

Abstract
In situ precipitation of magnetite and other minerals potentially sequesters dissolved arsenic (As) in contaminated aquifers. This study examines As retention and transport in aquifer sediments using a multistage column experiment in which magnetite and other minerals formed from added nitrate and ferrous iron (Fe). Sediments were collected from the Dover Municipal Landfill Superfund site. Prior to nitrate–Fe(II) addition, As was not effectively retained within the sediments in the column. The combination of nitrate (10 mM) and Fe(II) (4 mM), resulted in mineral precipitation and rapidly decreased effluent As concentrations to <10 μg L−1. Mineralogical analyses of sectioned replicate columns using sequential extractions, magnetic susceptibility and X-ray absorption spectroscopy indicate that magnetite and ferrihydrite formed in the column following nitrate–Fe(II) addition. This magnetite persisted in the column even as conditions became reducing, whereas ferrihydrite was transformed to more stable Fe oxides. This magnetite incorporated As into its structure during precipitation and subsequently adsorbed As. Adsorption to the minerals kept effluent As concentrations <10 μg L−1 for more than 100 pore volumes despite considerable Fe reduction. The results indicate that it should be feasible to produce an in situ reactive filter by nitrate–Fe(II) injection.
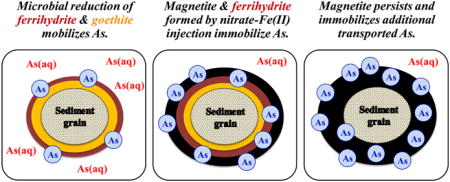
Graphical abstract
INTRODUCTION
As a result of natural processes and/or anthropogenic activities, unsafe levels of dissolved arsenic (As) in groundwaters are found in many parts of the world.1–3 Groundwater As contamination is also currently found at around 50% of National Priorities List (“Superfund”) sites in the U.S.4,5 Establishing effective strategies for groundwater As remediation has proven difficult. Traditional pump-and-treat is limited due to slow As desorption, aquifer heterogeneity and other factors.6,7 In situ remedial methods are aimed at immobilizing dissolved As within the aquifer matrix and usually involve transformation of iron (Fe) minerals to forms such as Fe(III) oxides and Fe(II) sulfides that retain As.4,8–12 The existing in situ remedial methods, however, are often ineffective due to instability of host mineral and/or changing flow paths. Many common Fe minerals are susceptible to microbially mediated redox changes,8,13–17 which hampers the long-term success of in situ As immobilization. Iron-based chemical amendments such as zerovalent iron (ZVI) have been used extensively within permeable barriers to intercept As from plumes.18 ZVI barrier methods can be effective when managed properly, but are plagued by decreasing barrier permeability and short residence times.18,19
Magnetite (Fe3O4) is a mixed-valence Fe oxide, which, unlike many common Fe minerals, is stable in a relatively wide range of conditions including both oxic and suboxic (Fe(III)-reducing) conditions where As is often solubilized,14,20,21 although magnetite formed under different in situ conditions may have different crystalline properties and different stability toward microbial Fe(III) reduction.22,23 Magnetite can sequester dissolved As through surface adsorption,9,24–27 and can coprecipitate As via incorporation into its crystal structure.16,28,29 In this study, we explore the potential of magnetite produced in situ within natural sediments to remediate groundwater As in columns under dynamic flow conditions typical of aquifer systems. Recently, a batch microcosm study using oxidized aquifer sediments from the Vineland Chemical Company Superfund site demonstrated that simultaneous addition of nitrate and Fe(II) produces an Fe oxide assemblage containing nanoparticulate magnetite that effectively immobilizes dissolved As(V).29 In situ mineral formation can be distinct from batch studies in part because aquifer systems are open, and have much higher solid-to-solution ratios than in batch microcosms. Nitrate and Fe(II) may not react together in the same way in open systems because nitrate travels more rapidly than Fe(II), which can be adsorbed on sediments. Aquifer hydrology also can be altered considerably by in situ precipitation, potentially leading to preferential flow, and affecting the capability of precipitated minerals to immobilize As and/or other ions. Additionally, As in contaminated aquifers is usually at least partly As(III), which reacts with magnetite and other minerals differently from As(V). These differences between batch studies and aquifer systems make it difficult to determine whether magnetite can successfully precipitate following nitrate–Fe(II) addition within real aquifers, and if it precipitates, whether this magnetite can effectively remediate groundwater As contamination.
Accordingly, the specific objectives of this column study were (i) to examine Fe mineral formation by nitrate–Fe(II) addition under continuous advective flow and realistic sediment-to-solution ratios, and (ii) to evaluate whether neoformed Fe minerals can retain As under long-term reducing conditions in an As(III)-rich environment. The temporal evolution of solution composition and mineralogical transformations were determined during the experiment. The results indicate that formation of magnetite and other Fe oxides by nitrate–Fe(II) addition can effectively immobilize dissolved As and capture additional advected As under prolonged reducing conditions. Thus, nitrate–Fe(II) injection can potentially be used to produce a reactive As filter and be established as an in situ remedial method.
MATERIALS AND METHODS
Site and Sediment Information
The Dover Municipal Landfill Superfund site (Dover, New Hampshire) was classified as a Superfund site in 1983, with the primary constituents of concern being volatile organic compounds (VOCs) and As.30 There is no known anthropogenic As source at the site, and this landfill site is similar to other landfill sites where elevated groundwater As concentrations were caused by the reducing conditions beneath the landfill mobilizing As from unconsolidated local sediments.14,20 Vapor extraction and pump-and-treat are the remedial strategies for VOCs and As, respectively, at this site.30 Groundwater VOC concentrations have decreased because of successful vapor extraction, along with natural attenuation and degradation processes associated with flushing. Groundwater As concentrations, however, have not decreased appreciably since remediation efforts began, and remain at around 150 μg L−1. Dover sediments used in this study were collected when extraction wells (part of the pump-and-treat system) EW1 and EW8b were installed in aquifer at the southwest and southeast toe of the landfill, respectively, by sonic vibration drilling. Immediately following collection, the sediments were sealed and refrigerated in steel cans with epoxy liners, and returned to laboratory for experiments. Detailed descriptions of the Dover Superfund site, and collection, handling and characterization of its sediments have been recently published elsewhere.6
Column Set-Up, Flow Condition, and Sampling
To conduct column experiment, Dover sediments were mixed 50%/50% w/w by dry weight with autoclaved silica sand (Acros Organics, 40–100 mesh) to improve flow properties. The sediments and sand were homogenized in an anaerobic glovebox filled with a 95%/5% N2(g)/H2(g) mixture, and then wet-packed into three identical 20 cm long polycarbonate columns with a 4 cm inner diameter and 0.5 cm walls. Before use, the column parts were washed with 70% ethanol and then autoclaved Milli-Q water. Glass wool was packed into each end of the columns to help distribute solution over the full cross sectional area. The three replicate columns are referred to hereafter as Dover columns. A fourth column with the exact same dimension was packed with autoclaved, pure silica sand, and is referred to hereafter as sand column. Using bromide breakthrough curves, effective porosity of three Dover columns and the sand column was estimated to be 0.28–0.30, similar to the Dover Superfund site (between 0.20 and 0.33). These four columns were run simultaneously and oriented vertically with upward flow as shown in Supporting Information (SI, Figure S1).
Four experimental stages, Stages I to IV, with varying influent compositions were designed and conducted to represent the stages of site remediation (details are in Table 1 and SI Table S1). Artificial groundwater (A-GW)8,16 was used, which mimics typical groundwater composition and consists of Milli-Q water amended with 0.02 mM NH4Cl, 1 mM KCl, 0.4 mM MgSO4, 0.4 mM CaCl2, and 10 mM PIPES buffer. The A-GW were added with additional amendments and adjusted to pH 7 in Stages I to IV (Table 1 and SI Table S1). Stage I used A-GW with 1 mM lactate and 100 μg L−1 As(III), to equilibrate the sediments and mimic As transport under landfill conditions. Lactate was used as a model organic carbon source and previously proved to be effective on stimulating microbial activity and reducing conditions within Dover sediments.6 Stage II used A-GW with 1 mM lactate, 100 μg L−1 As(III), 1 mg L−1 bromide, 4 mM FeSO4, and 10 mM NaNO3, to produce Fe minerals and test if they could immobilize As. The choices of the chemicals, FeSO4 and NaNO3, and their concentrations were based on findings from previous batch microcosms which were intended to induce Fe(II/III) mineral formation.29 Stage III used A-GW with 10 mM lactate, to further enhance microbial reduction and test if the neoformed Fe minerals are stable under reducing conditions that produce aqueous As. Stage IV used the same composition with Stage I, to return to reducing landfill condition with transported As and test if the neoformed Fe minerals could immobilize additional As. The influents were purged with N2(g) throughout Stages I to IV and injected into the columns by a peristaltic pump (ISMATEC). The injection rate was set at 2 pore volumes (PVs) day−1, equal to a flow velocity of 0.4 m day−1. The total duration of the column experiment was 4 months.
Table 1.
The design of the four-stage column experimenta
| Experimental Stage | Purpose | Amendments | # PVs |
|---|---|---|---|
| I: pre-equilibration | Mimic “landfill conditions” | 1 mM lactate and 100 μg L−1 As(III) | ~38b |
| II: mineral formation | Induce the formation of Fe minerals | 1 mM lactate, 100 μg L−1 As(III), 1 mg L−1 bromide, 10 mM nitrate and 4 mM Fe(II) | ~38 |
| Intermission: clean residual nitrate—Fe(II) amendment | 1 mM lactate | ~4 | |
| III: enhanced reduction | Further stimulate reduction | 10 mM lactate | ~45 |
| IV: reactive filter | Return to “landfill conditions” | 1 mM lactate and 100 μg L−1 As(III) | ~120 |
Three replicate Dover columns were sacrificed at the end of Stages II, III and IV, respectively. Sand column was sacrificed at the end of Stage V. More details, such as how the influents were prepared, are in SI Table S1.
We attempted to switch the experimental stage from Stage I to II at about PVs = 30, but switched back to Stage I after about 0.2 PV of injection (2 h) because precipitation was noticed in the Fe(II) influent. We therefore continued with Stage I for another 8 PVs (i.e., about 38 PVs in total) before successfully switching to Stage II.
Column effluents were collected and monitored for trace metals, anions, pH, and Eh. A fraction collector (LKB Bromma) was used to collect effluent samples. Trace metal samples were acidified to 1% HCl and analyzed by inductively coupled plasma mass spectrometry (ICP-MS). Anion samples were unacidified and analyzed by ion chromatography (IC). Reported data were from unfiltered effluent samples. This did not bias the results since curves of effluent concentration over PVs were smooth (suspended particles or colloids would produce concentration spikes) and were consistent with results from filtered samples (to 0.2 μm) that were collected occasionally. pH and Eh samples were measured using a calibrated electrode immediately following collection. Because sulfide smell was noticed in effluent from the sand column at Stage IV of the experiment, a few effluent samples from the columns were preserved with Zn-acetate31 and analyzed for dissolved sulfide immediately following collection.
Columns were sacrificed to retrieve solid samples for mineralogical characterization by sequential extraction, magnetic susceptibility, and X-ray absorption spectroscopy (XAS). The three replicate Dover columns were conducted such that they could be sacrificed at the end of Stages II, III, and IV of the experiment, respectively. The (one) sand column was sacrificed at the end of Stage IV. Each column was divided into 5 × 4 cm sections using a tube cutter and the solids were retrieved from the column sections immediately. Aliquots of the solids were immediately freeze-dried, and used in sequential extraction and digestion and in magnetic susceptibility measurement. Aliquots of the solids were also immediately coated in glycerol to prevent exposure to oxygen and stored at −20 °C for XAS analysis.
Analytical Procedures for Solid Samples
Sequential extractions were performed on 150 mg of ground freeze-dried solids and targeted seven different fractions (details are in SI Table S2). The procedure of sequential extractions was designed and validated in recent microcosm study,29 to differentiate magnetite from other Fe minerals, and to distinguish between As coprecipitated and adsorbed to magnetite. Elemental concentrations in the solid residue after the last extraction step were determined using microwave-assisted digestion with HNO3 and HF.32 Magnetic susceptibility was measured on freeze-dried solids with a Bartington MS2B instrument. Arsenic X-ray absorption near edge structure (XANES) spectra were collected on glycerol-coated solid samples from Dover columns at the Stanford Synchrotron Radiation Laboratory (SSRL) on beamline 11−2, configured with a Si(220) monochromator and a phi angle of 0 degrees. Sample spectra were collected with a 100-element Ge detector in fluorescence mode, in combination with a 6 μx Ge filter. The spectra were then processed in SIXpack.33 Normalized As XANES spectra were compared with reference spectra of Na-arsenate, Na-arsenite and orpiment (As2S3). Least-squares linear combination fitting was used to quantify the fraction of each reference in the sample. Iron extended X-ray absorption fine structure (EXAFS) spectra were also collected on the same beamline as described in SI Figure S2.
Analytical Procedures for Solution Samples
Dissolved trace metal concentrations in column effluents and extraction/digestion samples were determined by ICP-MS (Thermo Fisher Scientific Element XR) using previously published procedures.6,29 Dissolved bromide, nitrate and nitrite concentrations in column effluents were determined using a Dionex ICS-2000 IC system (Sunnyvale) with an IonPac AS15 analytical column and an AG15 guard column using a self-regenerating KOH eluent. Dissolved sulfide concentrations were analyzed using the methylene blue method.34
RESULTS
Dover Columns
Effluent Composition
Stage I of the experiment was designed to mimic landfill condition with 38 PVs of influents containing 1 mM lactate and 100 μg L−1 As(III), during which both As immobilization and release were observed in Dover columns (Figure 1A). Effluent As concentration changed rapidly, and coincided with changes in effluent Fe (Figure 1B). Eh value of the effluent was around +100 mV in the beginning of Stage I and then decreased to −90 mV (SI Figure S3). Additionally, when lactate-free influent was used, effluent As and Fe concentrations were consistently low, and Eh did not decrease over time (SI Figure S4). Stage II was designed to produce Fe minerals with 38 PVs of nitrate–Fe(II)-containing influents, during which effluent As concentration quickly decreased to and stayed below 10 μg L−1 (Figure 1A). Effluent nitrate and Fe concentrations were high due to the use of nitrate–Fe(II) amendment, but lower than amended levels (Figure 1B and 1C). Effluent nitrite was monitored by IC analysis but was never detected. The pH dropped half a pH unit during nitrate–Fe(II) injection (Figure 1D), while Eh stayed at around −90 mV (SI Figure S3). Once nitrate–Fe(II) amendment was discontinued, effluent nitrate and Fe concentrations decreased rapidly and pH returned to pH around 7. Stage III was designed to stimulate reduction with 45 PVs of influents containing 10 mM lactate, during which effluent As concentration remained below 10 μg L−1 (Figure 1A). Iron(III) reduction was apparent, with effluent Fe concentration staying between 0.2 and 0.4 mM (Figure 1B). Eh of the effluent stayed at around −60 mV during this stage (SI Figure S3). Stage IV was designed to return to “landfill condition” with 120 PVs of the same influent as Stage I, during which effluent As concentration remained below 10 μg L−1 for about 80 PVs and then slowly increased to 46 μg L−1 at the end of the stage (Figure 1A). Effluent Fe concentration was between 0.05 and 0.1 mM, indicating continued Fe(III) reduction (Figure 1B). Eh value during Stage IV stayed similar to Stage III, that is, at around −60 mV (SI Figure S3). Additional data show that effluent Ca concentration was variable over Stages I to IV probably due to cation exchange (SI Figure S3), effluent P (indicating phosphate) concentration was low to nondetect (data not shown), and effluent sulfide was nondetect. Replicate Dover columns, when available, behaved similarly.
Figure 1.
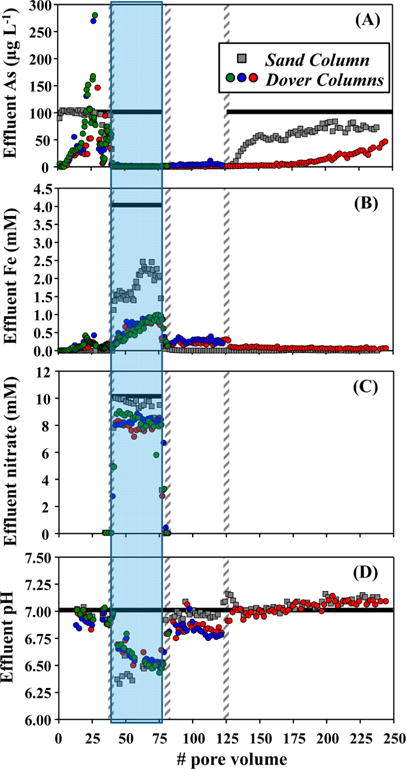
Effluent (A) As, (B) Fe and (C) nitrate concentrations, and (D) pH, as a function of pore volumes. Green, blue and red symbols represent replicate Dover columns which were sacrificed at different time, whereas grey symbols represent sand column. Vertical bars with diagonal lines represent the switches between experimental stages (Table 1) with nitrate–Fe(II) injection shown with shading. Horizontal black lines represent influent conditions.
Sediment Iron Mineralogy
Initial (unamended) Dover sediments used in this study had magnetic susceptibility of 40 (unitless, CGS units) before the column experiment began (Figure 2). Sequential extractions suggest that unamended Dover sediments did not contain recalcitrant Fe oxides, and contained primarily nonreactive Fe-bearing minerals (76 mol % of Fe in solid residues after sequential extractions) and lesser quantities of carbonates, (oxyhydr)oxides and sulfides. This Fe mineral composition is consistent with that determined by Fe EXAFS analysis (SI Figure S2).6 After Stage II with nitrate–Fe(II) injection, black-colored, magnetic particles was observed within Dover sediments near the influent end of the column (column inlet) (SI Figure S1). The amended sediments had a magnetic susceptibility of 2000 at the column inlet and decreased with distance in the column (Figure 2). Sequential extractions suggest significant quantities of recalcitrant Fe oxides and amorphous Fe(III) oxyhydroxides at the column inlet, which also decreased with distance in the column. After Stage III with enhanced Fe(III) reduction, magnetic susceptibility of the sediments and the quantity of recalcitrant Fe oxides remained similar to those before this stage (Figure 2). The quantity of amorphous Fe(III) (oxyhydr)oxides, on the other hand, decreased throughout the column. After Stage IV with continued Fe(III) reduction, magnetic susceptibility of the sediments and the quantity of recalcitrant Fe oxides still remained stable (Figure 2). The quantity of crystalline Fe(III) (oxyhydr)oxides decreased close to column outlet, Fe carbonates decreased close to column inlet, and Fe sulfides increased close to column inlet. Nonreactive residual Fe remained stable in Dover columns throughout the experiment.
Figure 2.
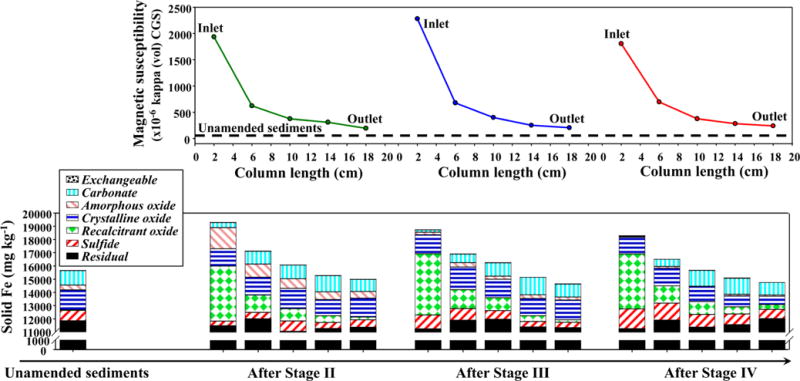
(Upper panel) Magnetic susceptibility measurements for the solid samples from Dover columns, which were sacrificed at the end of Stages II, III and IV, respectively. The thick black dash line represents unamended Dover sediments. (Lower panel) Results from sequential extractions and digestions on the solid samples for Fe.
Sediment Arsenic Speciation
Unamended Dover sediments contained 3.3 mg kg−1 of As, 61 ± 2% of which was As(III) (Figure 3). This As was almost completely extracted with hydroxylamine-hydrochloride and dithionite-citrate, suggesting that it was associated with amorphous and crystalline Fe(III) (oxyhydr)oxides. After Stages I and II with As(III) injection, the total concentration of As and the fraction of As(III) increased in the sediments, especially near the column inlet where As concentration increased to 7.0 mg kg−1, 78 ± 2% of which was As(III) (Figure 3). Sequential extractions suggest that the majority of this sediment As was associated with amorphous Fe(III) (oxyhydr)oxides (extracted with hydroxyl-amine-hydrochloride). After Stage III with extensive reduction, sediment As concentration remained similar to that before this stage, whereas the fraction of As(III) slightly increased (Figure 3). A large portion of the sediment As that was originally associated with amorphous Fe(III) (oxyhydr)oxides became associated with recalcitrant Fe oxides (extracted with ammonium oxalate) through a combination of coprecipitation and surface adsorption. Adsorption isotherms of As(III) were also conducted with Dover sediments retrieved after Stage III. Compared to the unamended sediments, As(III) adsorption capacity on the amended sediments significantly increased near the column inlet, and also seemed to slightly increase near the column outlet (SI Figure S5). After Stage IV with prolonged period of As(III) injection, sediment As concentration increased throughout the column (12.1 mg kg−1 near the column inlet), and the majority of that As was As(III) (89 ± 3% near the column inlet) (Figure 3). The amount of As coprecipitated within recalcitrant Fe oxides slightly increased, and the As on the surface of recalcitrant Fe oxides became the majority of the sediment As at the end of Stage IV. Crystalline Fe(III) (oxyhydr)oxides also were significant sinks of As at the end of Stage IV, and As associated with these phases were more prevalent near the column outlet. Despite of prolonged microbial reduction, no As-bearing sulfidic minerals were detected in Dover columns by As XANES or sequential extractions; and no significant As was associated with carbonates or residues after extractions (Figure 3).
Figure 3.
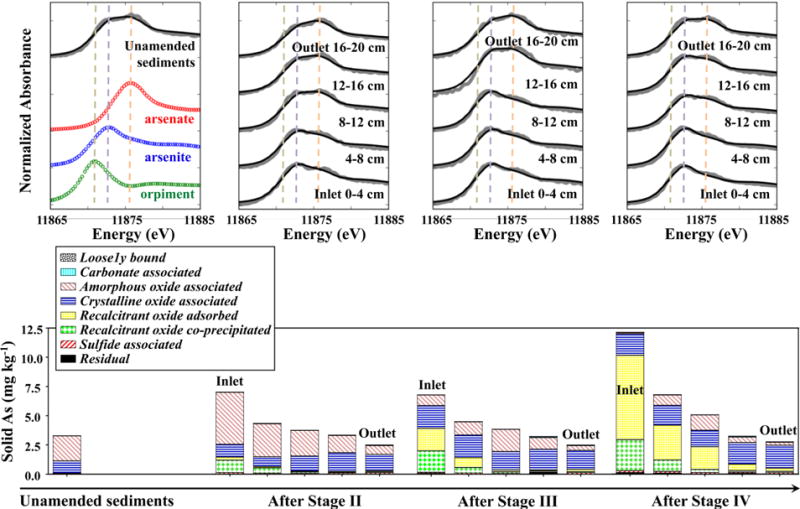
(Upper panel) Normalized As XANES spectra (thick grey lines) and linear combination fits (black lines) for the solid samples from Dover columns. Reference spectra are plotted for comparison. The spectra are vertically offset for clarity. Results of fitting are in SI Table S3. (Lower panel) Results from sequential extractions and digestions on the solid samples for As.
Sand Column
During Stage I, sand column quickly equilibrated with 100 μg L−1 As containing influent and effluent As concentration was around 100 μg L−1 (Figure 1A). Effluent pH stayed at around 7 (Figure 1D) and Eh stayed at around +130 mV (SI Figure S3). During Stage II, effluent As concentration quickly lowered to below 10 μg L−1. Effluent nitrate and Fe concentrations were lower than amended levels but higher than those in Dover columns (Figure 1B and C), indicating less Fe precipitation in sand column. Similar to Dover columns, effluent pH in sand column dropped to pH around 6.5 (Figure 1D) and effluent nitrite was nondetected. Effluent Eh was around −90 mV in the beginning of Stage II and then decreased to −140 mV at the end (SI Figure S3). During Stage III, effluent As and Fe concentrations were both low, pH returned to pH around 7, and Eh increased to +50 mV. During Stage IV, effluent As concentration increased to more than 10 μg L−1 after 10 PVs of 100 μg L−1 As containing influent and then stabilized at about 70 μg L−1 (Figure 1A). Concurrently, strong sulfide smell was noticed in the effluent and measured sulfide concentration was consistently about 1.2 mg L−1 (given that sulfide could escape as H2S(g) or be partially oxidized during collection, actual sulfide concentration was likely higher). Eh value of the effluent was around +15 mV in the beginning of Stage IV, sharply decreased to −50 mV after around 45 PVs of influent injection, and stayed at around −50 mV until the end (SI Figure S3). Effluent Fe concentration remained low in Stage IV (Figure 1B). Amended sand was retrieved from this column at the end of Stage IV for mineralogical analyses. Magnetic susceptibility increased from −0.5 in pure silica sand to more than 1200 at the column inlet (Figure 4A). Sequential extractions suggest that the amended sand contained recalcitrant Fe oxides and small quantity of Fe sulfides (Figure 4B), and that most of the As immobilized within the sand was associated with recalcitrant Fe oxides mainly as surface As and some structural As (Figure 4C).
Figure 4.
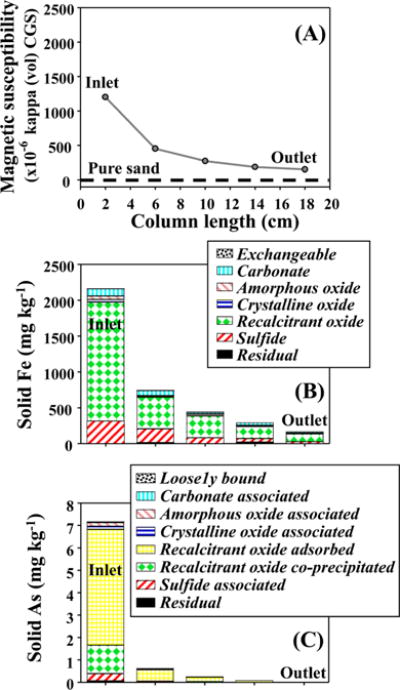
(A) Magnetic susceptibility measurements for the solid samples from sand column, which was sacrificed at the end of Stage IV. The thick black dash line represents pure sand. Results from sequential extractions and digestions on the solid samples for (B) Fe and (C) As.
DISCUSSION
Mechanisms of Magnetite Formation by Nitrate–Iron(II) Addition
Co-injection of nitrate and Fe(II) during Stage II of the column experiment produced black magnetic particles (SI Figure S1). Magnetic susceptibility, which is overwhelmingly sensitive to magnetite compared to other Fe(III) minerals, significantly increased in amended solids from both Dover columns (Figure 2) and sand column (Figure 4A) in Stage II, and then appeared to be stabilized in Stages III and IV. Sequential extractions also suggest that the amount of recalcitrant Fe oxides, which include magnetite, increased in Stage II and then stabilized in Stages III and IV (Figure 2 and Figure 4B). These observations consistently indicate that magnetite, and potentially other Fe minerals, precipitate from added nitrate and Fe(II) in these columns. Sequential extractions suggest that about 1700 mg kg−1 of the Fe retained at the column inlet at the end of Stage IV was probably present as magnetite in sand column, with more produced in Dover columns, about 4100 mg kg−1 or 20% of total Fe. Linear combination fitting of EXAFS spectra did not reveal significant mineralogical changes (SI Figure S2). We attribute this difference to the insensitivity of the EXAFS fitting to minor phases, and the combined errors in quantification (at least ±7% for magnetite for EXAFS fitting, probably similar in magnitude for extractions29,35), and possibly to problems with sample preservation prior to analysis. Structural differences between the precipitated magnetite in these columns and that used as a reference may also have biased EXAFS fitting.
As indicated by magnetic susceptibility and sequential extractions, most of the magnetite precipitated during nitrate–Fe(II) addition (Stage II). This magnetite can precipitate from (i) the partial oxidation of Fe(II) by nitrate; (ii) the partial oxidation of Fe(II) by nitrite; and/or (iii) the reductive recrystallization of amorphous ferrihydrite and presumably some other Fe(III) minerals.
The partial oxidation of dissolved Fe(II) by nitrate (Reaction 1) can be an abiotic (chemical) or a microbial process:36,37
| (1) |
Without catalysts (e.g., Cu2+), abiotic Fe(II) oxidation by nitrate is slow at circumneutral conditions.38,39 About half of the amended level of dissolved Fe(II) was eluted in the sand column, whereas significantly more of the Fe(II) was retained in Dover columns (Figure 1B). The faster Fe precipitation within Dover sediments reflects that the reaction(s) following nitrate–Fe(II) addition in Dover columns was microbially mediated. Microbes such as nitrate–dependent Fe(II) oxidizers can accelerate Fe(II) oxidation by nitrate and subsequent mineral precipitation. Such microbes have been encountered in numerous natural systems,40–43 with several being specifically reported to be important for the formation of biogenic magnetite.44–47 Indigenous nitrate–dependent Fe(II) oxidizers are likely present in natural Dover sediments.
Denitrifying microbes are also likely present in natural Dover sediments, which can use an electron donor such as organic matter and produce nitrite as an intermediate:39
| (2) |
Subsequently, magnetite can precipitate from the partial oxidation of Fe(II) by nitrite:
| (3) |
In contrast to nitrate, nitrite is an effective oxidant of Fe(II),39 making Reaction 3 a relatively rapid abiotic process. Nitrite was never detected in effluents from the columns in this study. The lack of nitrite is consistent with Reaction 3 being rapid and complete, or denitrification (Reaction 2) being inactive. The difference between Dover columns and sand column suggests that microbial processes are probably responsible for much of the magnetite formation. Identifying the specific microbial species within Dover sediments is a focus of ongoing research.
Other Fe minerals were also identified in amended Dover sediments, including a significant quantity of easily extracted amorphous Fe(III) oxyhydroxides (Figure 2). This amorphous Fe oxyhydroxide has been identified as ferrihydrite based on EXAFS results in these columns and in recent microcosm study.29 Ferrihydrite could be produced through a variety of pathways and then reductively transformed to some other Fe minerals.36,48 In Stage II with high concentration of dissolved Fe(II) (because of nitrate–Fe(II) addition, Figure 1B), Fe(II)-induced (re)crystallization of ferrihydrite is another mechanism that produces magnetite:36,37
| (4) |
Reactions 1, 3, and 4 all lead to the production of acidity, and are consistent with the slight decreases in pH observed during nitrate–Fe(II) addition (Figure 1D), even though 10 mM PIPES was used to buffer the system. Our ongoing research examines which metabolic pathway (Reaction 1, 3, or 4) plays the most significant role in producing magnetite following nitrate–Fe(II) addition. Regardless of how magnetite was produced, it was resistant to prolonged microbial reduction stimulated by addition of lactate (Figure 2). The thermodynamic stability of magnetite under reducing conditions in aquifers compared to other common Fe minerals makes it a better host for long-term As immobilization.
Arsenic Associated with Neoformed Iron Minerals
Before nitrate–Fe(II) amendment was used on Dover columns, effluent As concentration fluctuated rapidly (Figure 1A), which probably resulted from microbial reduction that was stimulated by exogenous organic carbon — lactate. Presumably due to changes in Fe mineralogy, nitrate–Fe(II) addition to Dover columns significantly enhanced As retention. Most of the dissolved As(III) injected into Dover columns was immobilized (Figure 1A). Sequential extractions suggested that some of the As was coprecipitated within recalcitrant oxides, including magnetite, during mineral formation stage, and this structural As persisted throughout subsequent reductive stages (Figure 3). Nitrate–Fe(II) addition produced ferrihydrite in parallel to magnetite in Dover columns (Figure 2) and in recent microcosm study.29 Ferrihydrite has a high adsorption capacity and fast adsorption kinetics for As oxyanions;7,38 extractions implied that significant quantities of As were also adsorbed on this ferrihydrite following nitrate–Fe(II) addition (Figure 3). Extensive transformation of ferrihydrite then occurred when lactate was added to stimulate microbial reduction, however, dissolved effluent As concentrations remained low even as As was a(Figure 1A). We attribute the continued retention of As under reducing conditions to the stability and sorption capacity of magnetite.29 Fresh magnetite could retain As released as ferrihydrite was reduced. In fact, extractions suggest that as additional dissolved As was injected into the columns in Stage IV, it adsorbed primarily to magnetite (Figure 3).
Although effluent As concentrations from the sand column were consistently lower than influent levels during Stage IV, the sand column had a much faster As breakthrough than Dover columns (Figure 1A). This faster As breakthrough might be related to less neoformed magnetite in the sand column than that in Dover columns (Figure 2 VS Figure 4). However, given the quantity and sorption capacity of magnetite that precipitated in the sand column (1700 mg kg−1 Fe as magnetite at the column inlet, suggested by sequential extraction), this breakthrough occurred in advance of sorption site saturation.10,27 We attribute the faster-than-expected breakthrough of As to the formation of thioarsenic solution complexes.13,49,50 Efforts to keep the sand column sterile were ineffective over the 4-month experiment, and obvious sulfate reduction was observed: dissolved sulfide was detected in the effluent, and Fe sulfide minerals were identified in the sand (Figure 4B). In principal, As(III) can precipitate following sulfate reduction, within As sulfide minerals such as As2S3 or some Fe sulfide minerals. However, when sulfide activities are higher than about 0.5 μM, As(III) can instead complex with dissolved sulfide.51,52 Consequently in the sand column, the formation of As-bearing sulfide minerals were limited (Figure 4C), and thioarsenic solution complexes were effectively advected and eluted.
Given that sulfate-reducing bacteria (SRBs) are ubiquitous in natural environments,50,53,54 sulfate reduction probably also occurred in Dover columns. Dover columns, however, contained excess reactive Fe(III) in forms like ferrihydrite, which reacts with dissolved sulfide:13,49,53
| (5) |
Reaction 5 is a fast, abiotic process which can effectively “buffer” dissolved sulfide concentrations at levels low enough to prevent thioarsenic complex formation. Even though Reaction 5 would dissolve ferrihydrite and release the ferrihydrite-adsorbed As, Dover columns contained enough other sorbents to retain As(III), including magnetite.
During Stages III and IV, one of the most apparent mineralogical changes is amorphous Fe(III) minerals including ferrihydrite being transformed to some more stable minerals including goethite (Figure 2). This change from ferrihydrite to goethite has the potential to significantly affect As retention. This change produces minerals that are more resistant to microbial reduction,8,13,36,55 but typically lowers the surface area and thus decreases adsorption capacity.10,27 Additionally, when reducing condition is prolonged and ferrihydrite is depleted, reductive dissolution/transformation of goethite would eventually become favorable.9,15,55 Consistent with these predictions, effluent As concentrations gradually increased for Dover columns at the end of Stage IV (Figure 1A).
IMPLICATIONS
The main goal of this study was to determine the efficacy of a novel magnetite forming As remedial method in dynamic flow-through columns that simulate in situ conditions at Superfund sites. This study demonstrated that coinjection of nitrate and Fe(II) enabled the formation of an Fe mineral assemblage that included significant amounts of magnetite and immobilized As even under prolonged Fe reduction. Magnetite can incorporate As into its structure during formation, in which case desorption (and As(V) reduction) are less likely. In natural aquifers with circumneutral pH and a microbial community, the neoformed biogenic magnetite can be nanoparticulate magnetite with large surface area29 and effectively immobilize additional As by surface adsorption. Injection of nitrate was suggested as a possible treatment route for As burdened Asian groundwater aquifers,12 by stimulating the formation of hydrous ferric oxides, which might not be stable within aquifers over the long-term. Co-injection of nitrate and Fe(II) to increase the formation of magnetite can potentially be an improvement on this route.
The nitrate–Fe(II) strategy would use direct injection rather than trenching. Although the Fe mineral assemblage would precipitate more near the injection point, it formed slowly enough to disperse through much of the column (Figure 2), and did not restrict the flow. Even where concentrated, the precipitation would not occupy more than 1% of the available pore space (based on porosity and the quantities and densities of magnetite and ferrihydrite). This implies that nitrate–Fe(II) injection(s) would less likely cause pore clogging issues or make the contaminated groundwater flow bypassing the amended zone, which are often associated with the physical presence of ZVI barrier and its voluminous corrosion products.18,56 For field implementation, amendment injections can also be designed to further disperse mineral precipitation and remediate larger volumes. Such nitrate–Fe(II) strategy, therefore, can potentially produce a stable, dispersed reactive filter within the aquifer and immobilize dissolved As from contaminated groundwater when it migrates through the amended zone.
Of course, continued efforts are warranted for the nitrate–Fe(II) strategy to be used at the field-scale. The composition of the nitrate–Fe(II) amendment itself probably needs to be refined according to varying field conditions, to increase the amount of neoformed magnetite, to limit the formation of thioarsenic complexes, and thus to further improve As retention. To better understand and utilize the reaction mechanisms behind this strategy, investigations on the synergistic effects between abiotic and microbial processes would be required. Laboratory experiments with real groundwater or a pilot field experiment, which contains phosphate, natural organic compounds and other ions that often impact the fate of As and potentially impact Fe mineralogical transformation,17,57–59 would be valuable. Reactive transport modeling can also be useful, to quantify the biogeochemical dynamics following nitrate–Fe(II) injection in laboratory columns, to scale laboratory results to field systems, and to design and optimize field operation of the strategy.60 Nevertheless, the findings in this column study with reduced sediments and As(III), together with the findings in recent microcosm study with somewhat oxidized sediments and As(V),29 suggest that coinjection of nitrate and Fe(II), which produces a Fe mineral assemblage containing magnetite, can potentially be a long-term As immobilization method under a relatively wide range of conditions.
Supplementary Material
Acknowledgments
This study was funded by National Institute of Environmental Health Sciences (grants ES010349 and ES009089). This is LDEO contribution number 8047. Some of the analyses were conducted at SSRL, a national user facility operated by Stanford University for the U.S. Department of Energy. We are grateful to D. Peschel, M. Webster and D. Luce for access to the Dover Municipal Landfill Superfund site, to D. Ntarlagiannis, L. Slater and A. Samuel for providing the column parts for the experiment, to J. Ross, M. Fleisher, K. Nguyen, T. Ellis, C. Lepre, and R. Davis for valuable assistance during laboratory work and analyses, and to A. van Geen and P. Schlosser for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.6b02362.
Tables S1: details on experimental stages; Tables S2: sequential extraction scheme; Tables S3: As XANES linear combination fitting results; Figures S1: digital pictures of column setup and black-colored magnetic particles from the amended Dover sediments; Figures S2: Fe EXAFS spectra collected on unamended and amended Dover sediments; Figures S3: additional solution data on column effluents; Figures S4: results from another Dover column with lactate-free influent; and Figure S5: As(III) adsorption isotherms with unamended and amended Dover sediments (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Fendorf S, Michael HA, van Geen A. Spatial and temporal variations of groundwater arsenic in South and Southeast Asia. Science. 2010;328(5982):1123–1127. doi: 10.1126/science.1172974. [DOI] [PubMed] [Google Scholar]
- 2.Smedley PL, Kinniburgh DG. A review of the source, behaviour and distribution of arsenic in natural waters. Appl Geochem. 2002;17(5):517–568. [Google Scholar]
- 3.Morin G, Calas G. Arsenic in soils, mine tailings, and former industrial sites. Elements. 2006;2(2):97–101. [Google Scholar]
- 4.U.S. EPA. Arsenic treatment technologies for soil, waste, and water. 2002 http://www.clu-in.org/download/remed/542r02004/arsenic_report.pdf.
- 5.Public Access U.S. EPA. CERCLIS Database. 2016 https://cumulis.epa.gov/supercpad/Cursites/srchsites.cfm.
- 6.Sun J, Bostick BC, Mailloux BJ, Ross JM, Chillrud SN. Effect of Oxalic Acid Treatment on Sediment Arsenic Concentrations and Lability under Reducing Conditions. J Hazard Mater. 2016;311:125–133. doi: 10.1016/j.jhazmat.2016.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.EPA, U. S. Improving Nationwide Effectiveness of Pump-and-Treat Remedies Requires Sustained and Focused Action to Realize Benefits Report. EPA Office of Inspector General, Memorandum Report 2003-P-000006. 2003 [Google Scholar]
- 8.Benner SG, Hansel CM, Wielinga BW, Barber TM, Fendorf S. Reductive dissolution and biomineralization of iron hydroxide under dynamic flow conditions. Environ Sci Technol. 2002;36(8):1705–1711. doi: 10.1021/es0156441. [DOI] [PubMed] [Google Scholar]
- 9.Pedersen HD, Postma D, Jakobsen R. Release of arsenic associated with the reduction and transformation of iron oxides. Geochim Cosmochim Acta. 2006;70(16):4116–4129. [Google Scholar]
- 10.Dixit S, Hering JG. Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: Implications for arsenic mobility. Environ Sci Technol. 2003;37(18):4182–4189. doi: 10.1021/es030309t. [DOI] [PubMed] [Google Scholar]
- 11.Kneebone PE, O’Day PA, Jones N, Hering JG. Deposition and fate of arsenic in iron- and arsenic-enriched reservoir sediments. Environ Sci Technol. 2002;36(3):381–386. doi: 10.1021/es010922h. [DOI] [PubMed] [Google Scholar]
- 12.Omoregie EO, Couture R-M, Van Cappellen P, Corkhill CL, Charnock JM, Polya DA, Vaughan D, Vanbroekhoven K, Lloyd JR. Arsenic bioremediation by biogenic iron oxides and sulfides. Appl Environ Microb. 2013;79(14):4325–4335. doi: 10.1128/AEM.00683-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burton ED, Johnston SG, Bush RT. Microbial sulfidogenesis in ferrihydrite-rich environments: Effects on iron mineralogy and arsenic mobility. Geochim Cosmochim Acta. 2011;75(11):3072–3087. [Google Scholar]
- 14.deLemos JL, Bostick BC, Renshaw CE, Sturup S, Feng XH. Landfill-stimulated iron reduction and arsenic release at the Coakley Superfund Site (NH) Environ Sci Technol. 2006;40(1):67–73. doi: 10.1021/es051054h. [DOI] [PubMed] [Google Scholar]
- 15.Postma D, Jessen S, Nguyen TMH, Mai TD, Koch CB, Pham HV, Pham QN, Larsen F. Mobilization of arsenic and iron from Red River floodplain sediments. Vietnam Geochim Cosmochim Acta. 2010;74(12):3367–3381. [Google Scholar]
- 16.Saalfield SL, Bostick BC. Changes in Iron, Sulfur, and Arsenic Speciation Associated with Bacterial Sulfate Reduction in Ferrihydrite-Rich Systems. Environ Sci Technol. 2009;43(23):8787–8793. doi: 10.1021/es901651k. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Schenkeveld WD, Kraemer SM, Giammar DE. Synergistic effect of reductive and ligand-promoted dissolution of goethite. Environ Sci Technol. 2015;49(12):7236–7244. doi: 10.1021/acs.est.5b01191. [DOI] [PubMed] [Google Scholar]
- 18.Noubactep C, Caré S, Crane R. Nanoscale metallic iron for environmental remediation: prospects and limitations. Water, Air, Soil Pollut. 2012;223(3):1363–1382. doi: 10.1007/s11270-011-0951-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wantanaphong J, Mooney S, Bailey E. Quantification of pore clogging characteristics in potential permeable reactive barrier (PRB) substrates using image analysis. J Contam Hydrol. 2006;86(3):299–320. doi: 10.1016/j.jconhyd.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Keimowitz AR, Simpson HJ, Stute M, Datta S, Chillrud SN, Ross J, Tsang M. Naturally occurring arsenic: Mobilization at a landfill in Maine and implications for remediation. Appl Geochem. 2005;20(11):1985–2002. [Google Scholar]
- 21.He YT, Fitzmaurice AG, Bilgin A, Choi S, O’Day P, Horst J, Harrington J, Reisinger HJ, Burris DR, Hering JG. Geochemical processes controlling arsenic mobility in groundwater: A case study of arsenic mobilization and natural attenuation. Appl Geochem. 2010;25(1):69–80. [Google Scholar]
- 22.Piepenbrock A, Dippon U, Porsch K, Appel E, Kappler A. Dependence of microbial magnetite formation on humic substance and ferrihydrite concentrations. Geochim Cosmochim Acta. 2011;75(22):6844–6858. [Google Scholar]
- 23.Muehe EM, Scheer L, Daus B, Kappler A. Fate of arsenic during microbial reduction of biogenic versus abiogenic As–Fe (III)– mineral coprecipitates. Environ Sci Technol. 2013;47(15):8297–8307. doi: 10.1021/es400801z. [DOI] [PubMed] [Google Scholar]
- 24.Wang YH, Morin G, Ona-Nguema G, Menguy N, Juillot F, Aubry E, Guyot F, Calas G, Brown GE. Arsenite sorption at the magnetite-water interface during aqueous precipitation of magnetite: EXAFS evidence for a new arsenite surface complex. Geochim Cosmochim Acta. 2008;72(11):2573–2586. [Google Scholar]
- 25.Chowdhury SR, Yanful EK, Pratt AR. Arsenic removal from aqueous solutions by mixed magnetite-maghemite nanoparticles. Environ Earth Sci. 2011;64(2):411–423. [Google Scholar]
- 26.Yean S, Cong L, Yavuz CT, Mayo JT, Yu WW, Kan AT, Colvin VL, Tomson MB. Effect of magnetite particle size on adsorption and desorption of arsenite and arsenate. J Mater Res. 2005;20(12):3255–3264. [Google Scholar]
- 27.Gimenez J, Martinez M, de Pablo J, Rovira M, Duro L. Arsenic sorption onto natural hematite, magnetite, and goethite. J Hazard Mater. 2007;141(3):575–580. doi: 10.1016/j.jhazmat.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 28.Coker VS, Gault AG, Pearce CI, van der Laan G, Telling ND, Charnock JM, Polya DA, Lloyd JR. XAS and XMCD evidence for species-dependent partitioning of arsenic during microbial reduction of ferrihydrite to magnetite. Environ Sci Technol. 2006;40(24):7745–7750. doi: 10.1021/es060990+. [DOI] [PubMed] [Google Scholar]
- 29.Sun J, Chillrud SN, Mailloux BJ, Stute M, Singh R, Dong H, Lepre CJ, Bostick BC. Enhanced and Stabilized Arsenic Retention in Microcosms through the Microbial Oxidation of Ferrous Iron by Nitrate. Chemosphere. 2016;144:1106–1115. doi: 10.1016/j.chemosphere.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.U.S. EPA. Final source control remedial action work plan, Dover Municipal Landfill Superfund Site. 2011 http://www.epa.gov/region1/superfund/sites/dover/481851.pdf.
- 31.Hare L, Carignan R, Huerta-Diaz MA. A field study of metal toxicity and accumulation by benthic invertebrates; implications for the acid-volatile sulfide (AVS) model. Limnol Oceanogr. 1994;39(7):1653–1668. [Google Scholar]
- 32.U.S. EPA. Method 3052: Microwave assisted acid digestion of siliceous and organically based matrices. Test Methods for Evaluating Solid Waste. 1995 [Google Scholar]
- 33.Webb SM. SIXpack: a graphical user interface for XAS analysis using IFEFFIT. Phys Scr. 2005;T115:1011–1014. [Google Scholar]
- 34.Rice EW, Bridgewater L, Association, A. P. H . Standard Methods for the Examination of Water and Wastewater. American Public Health Association; Washington, DC: 2012. [Google Scholar]
- 35.Poulton SW, Canfield DE. Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem Geol. 2005;214(3–4):209–221. [Google Scholar]
- 36.Hansel CM, Benner SG, Fendorf S. Competing Fe(II)-induced mineralization pathways of ferrihydrite. Environ Sci Technol. 2005;39(18):7147–7153. doi: 10.1021/es050666z. [DOI] [PubMed] [Google Scholar]
- 37.Schwertmann U, Cornell RM. Iron Oxides in the Laboratory. John Wiley & Sons; 2008. [Google Scholar]
- 38.Straub KL, Benz M, Schink B, Widdel F. Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl Environ Microb. 1996;62(4):1458–1460. doi: 10.1128/aem.62.4.1458-1460.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melton ED, Swanner ED, Behrens S, Schmidt C, Kappler A. The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle. Nat Rev Microbiol. 2014;12:797–808. doi: 10.1038/nrmicro3347. [DOI] [PubMed] [Google Scholar]
- 40.Hohmann C, Morin G, Ona-Nguema G, Guigner JM, Brown GE, Kappler A. Molecular-level modes of As binding to Fe(III) (oxyhydr)oxides precipitated by the anaerobic nitrate-reducing Fe(II)-oxidizing Acidovorax sp strain BoFeN1. Geochim Cosmochim Acta. 2011;75(17):4699–4712. [Google Scholar]
- 41.Pantke C, Obst M, Benzerara K, Morin G, Ona-Nguema G, Dippon U, Kappler A. Green Rust Formation during Fe(II) Oxidation by the Nitrate-Reducing Acidovorax sp. Strain BoFeN1. Environ Sci Technol. 2012;46:1439–1446. doi: 10.1021/es2016457. [DOI] [PubMed] [Google Scholar]
- 42.Senn DB, Hemond HF. Nitrate controls on iron and arsenic in an urban lake. Science. 2002;296(5577):2373–2376. doi: 10.1126/science.1072402. [DOI] [PubMed] [Google Scholar]
- 43.Xiu W, Guo H, Shen J, Liu S, Ding S, Hou W, Jie M, Dong H. Stimulation of Fe (II) oxidation, biogenic lepidocrocite formation and arsenic immobilization by Pseudogulbenkiania sp. strain 2002. Environ Sci Technol. 2016;50:6449. doi: 10.1021/acs.est.6b00562. [DOI] [PubMed] [Google Scholar]
- 44.Chaudhuri SK, Lack JG, Coates JD. Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Appl Environ Microb. 2001;67(6):2844–2848. doi: 10.1128/AEM.67.6.2844-2848.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dippon U, Pantke C, Porsch K, Larese-Casanova P, Kappler A. Potential function of added minerals as nucleation sites and effect of humic substances on mineral formation by the nitrate-reducing Fe (II)-oxidizer Acidovorax sp. BoFeN1. Environ Sci Technol. 2012;46(12):6556–6565. doi: 10.1021/es2046266. [DOI] [PubMed] [Google Scholar]
- 46.Miot J, Li JH, Benzerara K, Sougrati MT, Ona-Nguema G, Bernard S, Jumas JC, Guyot F. Formation of single domain magnetite by green rust oxidation promoted by microbial anaerobic nitrate-dependent iron oxidation. Geochim Cosmochim Acta. 2014;139:327–343. [Google Scholar]
- 47.Zhao LD, Dong HL, Kukkadapu R, Agrawal A, Liu D, Zhang J, Edelmann RE. Biological oxidation of Fe(II) in reduced nontronite coupled with nitrate reduction by Pseudogulbenkiania sp Strain 2002. Geochim Cosmochim Acta. 2013;119:231–247. [Google Scholar]
- 48.Zhu M, Frandsen C, Wallace AF, Legg B, Khalid S, Zhang H, Mørup S, Banfield JF, Waychunas GA. Precipitation pathways for ferrihydrite formation in acidic solutions. Geochim Cosmochim Acta. 2016;172:247–264. [Google Scholar]
- 49.Kocar BD, Borch T, Fendorf S. Arsenic repartitioning during biogenic sulfidization and transformation of ferrihydrite. Geochim Cosmochim Acta. 2010;74(3):980–994. [Google Scholar]
- 50.Keimowitz AR, Mailloux BJ, Cole P, Stute M, Simpson HJ, Chillrud SN. Laboratory investigations of enhanced sulfate reduction as a groundwater arsenic remediation strategy. Environ Sci Technol. 2007;41(19):6718–6724. doi: 10.1021/es061957q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Helz GR, Tossell JA, Charnock JM, Pattrick RAD, Vaughan DJ, Garner CD. Oligomerization in As(III) Sulfide Solutions - Theoretical Constraints and Spectroscopic Evidence. Geochim Cosmochim Acta. 1995;59(22):4591–4604. [Google Scholar]
- 52.Eary LE. The Solubility of Amorphous As2S3 from 25 to 90-Degrees-C. Geochim Cosmochim Acta. 1992;56(6):2267–2280. [Google Scholar]
- 53.Burton ED, Johnston SG, Kocar BD. Arsenic Mobility during Flooding of Contaminated Soil: The Effect of Microbial Sulfate Reduction. Environ Sci Technol. 2014;48(23):13660–13667. doi: 10.1021/es503963k. [DOI] [PubMed] [Google Scholar]
- 54.Onstott TC, Chan E, Polizzotto ML, Lanzon J, DeFlaun MF. Precipitation of arsenic under sulfate reducing conditions and subsequent leaching under aerobic conditions. Appl Geochem. 2011;26(3):269–285. [Google Scholar]
- 55.Kocar BD, Fendorf S. Thermodynamic constraints on reductive reactions influencing the biogeochemistry of arsenic in soils and sediments. Environ Sci Technol. 2009;43(13):4871–4877. doi: 10.1021/es8035384. [DOI] [PubMed] [Google Scholar]
- 56.Calabrò P, Moraci N, Suraci P. Estimate of the optimum weight ratio in zero-valent iron/pumice granular mixtures used in permeable reactive barriers for the remediation of nickel contaminated groundwater. J Hazard Mater. 2012;207:111–116. doi: 10.1016/j.jhazmat.2011.06.094. [DOI] [PubMed] [Google Scholar]
- 57.Larese-Casanova P, Haderlein SB, Kappler A. Biomineralization of lepidocrocite and goethite by nitrate-reducing Fe(II)-oxidizing bacteria: Effect of pH, bicarbonate, phosphate, and humic acids. Geochim Cosmochim Acta. 2010;74(13):3721–3734. [Google Scholar]
- 58.Porsch K, Dippon U, Rijal ML, Appel E, Kappler A. In-Situ Magnetic Susceptibility Measurements As a Tool to Follow Geomicrobiological Transformation of Fe Minerals. Environ Sci Technol. 2010;44(10):3846–3852. doi: 10.1021/es903954u. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Zhu M, Lan S, Ginder-Vogel M, Liu F, Feng X. Formation and secondary mineralization of ferrihydrite in the presence of silicate and Mn (II) Chem Geol. 2015;415:37–46. [Google Scholar]
- 60.Rawson J, Prommer H, Siade A, Carr J, Berg M, Davis JA, Fendorf S. Numerical modeling of arsenic mobility during reductive iron-mineral transformations. Environ Sci Technol. 2016;50:2459. doi: 10.1021/acs.est.5b05956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.