

Abstract
Caffeic acid phenethyl ester (CAPE) could ameliorate myocardial ischemia/reperfusion injury (MIRI) by various mechanisms, but there hadn’t been any reports on that CAPE could regulate silent information regulator 1 (SIRT1) and endothelial nitric oxide synthase (eNOS) to exert cardioprotective effect. The present study aimed to investigate the cardioprotective potential of caffeic acid o-nitro phenethyl ester (CAPE-oNO2) on MIRI and the possible mechanism based on the positive control of CAPE. The SD rats were subjected to left coronary artery ischemia /reperfusion (IR) and the H9c2 cell cultured in hypoxia/reoxygenation (HR) to induce the MIRI model. Prior to the procedure, vehicle, CAPE or CAPE-oNO2 were treated in the absence or presence of a SIRT1 inhibitor nicotinamide (NAM) and an eNOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME). In vivo, CAPE and CAPE-oNO2 conferred a cardioprotective effect as shown by reduced myocardial infarct size, cardiac marker enzymes and structural abnormalities. From immunohistochemical and sirius red staining, above two compounds ameliorated the TNF-α release and collagen deposition of IR rat hearts. They could agitate SIRT1 and eNOS expression, and consequently enhance NO release and suppress NF-κB signaling, to reduce the malondialdehyde content and cell necrosis. In vitro, they could inhibit HR-induced H9c2 cell apoptosis and ROS generation by activating SIRT1/eNOS pathway and inhabiting NF-κB expression. Emphatically, CAPE-oNO2 presented the stronger cardioprotection than CAPE both in vivo and in vitro. However, NAM and L-NAME eliminated the CAPE-oNO2-mediated cardioprotection by restraining SIRT1 and eNOS expression, respectively. It suggested that CAPE-oNO2 ameliorated MIRI by suppressing the oxidative stress, inflammatory response, fibrosis and necrocytosis via the SIRT1/eNOS/NF-κB pathway.
Keywords: Caffeic acid o-nitro phenethyl ester (CAPE-oNO2), Myocardial ischemia/reperfusion injury (MIRI), Reactive oxygen species (ROS), SIRT1/eNOS/NF-κB pathway
Graphical abstract
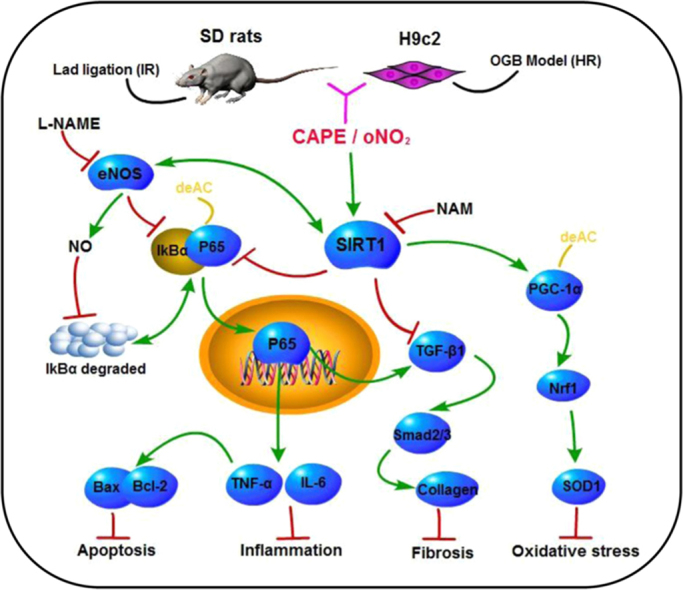
The Cardioprotection of CAPE-oNO2against MIRI via the SIRT1/ eNOS / NF-κB pathway. CAPE-oNO2 ameliorated MIRI by suppressing the oxidative stress, inflammatory response, fibrosis and necrocytosis via the SIRT1/ eNOS / NF-κB pathway. IR, ischemia/reperfusion; HR, hypoxia/reoxygenation; L-NAME, eNOS inhibitor Nω-nitro-L-arginine methyl ester; NAM, SIRT1 inhibitor nicotinamide; CAPE, caffeic acid phenethyl ester; oNO2, caffeic acid o-nitro phenethyl ester.
Highlights
-
•
CAPE-oNO2 exerting cardioprotective potential was firstly synthesized.
-
•
IR-induced ROS increase aggravates inflammation, fibrosis and necrocytosis.
-
•
The SIRT1/eNOS/NF-κB pathway is contributed to MIRI both in vivo and in vitro.
1. Introduction
Cardiac injury after ischemia/reperfusion (IR) contributes to high rates of morbidity and mortality in patients, which causes the myocardial cellular structure destruction and the myocardial cell energy metabolism disorder, following by myocardium tissue infarction [1]. Reactive oxygen species (ROS) generated through a series of interacting pathways in cardiomyocytes and endothelial cells is greatly increased in the post-ischemic heart and serves as a critical central mechanism of post-ischemic injury [2]. When ischemia or myocardial infarction occurs, oxygen free radical formation increases and scavenging system limits, the scavenging of superoxide anion (O2·−) and hydrogen peroxide (H2O2) reduce, transforming oxygen free radical into the strongest oxidation hydroxyl radical (HO·). ROS trigger the chain lipid peroxidation, altering the structural and functional integrity of cells and causing cell necrosis and apoptosis [3], [4], [5]. For another, ROS is a stimulatory signal of nuclear factor kB (NF-κB) activation which can trigger inflammation and damage though up-regulating tumor necrosis factor-α (TNF-α), Bcl-2 associated X Protein (Bax) and transforming growth factor β1 (TGF-β1) [6]. Oxidative stress and inflammatory response play vital roles in the processes of apoptosis and fibrosis, which are also involved in the pathogenesis of myocardial IR injury (MIRI) [7], [8], [9]. Therefore, the ROS generation and NF-κB activation are the main factors that determine the severity of myocardial damage.
Silent information regulator 1 (SIRT1), a multifunctional nicotinamide adenine dinucleotide (NAD+) dependence protein deacetylase, acts a pivotal part in the DNA damage repair [10], anti-apoptosis [11], energy metabolism [12], mitochondrial function protection [13], immunoregulation [14] and resistance to oxidative stress [15] by stimulating the deacetylation of its substrates such as NF-κB, endothelial nitric oxide synthase (eNOS), peroxisomal proliferators-activated receptor γ-coactivator-1α (PGC-1α). The deacetylation of NF-κB and PGC-1α inhibit the NF-κB transcription and improve PGC-1α activity [16], leading to ROS generation reduction, inflammatory cytokines inhibition and cell death decreasing. The cardioprotective effect of SIRT1 has been well demonstrated. Curcumin pretreatment attenuated MIRI by decreasing IR-induced mitochondrial oxidative damage through the SIRT1 signaling activation. Similarly, resveratrol (a SIRT1 agonist) treatment reduced MIRI via SIRT1 activation while Nicotinamide (NAM, a SIRT1 inhibitor) aggravated the myocardial cell apoptosis in dose dependent manner, indicating that SIRT1 can palliate myocardial cells apoptosis [17], [18], [19], [20].
Caffeic acid phenethyl ester (CAPE, Fig. 1A), a major phenolic active ingredient of propolis, has a wide range of pharmacological activities such as anti-microbial, anti-tumor, anti-oxidant, anti-inflammatory, and so on [21]. Due to its capabilities of scavenging ROS and immunomodulatory, CAPE has been reported to contribute to cardioprotection both in vitro and in vivo studies, including attenuation of cardiomyocyte apoptosis and reduction of myocardial infarction size [22]. Caffeic acid p-nitro phenethyl ester (CAPE-pNO2, Fig. 1A), a CAPE analogue with para nitro replace, was determined to be an anti-platelet agent and a protector of myocardial ischemia with more potent effects in our previous researches [23], [24]. According to the SYBYL molecular simulation software, nitro is the functional group in the cardioprotection, in addition, caffeic acid o-nitro phenethyl ester (CAPE-oNO2, Fig. 1A), synthesized by adding a nitro moiety to the ortho position in the present study has higher solubility and lower viscera toxicity (Suppl.Fig. 1). So we selected CAPE-oNO2 to investigate its potential cardioprotective effect and mechanism on MIRI in this study. As mentioned above, curcumin and resveratrol are polyphenol compounds and stimulate the SIRT1 activity to reduce MIRI. CAPE and CAPE-oNO2, also possessing polyphenol structure, have yet to be studied with regard as SIRT1 activation. Therefore, a specific SIRT1 inhibitor nicotinamide (NAM) had used to elucidate whether CAPE and CAPE-oNO2 can agitate SIRT1 signaling in this study.
Fig. 1.

Chemical structure of the compounds used in the present study, CAPE-oNO2synthetic routes and IR model establishment methods. (A) Chemical structure of CAPE, CAPE-pNO2 and CAPE-oNO2. (B) The synthetic routes of CAPE-oNO2. (C) The IR rat model was established by 30 min of LAD ligation and 2 h of reperfusion. (D) H9c2 cells were subjected to 2 h of hypoxia followed by 4 h of reoxygenation to induce MIRI. CAPE, caffeic acid phenethyl ester; CAPE-pNO2, caffeic acid p-nitro phenethyl ester; CAPE-oNO2, caffeic acid o-nitro phenethyl ester; IR, ischemia/reperfusion; LAD, left anterior descending coronary artery; MIRI, myocardial ischemia/reperfusion injury.
Nitric oxide (NO), a kind of endothelial diastolic factor, is important for maintaining the constant diastolic state of the cardiovascular system, regulating the coronary artery base tension and holding the perfusion of myocardial blood flow [25]. Mollaoglu H found that CAPE improved the level of NO to exert protective effect on Cd-induced hypertension mediated cardiac impairment in the rats [26]. eNOS, an enzyme responsible for NO generation, may be of tremendous value for MIRI. Following NOS inhibition with Nω-nitro-L-arginine methyl ester (L-NAME), coronary blood flow is prevented while over-expression of eNOS accelerates functional recovery [27], [28]. The results of the previously study indicated that nitro-substitution CAPE (CAPE-NO2) further increased myocardial survival, protected cardiac function following myocardial ischemia and increased NO content as compared with CAPE [24]. Since the only change of CAPE-NO2 structure is the introduction of nitro group, we hypothesized the reason of the stronger cardioprotection of CAPE-NO2 may be the more NO acceleration which is directly metabolized from CAPE-NO2 or indirectly derived from eNOS. In order to verify the source of NO and the contribution of eNOS on MIRI, we have investigated the eNOS expression and NO level by pretreatment with the eNOS inhibitor L-NAME in the present study.
2. Materials and methods
CAPE was purchased from Meilun Biotechnology (Dalian, China). CAPE-oNO2 was synthesized firstly in the present study. Triphenyltetrazolium chloride (TTC), trypsin, penicillin, streptomycin, dimethyl sulfoxide (DMSO), albumin from bovine serum (BSA), 3-[4, 5-dimethyl-2-thiazolyl]-2, 5-diphenyl-2-tetrazolium bromide (MTT) were purchased from Sigma chemicals (St. Louis, MO, USA). The lactate dehydrogenase (LDH), alpha-hydroxybutyrate dehydrogenase (HBDH), creatine Kinase (CK), creatine kinase isoenzymes (CK-MB) and ischemia modified albumin (IMA) assay kits were purchased from Maccura Biotechnology (Chengdu, China). The malondialdehyde (MDA), myeloperoxidase (MPO), total superoxide dismutase (T-SOD), NO assay kits and RIPA lysis buffer were purchased from Nanjing Jiancheng Biotechnology (Nanjing, China). H9c2 cardiomyocytes were obtained from the Cell Bank at the Chinese Academy of Sciences (Shanghai, China). Dulbecco's modified Eagle medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco/Invitrogen (Carlsbad, CA, USA). NAM and L-NAME were purchased form Selleck (USA). Acridine orange (AO) and 2′,7′-dichlorofluorescein-diacetate (DCFH-DA) were from Beyotime Biotechnology (Shanghai, China). Antibodies against silent information regulator 1 (SIRT1), endothelial nitric oxide synthase (eNOS), B-cell lymphoma-2 (Bcl-2), Bcl-2 associated X protein (Bax), peroxisomal proliferators-activated receptor γ coactivator-1α (PGC-1α), nuclear respiratory factor-2 (Nrf-2), superoxide dismutase 1 (SOD1), NF-kappa-B inhibitor (IκB), phosphorylated-IκB (p-IκB), nuclear factor kB P65 (P65), phosphorylated-P65 (p-P65), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), transforming growth factor β1 (TGF-β1), mothers against decapentaplegic homolog 2/3 (Smad23), phosphorylated-Smad23 (p-Smad23), Collagen, GAPDH and horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody were purchased from Proteintech Group Inc (Wuhan, China).
2.1. Synthesis and characterization of CAPE-oNO2
Firstly, a mixure of caffee acid (0.9 g, 5.0 mmol, Ⅰ) in thionyl chloride (SOCl2, 20 mL) was heated at 80 °C until the reaction fluid clarified and then distilled under vacuum to remove the excess SOCl2 completely to obtain the intermediate product (caffeoyl chloride, Ⅱ). Secondly, a solution of caffeoyl chloride in dichloromethane (DCM, 20 mL) was added o-nitrophenylethanol (0.8 g, 5.0 mmol, Ⅲ) which was dissolved in 20 mL DCM at room temperature. Next, a catalyst triethylamine TEA, 0.4 mL, 2.9 mmol)was added dropwise, and then the mixture was stirred at 80 °C for 3 h. Finally, the reaction mixture was evaporated under vacuum, and the crude product was purified by silica gel column chromatography (acetone/petroleum ether, 1:4, v/v). The final products were recrystallized from acetone to obtain pure crystals (Fig. 1B). 1H (CD3COCD3, 400 MHz) and 13C (CD3COCD3, 100 MHz) nuclear magnetic resonance (NMR) spectra were recorded on a spectrometer (Ascend 400, Bruker, USA) and liquid chromatography-mass spectrum (LC-MS) were determined on a mass spectrometer (LCMS-803010500, Shimadzu, Japan).
2.2. Preparation of myocardial IR model
Sprague-Dawley rats, weighting 200–250 g, were purchased from the Experimental Animal Centre of Chongqing Medical University [SCXK (Yu) 2015-001]. The animals were housed in a conditioned environment (22 ± 1 °C, 12 h light/darkness cycle, free access to food and water). All the animal protocols conformed to the National Institutes of Health (NIH) guidelines and were approved by the Ethical Committee for Animal of Southwest University and all efforts were made to minimize animal pain and suffering throughout the studies.
The myocardial IR rat model was induced by ligation/perfusing left anterior descending coronary artery (LAD) as described in our previous study [24]. Under anesthesia, the animals in a tracheotomy had an electrocardiogram record during the procedure. Then the chest was opened to expose the heart and the LAD was ligated using a 6-0 silk suture slipknot. After ischemia for 30 min, the LAD allowed to recover blood flow for 2 h with slipknot release. At the end of reperfusion, blood was collected and the heart was quickly removed. Rats were randomly divided into eight groups, 8 rats in each group: 1) the Con group, rats were treated with the vehicle (DMSO diluted with normal saline to a final concentration of 0.01%, 1 mL/kg) and a suture was passed under the LAD without occlusion; 2) the IR group: IR rats were treated with vehicle; 3) the CAPE group: IR rats with CAPE (0.1 mg/kg); 4) the oNO2 group: IR rats with CAPE-oNO2 (0.1 mg/kg); 5) the NAM + oNO2 group: IR rats were pretreated with NAM (1 mg/kg) before CAPE-oNO2 administration (0.1 mg/kg); 6) the NAM + Veh group: IR rats were pretreated with NAM (1 mg/kg) and vehicle (1 mL/kg); 7) the L-NAME + oNO2 group: IR rats with L-NAME (1 mg/kg) and CAPE-oNO2 (0.1 mg/kg); 8) the L-NAME + Veh group: IR rats with L-NAME (1 mg/kg) and vehicle (1 mL/kg).
CAPE and CAPE-oNO2 were dissolved in the vehicle to a final concentration of 0.1 mg/mL. CAPE, CAPE-oNO2 and vehicle were tail intravenously injected 15 min prior to ischemia, while NAM and L-NAME were given at 30 min before ischemia (Fig. 1C).
2.3. Quantification of myocardial infarct size
Myocardial infarct size was evaluated by TTC (1% in PBS) staining. The rat hearts were frozen at −20 °C then sliced into transverse 1-mm-thick section that was incubated in 1% TTC at 37 °C in the dark for 15 min. The area of the infarcted tissues which were stained white or pale was demarcated and measured digitally using Image-Pro Plus (IPP, Version 6.0, Media Cybernetics, Inc., Rockville, MD, USA). Infarct size was calculated as the ratio of infarcted myocardium to the risk region × 100%.
2.4. Determination of cardiac marker enzymes in serum
Serum samples were isolated from the blood samples by centrifugation (3000 rpm, 4 °C, 10 min). Serum cardiac marker enzymes including HBDH, LDH, CK, CK-MB and IMA were measured using spectrophotometric kits by automatic biochemical analyzer (Hitachi, Japan).
2.5. Determination of SOD, MPO activity and MDA, NO content in tissue
The heart tissues were homogenized (10,000 rpm, 4 °C, 5 min) in normal saline to obtain 10% homogenates. The homogenates were centrifuged (3000 rpm, 4 °C, 10 min), and the supernatants were collected for subsequently measurements. SOD, MPO, MDA and NO levels in the tissues were determined by commercially available kits as the respective manufacturer's protocols.
2.6. HE, SR and IHC staining of the heart tissues
The heart tissues were immersion-fixed in 4% paraformaldehyde and embedded in paraffin to prepare 5 µm slices. Deparaffinized and rehydrated slices were stained with haematoxylin-eosin (HE) and sirius-red (SR), respectively. Images of these stained sections were examined using a microscope (Olympus, Tokyo, Japan) and photographed with a digital camera. For immunohistochemical (IHC) analysis, heart tissue sections (5 µm) were incubated with TNF-α (1:200 dilution) at 4 °C for 12 h and subsequently incubated with anti-mouse HRP reagent for 1 h at room temperature. The sections were rinsed in PBS, added the diaminobenzidine and counterstained with hematoxylin. The images were acquired using a light microscopy and camera. The collagen deposition of SR staining and the area of TNF-α positive staining were measured by IPP.
2.7. Cell culture and Preparation of cardiomyocytes IR model
For normal cell culture, H9c2 cell was cultured in high glucose DMEM medium with 10% FBS, penicillin (100 U/mL) and streptomycin (100 μg/mL) in a humidified incubator (5% CO2, 95% air, 37 °C). Upon reaching 90% confluency, the cells were passaged with 0.25 mM trypsin/0.03% EDTA.
To establish cardiomyocytes IR model, H9c2 cell were subjected to 2 h of hypoxia followed by 4 h of reoxygenation. To simulate hypoxia, cells were cultured in a hypoxic incubator (5% CO2, 95% N2, 37 °C) and the culture medium was replaced with low glucose DMEM medium without FBS; Reoxygenation was conducted with the normal culture method. The control group cells kept in the normal incubator without the hypoxic/reoxygenation (HR) stimulus. CAPE and CAPE-oNO2 were dissolved in the vehicle (DMSO diluted with culture medium to a final concentration of 0.01%) and administered at the onset of reoxygenation; NAM and L-NAME were dissolved in the vehicle and given at the onset of hypoxia (Fig. 1D).
2.8. Cell toxicity and viability assay
H9c2 cells were seeded into 96-well plates at 1 × 104 cells/well and incubated for 24 h. To test the cell toxicity of CAPE-oNO2 and vehicle, cells were given CAPE-oNO2 or vehicle and cultured for 4, 8, 24 h, respectively. To evaluate the cell viability of CAPE and CAPE-oNO2, cells were subjected to HR and given CAPE and CAPE-oNO2. Next, the number of viabile cell was evaluated by MTT assay. Briefly, the cells were incubated with MTT at a final concentration of 0.5 mg/mL at the end of reoxygenation. After 4 h, the number of viable cells was measured by evaluating absorbance at 490 nm with a microplate reader (Tecan Infnite M1000, Austria).
2.9. AO and ROS staining
H9c2 cells were seeded into 6-well plates at 1 × 106 cells/well and incubated for 24 h followed by HR. For evaluation of apoptosis, cells were stained with AO dye solution (100 μg/mL) after reoxygenation. For measurement of ROS generation, cells were incubated with DCFH-DA (10 μmol/L) at 37 °C for 20 min. The apoptosis cells and ROS were detected by a fluorescence microscope (BD Biosciences, USA). The fluorescence intensity of ROS was quantified by IPP.
2.10. Western blot analysis
The heart tissues were homogenized in RIPA lysis buffer at 4 °C for 5 min to get 10% homogenates. The homogenates and H9c2 cells were lysed in RIPA buffer at 4 °C for 45 min, centrifuged (10,000 rpm, 4 °C, 5 min) and collected the supernatant for next experiment. The BCA protein assay kit was used to measure total protein concentration according to the manufacturer's protocol. Protein samples were separated on SDS-PAGE gels and transferred to PVDF membranes (Bio-Rad, U.S.). Following the membranes were blocked in tris-buffered saline and Tween 20 (TBST) containing 5% nonfat dry milk for 1.5 h and then blotted with SIRT1, GAPDH, eNOS, TGF-β1, P65, p-P65, IκB, p-IκB, PGC-1α, Nrf1, SOD1, IL-6, TNF-α, Bcl-2, Bax, Smad2/3, p-Smad2/3 and Collagen (1:2000) primary antibodies overnight at 4 °C. The membranes were washed with TBST and then incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies (1:5000) for 1.5 h at room temperature. After the membranes were washed again, bound antibodies were visualized with enhanced ECL reagent and quantifed via densitometric analysis using Quantity One software (Bio-Rad Laboratories, U.S.). Detection of GAPDH was used as a control.
2.11. Statistical analysis
Data were analyzed using Statistic Package for Social Science (SPSS, version 16.0) statistical analysis software. Data One-way analysis of variance (ANOVA) with the Student–Newman–Keuls post-test was used for comparisons of data between groups. P-values below 0.05 (p < 0.05) were considered indicative of statistical significance.
3. Results
3.1. Structural characterization of CAPE-oNO2
CAPE-oNO2: light brown solid, melting point 148–152 °C. 1H NMR (CD3COCD3, 400 MHz): δ 3.33(2 H, m, CH2), 4.43(2 H, t, J = 6.48, CH2), 6.23(1 H, d, J = 15.96, α-H), 7.50(1 H, d, J = 15.96, β-H), 6.87(1 H, d, J = 8.19, Ph-H), 7.03(1 H, dd, J = 8.09, 2.20, Ph-H), 7.15(1 H, dd, J = 4.15, 2.09, Ph-H), 7.52(1 H, d, J = 1.53, Ph-H), 7.61(1 H, d, J = 7.73, 1.53, Ph-H), 7.68(1 H, td, J = 7.51, 1.37, Ph-H), 7.96(1 H, dd, J = 8.16, 1.32, Ph-H). 13C NMR (CD3COCD3, 400 MHz): δ 31.7, 63.4, 114.3, 114.4, 114.5, 115.5, 121.7, 124.5, 126.7, 128.0, 132.8, 132.9, 133.0, 145.0, 145.4, 147.9, 166.2. LC-MS m/z (%): 328.0724[M-H]+ (calc. C17H15NO6: 329.02) (Suppl. Fig. 1).
3.2. CAPE and CAPE-oNO2 protected against myocardial injury in the IR rats
The infarct size of heart extended after the IR procedure, CAPE and CAPE-oNO2 caused a significant lessening in the infarct size, and the lessening degree of CAPE-oNO2 was more than that of CAPE (p < 0.05). However, pretreated with L-NAME and NAM could effectively abolish the alleviative effect of CAPE-oNO2 on the infarct size (p < 0.05, Fig. 2A). Furthermore, HE staining shows that the cardiac tissue exhibited a clear and well organized structure little inflammatory infiltration or cardiac necrosis in the control, but myocardial structural abnormalities histological changes including perinuclear vacuolization, necrosis and massive inflammatory infiltration were detected in the IR group, the myocardial structural abnormalities were almost undetectable in the CAPE and the CAPE-oNO2 group. In contrast, pretreated with NAM and L-NAME could neutralize the ameliorating effect of CAPE-oNO2 (Fig. 2B).
Fig. 2.

Effects of CAPE-oNO2on reducing infarct size and structural abnormality in the IR rat heart. (A) Results of TTC staining. Red-stained areas represent normal tissue and unstained pale areas indicate infarcted tissue. Infarct size as a percentage of total volume. Data are expressed as the mean ± standard deviation (n = 8). *p < 0.05 vs. the control group; #p < 0.05 vs. the IR group; &p < 0.05 vs. the CAPE group; +p < 0.05 vs. the oNO2 group. (B) Results of HE staining (×200). TTC, triphenyl tetrazolium chloride; HE, haematoxylin-eosin.
The measurement of cardiac enzymes as outlined in Table 1, the activities of LDH, HDBH, CK, CK-MB and MIA were significantly increased in the IR group (p < 0.05). Treating with CAPE-oNO2 decreased cardiac enzymes release much stronger than CAPE did (p < 0.05). Conversely, there were no marked differences among the L-NAME, NAM group and the IR group.
Table 1.
Effects of CAPE-oNO2 on reducing cardiac enzymes in IR rat serum.
| Con | IR | CAPE | oNO2 | NAM+ oNO2 | NAM+Veh | L-NAME+oNO2 | L-NAME+Veh | |
|---|---|---|---|---|---|---|---|---|
| CK (U/L) | 1119.33±91.50 | 2829.00±49.33* | 1800.40±89.11# | 1543.60±104#& | 2225.33±75.80+ | 2277.53±86.61+ | 2339.20±125.17+ | 2523±141.85+ |
| CK-MB (U/L) | 1224.33±47.12 | 1874.67±104.0* | 1358.20±50.52# | 1294.17±91.31# | 1707.67±116.60+ | 1673.33±60.70+ | 1638.43±55.30+ | 1655.00±55.00+ |
| LDH (U/L) | 1321.00±85.28 | 2226.67±94.52* | 1501.67±33.29# | 1351.00±101.8# | 1882.33±84.20+ | 1905.67±11.93+ | 1974.67±46.20+ | 1990.33±111.51+ |
| HBDH (U/L) | 666.67±30.55 | 1056.33±57.27* | 823.67±104.51# | 765.67±67.68#& | 970.67±61.65+ | 985.67±24.91+ | 1045.00±79.50+ | 1019.33±49.60+ |
| MIA (U/mL) | 45.73±2.88 | 70.00±6.26* | 60.88±0.98# | 59.13±3.00# | 63.85±2.07+ | 63.33±0.84+ | 63.06±0.93+ | 63.11±0.33+ |
LDH, lactate dehydrogenase; HBDH, alpha-hydroxybutyrate dehydrogenase; CK, creatine Kinase; CK-MB, creatine kinase isoenzymes; IMA, ischemia modified albumin. Data are expressed as the mean ± standard deviation (n = 8).
P<0.05 vs. the control group.
P<0.05 vs. the IR group.
P<0.05 vs. the CAPE group.
P<0.05 vs. the oNO2 group.
3.3. CAPE and CAPE-oNO2 motivated SIRT1 and eNOS expression in the IR rats
As shown in Fig. 3, the expression of SIRT1, eNOS and Bcl-2, and NO level were markedly decreased (p < 0.05). CAPE and CAPE-oNO2 increased significantly the content of SIRT1, eNOS and NO (p < 0.05), and the up-regulation of CAPE-oNO2 on SIRT, eNOS and NO was higher than those of CAPE (p < 0.05). Inversely, pretreated with NAM and L-NAME eliminated the CAPE-oNO2-mediated rise of SIRT1, eNOS, Bcl-2 and NO level, respectively (p < 0.01). The Bax expression was the opposite.
Fig. 3.

Effects of CAPE-oNO2on SIRT1, eNOS, Bcl-2 and Bax expression, and NO level of the IR rat heart. (A) Representative images of the Western blots are shown. GAPDH was used as an internal control. (B) The NO level in the heart tissue was determined by commercially available kits. Data are expressed as the mean ± standard deviation (n = 8). *p < 0.05 vs. the control group; #p < 0.05 vs. the IR group; &p < 0.05 vs. the CAPE group; +p < 0.05 vs. the oNO2 group. SIRT1, silent information regulator 1; eNOS, endothelial nitric oxide synthase; Bcl-2, B-cell lymphoma-2; Bax, Bcl-2 Associated X Protein.
3.4. CAPE and CAPE-oNO2 attenuated cardiac oxidative stress in the IR rat
After IR operation, the IR group had a significantly increasing in MDA content (Fig. 4A) and a significantly decreasing in T-SOD activity ((Fig. 4B, p < 0.05), and CAPE and CAPE-oNO2 administration markedly decreased these enzyme activity (p < 0.05). However, neither MDA content nor SOD activity had significant changes in the NAM and L-NAME group compared with the IR group. Western blot analysis showed that the expression of antioxidant responsive protein which including PGC-1α, Nrf-2 and SOD1 were decreased markedly after IR produce (p < 0.05) (Fig. 4C). Treatment with CAPE and CAPE-oNO2 activated PGC-1α, Nrf-2 and SOD1. However, CAPE-oNO2 didn’t increase the PGC-1α, Nrf-2 and SOD1 expression in the presence of NAM or L-NAME.
Fig. 4.

Effects of CAPE-oNO2on attenuating cardiac oxidative stress in the IR rat. (A) Production of MDA in heart tissue. (B) The activity of T-SOD in heart tissue. (C) The expression of PGC-1α, Nrf-2 and SOD1 were examined by Western blot assay and quantified by densitometric analysis. GAPDH was used as an internal control. Data are expressed as the mean ± standard deviation (n = 8). *p < 0.05 vs. the control group; #p < 0.05 vs. the IR group; &p < 0.05 vs. the CAPE group; +p < 0.05 vs. the oNO2 group. MDA, malondialdehyde; T-SOD, total superoxide dismutase; PGC-1α, peroxisomal proliferators-activated receptor γ coactivator-1α; Nrf-2, nuclear respiratory factor-2; SOD1, superoxide dismutase 1.
3.5. CAPE and CAPE-oNO2 remitted cardiac inflammation in the IR rat
In IHC staining (Fig. 5A), TNF-α level was meaningfully raised in the IR group (p < 0.05), and CAPE and CAPE-oNO2 settlement abated tissue TNF-α level (p < 0.05). L-NAME and NAM disposing elevated TNF-α level compared with CAPE-oNO2 group (p < 0.05). MPO activity was markedly fortified after the IR, treatment with CAPE and CAPE-oNO2 diminished tissue MPO levels (p < 0.05), whereas the MPO content wasn’t decreased by CAPE-oNO2 in the presence of L-NAME and NAM (Fig. 5B, p < 0.05). Moreover, western blot analysis indicated that p-P65/P65, p-IκB/IκB, IL6 and TNF-α dramatically lifted in the IR group (p < 0.05). Treatment with CAPE and CAPE-oNO2 decreased these proteins expression, and the reduced degree of CAPE-oNO2 was much heavier than that of CAPE (p < 0.05). However, the down-regulation effects of these proteins didn’t appear in the NAM and the L-NAME group (Fig. 5C).
Fig. 5.

Effects of CAPE-oNO2on remitting cardiac inflammation in the IR rat. (A) Results of IHC staining (×200). The integrated option density value was used to measure the expression level of TNF-α. (B) The activity of MPO in heart tissue. (C) The expression of IκB, p-IκB, P65, p-P65, TNF-α and IL-6 were examined by Western blot assay and quantified by densitometric analysis. GAPDH was used as an internal control. Data are expressed as the mean ± standard deviation (n = 8). *p < 0.05 vs. the control group; #p < 0.05 vs. the IR group; &p < 0.05 vs. the CAPE group; +p < 0.05 vs. the oNO2 group. IHC, immunohistochemistry; MPO, myeloperoxidase; TNF-α, tumor necrosis factor-α; IκB, NF-kappa-B inhibitor; p-IκB, phosphorylated-IκB; P65, nuclear factor kB P65, phosphorylated-P65 (p-P65), and IL-6, interleukin-6.
3.6. CAPE and CAPE-oNO2 attenuated cardiac fibrosis in the IR rat
In SR staining (Fig. 6A), significant collagen deposition in heart tissue was seen in IR group, and the collagen deposition could be obviously weakened by CAPE and CAPE-oNO2 therapy (p < 0.05). However, CAPE-oNO2 had little effects on inhibiting cardiac collagen deposition with NAM and L-NAME pretreatment.
Fig. 6.

Effects of CAPE-oNO2on attenuating cardiac fibrosis in the IR rat. (A) Collagen deposition was measured by SR staining (×200), followed by semi-quantitative analysis. (B) The expression of TGF-β, Smad23, p-Smad23 and collagen were examined by Western blot assay and quantified by densitometric analysis. GAPDH was used as an internal control. Data are expressed as the mean ± standard deviation (n = 8). *p < 0.05 vs. the control group; #p < 0.05 vs. the IR group; &p < 0.05 vs. the CAPE group; +p < 0.05 vs. the oNO2 group. SR, Sirius-red; TGF-β1, transforming growth factor β1; Smad23, mothers against decapentaplegic homolog 2/3; p-Smad23, phosphorylated-Smad23.
Meanwhile, the western blot showed that TGF-β1, p-Smad23/Smad23 and Collagen expression increased after the IR injury (p < 0.05). In the CAPE and CAPE-oNO2 group displayed a significant reduction of TGF-β1, p-Smad23/Smad23 and Collagen expression (p < 0.05). However, the expression of TGF-β1, p-Smad23/Smad23 and Collagen weren’t lessened in the NAM and L-NAME group (vs the CAPE-oNO2 group, p < 0.05, Fig. 6B).
3.7. CAPE and CAPE-oNO2 prevent HR injury in H9c2 cell model
MTT assay showed that there was no significant difference in cell viability between CAPE-oNO2 (0, 1, 10, 20, 40, 60, 80, 100 μM) groups and the control in spite of cultivation for 4, 8 and 24 h (Fig. 7A). After HR, a remarkable debasement in cell viability compared to control group were observed in MTT assay. Treatment with CAPE and CAPE-oNO2 at concentrations of 10–40 μM increased cell viability in a dose-dependent manner. However, the cell viability gradually decreased after treating with CAPE and CAPE-oNO2 at concentrations of 60 and 80 μM, compared with 40 μM CAPE and CAPE-oNO2 administration, respectively. Besides, the cell viability of CAPE group was lower than that of CAPE-oNO2 group (p < 0.05, Fig. 7B).
Fig. 7.

Effects of CAPE-oNO2on inhibiting cell apoptosis and ROS generation cell in the HR H9c2 cardiomyocytes. (A) To investigate the cytotoxicity of CAPE-oNO2, H9c2 cardiomyocytes were treated with 0, 5, 10, 20, 40, 60, 80 and 100 μM CAPE-oNO2 for 4, 8 and 24 h, followed by MTT analysis. After 2 h of hypoxia, H9c2 cell were treated with different concentrations of CAPE and CAPE-oNO2 in the absence or presence of NAM and L-NAME, followed by 4 h of reoxygenation. (B) The cell viability was measured with MTT assays. (C) The cell apotosis was detected with AO staining. (D) The ROS generation was stained with DCFH-DA. Data are expressed as the mean ± standard deviation (n = 8). *p < 0.05 vs. the control group; #p < 0.05 vs. the IR group; &p < 0.05 vs. the CAPE group; +p < 0.05 vs. the oNO2 group. ROS, reactive oxygen species; NAM, nicotinamide; L-NAME, Nω-nitro-L-arginine methyl ester, MTT, 3-[4, 5-dimethyl-2-thiazolyl]−2, 5-diphenyl-2-tetrazolium bromide; DCFH-DA, 2′,7′-dichlorofluorescein-diacetate.
And then, as shown in AO staining (Fig. 7C), a large number of normal H9c2 cell were stained green and the nucleus were clear and intact in the control, the number of viable cells decreased significantly and the characteristics of cell apoptosis, including chromosome fixation, fragmentation, and sparse cytoplasm were detected in the HR group. Treatment with CAPE and CAPE-oNO2 increased the viable cell quantity and remitted HR-induced cardiomyocytes apoptosis. However, the protection effect of CAPE-oNO2 was each partly suppressed by L-NAME and NAM. As ROS staining (Fig. 7D) shown that HR-induced ROS rise was prominently restrained by CAPE or CAPE-oNO2 treatment in a dose-dependent manner. But pretreatment with NAM and L-NAME, CAPE-oNO2 couldn’t excellently cut down the ROS production.
3.8. CAPE and CAPE-oNO2 agitated SIRT1 and eNOS signaling in the HR H9c2 cell
Western blot analysis (Fig. 8) showed that SIRT1, eNOS, PGC-1α, SOD1 and Bcl-2 protein level markedly decreased in the HR group (p < 0.05). CAPE and CAPE-oNO2 treatment increased the three proteins expression with dose-dependent. However pretreatment with NAM and L-NAME eliminated the rise of those proteins level, respectively (p < 0.05). The trend of p-P65/P65, p-IκB/IκB and Bax were the opposite.
Fig. 8.

Effects of CAPE-oNO2on SIRT1/eNOS pathway proteins expression in the HR H9c2 cells. The expression of SIRT1, eNOS,PGC-1α,p-IκB, IκB, p-P65, P65,SOD1, Bcl-2 and Bax were examined by Western blot assay and quantified by densitometric analysis. GAPDH was used as an internal control. Data are expressed as the mean ± standard deviation (n = 8). *p < 0.05 vs. the control group; #p < 0.05 vs. the IR group; &p < 0.05 vs. the CAPE group; +p < 0.05 vs. the oNO2 group.
4. Discussion
MIRI results in the cardiac contractile dysfunction, apoptotic and necrotic cell death, leading to fibrosis and irreversible myocyte damage [29]. The mechanism of MIRI is complex and involves that reperfusion exacerbates cell ROS generation, oxidative stress increase and various downstream transcription factors activations, including NF-κB, TNF-α and TGF-β1, to accelerate the pro-inflammatory and cell death pathways [30]. Therefore, suppression of oxidant stress and inflammation pathways holds great promise for alleviating cardiac fibrosis and cell death. Correspondingly, increasing evidence indicates that CAPE is an effective anti-inflammatory and antioxidant, and the two main properties of CAPE make it a promising cardioprotective efficacy. The proposed mechanism is seen as an attenuation of cardiomyocyte apoptosis via CAPE-inhibition of lipid peroxidation, NF-κB activation and Nrf2 inactivation, and ROS scavenging of CAPE is also involved in preventing the IR-induced apoptotic [31], [32], [33]. CAPE-oNO2, a new derivative of CAPE, was synthesized firstly and characterized in this study, and it might possess a variety of biological properties and potential cardiovascular protection on the basis of SYBYL analysis.
In this study, we have evaluated the effects of CAPE-oNO2 on the rat model of LAD ligation/reperfusion and the cell model of H9c2 cell cultured in the oxygen-glucose deprivation/reoxygenation (OGD/R) based on the positive control of CAPE. The results showed that CAPE-oNO2 treatment conferred a cardioprotective effect on the model rat and the model cell, as evidenced by decreased myocardial infarct size, ameliorated the structural abnormalities, reduced serum cardiac marker enzymes activities, increased oxidation resistance, decreased the inflammatory response, attenuated collagen deposition and improved cell viability. More importantly, CAPE-oNO2 exerted the much more capacity both in vivo and in vitro.
In vivo, the results of TTC staining, HE staining and cardiac enzymes measuring showed that CAPE-oNO2 was able to abolish the raise in infarct size, the structural abnormalities and the elevation of cardiac enzymes (CK, CK-MB, LDH, HBDH and MIA), increase the anti-apoptotic factor Bcl2 expression and reduce the pro-apoptotic factor Bax expression in the IR rats. These results indicated that CAPE-oNO2 increased myocardial survival and protected cardiac structure following myocardial ischemia, which was basically consistent with CAPE-pNO2 based on Duqin's research [24]. Moreover, CAPE-oNO2 could stimulate the activities of SIRT1 and eNOS more excellently than CAPE could do, resulting in the stronger cardioprotection of CAPE-oNO2. SIRT1 has an unequivocal protective demonstrated both in animal experiments and human studies [34]. The PGC-1α agitation of SIRT1 may protect against ischemia-induced oxidative damage [35], the level of P65 acetylation and TGF-β1 expression are inhibited by SIRT1 [36], and the phosphorylation of eNOS was significantly higher in an isolated myocardial IR model of SIRT1-overexpressing mice than in lack of SIRT1 mice [37]. Furthermore, eNOS that enhance NO release prior to ischemia protects against MIRI by preserving ischemic blood flow, attenuating platelet aggregation and leukocyte adherence [38]. Besides, NO has been reported to suppress NF-κB activation by stabilizing the NF-κB inhibitor IκB-α and inhibiting NF-κB DNA binding through S-nitrosylation of cysteine residue of the p50 subunit [39]. There is report that the resveratrol-mediated activation of SIRT1 increased MnSOD levels in cardiomyocytes and suppressed fibrosis, preserved cardiac function, and significantly improved the survival in TO-2 hamsters [40]. Likewise, our results illustrated that the CAPE-oNO2-mediated activation of SIRT1 increased anti-oxidative effect through PGC-1α system sensitization, decreased inflammatory response through NF-κB inactivation, suppressed fibrosis through TGF-β1 down-regulation in the IR rats.
Oxidative stress resulted from the IR-induced massive ROS generation is an important pathogenesis of MIRI [41], restraining the propagation of the oxidative chain reaction and direct decreasing the free radical levels could be significant to prevent IR-induced myocardial injury. The present study showed that IR could aggravate myocardial oxidative stress. CAPE-oNO2 significantly diminished the MDA level, intensified the T-SOD activity and increased the proteins expression of PGC-1α, Nrf1, SOD1 in the IR rat hearts. It implied that CAPE-oNO2 as a CAPE homolog also possess antioxidant activity that was involved in the cardio-protection on MIRI. MDA is an end-product of lipid peroxidation while SOD is an enzyme for scavenging ROS, they all could reflect the lipid peroxidation degree. Antioxidant responsive gene SOD1 is induced by the Nrf system sensitization of PGC-1α and the PGC-1α expression is incited by SIRT1 and inhibited by NF-κB [42].
CAPE-oNO2 could significantly rebate MPO and the major inflammatory gene expression including p-IκB/IκB, p-P65/P65, IL6 and TNF-α in the IR rats heart. ROS stimulates NF-κB activation by promoting IκB phosphorylation and degradation, which is one of the main ways of regulating cardiac inflammation and damage [43]. The disassembly of IκB and NF-κB allow -P65 subunit to translocate into nuclear and subsequently motivate the specific promoter sequence of the target genes, such as TNF-α, IL6, Bcl-2 and Bax, triggering inflammatory injury and apotosis [44]. Our results demonstrated that CAPE-oNO2 possessed the anti-inflammatory effect through inhabiting IκB/P65 pathway activation.
CAPE-oNO2 could remit collagen deposition through suppressing TGF-β1, p-Smad23/ Smad23 and Collagen. TGF-β1 whose expression is triggered by P65 and limited by SIRT1, makes myocardial fibroblasts to secrete copious collagen depositing in myocardial interstitium, causing the imbalance of collagen and fibrosis [45], [46], [47]. Moreover, ROS not only induce cardiomyocyte hypertrophy, apoptosis through TGF-β1 activation but also lead to vascular endothelial dysfunction through NO exhaustion, which aggravate the occurrence of myocardial fibers [48]. It indicated that the TGF-β1/Smad pathway was directly inhibited by the CAPE-oNO2-mediated SIRT1 activation and ROS scavenging, and that the P65 inactivation and eNOS activation of SIRT1 could also palliate the cardio fibrosis.
In vitro, The MTT assay testified that CAPE-oNO2 and vehicle had no cell toxicity at concentrations from 1 to 100 μM regardless of culturing for 4, 8 and 24 h. After HR, CAPE-oNO2 was able to increase cell viabilities in a dose-dependent manner at concentrations from 10 to 40 μM. However, the cell viabilities were gradually decreased at concentrations from 60 and 80 μM, it may because that when the concentration of CAPE-oNO2 overly increased, excessive NO was produced by eNOS overexpression to aggravate damage [36]. Therefore we used the concentrations from 10 to 40 μM for the next research. The results of AO staining and DCFH-DA staining showed that CAPE-oNO2 could reduce the HR-induced apoptosis and ROS generation, respectively. West-blot analysis exhibited the SIRT1, eNOS, Bcl-2, SOD1 up-regulations and p-IκB/IκB, p-P65/P65 down-regulations. These results denoted that CAPE-oNO2 increased survival, reduced apoptosis and ROS on HR H9c2 cell through SIRT1 agitation.
To further confirm that the SIRT1 and eNOS pathway play a critical role on the cardio-protective effect of CAPE-oNO2, we have pretreated with SIRT1 inhibitor NAM and the NOS inhibitor L-NAME before CAPE-oNO2 or vehicle administration, respectively. As anticipation, SIRT1 and eNOS expression were suppressed and the CAPE-oNO2-mediated cardioprotection was largely eliminated in the presence of the NAM and L-NAME both in vivo and in vitro. Interestingly, SIRT1 expression is only stifled by NAM while eNOS expression is suppressed by both two inhibitors, along with the alternation of P65, PGC-1α, TGF-β1downstream pathway, which manifested that SIRT1 may be an upstream of eNOS, and both SIRT1 and eNOS could down-regulate P65 signaling. After pretreatment of NAM and L-NAME, the CAPE-NO2-mediated NO increase was also eliminated, which indicated that NO may be derived from eNOS rather than directly metabolized from CAPE-NO2. The previous studies in our lab also showed that CAPE-NO2 couldn’t metabolize out NO. Lai XY used a mixture system of rat liver microsome and CAPE-NO2 to build a hepatic metabolism model in vitro and speculate that the main metabolic products of CAPE-NO2 included caffeic acid, ferulic acid, nitro phenylethyl alcohol and ferulic acid nitro phenethyl ester [49]. CAPE-NO2 was transformed into C14H16N2O6, C8H11O4P, C10H14N2O6S or C16H11NO7 rather than denitration metabolites in the HT-29 cells Xenografts athymic mice heart, liver, spleen and kidney [50]. What's more, Yao XF revealed that the major metabolic pathway of CAPE-NO2 may be a phase II reaction (biotransformed by glucuronidation, sulfonation or methylation), although a small quantity of them could be metabolized via a phase I reaction (hydrolysis) [51].
Summary, SIRT1 stimulates the deacetylation of NF-κB, PGC-1α and eNOS, leading to the inhibition of NF-κB transcription and the promotion of PGC-1α and eNOS activities. NO derived by eNOS suppresses NF-κB activation as well. Afterwards, the NF-κB inhibition, results in that oxidative stress reduces through PGC-1α/Nrf1 system sensitization, inflammatory response abates through TNF-α and IL-6 up-regulation, collagen deposition diminishes through TGF-β1/Samd signal inhibition, and necrocytosis lessens through Bcl-2 intensifying and Bax depressing. It signified that CAPE-oNO2 exert cardioprotective effect on anti-oxidatant, anti-inflammatory, collagen deposition and anti-apoptosis via the SIRT1/ eNOS/ NF-κB signaling agitation. The stronger cardioprotection of CAPE-oNO2 may attribute to the more powerful agitation of SIRT1/ eNOS/ NF-κB signaling (Fig. 9).
Fig. 9.

Cardioprotective effect of CAPE-oNO2. CAPE-oNO2 ameliorated MIRI by suppressing the oxidative stress, inflammatory response, fibrosis and necrocytosis via the SIRT1/eNOS/NF-κB pathway. IR, ischemia/reperfusion; HR, hypoxia/reoxygenation; L-NAME, eNOS inhibitor Nω-nitro-L-arginine methyl ester; NAM, SIRT1 inhibitor nicotinamide; CAPE, caffeic acid phenethyl ester; oNO2, caffeic acid o-nitro phenethyl ester.
5. Conclusion
Collectively, our findings using SD rat hearts and H9c2 cardiomyocytes suggest that CAPE-oNO2 exerts a profound cardioprotection against MIRI in the absence of NAM and L-NAME. In vivo, CAPE-oNO2 ameliorated MIRI by suppressing the oxidative stress, inflammatory response, fibrosis and necrocytosis via the SIRT1/eNOS/NF-κB/ pathway. In vitro, CAPE-oNO2 inhibits HR-induced cell apoptosis and ROS production by activating SIRT1/eNOS/NF-κB. Emphatically, CAPE-oNO2 presented a stronger cardioprotective effect than CAPE both in vivo and in vitro while NAM and L-NAME eliminates the CAPE-oNO2-mediated cardioprotection. These results reveal that CAPE-oNO2 may have tremendous therapeutic potential in the treatment of MIRI.
Acknowledgments
We would like to thank the Shanxi Zhaoyi Biological Co. Ltd., China (SWU2013130) and the Fundamental Research Funds for the Central Universities (XDJK2017A008) for financial support.
Acknowledgments
Disclosures
The authors have no disclosures to declare.
Author contributions
Li Z and Li D designed the project; Li D, Wang X and Li S performed the rat experiments; Li D, Zhou Y and Huang Q took charge the cell experiments; Li D and Li Z wrote the main manuscripts; H. T. Wang X analyzed and interpreted data; all authors reviewed the manuscript.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.11.023.
Appendix A. Supplementary material
Supplementary material
References
- 1.Heusch G., Musiolik J., Gedik N. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ. Res. 2011;109(11):1302–1308. doi: 10.1161/CIRCRESAHA.111.255604. [DOI] [PubMed] [Google Scholar]
- 2.Zweier J.L., Talukder M.A. The role of oxidants and free radicals in reperfusion injury. Cardiovasc. Res. 2006;70(2):181–190. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 3.Werns S.W., Shea M.J., Lucchesi B.R. Free radicals and myocardial injury: pharmacologic implications. Circulation. 1986;74(1):1–5. doi: 10.1161/01.cir.74.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Kurian G.A., Rajagopal R., Vedantham S. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxid. Med. Cell. Longev. 2016;2016(12):1656450. doi: 10.1155/2016/1656450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moens A.L., Claeys M.J., Timmermans J.P. Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int. J. Cardiol. 2005;100(2):179–190. doi: 10.1016/j.ijcard.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 6.Marin-Garcia J. Springer; New York: 2011. Cardioprotection and Signaling Pathways. [Google Scholar]
- 7.Ott M., Gogvadze V., Orrenius S. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 8.Zhao W., Zhao T., Chen Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol. Cell. Biochem. 2008;317(1–2):43–50. doi: 10.1007/s11010-008-9803-8. [DOI] [PubMed] [Google Scholar]
- 9.Pennathur S., Hecker L., Thannickal V.J. Humana Press; 2010. Oxidative Stress and Cardiovascular Fibrosis. In: Studies on Cardiovascular Disorders; pp. 425–441. [Google Scholar]
- 10.Song S.H., Lee M.O., Lee J.S. Sirt1 promotes DNA damage repair and cellular survival. Biomol. Ther. 2011;19(3):282–287. [Google Scholar]
- 11.Jiang W., Zhang X., Hao J. SIRT1 protects against apoptosis by promoting autophagy in degenerative human disc nucleus pulposus cells. Sci. Rep. 2014;4:7456. doi: 10.1038/srep07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawashima T., Inuzuka Y., Okuda J. SIRT1 overexpression modifies cardiac energy metabolism. J. Card. Fail. 2010;16(9):1026–1036. [Google Scholar]
- 13.Lagouge M., Argmann C., Gerharthines Z. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127(6):1109–1121. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 14.Salminen A., Kauppinen A., Suuronen T. SIRT1 longevity factor suppresses NF-kappaB -driven immune responses: regulation of aging via NF-kappaB acetylation? Bioessays. 2008;30(10):939–942. doi: 10.1002/bies.20799. [DOI] [PubMed] [Google Scholar]
- 15.Alcendor R.R., Gao S., Zhai P. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007;100(10):1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 16.Yang H., Zhang W., Pan H. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS One. 2012;7(9):e46364. doi: 10.1371/journal.pone.0046364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y., Duan W., Lin Y. SIRT1 activation by curcumin pretreatment attenuates mitochondrial oxidative damage induced by myocardial ischemia reperfusion injury. Free Radic. Biol. Med. 2013;65(4):667–679. doi: 10.1016/j.freeradbiomed.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Nadtochiy S.M., Redman E., Rahman I. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc. Res. 2011;89(3):643–649. doi: 10.1093/cvr/cvq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C.J., Yu W., Fu Y.C. Resveratrol protects cardiomyocytes from hypoxia-induced apoptosis through the SIRT1–FoxO1 pathway. Biochem. Biophys. Res. Commun. 2009;378(3):389–393. doi: 10.1016/j.bbrc.2008.11.110. [DOI] [PubMed] [Google Scholar]
- 20.Alcendor R.R., Kirshenbaum L.A., Imai S. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 2004;95(10):971-980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 21.Zhang P., Tang Y., Li N.G. Bioactivity and chemical synthesis of caffeic acid phenethyl ester and its derivatives. Molecules. 2014;19(10):16458–16476. doi: 10.3390/molecules191016458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parlakpinar H., Sahna E., Acet A. Protective effect of caffeic acid phenethyl ester (CAPE) on myocardial ischemia-reperfusion-induced apoptotic cell death. Toxicology. 2005;209(1):1–14. doi: 10.1016/j.tox.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 23.Zhou K., Li X., Du Q. A CAPE analogue as novel antiplatelet agent efficiently inhibits collagen-induced platelet aggregation. Pharmazie. 2014;69(8):615–620. [PubMed] [Google Scholar]
- 24.Du Q., Hao C., Gou J. Protective effects of p-nitro caffeic acid phenethyl ester on acute myocardial ischemia-reperfusion injury in rats. Exp. Ther. Med. 2016;11(4):1433. doi: 10.3892/etm.2016.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelm M., Schrader J. Control of coronary vascular tone by nitric oxide. Circ. Res. 1990;66(6):1561–1575. doi: 10.1161/01.res.66.6.1561. [DOI] [PubMed] [Google Scholar]
- 26.Mollaoglu H., Gokcimen A., Ozguner F. Caffeic acid phenethyl ester prevents cadmium-induced cardiac impairment in rat. Toxicology. 2006;227(1–2):15–20. doi: 10.1016/j.tox.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 27.Pabla R., Buda AJFlynn D.M., Blesse S.A. Nitric oxide attenuates neutrophil-mediated myocardial contractile dysfunction after ischemia and reperfusion. Circ. Res. 1996;78(1):65–72. doi: 10.1161/01.res.78.1.65. [DOI] [PubMed] [Google Scholar]
- 28.Ho Y.J., Lee A.S., Chen W.P. Caffeic acid phenethyl amide ameliorates ischemia/reperfusion injury and cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabeto. 2014;13(1):98–111. doi: 10.1186/1475-2840-13-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhalla N.S., Elmoselhi A.B., Hata T. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc. Res. 2000;47(3):446–456. doi: 10.1016/s0008-6363(00)00078-x. [DOI] [PubMed] [Google Scholar]
- 30.Neri M., Riezzo I., Pascale N. Ischemia/reperfusion injury following acute myocardial infarction: a critical issue for clinicians and forensic pathologists. Mediat. Inflamm. 2017;2017(4):7018393. doi: 10.1155/2017/7018393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan J., Ma Z., Han L. Caffeic acid phenethyl ester possesses potent cardioprotective effects in a rabbit model of acute myocardial ischemia-reperfusion injury. Am. J. Physiol.- Heart C. 2005;289(5):H2265–H2271. doi: 10.1152/ajpheart.01106.2004. [DOI] [PubMed] [Google Scholar]
- 32.Hassan N.A., El-Bassossy H.M., Mahmoud M.F. Caffeic acid phenethyl ester, a 5-lipoxygenase enzyme inhibitor, alleviates diabetic atherosclerotic manifestations: effect on vascular reactivity and stiffness. Chem.-Biol. Interact. 2014;213(3):28–36. doi: 10.1016/j.cbi.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 33.Lee Y., Shin D.H., Kim J.H. Caffeic acid phenethyl ester-mediated Nrf2 activation and IκB kinase inhibition are involved in NF-κB inhibitory effect: structural analysis for NF-κB inhibition. Eur. J. Pharmacol. 2010;643(1):21–28. doi: 10.1016/j.ejphar.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y., Duan W., Li Y. Novel role of silent information regulator 1 in myocardial ischemia. Circulation. 2013;128(20):2232–2240. doi: 10.1161/CIRCULATIONAHA.113.002480. [DOI] [PubMed] [Google Scholar]
- 35.Nemoto S., Fergusson M.M., Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J. Biol. Chem. 2005;280(16):16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 36.Fan Y., Hoberg J.E., Ramsey C.S. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23(12):2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nadtochiy S.M., Yao H., McBurney M.W. SIRT1-mediated acute cardioprotection. Am. J. Physiol.- Heart C. 2011;301(4):H1506–H1512. doi: 10.1152/ajpheart.00587.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulz R., Kelm M., Heusch G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004;61(3):402–413. doi: 10.1016/j.cardiores.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 39.Colasanti M., Persichini T. Nitric oxide: an inhibitor of NF-kappaB/Rel system in glial cells. Brain Res. Bull. 2000;52(3):155–161. doi: 10.1016/s0361-9230(00)00262-8. [DOI] [PubMed] [Google Scholar]
- 40.Tanno M., Kuno A., Yano T. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 2010;285(11):8375–8382. doi: 10.1074/jbc.M109.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westlin W., Mullane K. Does captopril attenuate reperfusion-induced myocardial dysfunction by scavenging free radicals? Circulation. 1988;77(2):30–39. [PubMed] [Google Scholar]
- 42.Coll T., Jové M., Rodríguezcalvo R. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes. 2006;55(10):2779–2787. doi: 10.2337/db05-1494. [DOI] [PubMed] [Google Scholar]
- 43.Song L., Yang H., Wang H.X. Inhibition of 12/15 lipoxygenase by baicalein reduces myocardial ischemia/reperfusion injury via modulation of multiple signaling pathways. Apoptosis. 2014;19(4):567–580. doi: 10.1007/s10495-013-0946-z. [DOI] [PubMed] [Google Scholar]
- 44.Rajendrasozhan S., Yang S.R., Edirisinghe I. Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid. Redox Sign. 2008;10(4):799–811. doi: 10.1089/ars.2007.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hangxiang Z., Jing W., Hailong D. Fibulin-2 deficiency attenuates angiotensin II-induced cardiac hypertrophy by reducing transforming growth factor-β signalling. Clin. Sci. 2014;126(4):275–288. doi: 10.1042/CS20120636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei C., Kim I.K., Kumar S. NF-κB mediated miR-26a regulation in cardiac fibrosis. J. Cell. Physiol. 2013;228(7):1433–1442. doi: 10.1002/jcp.24296. [DOI] [PubMed] [Google Scholar]
- 47.Chuang S.T., Kuo Y.H., Su M.J. Antifibrotic effects of KS370G, a caffeamide derivative, in renal ischemia-reperfusion injured mice and renal tubular epithelial cells. Sci. Rep. 2014;4:5814. doi: 10.1038/srep05814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saito K., Ishizaka N., Aizawa T. Iron chelation and a free radical scavenger suppress angiotensin II-induced upregulation of TGF-beta1 in the heart. Am. J. Physiol.- Heart C. 2005;288(4):H1836–H1843. doi: 10.1152/ajpheart.00679.2004. [DOI] [PubMed] [Google Scholar]
- 49.Lai X.Y. Southwest University; China: 2012. Research of p-nitro caffeic acid phenethyl ester in vitro metabolism, Thesis of M.Sc. in. [Google Scholar]
- 50.Tang H., Yao X., Yao C. Anti-colon cancer effect of caffeic acid p-nitro-phenethyl ester in vitro and in vivo and detection of its metabolites. Sci. Rep. 2017;7(1):7599. doi: 10.1038/s41598-017-07953-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yao X.F., Tang H., Ren Q. Inhibited effects of CAPE-pNO2 on cervical carcinoma in vivo and in vitro and its detected metabolites. Oncotarget. 2017;8(55):94197–94209. doi: 10.18632/oncotarget.21617. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material