal bovine serum (GIBCO) until reaching 70% - 90% confluence. Lentiviral gene delivery system was used to stably transduce cells with rtTA-M2 and MyoD genes. To improve transduction, hexadimethrine bromide (Sigma) was added to the medium at a concentration of 8 μg/mL. After transduction, doxycycline (Dox) (Sigma) at a concentration of 3 μg/mL was added to MIX differentiation medium containing 2% house serum in high-glucose DMEM. Cells were cultured in the differentiation medium for 12 days.
Electrical stimulation
Polydimethylsiloxane (PDMS) (Dow Corning) was used to fabricate bioreactor cassettes designed with 5 rows of 6 culture wells, 1 cm × 1 cm in size. Carbon rods (0.120 inch in diameter, Ladd Research) spanned the two ends of each row, such that the four wells in each row received the same stimulation. The bioreactor was sealed to a 10 cm × 15 cm glass (Thermo Fisher) using PDMS. For electrical stimulation of motoneurons and muscle/motorneuron cocultures, stimulation plates were constructed from 12-well plates inserted with 4 pairs of carbon rods, such that three wells in each row received the same electrical stimulation. Carbon electrodes were spaced 1 cm apart for both bioreactors. Before plating cells, the stimulation plates were coated with poly-L-ornithine and laminin. After 2 days of pre-culture of motoneurons and transduction of myocytes, carbon rods of bioreactors were connected to leads from an S88X Dual Output Square Pulse Stimulator (Grass Technologies) generating continuous pulses of 3 V, 1 Hz, and 2 ms duration. Myocytes were stimulated 6 hours per day for 10 days and motoneurons were stimulated 6 hours per day for 6 days before samples were collected for analysis. The control groups in the bioreactors did not receive electrical stimulation. For an additional control group in stimulation experiments with motoneurons, cells received electrical stimulation, but 0.5 μM of TTX was added to the culture medium. After each experiment, the bioreactor cassettes were thoroughly cleaned. Specifically, to avoid protein accumulation on the carbon electrodes, the bioreactor culture surface and electrodes were scrubbed with a soft brush and then washed with a water jet. Afterwards, the plates were soaked in 70% ethanol for 1-2 hours, dried in the hood and autoclaved.
Coculture of myocytes and motoneurons
Reprogrammed myocytes from MEFs were obtained by a slight modification of a previously established protocol14. Two days after transduction, cells cultured in differentiation media supplemented with Dox were electrically stimulated for 3 days, over 6 hours per day using pulses of 3 V, 1 Hz and 2 ms in duration. After 3 days, motoneurons were added to the culture and cells were cultured in the CMHS coculture media supplement with Dox for 1 day before initiating another round of electrical stimulation. The coculture was stimulated using the same regime of stimulation for 6 hours per day for 6 days before analysis.
Pharmacological study
Pharmacological testing was performed at the end of coculture. Drugs were prepared as stock solutions in water or 100% dimethylsulphoxide (DMSO) and sonicated if necessary to dissolve the powder. The stock solutions were further diluted to the appropriate concentration using coculture medium. The DMSO concentration in the culture wells with the highest drug concentration did not exceed 1%. Cocultures were exposed to neostigmine (Sigma) and pancuronium (Sigma) for 10 minutes, and to acetylethylcholine mustard hydrochloride (Sigma), BoT-A (List Biological Laboratories) and vesamicol (Sigma) for 12 hours before being stimulated by glutamate (Sigma) at final concentration of 0.75 mM. After 5 minutes, coculture activities were recorded for contraction analysis.
Immunostaining
For acetylcholine receptor (AChR) staining, cells were incubated with 5×10-8 M of α-bungarotoxin (Life Technologies) for 1.5 hours at 37 °C prior to washing and fixing. Cells were then washed with PBS, fixed in PBS containing 4% paraformaldehyde (Sigma) for 15 min, permeabilized using 0.1% Triton-X (Sigma) for 15 min, and blocked with 5% goat serum (Sigma) in PBS for 20 minutes. Cells were stained with primary antibodies for 2-3 hours using monoclonal anti-α-actinin (sarcomeric) antibody (1:400, Sigma). After 3 washes with PBS, cells were stained for one hour with the following secondary antibodies: goat anti-mouse Alexa Fluor 488 (1:500, Life Technologies), anti-GFP antibody (1:500, Abcam), Alexa Fluor 546 phalloidin (1:500, Life Technologies) for actin and 4’,6-Diamidino-2-Phenylindole (DAPI) (1:1000, Life Technologies) for nuclear staining.
Quantitative real-time polymerase chain reaction (qRT-PCR)
RNA was extracted from cells with TRIzol reagent (Life Technologies) and quantified for yield and purity using NanoDrop Spectrophotometer (Thermo Scientific). 10 ng was used for reverse transcription with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR was performed on the StepOnePlus Real-Time PCR system (Applied Biosystems). The following TaqMan Gene Expression Assays were used for detection of important muscular and motoneuron transcription factors: Agrn (Mm01545840_m1), Musk (Mm01346929_m1), Gdnf (Mm00599849_m1), Bdnf (Mm01334047_m1), Actn2(Mm00473657_m1), Chrnb1 (Mm00680412_m1), Ntf5 (Mm01701591_m1), Myog (Mm00446195_g1), Syn2 (Mm00449780_m1). TNNT3 (Mm01268863_m1), Ntrk2(Mm00435422_m1) and Myh15 (Mm01170207_m1). Gene expression values were normalized to Gapdh (Mm99999915_g1) expression and presented as an expression relative to the control group by the 2-ΔCt method15. All reactions were performed in triplicates.
Electrophysiology
Motoneurons were seeded on glass coverslips double-coated with poly-L-ornithine and laminin. Astrocytes were added to promote motoneuron survival and maturation. A few days after cell plating, motoneurons were identified based on GFP expression and action potentials recorded using whole-cell current-clamp technique. Patch pipettes were fabricated from 1.5 mm outer diameter, 1.28 mm inner diameter filamented capillary glass (World Precision Instruments) and had a 2-5 MΩ resistance when filled with the pipette solution described below. Current protocol generation and data acquisition were performed using pClamp10 software and a Digidata 1550 digital to analog converter (Molecular Devices). Traces were filtered at 10 kHz using a low-pass Bessel filter, and digitized at 10 kHz. All recordings were performed at room temperature and are corrected for a calculated liquid junction potential of -14 mV. Internal solution contained the following: 130 mM potassium-methanesulfonate, 10 mM Na-methanesulfonate, 10 mM HEPES, 10 mM EGTA, 1 mM CaCl2, 5 mM MgATP, and 0.3 mM NaGTP. pH was adjusted to 7.4 using KOH. External solution was Ringer’s solution containing 145 mM NaCl, 5 mM KCl, 10 mM HEPES, 2 mM glucose, 2 mM CaCl2, 2 mM MgCl2. pH was adjusted to 7.4 using NaOH. Osmolality adjusted to 325 mOsm using sucrose.
Calcium imaging
MEF-transdifferentiated myocytes and motoneurons were assessed for functionality and response to electrical stimulation using calcium imaging. Cells were stained with 5 μg/mL Fluo-4 AM (ThermoFisher) diluted in pre-warmed Tyrodes Solution (Sigma) for 30 minutes and washed twice with Tyrodes solution prior to imaging. Cells were electrically stimulated every 10 seconds by 2 stimuli of 2 millisecond duration and a gap of 300 milliseconds between the two stimuli. A concentric electrode (FHC, Inc) was positioned about 100 μm away from an area on interest, and the amplitude of the electrical signal was set to 0.5 V. For motoneuron stimulation study, Concentration of TTX was 0.5 μM. Concentrations of glutamate and nicotine used in the control experiment for the coculture were 0.1 mM and 10 μM, respectively. Videos were taken on the Olympus IX81 using the Pike camera at 100 frames per second via the SPLASSH software16. Videos were imported into a custom MATLAB R2016b software that allowed users to measure average fluorescence intensity in an area of interest for each frame, yielding calcium intensity over time.
Contraction frequency measurement
Contraction in coculture was recorded after drug exposure on the Olympus IX81 using the Pike camera at 75 frames per second via the SPLASSH software. The duration of a single recording was 30 seconds. At the beginning of each recording session, contrast and brightness of the video image were optimized and the heated stage was kept at 37 °C during video recording. Videos were processed by a custom MATLAB software that correlated the changes of brightness between subsequent video frames in the region of interest with the frequency of muscle contractions17.
Data analysis and statistics
Contraction frequency of the coculture was quantified using MATLAB R2016b. Gene expression levels and pharmacological potency were analyzed using GraphPad Prism 7. Results were represented as mean ± standard deviation. Statistical significance levels were set to p = 0.05.
Results
To form the NMJ (Figure 1), the motoneuron component was derived from mouse embryonic stem cells (ESCs)13 that were genetically engineered to express (i) green fluorescence protein (GFP) for easy identification in coculture, and (ii) cluster of differentiation 2 (CD2) for enabling magnetic-activated cell sorting (MACS). The muscle component was derived by direct reprogramming of mouse embryonic fibroblasts (MEFs) into myocytes18. Electrical stimulation was applied using carbon electrodes that delivered signals with a 3 V/cm amplitude at 1 Hz frequency and 2-millisecond duration for 6 hours per day. Cells were maintained in the coculture media for 7 days before analysis.
Figure 1. Experimental diagram.
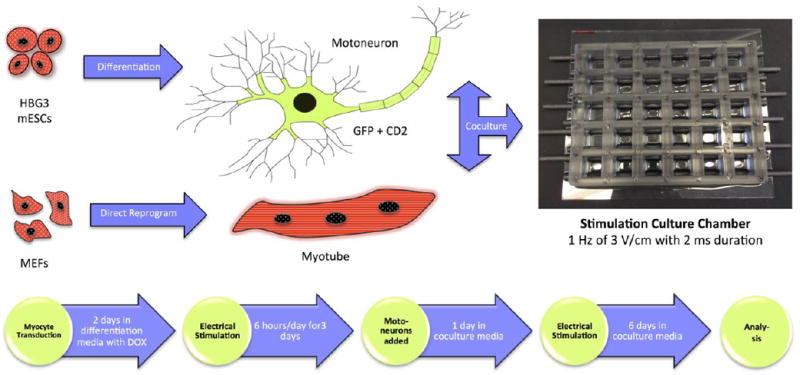
GFP+, CD2+ motoneurons derived from mouse ES cell line (HBG3) and myotubes transdifferentiated from MEFs were cocultured in a culture chamber integrated with electrical stimulation with the timeline shown above.
Mouse ESC-derived motoneurons functionally matured over 4 days in culture
Motoneurons used in the study were derived from HBG3 ESC line through the differentiation pathway shown in Figure 2A. Because GFP and CD2 were genetically engineered to express under the control of the motoneuron specific promoter HB9, they allowed simple identification of motoneurons in the culture and the use of MACS to isolate the cells. The functional maturity of motoneurons was assessed by whole-cell current clamp in co-culture with astrocytes (Figure 2B). Motoneurons were able to fire APs as early as on day 2 of the culture (Figure 2C), but these initial signals were weak and cells could not produce continuous trains of APs. By day 4, motoneurons could consistently generate a robust firing of APs, indicating functional development of motoneurons.
Figure 2. Derivation and characterization of motoneuron.
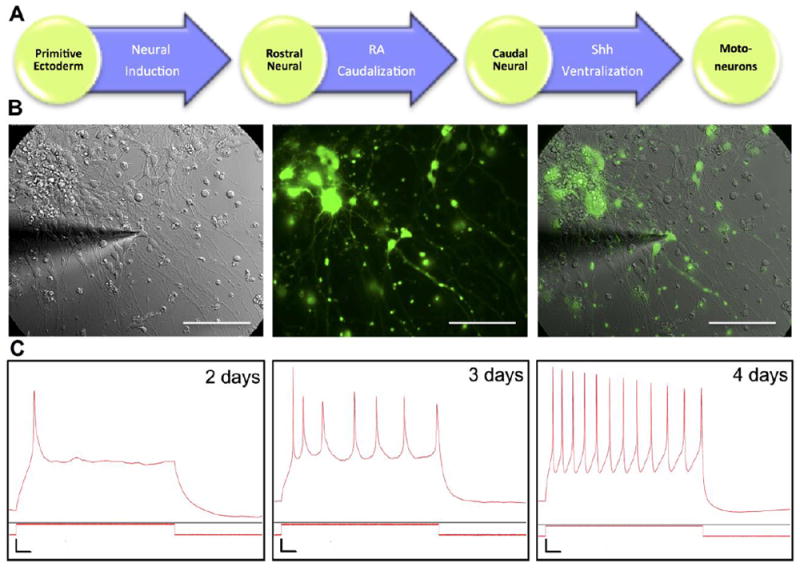
(A) Motoneurons were derived from ES cell line (HBG3) with GFP under HB9 promoter. Differentiation process started from neural induction of primitive ectoderm into rostral neural cells. Retinoic acid (RA) subsequently induced caudal neural fate, followed by Sonic hedgehog (Shh) signaling, converting cells to motoneurons. (B) Overlay of brightfield and fluorescence images of GFP+ motoneuron and astrocyte coculture during whole-cell current clamp experiment. Scale bar: 100 μm. (C) Representative traces of action potential (AP) measurements of motoneurons after a few days in culture. Weak single AP was observed on day 2. A weak train of APs was observed on day 3 and motoneurons were functionally mature on day 4 as evidenced by a robust firing of APs. Vertical scale bar: Vm = 10 mV, Im = 200 pA. Horizontal scale bar: 0.1 seconds.
Direct reprogramming of MEFs into myocytes resulted in increased muscular gene expressions and structurally and functionally matured myotubes
MyoD is a key regulatory factor in skeletal muscle and its ectopic overexpression induces transdifferentiation of MEFs into myocytes18, 19. MEFs were transduced with a Tet operator/CMV promoter-MyoD lentiviral construct (TetO/CMV-MyoD) and a lentivirus containing reverse tetracycline transactivator (rtTA-M2). The introduction of Dox into the culture activated rtTA-M2 to bind to TetO and initiate transcription of MyoD (Figure 3A).
Figure 3. Derivation and characterization of direct reprogramming of MEFs into skeletal myocytes.
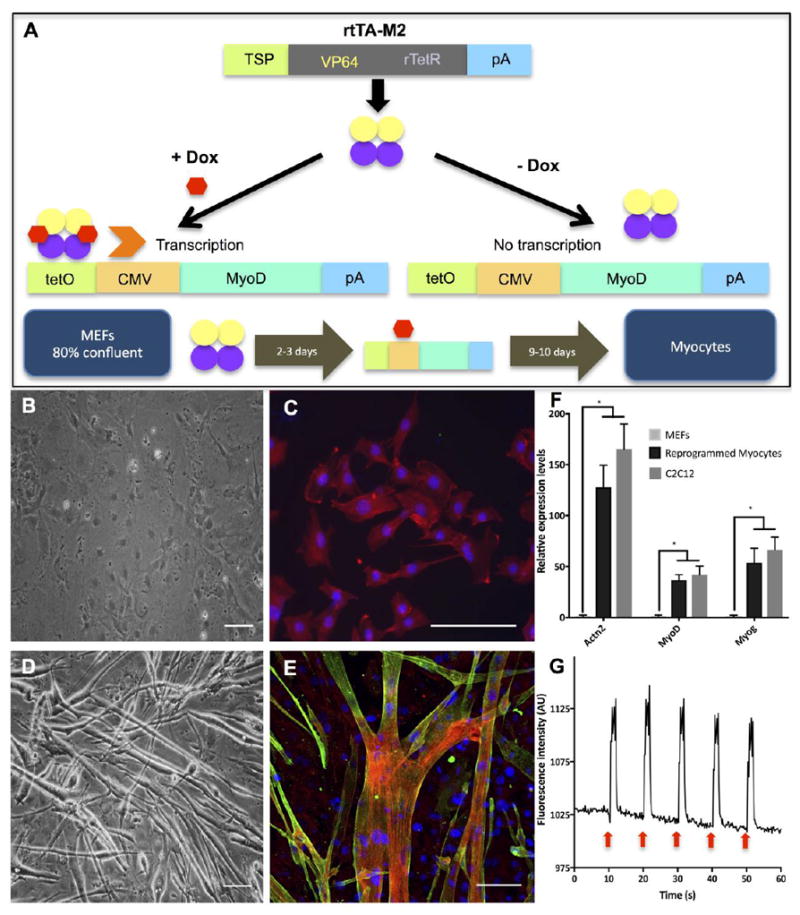
(A) The diagram showing rtTA-M2 lentiviral construct and a lentiviral construct containing TetO/CMV-MyoD. A tetracycline inducible system via rtTA-M2 was activated by doxycycline and initiated MyoD transcription. Overexpression of MyoD induced reprogramming of MEFs into skeletal myocytes. (B), (C) Brightfield and fluorescence images of MEFs after 12 days in MIX differentiation media. Actinin was not found in MEFs. Red, green and blue represent actin, actinin and DAPI, respectively. (D), (E) Brightfield and fluorescence images of MEF-transdifferentiated myocytes showed multinucleated myotubes and the presence of actinin protein after 12 days in MIX differentiation media. Red, green and blue represent actin, actinin and DAPI, respectively. Scale bar: 100 μm. (F) qRT-PCR analysis showed relative expressions of important muscular genes found in MEFs, reprogrammed myocytes and C2C12 myoblasts. Expression levels of each gene were normalized by the expression levels found in MEFs. Quantitative data presented as mean ± SD. Statistical significance was evaluated with the Student’s t-test (*p < 0.05, n = 5 biological replicates) (G) A diagram showing calcium transients of MEF-transdifferentiated myocytes being electrically stimulated every 10 seconds as shown by red arrows. Myocytes showed significant increase of fluorescence intensity corresponding to electrical stimulation. (See Supplementary Video S1)
At the end of differentiation process, we compared the morphology, protein and gene expressions levels of MEFs to those of MEF-transdifferentiated myocytes. MEFs had round shape (Figure 3B) and stained positive for actin (Figure 3C), while MEF-transdifferentiated myocytes fused to form elongated and multinucleated myotubes (Figure 3D) that showed the presence of actin and α-actinin, proteins found exclusively in muscle (Figure 3E). Direct reprogramming led to significantly higher expression levels of important muscular genes, including actinin, MyoD and Myogenin (Figure 3F), suggesting the effectiveness of the tetracycline inducible system and transdifferentiation. Gene expression levels in reprogrammed myocytes were comparable to those of C2C12 mouse myoblasts, while the expression of the same genes in the control MEFs was barely detectable (Figure 3F).
To demonstrate the functionality and maturity of MEF-transdifferentiated myocytes, muscle cells were electrically stimulated every 10 seconds using a concentric electrode with 2 stimuli of 0.5 V, 2 millisecond duration and 300 milliseconds between the two stimuli, and calcium transients were recorded. The spikes of intracellular calcium flux in reprogrammed myocytes assumed the frequency of electrical stimulation, indicating that myocytes derived from MEFs were excitable and functional (Figure 3G, Supplementary Video S1). The myotubes formed from reprogrammed myocytes also responded to chemical stimulation using nicotine (Supplementary Video S2). These results demonstrated that myocytes transdifferentiated from MEFs were structurally and functionally mature.
Electrical stimulation upregulated the expression of neuronal and muscular genes and improved the skeletal muscle morphology
After 6 days of electrical stimulation, expression neuronal genes in stimulated motoneurons was compared to the control group receiving no stimulation and to the motoneurons receiving stimulation in the presence of tetrodotoxin (TTX) in culture medium. qRT-PCR showed significant increases in the levels of brain-derived neurotrophic factor (BDNF) and tropomyosin receptor kinase B (Ntrk2) genes in the stimulated group relative to the control group of motoneurons. The fold change expressions of those genes were more than twice as high as expressions measured for the control group (Figure 4A). The levels of synapsin, a gene involved in the regulation of neurotransmitter release and neuromuscular synapse maturation20, also showed a trend of increase over levels measured for unstimulated neurons, but the difference was not statistically significant. The presence of TTX in the culture suppressed the effect of electrical stimulation on these genes.
Figure 4. Effects of electrical stimulation on motoneurons and skeletal myocytes.
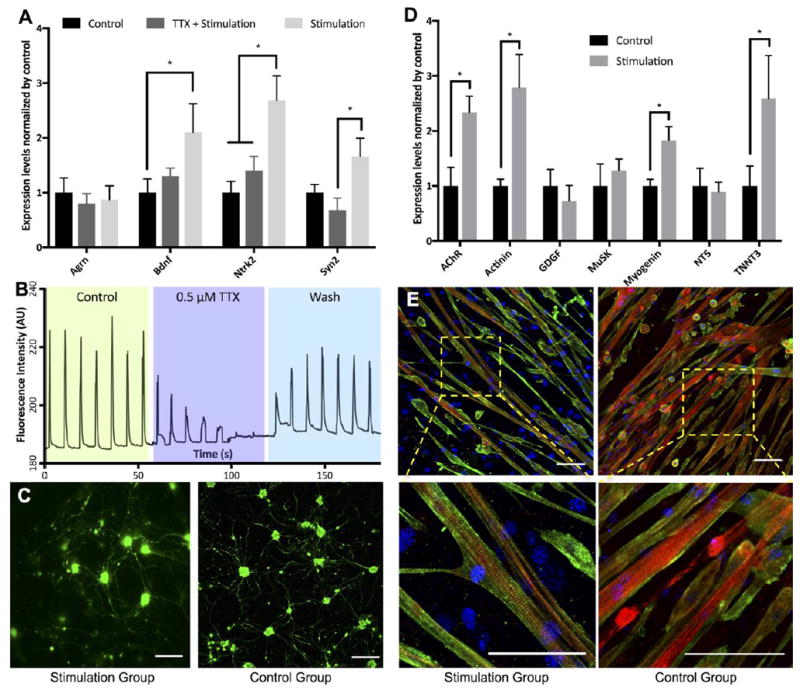
(A) qRT-PCR analysis showed normalized expression levels of important neuronal genes of motoneurons found in control, electrical stimulation with TTX in culture medium and electrical stimulation groups. (B) Calcium transients during electrical stimulation. The trace consisted of 3 × ~60 seconds recording spliced together. Two electrical stimuli of 0.2 seconds apart were delivered every 10 seconds. The first section was the control, second section was in the presence of TTX and the last section was after 5 minute wash. (C) Florescence images showed the morphology of GFP+ motoneurons in culture. (D) qRT-PCR analysis presented elevated relative expressions of important muscular genes of stimulated myocytes compared to those of control group after 12 days in culture. The expression levels of Chrnb1, Actn2, Myog and Tnnt3 found in stimulated myocytes were significantly increased compared to those of the control group. (E) Florescence images showed the morphology of myocytes in both study groups. Striated sarcomeres indicating mature myocytes were commonly found in myotubes from stimulation group. Red, green and blue represent actin, actinin and DAPI, respectively. Scale bar: 100 μm. Quantitative data presented as mean ± SD. Statistical significance was evaluated with the Student’s t-test (*p < 0.05, n = 5 biological replicates).
Calcium imaging revealed calcium transients during electrical stimulation in different conditions (Figure 4B). Motoneurons responded well to electrical stimulation as evidenced by calcium fluxes synchronizing with electrical stimulation. After adding TTX to the culture, calcium activities due to electrical stimulation were gradually blocked and they were completely suppressed after about 40 seconds. Following the washout of TTX and medium replacement, motoneurons responded to electrical stimulation at the pre-TTX treatment level. Fluorescence images showed comparable degrees of growth and axonal spreading of stimulated and unstimulated motoneurons (Figure 4C).
Electrical stimulation upregulated important muscular genes in myocytes, including AChR (Chrnb1), actinin (Actn2), myogenin (Myog) and troponin (Tnnt3) (Figure 4D). Expression levels of these genes were more than twice as high as in unstimulated myocytes. In addition, striated sarcomeres, indicators of skeletal muscle organization and maturation, were routinely found in myotubes formed from electrically stimulated myocytes after 10 days of culture (Figure 4E). Spontaneous contractions were observed by day 9 of culture for electrically stimulated myotubes, and only after more than 2 weeks of culture for unstimulated myotubes (Supplementary Videos S3, S4).
Clusterization of AChRs in the vicinity of motoneurons and glutamate-induced contractions indicate the formation of functional NMJ
After 8 days of coculture, motoneurons extended their axons to myotubes to make synaptic contacts with skeletal plasma membrane (Figure 5A). The muscle/motorneuron interactions resulted in the formation of NMJ and in muscle contractions (Supplementary Video S5). In addition, cells were immunolabeled to examine cellular and protein colocalization in the coculture. Myocytes were stained with α-actinin and α-bungarotoxin for actinin and AChRs, respectively. Motoneurons formed neuronal networks in coculture and AChRs aggregated near motoneurons, indicating maturation of AChR clusters and NMJ formation (Figure 5B)
Figure 5. NMJ formation and function.
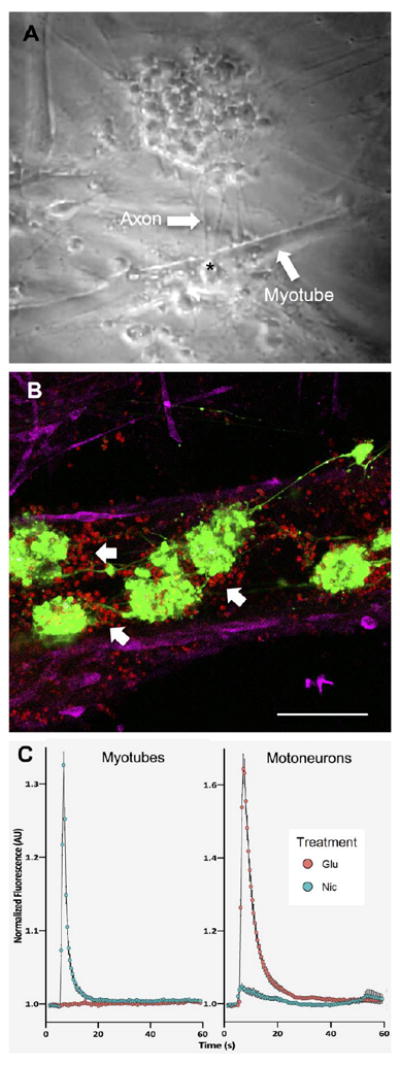
(A) A screenshot from a contraction video (See Supplementary Video S5) showed the interaction between myotubes and axons of motoneurons via NMJ after 8 days of coculture. An asterisk indicated a location of NMJ formation between axons of motoneurons and plasma membrane of myotubes. (B) Fluorescence image showed the localization of motoneurons and myotubes in coculture. Motoneurons formed a neuronal network and induced the clusterization of AChRs (showed by white arrows) in their vicinity, indicating properly developed myotubes and formation of NMJ. Red, green and magenta represent AChRs, motoneurons and actinin, respectively. Scale bar: 100 μm. (C) Normalized nicotine and glutamate-evoked calcium traces from calcium imaging on myotubes and motoneurons (See Supplementary Videos S7, S8). Nicotine could stimulate only myotubes while glutamate could trigger calcium flux only in motoneurons. Quantitative data presented as mean ± SD (n = 399 ROIs for myotubes and n = 105 ROIs for motoneurons).
To prove that the contractions were caused by synaptic transmission, the cocultured cells were stimulated using glutamate. Muscle contractions were observed shortly after adding glutamate to the system, suggesting that functional NMJ was formed (Supplementary Video S6). The contractions of myotubes could only have resulted from neurotransmitter release at the NMJ due to motoneuron activation by glutamate. Glutamate could not directly stimulate myotubes as shown by calcium traces (Figure 5C), calcium transient recordings (Supplementary Videos S7, S8), and contraction videos (Supplementary Videos S9, S10).
The NMJ model responded to drugs affecting neurotransmission in a dose-dependent manner
To demonstrate utility of the NMJ model, the cocultures were used to study the potency of pharmacological agents involved in NMJ signal transmission and function. To this end, we examined all five interconnected mechanisms including ACh synthesis, ACh vesicular storage, ACh synaptic release, AChR activation, and ACh inactivation in the synaptic cleft (Figure 6A). Different concentrations of drugs were added to the coculture, with a specific exposure time set for each drug, and contraction videos were recorded and analyzed.
Figure 6. Pharmacokinetic studies of various drugs affecting NMJ.
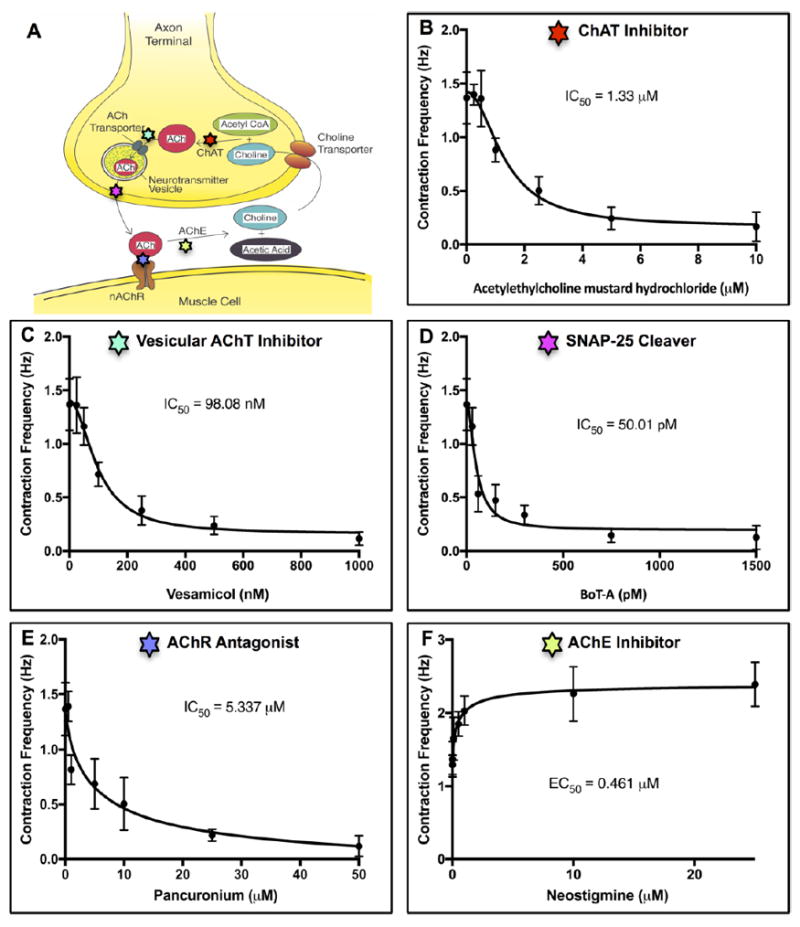
(A) A schematic shows the different cellular mechanisms involved in neuromuscular transmission. (B), (C), (D), (E), (F) Dose-dependent responses of coculture contraction frequency exposed to different drugs. (B) Acetylethylcholine mustard hydrochloride blocked the synthesis of acetylcholine (ACh) by irreversibly inhibiting choline acetyltransferase (ChAT). A half maximal inhibitory concentration (IC50) was 1.22 μM. (C) Vesamicol inhibited vesicular acetylcholine transporter (AChT) and prevented filling of synaptic vesicles with ACh. Its IC50 was 98.08 nM. (D) Botulinum toxin A (BoT-A) prevented ACh release by cleaving SNAP-25, a t-SNARE protein essential for fusion of neurotransmitter vesicles with the plasma membrane of motoneuron terminals. Its IC50 was 50.01 pM. (E) Pancuronium blocked neurotransmission by reversibly and competitively binding to acetylcholine receptors (AChRs). Its IC50 was 0.501 μM. (F) Neostigmine increased neuromuscular activities by inhibiting acetylcholinesterase (AChE). Its half maximal effective concentration (EC50) was 0.0461 μM. Quantitative data presented as mean ± SD, n = 6 biological replicates.
Dose–response curves exhibited concentration-dependent responses of NMJ to pharmacological agents. Specifically, acetylethylcholine mustard hydrochloride, a precursor for ethylcholine mustard aziridinium ion, blocked the synthesis of ACh and decreased the contraction frequency of the myotubes, with the half maximal inhibitory concentration (IC50) of 1.22 μM (Figure 6B). Vesamicol, a vesicular acetylcholine transporter (AChT) inhibitor, prevented filling of synaptic vesicles with ACh and inhibited neuromuscular transmission, with IC50 of 98.08 nM (Figure 6C). Botulinum toxin A (BoT-A) inhibited neurotransmission by preventing ACh release. BoT-A cleaved SNAP-25, a t-SNARE protein essential for neurotransmitter vesicle fusion with the plasma membrane of motoneuron terminals, with IC50 of 50.01 pM (Figure 6D). Pancuronium interfered with NMJ function by reversibly and competitively binding to AChRs and thereby reducing the contraction frequency of skeletal muscle, with IC50 of 0.501 μM (Figure 6E). Neostigmine enhanced neuromuscular activities by inhibiting acetylcholinesterase (AChE), a potent enzyme that catalyzed ACh, and terminated synaptic transmission, with the half maximal effective concentration (EC50) of 0.0461 μM (Figure 6F). Similar experiments were performed to assess the potency of these drugs using motoneurons derived from mouse ESCs and skeletal muscle from mouse myoblast cell line (C2C12) (Supplementary Table S1). The data were used to compare the potency values between the two cocultures.
Discussion
The goal of the present study was to create a stable and functional in vitro model of NMJ for physiologic and pharmacologic studies. To this end, we established a protocol for co-culturing motoneurons and myocytes with the application of developmentally relevant electrical signals. Over just 12 days of bioreactor cultivation, electrical simulation led to improved morphological development, upregulated expression of neuronal and muscular genes, the aggregation of AChRs in the vicinity of motoneurons, and the response of the coculture to glutamate stimulation. In this model, we used drug-induced contraction frequency of the muscle to quantify drug potency. As expected, the model responded to the drugs involved in different pathways during synaptic communication in a concentration-dependent manner.
Motoneurons derived from HBG3 ESC line were genetically engineered to express GFP for easy cell tracking and CD2 to allow the use of MACS for cell purification. CD2, a surface antigen of the T-lymphocytes, was chosen because it is a well-described epitope that is immunologically tractable. CD2 is not trypsin sensitive, which is important since trypsin was used during dissociation of embryoid bodies (EBs). Moreover, CD2 is not expressed in neural cells21, which were the target cells to purify. Importantly, cells expressing CD2 have not showed any detectable phenotypes when compared to parental lines22. Under the conditions established in this study, motoneurons were functionally mature after only 4 days of culture, as indicated by robust firing of APs (Figure 2C). The conditions and duration of neural maturation were appropriate, because motoneurons would be mature by the middle of the experiment, allowing sufficient time for functional coupling of skeletal muscle and motoneurons during NMJ formation.
Direct reprogramming of MEFs into functional myocytes via a tetracycline inducible system proved to be an effective derivation process. Ectopic overexpression of MyoD was sufficient to induce transdifferentiation. Reprogrammed myocytes could form multinucleated myotubes with colocalization of actin and actinin, indicating mature sarcomere structure (Figure 3E). The expression levels of important muscular genes were benchmarked using myotubes formed from C2C12 mouse myoblasts (Figure 3F). A possible explanation for slight discrepancy between gene expression levels of reprogrammed myocytes and C2C12 groups was that there were some undifferentiated MEFs after the reprogramming process.
An advantage of direct reprogramming is that the technique not only allows autologous cell generation, but also bypasses the pluripotent ESC/induced pluripotent stem cell (iPSC) stage, minimizing the risk of teratoma. The transdifferentiation process takes shorter time compared to iPSC generation23. Direct reprogramming is, therefore, a viable option for cell derivation in regenerative and personalized medicine application.
Because myocytes and motoneurons are both electrically excitable and the development is closely linked to their electrical activity, it was hypothesized that the implementation of electrical stimulation will be beneficial for maturation and function of motoneurons and muscle cells. Many studies have shown that electrical stimulation can promote muscle cell maturation, orientation, and differentiation leading to greater contraction of myotubes24, and to benefit neural development by guiding axonal growth25, improving synaptic connectivity26, and enhancing neural regeneration and reinnervation27. An optimal electrical stimulation should attain a desirable physiological response with minimal damage to the stimulated tissue. The stimulating voltage of 3V was chosen as the amplitude that resulted in favourable stimulation effects without causing cell death that was observed at higher voltages (5V and 7V, Supplementary Figure S1). In addition, a frequency of 1 Hz and a pulse duration of 2 milliseconds were utilized because they were adequately long to depolarize cells and also sufficient for the double layers on stimulation electrodes to dissipate between subsequent pulses28. Monophasic square-wave pulses were chosen due to the simplicity and compatibility with carbon rod electrodes29. We confirmed that electrical stimulation leads to upregulation of motoneuron- and muscle-specific genes and improves cellular structures. Adding TTX, a voltage-gated sodium channel antagonist, to the culture medium of motoneurons diminished calcium transients induced by electrical stimulation and abolished the effects of electrical stimulation on important neuronal gene expressions. Notably, myotubes exhibited striated sarcomeres and started to contract after 9 days of electrical stimulation, compared to up to 3 weeks for unstimulated cells18. Consistently, striations appeared sooner in stimulated myotubes than in the control group (8 days vs. 13 days, Supplementary Figure S2). A possible explanation for the beneficial effects of electrical stimulation is that the sub-threshold stimulation provided electrical activities that were absent in an in vitro culture environment, but needed in the activity-dependent development of motoneurons and myotubes.
This study is the first to create a functional in vitro NMJ model using ESC-derived motoneurons and MEF-transdifferentiated myocytes, and to demonstrate potential application of the NMJ model for testing potency of pharmacological agents that affect key mechanisms of neurotransmission. The coculture effectively captured the drug-induced response and functionality during signal transduction, and it exhibited a dose-dependent response to the applied drugs. The model was also very sensitive to drug concentrations as it could detect the concentration less than 1 nM in case of BoT-A. However, comparing the potency results from the NMJ model to the values found in literature was challenging because there are not many available data of drug potency testing in in vitro models and the data obtained from other models still have sizeable ranges of potency values. In general, the results were within the cited ranges and when compared to the results obtained from an in vitro model using myotubes derived from C2C12 and ESC-derived motoneurons (Supplementary Table S1), potency values were comparable.
Limitations of the study include the use of mouse cells, which might not adequately represent human native environment. The results from mouse models, therefore, might not be accurately predictive of phenomena or responses found in human tissues. Potential factors contributing to possible discrepancy include interspecies variations in gene expression, anatomical and physiological differences.
Nonetheless, mouse models should be able to recapitulate the general characteristics of NMJ and provide valuable insights regarding synaptogenesis and neuromuscular transmission. To better mimic the intrinsic NMJ milieu, other beneficial factors in addition to electrical stimulation can be incorporated, for example, mechanical stimulation, topological manipulation and substrate stiffness. Even though 2D culture offers simple and high-throughput platform for drug testing, 3D in vitro models can encompass these additional features to further replicate the in vivo mechanical and chemical microenvironment capable of facilitating cell maturation and neuromuscular function30-32. With improved physiological relevance, biomimetic 3D NMJ models, therefore, can potentially provide optimal platforms for drug screening.
In summary, we established an in vitro NMJ model that incorporated electrical stimulation and performed systematic analysis to show the formation and function of the NMJ. The model provides a simple and robust method for analyzing pharmacological potency using drug-induced contraction frequency to quantify cell response to different concentration of applied drugs. The proposed NMJ model also offers application in therapeutic agent screening, cytotoxicity testing, and disease modelling to study the relative roles of motoneuron and muscle in neurodegenerative diseases.
Supplementary Material
Insight, innovation, integration.
We created a stable and functional in vitro model of neuromuscular junction (NMJ) using reprogrammed myocytes and stem cell-derived motoneurons. Electrical stimulation was introduced to the muscle/neuron culture to mimic the native NMJ niche, where electrical signals are vital for NMJ development and function. Electrical stimulation enhanced cell morphology, expression of important muscular/neuronal genes, and the formation and function of NMJ. To validate the model, we examined drugs that affect the individual steps of neurotransmission, and quantified the dose-dependent drug responses. The optimized coculture medium implemented in conjunction with electrical stimulation resulted in a biomimetic model that effectively captured the critical characteristics and functions of the NMJ.
Acknowledgments
We thank Barbara Corneo, Dosh Whye, and Damian Williams from Columbia Stem Cell Core Facility for expert help with motoneuron derivation and electrophysiology; Syandan Chakraborty from Professor Kam Leoung’s lab for help with reprogramming of MEFs into myocytes; Theresa Swayne from Herbert Irving Comprehensive Cancer Center for help with confocal & specialized microscopy, and members of the Laboratory for Stem Cells and Tissue Engineering for advice and feedback. We gratefully acknowledge funding support of this work by NIH (grant EB002520).
Footnotes
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/x0xx00000x
References
- 1.Bloch-Gallego E. Cellular and molecular life sciences : CMLS. 2015;72:1029–1043. doi: 10.1007/s00018-014-1768-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferraro E, Molinari F, Berghella L. J Cachexia Sarcopenia Muscle. 2012;3:13–23. doi: 10.1007/s13539-011-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyer JG, Ferrier A, Kothary R. Frontiers in physiology. 2013;4:356. doi: 10.3389/fphys.2013.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez-Freire M, de Cabo R, Studenski SA, Ferrucci L. 2014 doi: 10.3389/fnagi.2014.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marteyn A, Maury Y, Gauthier MM, Lecuyer C, Vernet R, Denis JA, Pietu G, Peschanski M, Martinat C. Cell Stem Cell. 2011;8:434–444. doi: 10.1016/j.stem.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida M, Kitaoka S, Egawa N, Yamane M, Ikeda R, Tsukita K, Amano N, Watanabe A, Morimoto M, Takahashi J. Stem cell reports. 2015;4:561–568. doi: 10.1016/j.stemcr.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanes JR, Lichtman JW. Annual review of neuroscience. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 8.Shi L, Fu AK, Ip NY. Trends in neurosciences. 2012;35:441–453. doi: 10.1016/j.tins.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Das M, Rumsey JW, Bhargava N, Stancescu M, Hickman JJ. Biomaterials. 2010;31:4880–4888. doi: 10.1016/j.biomaterials.2010.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo X, Das M, Rumsey J, Gonzalez M, Stancescu M, Hickman J. Tissue Eng Part C Methods. 2010;16:1347–1355. doi: 10.1089/ten.tec.2010.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Umbach JA, Adams KL, Gundersen CB, Novitch BG. PloS one. 2012;7:e36049. doi: 10.1371/journal.pone.0036049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demestre M, Orth M, Föhr K, Achberger K, Ludolph A, Liebau S, Boeckers T. Stem cell research. 2015;15:328–336. doi: 10.1016/j.scr.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Wichterle H, Lieberam I, Porter JA, Jessell TM. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 14.Chakraborty S, Christoforou N, Fattahi A, Herzog RW, Leong KW. PloS one. 2013;8:e64342. doi: 10.1371/journal.pone.0064342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmittgen TD, Livak KJ. Nature protocols. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 16.Sun R, Bouchard MB, Hillman EM. Biomedical optics express. 2010;1:385–397. doi: 10.1364/BOE.1.000385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drexler B, Seeger T, Grasshoff C, Thiermann H, Antkowiak B. Toxicology letters. 2011;206:89–93. doi: 10.1016/j.toxlet.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Chakraborty S, Ji H, Kabadi AM, Gersbach CA, Christoforou N, Leong KW. Stem cell reports. 2014;3:940–947. doi: 10.1016/j.stemcr.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bichsel C, Neeld D, Hamazaki T, Chang L-J, Yang L-J, Terada N, Jin S. Cellular reprogramming. 2013;15:117–125. doi: 10.1089/cell.2012.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu B, Czernik A, Popov S, Wang T, Poo M-M, Greengard P. Neuroscience. 1996;74:1087–1097. doi: 10.1016/0306-4522(96)00187-x. [DOI] [PubMed] [Google Scholar]
- 21.Sewell WA, Brown MH, Dunne J, Owen MJ, Crumpton MJ. Proceedings of the National Academy of Sciences. 1986;83:8718–8722. doi: 10.1073/pnas.83.22.8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikiz B, Alvarez MJ, Ré DB, Le Verche V, Politi K, Lotti F, Phani S, Pradhan R, Yu C, Croft GF. Cell reports. 2015;12:335–345. doi: 10.1016/j.celrep.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelaini S, Cochrane A, Margariti A. Stem Cells Cloning. 2014;7:19–29. doi: 10.2147/SCCAA.S38006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondo H, Hashimoto S, Yamasaki K, Ono K, Okada M, Fujisato T, Kobayashi H, Mochizuki S, Ohsuga M, Yoshiura M. 2008 [Google Scholar]
- 25.Yao L, Pandit A, Yao S, McCaig CD. Tissue Engineering Part B: Reviews. 2011;17:143–153. doi: 10.1089/ten.TEB.2010.0561. [DOI] [PubMed] [Google Scholar]
- 26.Kalb RG, Hockfield S. Brain research reviews. 1992;17:283–289. doi: 10.1016/0165-0173(92)90020-m. [DOI] [PubMed] [Google Scholar]
- 27.Udina E, Furey M, Busch S, Silver J, Gordon T, Fouad K. Experimental neurology. 2008;210:238–247. doi: 10.1016/j.expneurol.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Tandon N, Marsano A, Maidhof R, Wan L, Park H, Vunjak-Novakovic G. Journal of tissue engineering and regenerative medicine. 2011;5 doi: 10.1002/term.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tandon N, Cannizzaro C, Figallo E, Voldman J, Vunjak-Novakovic G. 2006 doi: 10.1109/IEMBS.2006.259747. [DOI] [PubMed] [Google Scholar]
- 30.Morimoto Y, Kato-Negishi M, Onoe H, Takeuchi S. Biomaterials. 2013;34:9413–9419. doi: 10.1016/j.biomaterials.2013.08.062. [DOI] [PubMed] [Google Scholar]
- 31.Cvetkovic C, Rich MH, Raman R, Kong H, Bashir R. Microsystems & Nanoengineering. 2017;3 doi: 10.1038/micronano.2017.15. micronano201715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larkin LM, Van Der Meulen JH, Dennis RG, Kennedy JB. In Vitro Cellular & Developmental Biology-Animal. 2006;42:75–82. doi: 10.1290/0509064.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.