

Abstract
Propionyl-CoA carboxylase (PCC) is the enzyme which catalyzes the carboxylation of propionyl-CoA to methylmalonyl-CoA and is encoded by the genes PCCA and PCCB to form a hetero-dodecamer. Dysfunction of PCC leads to the inherited metabolic disorder propionic acidemia, which can result in an affected individual presenting with metabolic acidosis, hyperammonemia, lethargy, vomiting and sometimes coma and death if not treated. Individuals with propionic acidemia also have a number of long term complications resulting from the dysfunction of the PCC enzyme. Here we present an overview of the current knowledge about the structure and function of PCC. We review an updated list of human variants which are published and provide an overview of the disease.
Keywords: Propionyl-CoA Carboxylase, Propionic acidemia, methylcitrate, 3-hydroxypropionate
INTRODUCTION
Propionyl-CoA carboxylase (PCC, E.C. 6.4.1.3) catalyzes the carboxylation of propionyl-CoA with bicarbonate producing methylmalonyl-CoA which is then converted to succinyl-CoA, an intermediate in the tricarboxylic acid cycle (TCA) (Figure 1A). Bi-allelic variants that diminish or destroy the function of the PCCA or PCCB subunits result in Propionic Acidemia (PA, OMIM 606054). PA is a devastating inborn error of metabolism which causes substantial morbidity and mortality [1]. This review aims to provide an in depth discussion of known structure, function, biochemistry and pathology of PCC.
Figure 1.
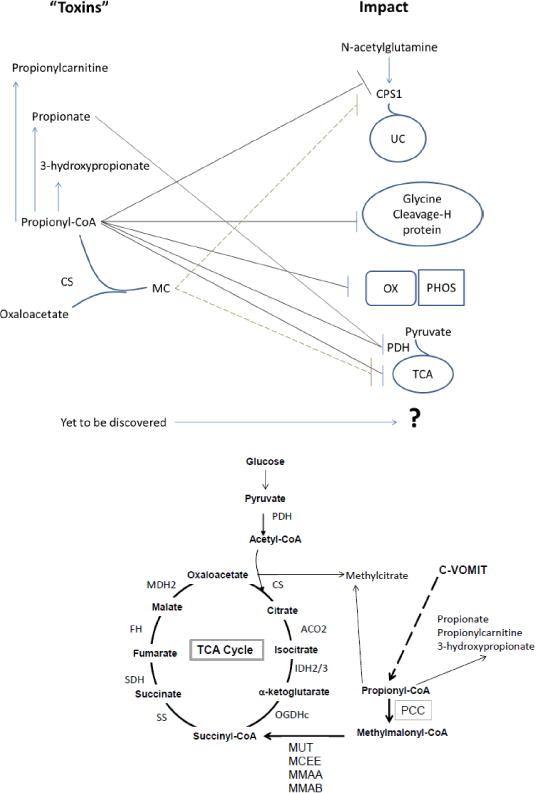
A. Diagram of TCA cycle and Propionate pathways with toxic intermediate sources PDH: pyruvate dehydrogenase complex, CS: citrate synthase, ACO2: mitochondrial aconitase, IDH2/3: mitochondrial isocitrate dehydrogenase 2 and 3 (NAD/NADP), OGDHc: 2- oxoglutarate dehydrogenase complex, C-VOMIT: cholesterol, odd chain fatty acids, methionine, isoleucine, threonine, PCC: propionyl-CoA carboxylase, MUT: methylmalonyl-CoA mutase, MCEE: methylmalonyl-CoA epimerase, MMAA: Cobalamin A, MMAB: Cobalamin B, SS: succinate synthase (succinyl-CoA ligase), SDH: succinate dehydrogenase, FH: fumarase, MDH2: mitochondrial malate dehydrogenase 2. B. Schematic illustrating current knowledge about the metabolic intermediates of dysfunctional PCC and their impact on other pathways. UC: urea cycle, MC: methylcitrate, CPS1: carbamoyl phosphate synthase 1, OX PHOS: oxidative phosphorylation system, PDH: pyruvate dehydrogenase complex.
MATERIALS AND METHODS
PubMed was searched for the term “Propionyl-CoA Carboxylase”. As of July 31, 2017, 689 papers were identified and reviewed for relevance. Additional papers were added to support the discussion as needed since they were not identified by the search term but clearly have a role in its dysfunction, for example enzymatic inhibition by 3-hydroxypropionic acid. Published variants were compiled from the literature and confirmed in ClinVar.
REVIEW
Propionyl-CoA Carboxylase Structure
PCC is 750 kDa heterododecamer composed of 6 propionyl-CoA carboxylase, alpha (PCCA, OMIM 232000) and 6 propionyl-CoA carboxylase, beta subunits (PCCB, OMIM 232050). Biotin is an obligate co-factor [2–5]. PCC is part of a subgroup of small acyl CoA carboxylases (YCC) which also includes acetyl CoA carboxylase, 3-methylcrotonyl-CoA carboxylase, and geranyl- CoA carboxylase [6]. This family of enzymes shares a common structure consisting of a biotin carboxylase domain (BC), carboxytransferase domain (CT), and biotin-carboxyl carrier protein (BCCP) domain (Figure 2) [6]. The structure of PCC appears to differ from that of other enzymes in the YCC family: PCC has a previously unrecognized component in the alpha subunit (PCCA), the biotin transfer, BT domain, formed by residues between the BC and BCCP. This segment is crucial for interactions with the beta subunit (PCCB) [7].
FIGURE 2.
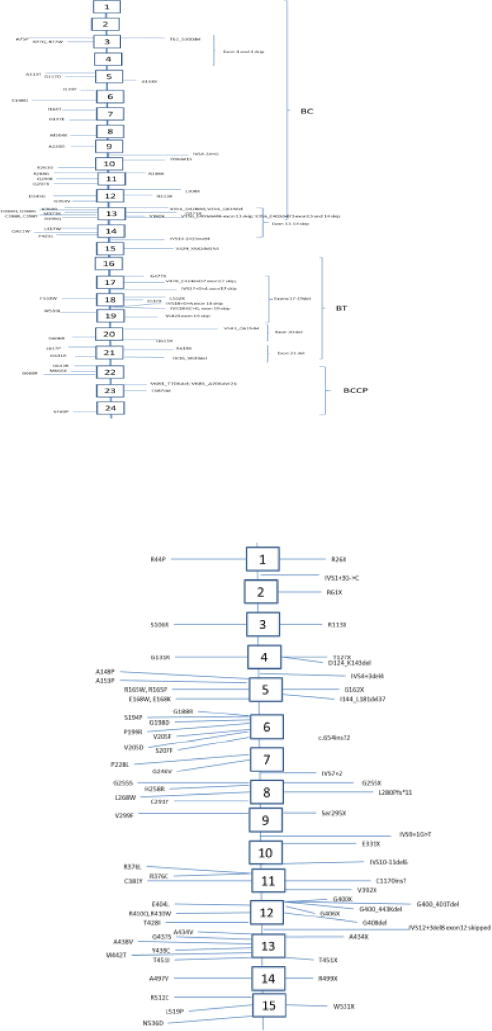
A. Schematic of PCCA variants which are also listed in Appendix 1 Regions of the protein are also delineated. The transcript used for exon-intronic boundaries was NM_000282.3 which is the longest known isoform. B: Schematic of PCCB variants, exons and introns as listed in Appendix 2. Intronic-exonic boundaries as defined by Rodriguez- Pombo [35]. Abbreviations: A: Alanine; R: arginine; N:asparagine; D: aspartic acid; C: cysteine; E: glutamic acid; Q: glutamine; G: glycine; H: histidine; I: isoleucine; L: leucine; K: lysine; M: methionine; F: phenylalanine; P: proline; T: threonine; Y: tyrosine; W: tryptophan; V: valine; X: frameshift or stop; BCCP: biotin-carboxyl carrier protein; BC: biotin carboxylase domain; BT: biotin transferase region
The PCC enzyme has been localized and is loosely bound to the inner membrane-matrix subcellular fraction [8]. It is often described as a matrix enzyme because it can dissociate with sonication.
The PCCA precursor does not contain biotin until imported into the mitochondrion and cleaved [2, 3, 9]. Biotin is loaded onto lysine 669 by holocarboxylase synthase (*60918; E.C. 6.3.4.10) and must be present for normal function [10, 11]. PCCA also contains 3 glycines and one valine which are conserved in other members of the YCC family of biotinylated mitochondrial enzymes [10]. PCCA may also be biotinylated in the cytosol but is likely not functional [10].
PCCB has also been crystalized and characterized [12]. PCCB requires PCCA for stability [13] and so is often absent in individuals who have no functional PCCA present. It also acts as a chaperonin to help assemble the human enzyme when studied in E. coli [11]. PCCB requires post-translational modification by Protease 1 that removes 7.5 kDa upon translocation into the mitochondrial matrix [14, 15].
Propionyl-CoA Carboxylase (PCC) Function
PCC’s primary function is to catalyze the carboxylation of propionyl-CoA to produce methylmalonyl-CoA (Figure 1A). Propionyl-CoA is produced by catabolism of cholesterol, valine, odd chain fatty acids, methionine, isoleucine and threonine (c-VOMIT) [2, 16–18]. Like other carboxylases, PCC is promiscuous and can carboxylate several other acyl-CoAs, but has greatest affinity for propionyl-CoA (Km = 0.29 mM). It catalyzes the reaction with acetyl-CoA at a rate about 1.5% of that of propionyl-CoA in experimental conditions [19]. The Km for bicarbonate, the usual source of the additional carboxylic group, is 3.0 mM [19]. Additionally, the catalysis requires ATP at a concentration of at least 3 mM [19].
Enzyme function requires biotin bound to biotin-carboxyl carrier protein (BCCP) in the active site. The biotin carboxylase domain (BC) first catalyzes the MgATP-dependent carboxylation on the N1 nitrogen of biotin [20, 21]. This step is readily reversible [16, 22]. The carboxy-biotin is then translocated to allow interaction with the carboxytransferase domain (CT), [6], after which the carboxyl group is transferred from biotin to the alpha-carbon of propionyl-CoA. Interestingly, the maximum arc of the N1 of biotin bound to a stationary BCCP is shorter than the distance between the BC and CT domains observed in holoenzymes, therefore BCCP has been proposed to be a swinging domain, also translocating in order to transport carboxy-biotin from BC to CT [6].
During the carboxylation process, hydrogen release is not in concert with CO2 binding through biotin, but rather concomitant with the formation of CO2 (through a carbanion). Thus, no base at the reaction center is needed [20, 21].
If biotin deficient, it takes 24 hours to restore activity following treatment with oral biotin in rats and so short trials of biotin are not useful to determine responsiveness [23]. Interestingly, the potentially toxic products of PCC deficiency, 3-hydroxypropionic acid (3OHPA) and methylcitrate (MC) are not good markers of biotin deficiency in humans [24].
As illustrated in PCCA-knock out mice that are rescued by a transgene directed to the liver, little enzyme activity is needed in the neonatal/infant period, and the need for functional PCC increases with age [25].
PCC activity may be assayed via several different methods. It can be measured in phytohemagglutinin-stimulated lymphocytes by high performance liquid chromatography (HPLC) by measuring amount of methylmalonyl-CoA produced from unlabeled propionyl-CoA [26]. In several types of tissues, enzyme assays look for the production of methylmalonyl-CoA or succinyl-CoA by non-radiometric assays using HPLC or radiometric assays following carboxylation by addition of radiolabeled CO2 [27–29]. Activity is inhibited by avidin and so the enzyme is difficult to isolate after death [30].
Human Variants in PA
Bi-allelic variants in PCCA or PCCB which abrogate enzyme function result in PA (OMIM 606054). Initial studies by Gravel et al. described complementation early (group A and BC) so 2 loci were suspected [31]. The genes PCCA and PCCB were later identified.
PCCA is located at 13q32.3 encoding a protein which is 72–80 kDa, having three splice isomers, but impact of these three isoforms to function has not been well characterized. Isoform A is the longest isoform (NM_000282.3) which has 729 amino acids. This is the typical isomer described and has 24 exons. Isoform B is shorter, contains 23 exons and 702 amino acids. Isoform C is shorter still, lacking an in-frame exon and so contains 23 exons and is 681 amino acids long. The biotin binding site is at the N-terminus [4] and the C-terminus is the location of the biotin carboxylase. An updated variant list may be found in Figure 2A and APPENDIX 1. PCCA null variants, such as R288X and S537X, result in the most severe phenotype [32]. Interpreting the severity of missense variants is difficult without expression assays since in some cases abnormal folding will lead to early degradation and loss of protein [32]. Splice site variants are also seen and in general, result in milder disease [33]. Of note, both N and C terminal of PCCA are necessary for holocarboxylase synthase interaction and variants in these domains can perturb this interaction and cause disease [34].
PCCB is located at 3q22.3 and encodes a protein of 58 kDa, 539 amino acids long encoded in fifteen exons (57 to 83 base pairs each) [35]. The currently published variants are listed in Figure 2B and Appendix 2 as well as the populations in which those variants have been seen. PCCB gene variants (A497V, R512C and L519P as well as W531X) appear to disturb the interaction between PCCA and PCCB, illustrating that these locations are important to this dimeric association [7, 36].
Unfortunately, phenotype-genotype correlation is hindered since most affected individuals are compound heterozygotes and so the severity of their PA becomes more complicated to predict [1, 37].
Dysfunction of PCC leads to secondary biochemical disturbances
Disturbances in the function of PCC lead to accumulation of propionyl-CoA (FIGURE 1A and B). In humans, it is difficult but possible (unpublished data) to detect elevations in propionyl- CoA. However, the levocarnitine ester of propionyl-CoA, propionylcarnitine (C3), is readily detectable in plasma, serum, and urine [38]. In addition, individuals have elevations in methylcitrate (MC) and 3-hydroxypropionate (3OHPA) which are usually assayed from urine clinically [39, 40]. MC is produced by conjugation of propionyl-CoA to the TCA intermediate oxaloacetate, a reaction thought to be catalyzed by citrate synthase (Figure 1A and 1B) [41]. 3OHPA is thought to be produced by beta-oxidation of propionic acid [39]. Intracellular accumulation of propionyl-CoA also inhibits mitochondrial metabolism and reduces the synthesis of citrate, GTP and ATP (Figure 1B) [42].
PCC participates in anaplerotic replenishment of TCA intermediates as its product, methylmalonyl-CoA, ultimately contributes to the succinyl-CoA pool (Figure 1A). In the brain, PCC has an important role in anaplerosis, given that the alpha-ketoglutarate pool contributes to production of GABA and glutamine. MR spectroscopy in humans with PA demonstrated decreased glutamine/glutamate which may suggest deamination of those compounds to supply TCA substrates and support TCA function [43].
Other than the direct contribution of PCC to anaplerosis, there is a complex interaction between PCC and the TCA cycle which is influenced by the tissue type and intermediate levels. For this reason, disturbance in one influences the other. This appears to not only be a consequence of potential “toxins” produced by a dysfunctional PCC impacting other enzymes such as pyruvate dehydrogenase (PDH) and alpha-ketoglutarate dehydrogenase (aKGDH or OGDH), but also direct impact of PCC dysfunction on TCA intermediate pool resulting in secondary effects on other pathways (Figure 1B). PCC dysfunction can alter alanine and aspartate aminotransferase activity, thus altering oxaloacetate, malate, and pyruvate concentrations [44], and secondarily the concentration of TCA intermediates.
Propionyl-CoA itself was showed to inhibit isolated aKGDH activity (Figure 1B) [45]. The muscle biopsies from individuals with PA also demonstrated decreased oxidation of pyruvate, malate and succinate, intermediates in the TCA cycle in addition to decreased enzyme activity from all the respiratory chain enzymes, COXI-IV (Figure 1B) [45]. Other studies have shown that propionate inhibits succinate ligase (GDP), another important TCA enzyme [46]. Methylcitrate has been implicated in dysfunction of citrate synthase and isocitrate dehydrogenase altering TCA cycle function [47].
Additionally, alterations in substrate availability may also affect TCA function. For example, carboxylation of propionyl-CoA is reduced in the absence of glutamate (which can be produced from alpha-ketoglutarate) in skeletal muscle. In heart muscle, a block in aKGDH resulting in increased glutamate leads to a decrease in contractile function and this is improved with contribution from the propionate pathway through PCC activity [48]. At rest, if acetyl-CoA is present, there is less of a contribution from PCC to TCA, but during exercise, there is an increase in PCC contribution to TCA [49]. Similarly, if glucose is available, the heart does not use PCC, but it can be induced to do so if fat or propionate is added to the system [44]. Glycine cleavage is decreased leading to elevations in plasma glycine thus the historical name: ketotic hyperglycinemia for PA. PA individuals have decrease H protein of the glycine cleavage system, which is suspected to be due to propionyl-CoA accumulation [50, 51]. Glycine cleavage may also be impacted by accumulation of other similar intermediates including isobutryl-CoA and 2-oxo-isovaleryl-CoA [52].
Propionyl-CoA inhibits the PDH (E.C. 1.2.4.1, 2.3.1.12, and 1.8.1.4), presumed to be due to alterations in propionyl-CoA/CoA ratios [42, 53]. Propionyl-CoA and propionate have been demonstrated to inhibit function of PDH in kidney, pig heart and liver respectively [45, 53–55]. Propionyl-CoA appears to directly inhibit carbamoyl phosphate synthase 1 (CPS1, E.C. 6.3.4.16) resulting in urea cycle dysfunction [53], but other studies implicate N-acetylglutamate synthase (NAGS, E.C. 2.3.1.1) as the cause [56, 57]. Elevated ammonium can also be seen in PCC models treated with methylcitrate [58]. Either CPS1 or NAGS dysfunction appears to be the cause of hyperammonemia [59]. Our laboratory has demonstrated the inhibition of CPS1/NAGS leading to high ammoniaand low urea in hepatocytes fromPA individuals [60].
PCC is highly expressed in rat brain and so accumulation of toxins is expected to cause cerebral damage [61]. For example propionic acid in rats have increases of free radicals and carbonylation in their striatum as well as it induces seizures [62]. Propionic acid in glia increases glutamine and so the glia may be at increased risk for damage. In the presence of propionic acid, cerebellar neurons and astrocytes increase histone acetylation, another potential source of injury [63].
Disruption of PCC leads to accumulation of odd-chain fatty acids (FA), as propionyl-CoA is the end product of beta oxidation of odd-numbered FA. Similarly, accumulation of propionyl-CoA may affect FA metabolism because it is the primer for the synthesis of odd-numbered FA [64]. In individuals with PA, pentadecanoic (C15:0), heptadecanoic (C17:0) and pentadecenoic (C15:1) acid can be assayed in plasma, are observed in erythrocyte membrane lipids, and their concentrations may serve as a biochemical marker of metabolic control, but currently are not used clinically [65, 66]. Of note, although odd chain fats are elevated, there are no differences in saturated and unsaturated FA in PA [67]. It has been noted that there is slower metabolism of pristanic acid by skin fibroblasts in cells from PA individuals [68]. As well, short chain FA like propionic acid upregulate fetal hemoglobin [69].
There are some additional impacts of accumulation of the “toxic” intermediates produced by dysfunction of PCC. Propionic acid, propionylcarnitine and MC can prevent normal function of the potassium channels of the heart (like KCNH2) resulting in delay re-polarization which can manifest as prolonged QTc [70]. This dysfunction has been postulated to explain the cardiac arrhythmias observed in PA. Elevated propionyl-CoA intermediates and oxidative stress molecules which impact TCA and oxidative phosphorylation function are also implicated in the cardiomyopathy seen [71, 72]. Other studies have shown that there appears to be tissue specific deficiencies in CoQ10 and carnitine [73, 74]. In addition, propionyl-CoA and propionylcarnitine are essential energy sources for the ischemic heart and so deficiency of succinyl-CoA from PCC dysfunction cannot be fully eliminated as a possible cause of cardiomyopathy [75, 76]. Improvement of cardiomyopathy following liver transplant which results in decreased circulating propionyl-CoA metabolites implies that the cause may be more than one of these possibilities [77, 78]. Finally PCC can help with purine metabolism during exercise by helping replenish the adenosine and guanosine stores needed to produce ATP and GTP through its role in cataplerosis [44].
Oxidative phosphorylation disruption
The mitochondrial respiratory chain oxidative phosphorylase enzymes (OXPHOS enzymes or cytochrome oxidases) have been demonstrated to have some dysfunction in explanted organs from individuals with PA [72]. In one of two PA individuals, assays of the activity of the OXPHOS enzymes in muscle, heart and liver showed decreased COXIII:citrate synthase and COXIV:citrate synthase ratios [72]. The second individual had normal ratios in all the COXs. The first PA individual also had decreased nitric oxide, superoxide anions, hydrogen peroxide, superoxide dismutase, and catalase levels in their fibroblasts. Liver from first PA individual had normal succinyl-CoA ligase (both GDP/ADP subunits), succinate dehydrogenase (SDH or COX2) and fumarase activity [72].
Inflammation and reactive oxygen species (ROS) appear to be elevated in individuals with PA and their tissues [71, 79–81]. ROS levels assayed in fibroblasts from individuals with PA after antioxidant use, showed some improvement. The antioxidant, tiron (scavenges superoxides) showed a decrease of 50–80% of ROS. Similarly, MitoQ (prevents disruption of mitochondrial function by acting as an antioxidant) decreased ROS by 25–50%. Trolox (a vitamin E analog which prevents lipid derived oxidant progression) also decreased ROS by 15–30%. In addition, resveratrol (inhibits lipid perioxidation) also showed a 30–40% decrease in ROS [79]. However, the traditional ROS scavengers including N-acetylcysteine (which re-generates glutathione) and melatonin (a direct free radical scavenger) were not successful in decreasing ROS implying that the best benefits are from stabilizers of mitochondrial function or preventers of lipid peroxidation and progression [79]. Moreover, free radical production in PA-derived fibroblast appears to be associated JNK and p38 signaling pathways [80]. These pathways may also be impacted by miRNAs which are dysregulated in PCC dysfunction further impacting the oxidative stress spiral [82]. Treatment of PA using currently available standard therapies decreases elevation of systemic inflammatory markers including di-tyrosine and isoprostanes [81].
Potential for Moonlighting
Moonlighting is the process by which a compound, in this case the PCC enzyme, does actions other than its major role. Several enzymes are thought to moonlight often by impacting transcription. These include the molybdenum co-factor enzyme mARC [83], holocarboxylase synthase [84], pyruvate kinase, lactate dehydrogenase, and succinate dehydrogenase [85]. PCCB mRNA accounts for 0.02% total hepatic mRNA [86] and this may reflect moonlighting roles. One such role is seen in the pancreas in which the PCCB precursor protein may interact with glucokinase for regulation of glycolysis [87]. Mineralocorticoid receptor interacts with PCCB precursor in the cytosol [88].
In the presence of dysfunctional PCC, there is increase in propionylation rather than acylation of lysines. Since acylation is important to the regulation of cellular processes, this may result in broader effects that are not directly related to the “toxic” intermediates or its impact on energy metabolism [89].
Propionic acidemia
Individuals with classical PA are usually asymptomatic at birth, but present in the first few hours to days of life due to symptoms of metabolic decompensation including poor feeding, vomiting, hyper- or hypotonia, temperature instability, irritability, and lethargy [90–92]. Laboratory abnormalities at presentation may reveal severe, persistent wide anion gap metabolic acidosis with ketosis, hyperlactatemia and hyperammonemia [90, 91, 93]. Since the clinical presentations of PA individuals are non-specific, mistreatment or inadequate treatment cause deterioration of metabolic decompensation and, if left untreated, lead to death.
As newborn screening has become more available, many individuals are being ascertained with elevated blood C3 (propionylcarnitine), often prior to clinical presentation. Newborn screening leads to early diagnosis and treatment, however, PA individuals, either diagnosed by newborn screening or those who survived from neonatal episode of metabolic decompensation, may still have many sequelae of the disorder [37, 94–97].
Neurodevelopmental sequelae are among the most important concerns for PA individuals and their families since more than 70% of the individuals are reported to have cognitive deficit and developmental delay in gross motor skill, fine motor skill and language skill [94]. Attention deficit-hyperactivity disorder and autism are also reported [94, 98]. Some individuals also suffered from acute psychotic episodes [99]. Neurological complications in individuals with PA include seizures of which generalized tonic-clonic is the most common type, metabolic stroke- like episode that mainly affects basal ganglia and optic nerve atrophy [100, 101]. Some individuals present with only movement disorders without metabolic decompensations [102].
Leukopenia is the most common hematologic complication but thrombocytopenia and anemia have also been described [37]. Immunological deficit was reported in some individuals but is still controversial [37, 94, 103]. Cardiomyopathy, both dilated and hypertrophic [77, 104], arrhythmia [105], pancreatitis [106] and failure to thrive [37] are also seen.
Apart from classical type, some individuals presented with unusual symptoms. Late-onset PA may present after infancy with the symptoms similar to acute neonatal metabolic decompensation that are triggered by physical stress, mostly infections, but may also present with any of the above complications [107]. A previous study has reported a previously healthy adolescent individual suffering from isolated dilated cardiomyopathy without history of metabolic decompensation also had reduced PCC enzyme activity and pathogenic variants in PCCB gene [108]. Recently, a male neonate born with nephromegaly and acute kidney injury with unknown cause was later found to have PA following a metabolic decompensation episode at the age of three months [109]. It remains unclear whether the congenital renal anomalies are related to this individual’s PA diagnosis. These uncommon presentations remind the physicians that clinical characteristics of individuals with PA are not totally elucidated and some individuals may be undiagnosed.
For individuals with suspected PA, urine organic acid and plasma amino acid analysis, serum acylcarnitine profile should be performed to differentiate PA from other metabolic diseases, such as, methylmalonic acidemia [1, 110]. However, definitive diagnosis of PA relies predominantly on DNA analysis and occasionally on enzyme assay [1]. Residual enzyme activity was shown to be correlated with milder diseases. However, molecular genetics studies should be undertaken to attempt to predictt disease severity, but also genetic testing is useful for carrier detection among family members and precise genetic counselling [1]. A marker of oxidation, exhaled carbon dioxide (CO2) when using stable isotope labeled propionate has been noted to be lower in individuals with severe PA, but this does not have any clinical utility at this time since few centers can do these studies currently [111, 112].
PA individuals who are in acute metabolic decompensation, triggered by a hyper-catabolic state, require sufficient calories and protein intake to prevent further protein catabolism [1, 90]. They may require dietary restriction of amino acids and odd-chain fatty acids which are precursors to propionic acid. Acutely hyperammonemic individuals also require a detoxification process, either pharmacological or extracorporeal, together with carnitine supplement. Individuals who experience multiple decompensation episodes despite adequate fluid and medical treatments are candidates for orthotopic liver transplantation [1, 96, 113, 114]. Long-term management includes dietary restriction, carnitine supplementation, and surveillance to prevent or early detect possible complications, for example, growth and developmental progress, neurologic evaluation, ophthalmologic evaluation, echocardiography and relevant cardiologic evaluation, metabolic, nutritional and basic hematologic and biochemical studies [1, 96]. The parents of a child with PA should be informed to be cautiously aware when the individuals have illnesses, even with mild symptoms, that these may lead to hypercatabolic state and metabolic decompensation. Apart from management of the individuals themselves, genetic counselling should be performed in order to help individuals’ families cope with the disease and also carrier detection, family planning and prenatal diagnosis [1, 110, 115].
CONCLUSION
PCC is a complicated, large mitochondrial enzyme which when dysfunctional, usually leads to a severe metabolic disorder demonstrating significant morbidity and mortality.
Enzyme functional studies have identified and explained a number of the elevated intermediates including MC, 3OHPA, propionic acid, propionylcarnitine, ammonia and propionyl-CoA which are used as diagnostic markers in individuals, but also have been studied to understand the pathophysiology of the disease. Studies have also illustrated the impact on TCA and oxidative phosphorylation of both these abnormal intermediates, but also the implications to function by a limitation in succinyl-CoA. The impact on TCA and OXPHOS probably explains the longer term complications which resemble energy deficiencies like primary mitochondrial disorders.
A greater understanding of the impact and pathophysiology of PCC will enable development of improved therapies for individuals. Active preclinical research is ongoing looking at enzyme replacement using mitochondrial matrix targeting and cell penetrating peptides [116]. Moreover, understanding of the gene and enzyme structure has brought to our attention a number of possible gene therapies which are currently being studied in mice [117–119]. The field continues to explore the impact of antioxidants [79] and anaplerotic supplementation [120].
Current therapy continues to be inadequate in preventing long term complications but insight has been gained by various therapies including carnitine supplementation which increases propionylcarnitine excretion, [121], liver transplant to attenuate disease, and other toxin scavengers (e.g., ammonia scavengers) are being used to mitigate toxin complications. An important endeavor for the community is to continue to develop new therapies for PA and understanding the pathophysiology and biochemistry of PCC is essential.
Supplementary Material
Highlights.
Review of Propionyl-CoA Carboxylase (PCC) structure and how it relates to function.
Review of currently know human variants in PCCA and PCCB.
Review of known toxic intermediates produced by PCC dysfunction and impact on other biochemical pathways
General review of propionic academia
Acknowledgments
We would like to thank Dr. Marshall Summar for his encouragement and support. K.A.C. is funded by NIH grant K08DK105233. The work of N.A.M. is generously supported by the Rashid Family Fund
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baumgartner MR, Horster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, Huemer M, Hochuli M, Assoun M, Ballhausen D, Burlina A, Fowler B, Grunert SC, Grunewald S, Honzik T, Merinero B, Perez-Cerda C, Scholl-Burgi S, Skovby F, Wijburg F, MacDonald A, Martinelli D, Sass JO, Valayannopoulos V, Chakrapani A. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia Orphanet. journal of rare diseases. 2014;9:130. doi: 10.1186/s13023-014-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lau EP, Cochran BC, Munson L, Fall RR. Bovine kidney 3-methylcrotonyl-CoA and propionyl-CoA carboxylases: each enzyme contains nonidentical subunits. Proc Natl Acad Sci USA. 1979;76:214–218. doi: 10.1073/pnas.76.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lam Hon Wah AM, Lam KF, Tsui F, Robinson B, Saunders ME, Gravel RA. Assignment of the alpha and beta chains of human propionyl-CoA carboxylase to genetic complementation groups. Am J Hum Genet. 1983;35:889–899. [PMC free article] [PubMed] [Google Scholar]
- 4.Lamhonwah AM, Barankiewicz TJ, Willard HF, Mahuran DJ, Quan F, Gravel RA. Isolation of cDNA clones coding for the alpha and beta chains of human propionyl-CoA carboxylase: chromosomal assignments and DNA polymorphisms associated with PCCA and PCCB genes. Proc Natl Acad Sci USA. 1986;83:4864–4868. doi: 10.1073/pnas.83.13.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamhonwah AM, Gravel RA. Propionicacidemia: absence of alpha-chain mRNA in fibroblasts from patients of the pccA complementation group. Am J Hum Genet. 1987;41:1124–1131. [PMC free article] [PubMed] [Google Scholar]
- 6.Tong L. Structure and function of biotin-dependent carboxylases Cellular and molecular life sciences. CMLS. 2013;70:863–891. doi: 10.1007/s00018-012-1096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang CS, Sadre-Bazzaz K, Shen Y, Deng B, Zhou ZH, Tong L. Crystal structure of the alpha(6)beta(6) holoenzyme of propionyl-coenzyme A carboxylase. Nature. 2010;466:1001–1005. doi: 10.1038/nature09302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frenkel EP, Kitchens RL. Intracellular localization of hepatic propionyl-CoA carboxylase and methylmalonyl-CoA mutase in humans and normal and vitamin B12 deficient rats. Br J Haematol. 1975;31:501–513. doi: 10.1111/j.1365-2141.1975.tb00885.x. [DOI] [PubMed] [Google Scholar]
- 9.Browner MF, Taroni F, Sztul E, Rosenberg LE. Sequence analysis, biogenesis, and mitochondrial import of the alpha-subunit of rat liver propionyl-CoA carboxylase. J Biol Chem. 1989;264:12680–12685. [PubMed] [Google Scholar]
- 10.Taroni F, Rosenberg LE. The precursor of the biotin-binding subunit of mammalian propionyl-CoA carboxylase can be translocated into mitochondria as apo- or holoprotein. J Biol Chem. 1991;266:13267–13271. [PubMed] [Google Scholar]
- 11.Kelson TL, Ohura T, Kraus JP. Chaperonin-mediated assembly of wild-type and mutant subunits of human propionyl-CoA carboxylase expressed in Escherichia coli. Hum Mol Genet. 1996;5:331–337. doi: 10.1093/hmg/5.3.331. [DOI] [PubMed] [Google Scholar]
- 12.Diacovich L, Mitchell DL, Pham H, Gago G, Melgar MM, Khosla C, Gramajo H, Tsai SC. Crystal structure of the beta-subunit of acyl-CoA carboxylase: structure-based engineering of substrate specificity. Biochemistry. 2004;43:14027–14036. doi: 10.1021/bi049065v. [DOI] [PubMed] [Google Scholar]
- 13.Ohura T, Kraus JP, Rosenberg LE. Unequal synthesis and differential degradation of propionyl CoA carboxylase subunits in cells from normal and propionic acidemia patients. Am J Hum Genet. 1989;45:33–40. [PMC free article] [PubMed] [Google Scholar]
- 14.Kraus JP, Kalousek F, Rosenberg LE. Biosynthesis and mitochondrial processing of the beta subunit of propionyl coenzyme A carboxylase from rat liver. J Biol Chem. 1983;258:7245–7248. [PubMed] [Google Scholar]
- 15.Kalousek F, Hendrick JP, Rosenberg LE. Two mitochondrial matrix proteases act sequentially in the processing of mammalian matrix enzymes. Proc Natl Acad Sci USA. 1988;85:7536–7540. doi: 10.1073/pnas.85.20.7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez-Melendez R, Cano S, Mendez ST, Velazquez A. Biotin regulates the genetic expression of holocarboxylase synthetase and mitochondrial carboxylases in rats. The Journal of nutrition. 2001;131:1909–1913. doi: 10.1093/jn/131.7.1909. [DOI] [PubMed] [Google Scholar]
- 17.Branched-Chain Organic Acidurias/Acidemias; Fernandez J, Saudubray JM, van den Berghe G, Walter JH, editors. Inborn Metabolic Diseases. Springer; Wurzburg, Germany: 2006. [Google Scholar]
- 18.Revsin B, Lebowitz J, Morrow G., III Effect of valine on propionate metabolism in control and hyperglycinemic fibroblasts and in rat liver. Pediatr Res. 1977;11:749–753. doi: 10.1203/00006450-197706000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Kalousek F, Darigo MD, Rosenberg LE. Isolation and characterization of propionyl-CoA carboxylase from normal human liver. Evidence for a protomeric tetramer of nonidentical subunits. J Biol Chem. 1980;255:60–65. [PubMed] [Google Scholar]
- 20.Stubbe J, Abeles RH. Biotin carboxylations–concerted or not concerted? That is the question! J Biol Chem. 1977;252:8338–8340. [PubMed] [Google Scholar]
- 21.Stubbe J, Fish S, Abeles RH. Are carboxylations involving biotin concerted or nonconcerted? J Biol Chem. 1980;255:236–242. [PubMed] [Google Scholar]
- 22.Kaziro Y, Grossman A, Ochoa S. METABOLISM OF PROPIONIC ACID IN ANIMAL TISSUES. XI. FURTHER STUDIES ON CRYSTALLINE PROPIONYL COENZYME A CARBOXYLASE. The Journal of biological chemistry. 1965;240:64–67. [PubMed] [Google Scholar]
- 23.Rodriguez-Melendez R, Perez-Andrade ME, Diaz A, Deolarte A, Camacho-Arroyo I, Ciceron I, Ibarra I, Velazquez A. Differential effects of biotin deficiency and replenishment on rat liver pyruvate and propionyl-CoA carboxylases and on their mRNAs. Mol Genet Metab. 1999;66:16–23. doi: 10.1006/mgme.1998.2777. [DOI] [PubMed] [Google Scholar]
- 24.Mock DM, Henrich-Shell CL, Carnell N, Stumbo P, Mock NI. 3-Hydroxypropionic acid and methylcitric acid are not reliable indicators of marginal biotin deficiency in humans. J Nutr. 2004;134:317–320. doi: 10.1093/jn/134.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazaki T, Ohura T, Kobayashi M, Shigematsu Y, Yamaguchi S, Suzuki Y, Hata I, Aoki Y, Yang X, Minjares C, Haruta I, Uto H, Ito Y, Muller U. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J Biol Chem. 2001;276:35995–35999. doi: 10.1074/jbc.M105467200. [DOI] [PubMed] [Google Scholar]
- 26.Liu YN, Liu TT, Fan YL, Niu DM, Chien YH, Chou YY, Lee NC, Hsiao KJ, Chiu YH. Measuring propionyl-CoA carboxylase activity in phytohemagglutinin stimulated lymphocytes using high performance liquid chromatography Clinica chimica acta; international. journal of clinical chemistry. 2016;453:13–20. doi: 10.1016/j.cca.2015.11.023. [DOI] [PubMed] [Google Scholar]
- 27.Tajima G, Yofune H, Bahagia Febriani AD, Nishimura Y, Ono H, Sakura N. A simple and rapid enzymatic assay for the branched-chain alpha-ketoacid dehydrogenase complex using high-performance liquid chromatography. Journal of inherited metabolic disease. 2004;27:633–639. doi: 10.1023/b:boli.0000042988.31581.ed. [DOI] [PubMed] [Google Scholar]
- 28.Gaire D, Sponne I, Droesch S, Charlier A, Nicolas JP, Lambert D. Comparison of two methods for the measurement of rat liver methylmalonyl-coenzyme A mutase activity: HPLC and radioisotopic assays. The Journal of nutritional biochemistry. 1999;10:56–62. doi: 10.1016/s0955-2863(98)00083-7. [DOI] [PubMed] [Google Scholar]
- 29.Gibson KM, Ugarte M, Fukao T, Mitchell GA. Molecular and enzymatic methods for detection of genetic defects in distal pathways of branched-chain amino acid metabolism Methods. Enzymol. 2000;324:432–453. doi: 10.1016/s0076-6879(00)24252-3. [DOI] [PubMed] [Google Scholar]
- 30.Giorgio AJ, Whitaker TR. Some properties of propionyl CoA carboxylase partially purified from human liver. Biochem Med. 1973;7:473–478. doi: 10.1016/0006-2944(73)90069-0. [DOI] [PubMed] [Google Scholar]
- 31.Gravel RA, Lam KF, Scully KJ, Hsia Y. Genetic complementation of propionyl-CoA carboxylase deficiency in cultured human fibroblasts. Am J Hum Genet. 1977;29:378–388. [PMC free article] [PubMed] [Google Scholar]
- 32.Perez-Cerda C, Merinero B, Rodriguez-Pombo P, Perez B, Desviat LR, Muro S, Richard E, Garcia MJ, Gangoiti J, Ruiz Sala P, Sanz P, Briones P, Ribes A, Martinez-Pardo M, Campistol J, Perez M, Lama R, Murga ML, Lema-Garrett T, Verdu A, Ugarte M. Potential relationship between genotype and clinical outcome in propionic acidaemia patients. European journal of human genetics: EJHG. 2000;8:187–194. doi: 10.1038/sj.ejhg.5200442. [DOI] [PubMed] [Google Scholar]
- 33.Richard E, Desviat LR, Perez B, Perez-Cerda C, Ugarte M. Three novel splice mutations in the PCCA gene causing identical exon skipping in propionic acidemia patients. Hum Genet. 1997;101:93–96. doi: 10.1007/s004390050593. [DOI] [PubMed] [Google Scholar]
- 34.Hassan YI, Moriyama H, Olsen LJ, Bi X, Zempleni J. N- and C-terminal domains in human holocarboxylase synthetase participate in substrate recognition. Mol Genet Metab. 2009;96:183–188. doi: 10.1016/j.ymgme.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez-Pombo P, Hoenicka J, Muro S, Perez B, Perez-Cerda C, Richard E, Desviat LR, Ugarte M. Human propionyl-CoA carboxylase beta subunit gene: exon-intron definition and mutation spectrum in Spanish and Latin American propionic acidemia patients. Am J Hum Genet. 1998;63:360–369. doi: 10.1086/301970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muro S, Perez B, Rodriguez-Pombo P, Desviat LR, Perez-Cerda C, Ugarte M. Mutations affecting the beta-beta homomeric interaction in propionic acidaemia: an approach to the determination of the beta-propionyl-CoA carboxylase functional domains. J Inherit Metab Dis. 2000;23:300–304. doi: 10.1023/a:1005617420460. [DOI] [PubMed] [Google Scholar]
- 37.Pena L, Franks J, Chapman KA, Gropman A, Ah MN, Chakrapani A, Island E, MacLeod E, Matern D, Smith B, Stagni K, Sutton VR, Ueda K, Urv T, Venditti C, Enns GM, Summar ML. Natural history of propionic acidemia. Mol Genet Metab. 2012;105:5–9. doi: 10.1016/j.ymgme.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 38.Kurczynski TW, Hoppel CL, Goldblatt PJ, Gunning WT. Metabolic studies of carnitine in a child with propionic acidemia. Pediatr Res. 1989;26:63–66. doi: 10.1203/00006450-198907000-00018. [DOI] [PubMed] [Google Scholar]
- 39.Ando T, Rasmussen K, Nyhan WL, Hull D. 3-hydroxypropionate: significance of -oxidation of propionate in patients with propionic acidemia and methylmalonic acidemia. Proc Natl Acad Sci USA. 1972;69:2807–2811. doi: 10.1073/pnas.69.10.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ando T, Rasmussen K, Wright JM, Nyhan WL. Isolation and identification of methylcitrate, a major metabolic product of propionate in patients with propionic acidemia. J Biol Chem. 1972;247:2200–2204. [PubMed] [Google Scholar]
- 41.Weidman SW, Drysdale GR. The biosynthesis of methylcitrate. The Biochemical journal. 1979;177:169–174. doi: 10.1042/bj1770169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fenton WA, Gravel RA, Rosenblatt DS. Disorders of Propionate and Methylmalonate Metabolism. In: Scriver CR, Beaudert AL, Sly William S, Valle David, editors. The Metabolic & Molecular Basis of Inherited Disease. McGraw-Hill, New York: 2001. pp. 2165–2193. [Google Scholar]
- 43.Davison JE, Davies NP, Wilson M, Sun Y, Chakrapani A, McKiernan PJ, Walter JH, Gissen P, Peet AC. MR spectroscopy-based brain metabolite profiling in propionic acidaemia: metabolic changes in the basal ganglia during acute decompensation and effect of liver transplantation. Orphanet journal of rare diseases. 2011;6:19. doi: 10.1186/1750-1172-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peuhkurinen KJ. Regulation of the tricarboxylic acid cycle pool size in heart muscle. Journal of molecular and cellular cardiology. 1984;16:487–495. doi: 10.1016/s0022-2828(84)80637-9. [DOI] [PubMed] [Google Scholar]
- 45.Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, Drose S, Brandt U, Hoffmann GF, Ter LH, Kolker S, Smeitink JA. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J. 2006;398:107–112. doi: 10.1042/BJ20060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stumpf DA, McAfee J, Parks JK, Eguren L. Propionate inhibition of succinate:CoA ligase (GDP) and the citric acid cycle in mitochondria Pediatric research. 1980;14:1127–1131. doi: 10.1203/00006450-198010000-00008. [DOI] [PubMed] [Google Scholar]
- 47.Cheema-Dhadli S, Leznoff CC, Halperin ML. Effect of 2-methylcitrate on citrate metabolism: implications for the management of patients with propionic acidemia and methylmalonic aciduria. Pediatr Res. 1975;9:905–908. doi: 10.1203/00006450-197512000-00008. [DOI] [PubMed] [Google Scholar]
- 48.Gibala MJ, Young ME, Taegtmeyer H. Anaplerosis of the citric acid cycle: role in energy metabolism of heart and skeletal muscle. Acta Physiol Scand. 2000;168:657–665. doi: 10.1046/j.1365-201x.2000.00717.x. [DOI] [PubMed] [Google Scholar]
- 49.Davis EJ, Spydevold O, Bremer J. Pyruvate carboxylase and propionyl-CoA carboxylase as anaplerotic enzymes in skeletal muscle mitochondria. Eur J Biochem. 1980;110:255–262. doi: 10.1111/j.1432-1033.1980.tb04863.x. [DOI] [PubMed] [Google Scholar]
- 50.Hayasaka K, Narisawa K, Satoh T, Tateda H, Metoki K, Tada K, Hiraga K, Aoki T, Kawakami T, Akamatsu H, Matsuo N. Glycine cleavage system in ketotic hyperglycinemia: a reduction of H-protein activity. Pediatr Res. 1982;16:5–7. doi: 10.1203/00006450-198201001-00002. [DOI] [PubMed] [Google Scholar]
- 51.Nishimura Y, Tada K, Arakawa T. Coexistence of defective activity in glycine-cleavage reaction and propionyl-CoA carboxylase in the liver of a hyperglycinemic child Tohoku. J Exp Med. 1974;113:267–271. doi: 10.1620/tjem.113.267. [DOI] [PubMed] [Google Scholar]
- 52.Kolvraa S. Inhibition of the glycine cleavage system by branched-chain amino acid metabolites. Pediatr Res. 1979;13:889–893. doi: 10.1203/00006450-197908000-00004. [DOI] [PubMed] [Google Scholar]
- 53.Martin-Requero A, Corkey BE, Cerdan S, Walajtys-Rode E, Parrilla RL, Williamson JR. Interactions between alpha-ketoisovalerate metabolism and the pathways of gluconeogenesis and urea synthesis in isolated hepatocytes. The Journal of biological chemistry. 1983;258:3673–3681. [PubMed] [Google Scholar]
- 54.Gregersen N. The specific inhibition of the pyruvate dehydrogenase complex from pig kidney by propionyl-CoA and isovaleryl-Co-A Biochemical medicine 26. 1981:20–27. doi: 10.1016/0006-2944(81)90026-0. [DOI] [PubMed] [Google Scholar]
- 55.Patel TB, DeBuysere MS, Olson MS. The effect of propionate on the regulation of the pyruvate dehydrogenase complex in the rat liver Archives of biochemistry and biophysics 220. 1983:405–414. doi: 10.1016/0003-9861(83)90430-7. [DOI] [PubMed] [Google Scholar]
- 56.Coude FX, Sweetman L, Nyhan WL. Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemia. J Clin Invest. 1979;64:1544–1551. doi: 10.1172/JCI109614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dercksen M, IJ L, Duran M, Mienie LJ, van Cruchten A, van der Westhuizen FH, Wanders RJ. Inhibition of N-acetylglutamate synthase by various monocarboxylic and dicarboxylic short-chain coenzyme A esters and the production of alternative glutamate. esters Biochimica et biophysica acta. 2014;1842:2510–2516. doi: 10.1016/j.bbadis.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 58.Jafari P, Braissant O, Zavadakova P, Henry H, Bonafe L, Ballhausen D. Brain damage in methylmalonic aciduria: 2-methylcitrate induces cerebral ammonium accumulation and apoptosis in 3D organotypic brain cell cultures. Orphanet journal of rare diseases. 2013;8:4. doi: 10.1186/1750-1172-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harris DJ, Yang BI, Wolf B, Snodgrass PJ. Dysautonomia in an infant with secondary hyperammonemia due to propionyl coenzyme. A carboxylase deficiency Pediatrics. 1980;65:107–110. [PubMed] [Google Scholar]
- 60.Chapman KA, Collado MS, Figler RA, Hoang SA, Armstrong AJ, Cui W, Purdy M, Simmers MB, Yazigi NA, Summar ML, Wamhoff BR, Dash A. Recapitulation of metabolic defects in a model of propionic acidemia using patient-derived primary hepatocytes. Molecular genetics and metabolism. 2016;117:355–362. doi: 10.1016/j.ymgme.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ballhausen D, Mittaz L, Boulat O, Bonafe L, Braissant O. Evidence for catabolic pathway of propionate metabolism in CNS: expression pattern of methylmalonyl-CoA mutase and propionyl-CoA carboxylase alpha-subunit in developing and adult rat brain. Neuroscience. 2009;164:578–587. doi: 10.1016/j.neuroscience.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 62.Rigo FK, Pasquetti L, Malfatti CR, Fighera MR, Coelho RC, Petri CZ, Mello CF. Propionic acid induces convulsions and protein carbonylation in rats. Neurosci Lett. 2006;408:151–154. doi: 10.1016/j.neulet.2006.08.075. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen NH, Morland C, Gonzalez SV, Rise F, Storm-Mathisen J, Gundersen V, Hassel B. Propionate increases neuronal histone acetylation, but is metabolized oxidatively by glia. Relevance for propionic acidemia. J Neurochem. 2007;101:806–814. doi: 10.1111/j.1471-4159.2006.04397.x. [DOI] [PubMed] [Google Scholar]
- 64.Lynen F, Domagk GF, Goldmann M, Kessel I. On the biosynthesis of fatty acids. I. Demonstration of malonyl-CoA as an intermediate product of fatty acid synthesis in yeast extracts. Biochemische Zeitschrift. 1962;335:519–539. [PubMed] [Google Scholar]
- 65.Wendel U. Abnormality of odd-numbered long-chain fatty acids in erythrocyte membrane lipids from patients with disorders of propionate metabolism. Pediatr Res. 1989;25:147–150. doi: 10.1203/00006450-198902000-00014. [DOI] [PubMed] [Google Scholar]
- 66.Wendel U, Eissler A, Sperl W, Schadewaldt P. On the differences between urinary metabolite excretion and odd-numbered fatty acid production in propionic and methylmalonic acidaemias. Journal of inherited metabolic disease. 1995;18:584–591. doi: 10.1007/BF02436003. [DOI] [PubMed] [Google Scholar]
- 67.Decsi T, Sperl W, Koletzko B. Essential fatty acids in clinically stable children with propionic acidaemia. J Inherit Metab Dis. 1997;20:778–782. doi: 10.1023/a:1005315717106. [DOI] [PubMed] [Google Scholar]
- 68.Poulos A, Johnson D, Singh H. Defective oxidation of pristanic acid by fibroblasts from patients with disorders in propionic acid metabolism. Clin Genet. 1990;37:106–110. doi: 10.1111/j.1399-0004.1990.tb03486.x. [DOI] [PubMed] [Google Scholar]
- 69.Bhatia H, Hallock JL, Dutta A, Karkashon S, Sterner LS, Miyazaki T, Dean A, Little JA. Short-chain fatty acid-mediated effects on erythropoiesis in primary definitive erythroid cells. Blood. 2009;113:6440–6448. doi: 10.1182/blood-2008-09-171728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bodi I, Grunert SC, Becker N, Stoelzle-Feix S, Spiekerkoetter U, Zehender M, Bugger H, Bode C, Odening KE. Mechanisms of acquired long QT syndrome in patients with propionic academia. Heart rhythm: the official journal of the Heart Rhythm Society. 2016;13:1335–1345. doi: 10.1016/j.hrthm.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 71.Gallego-Villar L, Rivera-Barahona A, Cuevas-Martin C, Guenzel A, Perez B, Barry MA, Murphy MP, Logan A, Gonzalez-Quintana A, Martin MA, Medina S, Gil-Izquierdo A, Cuezva JM, Richard E, Desviat LR. In vivo evidence of mitochondrial dysfunction and altered redox homeostasis in a genetic mouse model of propionic acidemia: Implications for the pathophysiology of this disorder. Free radical biology & medicine. 2016;96:1–12. doi: 10.1016/j.freeradbiomed.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 72.de Keyzer Y, Valayannopoulos V, Benoist JF, Batteux F, Lacaille F, Hubert L, Chretien D, Chadefeaux-Vekemans B, Niaudet P, Touati G, Munnich A, de Lonlay P. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr Res. 2009;66:91–95. doi: 10.1203/PDR.0b013e3181a7c270. [DOI] [PubMed] [Google Scholar]
- 73.Baruteau J, Hargreaves I, Krywawych S, Chalasani A, Land JM, Davison JE, Kwok MK, Christov G, Karimova A, Ashworth M, Anderson G, Prunty H, Rahman S, Grunewald S. Successful reversal of propionic acidaemia associated cardiomyopathy: evidence for low myocardial coenzyme Q10 status and secondary mitochondrial dysfunction as an underlying pathophysiological mechanism. Mitochondrion. 2014;17:150–156. doi: 10.1016/j.mito.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 74.Mardach R, Verity MA, Cederbaum SD. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol Genet Metab. 2005;85:286–290. doi: 10.1016/j.ymgme.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 75.Ferrari R, Ceconi C, Curello S, Pasini E, Visioli O. Protective effect of propionyl-L-carnitine against ischaemia and reperfusion-damage. Mol Cell Biochem. 1989;88:161–168. doi: 10.1007/BF00223438. [DOI] [PubMed] [Google Scholar]
- 76.Ferrari R, Merli E, Cicchitelli G, Mele D, Fucili A, Ceconi C. Therapeutic effects of L-carnitine and propionyl-L-carnitine on cardiovascular diseases: a review. Ann NY Acad Sci. 2004;1033:79–91. doi: 10.1196/annals.1320.007. [DOI] [PubMed] [Google Scholar]
- 77.Romano S, Valayannopoulos V, Touati G, Jais JP, Rabier D, de KY, Bonnet D, de LP. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr. 2010;156:128–134. doi: 10.1016/j.jpeds.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 78.Arrizza C, De Gottardi A, Foglia E, Baumgartner M, Gautschi M, Nuoffer JM. Reversal of cardiomyopathy in propionic acidemia after liver transplantation: a 10-year follow-up Transplant international: official journal of the. European Society for Organ Transplantation. 2015;28:1447–1450. doi: 10.1111/tri.12677. [DOI] [PubMed] [Google Scholar]
- 79.Gallego-Villar L, Perez B, Ugarte M, Desviat LR, Richard E. Antioxidants successfully reduce ROS production in propionic acidemia fibroblasts. Biochemical and biophysical research communications. 2014;452:457–461. doi: 10.1016/j.bbrc.2014.08.091. [DOI] [PubMed] [Google Scholar]
- 80.Gallego-Villar L, Perez-Cerda C, Perez B, Abia D, Ugarte M, Richard E, Desviat LR. Functional characterization of novel genotypes and cellular oxidative stress studies in propionic acidemia. Journal of inherited metabolic disease. 2013;36:731–740. doi: 10.1007/s10545-012-9545-3. [DOI] [PubMed] [Google Scholar]
- 81.Ribas GS, Biancini GB, Mescka C, Wayhs CY, Sitta A, Wajner M, Vargas CR. Oxidative stress parameters in urine from patients with disorders of propionate metabolism: a beneficial effect of L:-carnitine supplementation. Cellular and molecular neurobiology. 2012;32:77–82. doi: 10.1007/s10571-011-9736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rivera-Barahona A, Fulgencio-Covian A, Perez-Cerda C, Ramos R, Barry MA, Ugarte M, Perez B, Richard E, Desviat LR. Dysregulated miRNAs and their pathogenic implications for the neurometabolic disease propionic acidemia. Scientific reports. 2017;7:5727. doi: 10.1038/s41598-017-06420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Llamas A, Chamizo-Ampudia A, Tejada-Jimenez M, Galvan A, Fernandez E. The molybdenum cofactor enzyme mARC: Moonlighting or promiscuous enzyme? Biofactors. 2017;43:486–494. doi: 10.1002/biof.1362. [DOI] [PubMed] [Google Scholar]
- 84.Leon-Del-Rio A, Valadez-Graham V, Gravel RA. Holocarboxylase Synthetase: A Moonlighting Transcriptional Coregulator of Gene Expression and a Cytosolic Regulator of Biotin. Utilization Annual review of nutrition. 2017;37:207–223. doi: 10.1146/annurev-nutr-042617-104653. [DOI] [PubMed] [Google Scholar]
- 85.Boukouris AE, Zervopoulos SD, Michelakis ED. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription Trends in biochemical. sciences. 2016;41:712–730. doi: 10.1016/j.tibs.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 86.Kraus JP, Rosenberg LE. Purification of low-abundance messengerRNAs fromrat liver by polysome immunoadsorption. Proc Natl Acad Sci USA. 1982;79:4015–4019. doi: 10.1073/pnas.79.13.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shiraishi A, Yamada Y, Tsuura Y, Fijimoto S, Tsukiyama K, Mukai E, Toyoda Y, Miwa I, Seino Y. A novel glucokinase regulator in pancreatic beta cells: precursor of propionyl-CoA carboxylase beta subunit interacts with glucokinase and augments its activity. J Biol Chem. 2001;276:2325–2328. doi: 10.1074/jbc.C000530200. [DOI] [PubMed] [Google Scholar]
- 88.Weber M, Wehling M, Losel R. Proteins interact with the cytosolic mineralocorticoid receptor depending on the ligand. Am J Physiol Heart Circ Physiol. 2008;295:H361–H365. doi: 10.1152/ajpheart.00825.2007. [DOI] [PubMed] [Google Scholar]
- 89.Pougovkina O, Te Brinke H, Wanders RJ, Houten SM, de Boer VC. Aberrant protein acylation is a common observation in inborn errors of acyl-CoA metabolism. Journal of inherited metabolic disease. 2014;37:709–714. doi: 10.1007/s10545-014-9684-9. [DOI] [PubMed] [Google Scholar]
- 90.Chapman KA, Gropman A, MacLeod E, Stagni K, Summar ML, Ueda K, Ah MN, Franks J, Island E, Matern D, Pena L, Smith B, Sutton VR, Urv T, Venditti C, Chakrapani A. Acute management of propionic acidemia. Mol Genet Metab. 2012;105:16–25. doi: 10.1016/j.ymgme.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kolker S, Cazorla AG, Valayannopoulos V, Lund AM, Burlina AB, Sykut-Cegielska J, Wijburg FA, Teles EL, Zeman J, Dionisi-Vici C, Baric I, Karall D, Augoustides-Savvopoulou P, Aksglaede L, Arnoux JB, Avram P, Baumgartner MR, Blasco-Alonso J, Chabrol B, Chakrapani A, Chapman K, EC IS, Couce ML, de Meirleir L, Dobbelaere D, Dvorakova V, Furlan F, Gleich F, Gradowska W, Grunewald S, Jalan A, Haberle J, Haege G, Lachmann R, Laemmle A, Langereis E, de Lonlay P, Martinelli D, Matsumoto S, Muhlhausen C, de Baulny HO, Ortez C, Pena-Quintana L, Ramadza DP, Rodrigues E, Scholl-Burgi S, Sokal E, Staufner C, Summar ML, Thompson N, Vara R, Pinera IV, Walter JH, Williams M, Burgard P. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. Journal of inherited metabolic disease. 2015 doi: 10.1007/s10545-015-9839-3. [DOI] [PubMed] [Google Scholar]
- 92.Hsia YE, Scully KJ, Rosenberg LE. Inherited propionyl-Coa carboxylase deficiency in “ketotic hyperglycinemia”. J Clin Invest. 1971;50:127–130. doi: 10.1172/JCI106466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McCrory NM, Edick MJ, Ahmad A, Lipinski S, Scott Schwoerer JA, Zhai S, Justice K, Cameron CA, Berry SA, Pena LD. Comparison of Methods of Initial Ascertainment in 58 Cases of Propionic Acidemia Enrolled in the Inborn Errors of Metabolism Information System Reveals Significant Differences in Time to Evaluation and Symptoms at Presentation. The Journal of pediatrics. 2017;180:200–205.e208. doi: 10.1016/j.jpeds.2016.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pena L, Burton BK. Survey of health status and complications among propionic acidemia patients. Am J Med Genet A. 2012;158A:1641–1646. doi: 10.1002/ajmg.a.35387. [DOI] [PubMed] [Google Scholar]
- 95.Grunert SC, Mullerleile S, de SL, Barth M, Walter M, Walter K, Meissner T, Lindner M, Ensenauer R, Santer R, Bodamer OA, Baumgartner MR, Brunner-Krainz M, Karall D, Haase C, Knerr I, Marquardt T, Hennermann JB, Steinfeld R, Beblo S, Koch HG, Konstantopoulou V, Scholl-Burgi S, van Teeffelen-Heithoff A, Suormala T, Sperl W, Kraus JP, Superti-Furga A, Schwab KO, Sass JO. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J Rare Dis. 2013;8:6. doi: 10.1186/1750-1172-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sutton VR, Chapman KA, Gropman AL, MacLeod E, Stagni K, Summar ML, Ueda K, Ah Mew N, Franks J, Island E, Matern D, Pena L, Smith B, Urv T, Venditti C, Chakarapani A. Chronic management and health supervision of individuals with propionic acidemia. Mol Genet Metab. 2012;105:26–33. doi: 10.1016/j.ymgme.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 97.Grunert SC, Mullerleile S, de SL, Barth M, Walter M, Walter K, Meissner T, Lindner M, Ensenauer R, Santer R, Bodamer OA, Baumgartner MR, Brunner-Krainz M, Karall D, Haase C, Knerr I, Marquardt T, Hennermann JB, Steinfeld R, Beblo S, Koch HG, Konstantopoulou V, Scholl-Burgi S, van Teeffelen-Heithoff A, Suormala T, Sperl W, Kraus JP, Superti-Furga A, Schwab KO, Sass JO. Propionic acidemia: neonatal versus selective metabolic screening. J Inherit Metab Dis. 2012;35:41–49. doi: 10.1007/s10545-011-9419-0. [DOI] [PubMed] [Google Scholar]
- 98.Witters P, Debbold E, Crivelly K, Vande Kerckhove K, Corthouts K, Debbold B, Andersson H, Vannieuwenborg L, Geuens S, Baumgartner M, Kozicz T, Settles L, Morava E. Autism in patients with propionic acidemia. Molecular genetics and metabolism. 2016;119:317–321. doi: 10.1016/j.ymgme.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 99.Dejean de la Batie C, Barbier V, Valayannopoulos V, Touati G, Maltret A, Brassier A, Arnoux JB, Grevent D, Chadefaux B, Ottolenghi C, Canoui P, de Lonlay P. Acute psychosis in propionic acidemia: 2 case reports. Journal of child neurology. 2014;29:274–279. doi: 10.1177/0883073813508812. [DOI] [PubMed] [Google Scholar]
- 100.Schreiber J, Chapman KA, Summar ML, Ah MN, Sutton VR, MacLeod E, Stagni K, Ueda K, Franks J, Island E, Matern D, Pena L, Smith B, Urv T, Venditti C, Chakarapani A, Gropman AL. Neurologic considerations in propionic acidemia. Mol Genet Metab. 2012;105:10–15. doi: 10.1016/j.ymgme.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 101.Arias C, Raimann E, Peredo P, Cabello JF, Castro G, Valiente A, de la Parra A, Bravo P, Okuma C, Cornejo V. Propionic acidemia and optic neuropathy: a report of two cases. JIMD reports. 2014;12:1–4. doi: 10.1007/8904_2013_234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nyhan WL, Bay C, Beyer EW, Mazi M. Neurologic nonmetabolic presentation of propionic acidemia. Arch Neurol. 1999;56:1143–1147. doi: 10.1001/archneur.56.9.1143. [DOI] [PubMed] [Google Scholar]
- 103.Ozand PT, Rashed M, Gascon GG, Youssef NG, Harfi H, Rahbeeni Z, al Garawi S, al Aqeel A. Unusual presentations of propionic acidemia. Brain & development. 1994;16(Suppl):46–57. doi: 10.1016/0387-7604(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 104.Massoud AF, Leonard JV. Cardiomyopathy in propionic acidaemia. Eur J Pediatr. 1993;152:441–445. doi: 10.1007/BF01955907. [DOI] [PubMed] [Google Scholar]
- 105.Baumgartner D, Scholl-Burgi S, Sass JO, Sperl W, Schweigmann U, Stein JI, Karall D. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J Pediatr. 2007;150:192–197. 197. doi: 10.1016/j.jpeds.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 106.Burlina AB, Dionisi-Vici C, Piovan S, Saponara I, Bartuli A, Sabetta G, Zacchello F. Acute pancreatitis in propionic acidaemia. J Inherit Metab Dis. 1995;18:169–172. doi: 10.1007/BF00711758. [DOI] [PubMed] [Google Scholar]
- 107.Lucke T, Perez-Cerda C, Baumgartner M, Fowler B, Sander S, Sasse M, Scholl S, Ugarte M, Das AM. Propionic acidemia: unusual course with late onset and fatal outcome. Metabolism. 2004;53:809–810. doi: 10.1016/j.metabol.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 108.Lee TM, Addonizio LJ, Barshop BA, Chung WK. Unusual presentation of propionic acidaemia as isolated cardiomyopathy. J Inherit Metab Dis. 2009 doi: 10.1007/s10545-009-1084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bernheim S, Deschenes G, Schiff M, Cussenot I, Niel O. Antenatal nephromegaly and propionic acidemia: a case report. BMC nephrology. 2017;18:110. doi: 10.1186/s12882-017-0535-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shchelochkov OA, Carrillo N, Venditti C. Propionic Acidemia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R), University of Washington, Seattle University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved; Seattle (WA): 1993. [Google Scholar]
- 111.Barshop BA, Yoshida I, Ajami A, Sweetman L, Wolff JA, Sweetman FR, Prodanos C, Smith M, Nyhan WL. Metabolism of 1–13C-propionate in vivo in patients with disorders of propionate metabolism. Pediatr Res. 1991;30:15–22. doi: 10.1203/00006450-199107000-00004. [DOI] [PubMed] [Google Scholar]
- 112.Thompson GN, Walter JH, Bresson JL, Ford GC, Lyonnet SL, Chalmers RA, Saudubray JM, Leonard JV, Halliday D. Sources of propionate in inborn errors of propionate metabolism. Metabolism. 1990;39:1133–1137. doi: 10.1016/0026-0495(90)90084-p. [DOI] [PubMed] [Google Scholar]
- 113.Yorifuji T, Muroi J, Uematsu A, Nakahata T, Egawa H, Tanaka K. Living-related liver transplantation for neonatal-onset propionic acidemia. J Pediatr. 2000;137:572–574. doi: 10.1067/mpd.2000.108391. [DOI] [PubMed] [Google Scholar]
- 114.Charbit-Henrion F, Lacaille F, McKiernan P, Girard M, de Lonlay P, Valayannopoulos V, Ottolenghi C, Chakrapani A, Preece M, Sharif K, Chardot C, Hubert P, Dupic L. Early and late complications after liver transplantation for propionic acidemia in children: a two centers study American journal of transplantation : official journal of the American Society of Transplantation and the. American Society of Transplant Surgeons. 2015;15:786–791. doi: 10.1111/ajt.13027. [DOI] [PubMed] [Google Scholar]
- 115.Alberola TM, Bautista-Llacer R, Vendrell X, Garcia-Mengual E, Pardo M, Vila M, Calatayud C. Case report: birth of healthy twins after preimplantation genetic diagnosis of propionic acidemia. Journal of assisted reproduction and genetics. 2011;28:211–216. doi: 10.1007/s10815-010-9514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Darvish-Damavandi M, Ho HK, Kang TS. Towards the development of an enzyme replacement therapy for the metabolic disorder propionic acidemia. Molecular genetics and metabolism reports. 2016;8:51–60. doi: 10.1016/j.ymgmr.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chandler RJ, Chandrasekaran S, Carrillo-Carrasco N, Senac JS, Hofherr S, Barry MA, Venditti CP. Adeno-associated virus serotype 8 (AAV8) Gene Transfer Rescues a Neonatal Lethal Murine Model of Propionic. Acidemia Hum Gene Ther. 2010 doi: 10.1089/hum.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chandler RJ, Chandrasekaran S, Carrillo-Carrasco N, Senac JS, Hofherr SE, Barry MA, Venditti CP. Adeno-associated virus serotype 8 gene transfer rescues a neonatal lethal murine model of propionic acidemia. Human gene therapy. 2011;22:477–481. doi: 10.1089/hum.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hofherr SE, Senac JS, Chen CY, Palmer DJ, Ng P, Barry MA. Short-term rescue of neonatal lethality in a mouse model of propionic acidemia by gene therapy. Hum Gene Ther. 2009;20:169–180. doi: 10.1089/hum.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Longo N, Price LB, Gappmaier E, Cantor NL, Ernst SL, Bailey C, Pasquali M. Anaplerotic therapy in propionic acidemia. Molecular genetics and metabolism. 2017 doi: 10.1016/j.ymgme.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Roe CR, Millington DS, Maltby DA, Bohan TP, Hoppel CL. L-carnitine enhances excretion of propionyl coenzyme A as propionylcarnitine in propionic acidemia. J Clin Invest. 1984;73:1785–1788. doi: 10.1172/JCI111387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.