

Abstract
Background
Cycles of alcohol and stress are hypothesized to contribute to alcohol use disorders. How this occurs is poorly understood, although both alcohol and stress activate the neuroimmune system—the immune molecules and cells that interact with the nervous system. The effects of alcohol and stress on the neuroimmune system are mediated in part by peripheral signaling molecules. Alcohol and stress both enhance immunomodulatory molecules such as corticosterone and endotoxin to impact neuroimmune cells, such as microglia, and may subsequently impact neurons. In this study, we therefore examined the effects of acute and chronic ethanol (EtOH) on the corticosterone, endotoxin, and microglial and neuronal response to acute stress.
Methods
Male Wistar rats were treated intragastrically with acute EtOH and acutely stressed with restraint/water immersion. Another group of rats was treated intragastrically with chronic intermittent EtOH and acutely stressed following prolonged abstinence. Plasma corticosterone and endotoxin were measured, and immunohistochemical stains for the microglial marker CD11b and neuronal activation marker c‐Fos were performed.
Results
Acute EtOH and acute stress interacted to increase plasma endotoxin and microglial CD11b, but not plasma corticosterone or neuronal c‐Fos. Chronic EtOH caused a lasting sensitization of stress‐induced plasma endotoxin, but not plasma corticosterone. Chronic EtOH also caused a lasting sensitization of stress‐induced microglial CD11b, but not neuronal c‐Fos.
Conclusions
These results find acute EtOH combined with acute stress enhanced plasma endotoxin, as well as microglial CD11b in many brain regions. Chronic EtOH followed by acute stress also increased plasma endotoxin and microglial CD11b, suggesting a lasting sensitization to acute stress. Overall, these data suggest alcohol and stress interact to increase plasma endotoxin, resulting in enhanced microglial activation that could contribute to disease progression.
Keywords: Alcohol, Stress, Microglia, Neurons, Endotoxin
Alcohol use disorders (AUDs) are widespread mental health conditions that contribute to significant morbidity and mortality. Many studies have identified a relationship between AUDs and stress. Both alcohol and stress increase stress hormone levels and activity of brain stress systems (Koob et al., 2004). Stress and the release of stress hormones by the hypothalamic–pituitary–adrenal (HPA) axis have been implicated in binge/heavy drinking, motivation to drink, and the chronic relapsing nature of AUD (Blaine and Sinha, 2017; Stephens and Wand, 2012). Release of stress hormones is also linked to increased alcohol drinking, responses to alcohol cues, risk of relapse, and positive subjective responses to alcohol (Blaine and Sinha, 2017; Stephens and Wand, 2012). Interestingly, alcohol and stress both impact innate immune signaling, which has been suggested to contribute to AUDs (Crews et al., 2017; de Timary et al., 2017). Studies in rats find that acute ethanol (EtOH) dose dependently increases intestinal permeability (Ferrier et al., 2006), thereby increasing leakage of bacterial products such as endotoxin from the gut to the periphery and activating systemic and brain innate immune signaling (Crews et al., 2017; Qin et al., 2007; Rivera et al., 1998; de Timary et al., 2017). Other studies find both acute and chronic alcohol increase neuroimmune signaling in the brain (Vetreno and Crews, 2012; Walter and Crews, 2017). Acute stress also increases brain expression of cytokines such as TNF‐α, IL‐1β, and Ccl2 (Knapp et al., 2016) and the danger signaling molecule, high mobility group box‐1 protein (HMGB1) (Frank et al., 2015; Weber et al., 2015). Stress sensitizes neuroimmune responses (Frank et al., 2015), and stress hormones that are generally anti‐inflammatory have been hypothesized to have the opposite effect of enhancing brain innate immune signaling under some circumstances (Frank et al., 2015; Sorrells et al., 2009). Further, studies in rats find partial restraint stress increases blood endotoxin and stress hormones, such as ACTH and corticosterone (CORT), as well as brain proinflammatory cytokines. However, reducing endotoxin release with antibiotics, probiotics, and drugs resulted in a loss of the stress hormone response, suggesting stress‐induced changes in blood endotoxin contribute to stress hormone responses (Ait‐Belgnaoui et al., 2012). Innate immune signaling cytokines also alter EtOH consumption (Marshall et al., 2016, 2017), and the cytokine receptors IL1R1 and TNFR1 contribute to stress‐induced EtOH consumption (Karlsson et al., 2017), suggesting the neuroimmune system may contribute to addiction. Cytokines also enhance stressful emotional states following alcohol withdrawal (Breese et al., 2008). Although alcohol and stress both induce stress hormones and innate immune signaling, the mechanisms of interaction of these 2 responses and how they contribute to AUDs are poorly understood.
Neuroimmune signaling in the brain involves innate immune signaling molecules such as cytokines and chemokines that are constitutively expressed in brain microglia, that is, the brain tissue–specific monocytes (Kettenmann et al., 2011). Microglia are activated by both alcohol (Qin and Crews, 2012) and stress (Sugama et al., 2009). It is possible this may occur through alcohol‐ and stress‐induced increases in stress hormones and endotoxin. Furthermore, microglia can also modulate neuronal function. Studies find that microglia release cytokines, such as TNF‐α (Lewitus et al., 2016), and neurotrophins, such as brain‐derived neurotrophic factor (Parkhurst et al., 2013), that can impact neuronal activity. These data suggest alcohol and stress may impact neurons through microglia. However, whether alcohol and stress interact to impact plasma CORT, plasma endotoxin, microglia, or neurons is currently unknown. In this study, we investigated the effects of EtOH and stress on plasma CORT and endotoxin, as well as microglial and neuronal activation. We performed 2 experiments: the first studying acute EtOH and acute stress and the combination of both. In the second experiment, we investigated the effect of adolescent chronic EtOH on long‐lasting changes in the adult stress response to determine persistence of potential stress–alcohol interactions. In humans, adolescent stress, trauma, and binge drinking have been hypothesized to contribute to persistent dysfunctional HPA responses that increase risks of AUD (Schepis et al., 2011). We studied a rat adolescent intermittent EtOH (AIE) model that has been shown to cause a persistent increase adult brain neuroimmune signaling cytokines and chemokines to determine whether there were persistent changes in microglial or neuronal responses to stress (Crews and Vetreno, 2016; Vetreno and Crews, 2012; Vetreno et al., 2013). We examined plasma levels of CORT and endotoxin, and assessed microglial and neuronal activation by performing immunohistochemical stains for cluster of differentiation molecule 11B (CD11b) and c‐Fos, respectively. We hypothesized that acute EtOH and acute stress would interact to enhance plasma CORT and endotoxin levels, as well as microglial and neuronal activation. We further hypothesized that chronic EtOH would persistently enhance the response of CORT, endotoxin, microglia, and neurons to acute stress.
Materials and Methods
Animals
For the acute EtOH and acute stress studies, male Wistar rats (P75; Harlan Sprague‐Dawley; Indianapolis, IN) were acclimated to the animal facility for 3 weeks prior to experimental procedures. Subjects were group‐housed (n = 2/cage). For the chronic EtOH and acute stress studies, young pregnant female Wistar rats (embryonic day 17; Harlan Sprague‐Dawley) were acclimated to the animal facility prior to birthing. On postnatal day (P) 1, 24 hours after birth, litters were culled to 10 pups per dam and housed with their mother until group housing with same‐sex littermates at weaning on P21. All animals were housed in a temperature (20°C)‐ and humidity‐controlled vivarium on a 12‐h/12‐h light/dark cycle (light onset at 0700 hour) and provided ad libitum access to food and water. Experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill and conducted in accordance with National Institutes of Health regulations for the care and use of animals.
Acute EtOH and Acute Stress Experiment
Male Wistar rats (P96) were treated with EtOH (5.0 g/kg, 25% v/v, intragastric [i.g.]) or a comparable volume of water. One hour later, the rats were either acutely stressed or left in their home cage, resulting in a total of 4 treatment groups: water‐gavaged, nonstressed (CON); EtOH‐gavaged, nonstressed (EtOH); water‐gavaged, stressed (Stress); and EtOH‐gavaged, stressed (EtOH+Stress). Animals in the Stress groups received an acute stressor consisting of restraint/partial water immersion as previously described (Sugama et al., 2009). Briefly, the rats were restrained in DecapiCone restrainers (Cat. # DCL‐200; Braintree Scientific, Braintree, MA) and immersed up to their xyphoid processes in chilled water (21 ± 2°C) for 2 hours. The animals were sacrificed immediately following the termination of the stressor (see Fig. 1 A). Nonstressed animals remained in their home cages until sacrifice. Trunk blood was collected at sacrifice, and blood EtOH concentrations (BECs) were calculated using a GM7 Analyzer (Analox, London, UK).
Figure 1.
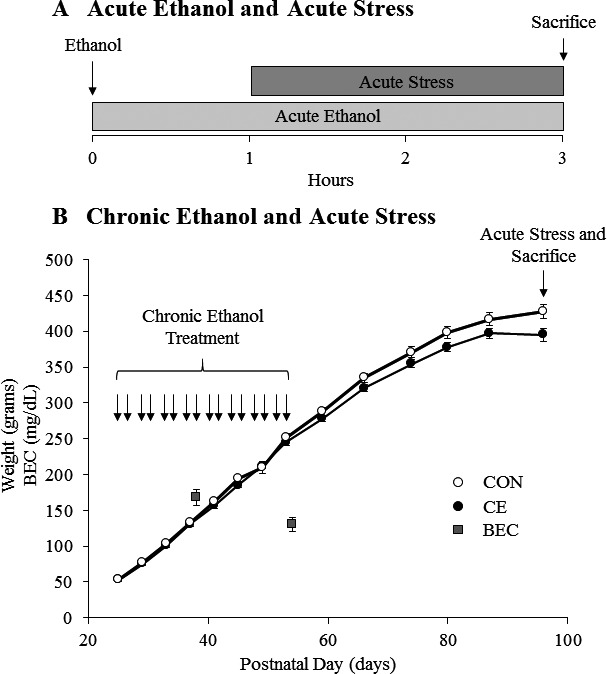
Graphical representation of the experimental protocols. (A) Rats were treated with acute ethanol (EtOH) (5 g/kg, 25% v/v, i.g.) or a comparable volume of water. The arrow represents the time of acute EtOH administration. One hour later, rats were acutely stressed for 2 hours using a restraint/water immersion procedure or were left in their home cage. The rats were sacrificed immediately following the conclusion of the stressor. (B) Body weights of control (CON—white dots) and chronic EtOH (CE—black dots)‐treated rats are shown over time. Rats were treated from P25 to P54 with either EtOH (5 g/kg, 20 to 30% v/v, i.g.) or water on a 2‐d on/2‐d off cycle. Arrows designate days the rats were treated. Blood EtOH concentrations (BECs) were measured 1 hour after EtOH treatment on P38 and P54 and are represented by gray squares. Rats were acutely stressed on P96 or P97 with a 2‐hour restraint/water immersion procedure or left in their home cage. The rats were sacrificed 2 hours following the conclusion of the stressor. Data are presented as mean ± SEM.
Chronic EtOH and Acute Stress Experiment
On P21, male subjects were weaned and randomly assigned to either chronic EtOH or water control (CON) treatment, as described previously (Vetreno and Crews, 2012). Beginning on P25, a standard AIE protocol was started as described previously (Vetreno and Crews, 2012). In brief, rats received i.g. administration of EtOH (5.0 g/kg, 20 to 30% v/v) on a 2‐d on/2‐d off schedule until P54 (see Fig. 1 B). Animals in the control condition received comparable volumes of water on an identical schedule. Tail blood was collected 1 hour after EtOH administration on P38 and P54 for the assessment of BECs. Following the conclusion of AIE treatment, subjects were pair‐housed (n = 2/cage) and maintained on ad libitum access to food and water for the duration of experiments. One group of animals was sacrificed on P55, 24 hours after the last EtOH exposure. The remaining animals continued to mature until P96 or P97. This was done to determine the effects of adolescent EtOH treatment on the adult brain response to stress. Therefore, a period of prolonged abstinence was included in between the chronic EtOH treatment and the acute stress. On P96/P97, animals were randomly assigned to either a stress or control treatment group, for a total of 4 groups: water‐gavaged, nonstressed (CON); EtOH‐gavaged, nonstressed (CE); water‐gavaged, stressed (Stress); and EtOH‐gavaged, stressed (CE+Stress). Stressed animals received a 2‐hour restraint/water immersion stressor as described above. The rats were sacrificed 2 hours after the conclusion of the 2‐hour acute stressor. Nonstressed animals remained in their home cages until sacrifice.
Perfusion and Brain Tissue Preparation
Rats were anesthetized with sodium pentobarbital (100 mg/kg, intraperitoneally) and transcardially perfused with 0.1 M phosphate‐buffered saline (PBS) (pH 7.4) followed by 4.0% paraformaldehyde in PBS. Brains were extracted, postfixed in 4.0% paraformaldehyde/PBS solution for 24 hours, and then placed in a 30% sucrose solution in PBS. Brain tissue was sectioned coronally at a thickness of 40 μm on a sliding microtome (MICROM HM450; Thermo Scientific, Austin, TX). Sections were sequentially collected into well plates and stored at −20°C in a cryoprotectant solution consisting of 30% glycol/30% ethylene glycol in PBS for immunohistochemistry.
CORT Measurements
Trunk blood was collected during perfusion. Blood was drawn from the left ventricle of the heart into a syringe, placed in BD Microtainer™ Plastic Capillary Blood Collector with lithium heparin (Cat. # 02‐668‐75; Fisher Scientific, Hampton, NH), and placed on ice. Plasma was collected by spinning down the whole blood twice at 4°C for 15 minutes at 13,800×g. CORT measurements were taken using an RIA kit (Cat. # 07120102; MP Biomedical, Santa Ana, CA). Briefly, rat plasma was diluted 1:200 in 10 × 75 mm glass tubes with the diluent provided in the kit as per the kit instructions. Radioactive CORT125I followed by anti‐CORT antibody provided in the kit was then added to the tubes as per the kit instructions. All tubes were vortexed and incubated at room temperature for 2 hours. A precipitant solution provided in the kit was added to the tubes, which were then centrifuged at 1,000×g for 15 minutes as per the kit instructions. The supernatant was aspirated, and radioactivity of the precipitate was measured by a gamma counter. The amount of bound radioactive CORT 125I was calculated from the counts and used with a standard curve to calculate the concentration of CORT in each sample.
Endotoxin Measurements
Trunk blood was collected during perfusion. Blood was drawn from the left ventricle of the heart into a syringe, place in BD Microtainer™ Plastic Capillary Blood Collector with lithium heparin (Cat. # 02‐668‐75), and placed on ice. Plasma was collected by spinning down the whole blood twice at 4°C for 15 minutes at 13,800×g. Endotoxin was measured using an Endotoxin Detection kit (Cat. # 50‐647U; Lonza, Basel, Switzerland). Briefly, plasma samples were diluted 1:3 in microcentrifuge tubes with endotoxin‐free water provided in the kit. The tubes were then heated for 10 minutes at 70°C in a heating block to denature plasma proteins that interfere with the assay. Fifty microliters of diluted plasma samples was then transferred to a 96‐well plate, and an equivalent volume of limulus amebocyte lysate provided in the kit was added to each well. The plate was incubated at 37°C for 10 minutes, and then, substrate solution provided in the kit was added to each well. After 30 to 120 minutes of further incubation at 37°C, stop reagent was added to each well, and the absorbance of each well was measured at 405 nm using a plate reader. The concentration of endotoxin in each well was calculated using the absorbance and a standard curve developed in the same plate.
Immunohistochemistry
Free‐floating sections were washed in 0.1 M PBS, incubated in 0.3% H2O2 for 30 minutes, washed again in PBS, and blocked for 1 hour at room temperature in 0.25% Triton X‐100/5% normal serum (Cat. # 19135680; MP Biomedicals, Solon, OH). Sections were transferred directly from the block to primary antibody diluted in blocking solution and were incubated overnight at 4°C. For the details of staining with each primary antibody, see Table 1. Sections were then washed in PBS, incubated for 1 hour in biotinylated secondary antibody (1:200; Vector Laboratories, Burlingame, CA), washed, and incubated for 1 hour in avidin–biotin complex solution (Cat. # PK6100; Vectastain ABC Kit; Vector Laboratories). The chromogen, nickel‐enhanced diaminobenzidine (Cat. # D5637; Sigma‐Aldrich, St. Louis, MO), was used to visualize immunoreactivity (IR). Tissue was mounted onto slides, dehydrated, and coverslipped.
Table 1.
List of Antibodies Used for Immunohistochemistry
| Antibody | Host | Dilution | Company | Catalog Number |
|---|---|---|---|---|
| c‐Fos | Rabbit | 1:10,000 | Calbiochem | PC38 |
| EGR1 | Rabbit | 1:150 | Cell Signaling | 15F7 |
| CD11b | Mouse | 1:1,000 | Serotec | MCA275R |
| Iba1 | Rabbit | 1:1,000 | Wako | 019‐19741 |
| CD68 | Mouse | 1:1,000 | Serotec | MCA341R |
| MHCII | Mouse | 1:500 | Serotec | MCA46R |
| iNOS | Mouse | 1:1,000 | Santa Cruz | sc‐7271 |
Microscopic Quantification and Image Analysis
For staining in the various brain regions, tissue was drawn as follows using the atlas of Paxinos and Watson (1998): for the prelimbic (PrL) and infralimbic (IL) cortex, a 1:6 series from approximately bregma +3.70 to +2.20 mm (~4 slices per animal); for the nucleus accumbens (NAc) core and shell, a 1:6 series from approximately bregma +1.70 to +0.70 mm (~4 slices per animal); for the dorsal and ventral bed nucleus of the stria terminalis (dBNST and vBNST), a 1:4 series from approximately bregma −0.26 to −0.40 mm (~3 slices per animal); for the paraventricular nucleus of the hypothalamus (PVN), a 1:3 series from approximately bregma −1.40 to −2.12 mm (~3 slices per animal); for the CA1 region, CA3 region, and dentate gyrus (DG) of the hippocampus, a 1:6 series from approximately bregma −2.56 to −3.60 mm (~4 slices per animal); for the central nucleus of the amygdala (CeA) and basolateral nucleus of the amygdala (BLA), a 1:3 series from approximately bregma −1.88 to −2.80 mm (~3 slices per animal); and for the lateral periaqueductal gray (lPAG), a 1:6 series from approximately bregma −5.80 to −6.72 mm (~3 slices per animal) (see Fig. S1). The only exception was staining in the PVN, for which 1 slice per animal was used due to limited tissue. A modified stereological profile quantification method was used to quantify immunopositive cell counts and pixel density within the regions of interest. We have previously published that a comparison of this method with unbiased stereological methodology yielded nearly identical values relative to control subjects (Crews et al., 2004). Images of the regions of interest were captured using an Olympus BX50 microscope (Olympus Corporation, Tokyo, Japan) and Sony DXC‐390 video camera (Sony Corporation, Tokyo, Japan) linked to a computer (see Fig. S1). BioQuant Nova Advanced Image Analysis (R&M Biometric, Nashville, TN) was used for image analysis. The threshold was rigorously determined by calculating the average of the darkest and lightest values from each region of interest from control subjects. Sections were imaged under identical conditions (Beynon and Walker, 2012). Immunopositive cell count or pixel density was then determined by the BioQuant program. The area of the outlined regions of interest was determined, and IR was calculated by dividing either cell counts or pixel density by the overall area (mm2).
Statistical Analyses
The Statistical Package for the Social Sciences (SPSS; Chicago, IL) was used for all statistical analyses. The data from each experiment was analyzed using a 2‐by‐2 analysis of variance. The statistical significance of main effects and interactions was determined and is reported in a Table S1. Significant differences between individual groups were further investigated using Tukey's post hoc test for multiple comparisons, unless otherwise noted. All values are reported as mean ± SEM, and significance was defined at a level of p ≤ 0.05.
Results
Effects of Acute EtOH and Acute Stress on Plasma CORT and Endotoxin Levels
Previous studies find that acute EtOH (Ellis, 1966) and acute stress increase plasma CORT. Furthermore, acute EtOH (Rivera et al., 1998) and acute stress (Ait‐Belgnaoui et al., 2012) also increase plasma endotoxin. However, whether EtOH and stress interact to impact plasma CORT or plasma endotoxin is unknown. To investigate this, rats were treated with acute EtOH (5 g/kg, 25% v/v) and 1 hour later treated with a 2‐hour restraint/water immersion stressor (Fig. 1 A), as described previously (Sugama et al., 2009). This stress protocol has been widely used to study the effects of stress, including ulcers (Kato et al., 1998) and memory impairment (Mizoguchi et al., 2000). The rats were sacrificed immediately following the conclusion of the stressor, and trunk blood was collected. Analysis of BECs found that rats in the EtOH and EtOH+Stress groups had comparable BECs (191 ± 33 mg/dl in the EtOH group vs. 221 ± 21 mg/dl in the EtOH+Stress group). Acute EtOH increased plasma CORT 41‐fold (p < 0.05), while 2 hours of acute restraint/water immersion stress increased plasma CORT a marked 78‐fold (p < 0.05) (Fig. 2 A). However, the combination of acute EtOH and acute stress did not further increase plasma CORT beyond the levels induced by acute stress alone (Fig. 2 A). There was a trend for acute EtOH and acute stress to increase plasma endotoxin individually (Fig. 2 B). Interestingly, the combination of acute EtOH and acute stress caused a 50‐fold increase in plasma endotoxin that was significantly higher (p < 0.05) than endotoxin levels in the Stress group (Fig. 2 B). Overall, these results show plasma CORT is markedly increased by acute EtOH and acute stress, while plasma endotoxin shows a modest increase with EtOH or stress alone, and a marked increase when EtOH and stress are combined.
Figure 2.
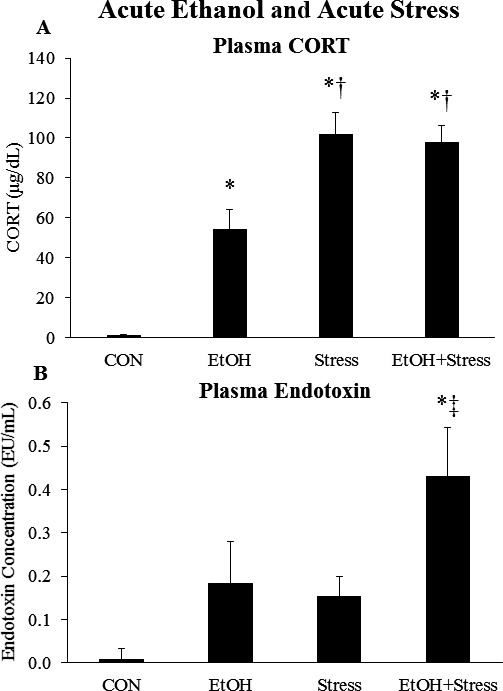
Effects of acute ethanol (EtOH) and acute stress on plasma corticosterone and endotoxin. Rats were treated with EtOH (5.0 g/kg, 25% v/v, i.g.) and/or a 2‐hour restraint/water immersion stressor and sacrificed immediately following the conclusion of the stressor. (A) Plasma corticosterone levels were measured. Data are presented as mean ± SEM. *p < 0.05 compared to CON, † p < 0.05 compared to EtOH (Tukey's post hoc test). n = 4 to 10/group. (B) Plasma endotoxin levels were measured. Data are presented as mean ± SEM. *p < 0.05 compared to CON, ‡ p < 0.05 compared to Stress (least significant difference post hoc test). n = 4 to 10/group.
Effects of Acute EtOH and Acute Stress on Microglia and Neurons in the Prefrontal Cortex
Previous studies show that acute EtOH and acute stress individually activate microglia (Ahlers et al., 2015; Sugama et al., 2009) and neurons (Vilpoux et al., 2009; Watanabe et al., 1994) across multiple brain regions. However, whether the 2 interact to affect microglial or neuronal activation is unknown. To investigate this, we treated rats with acute EtOH and acute stress as described above and examined several brain regions using CD11b and c‐Fos as markers of microglial and neuronal activation, respectively (Tables 2 and 3). We first examined the medial prefrontal cortex (mPFC), a brain region affected by both EtOH and stress. In the prelimbic mPFC (PrL), neither acute EtOH nor acute stress increased CD11b+ IR; however, the 2 interacted to increase CD11b+ IR 67% (p < 0.05) (Fig. 3 A). A similar result was seen in the infralimbic mPFC (IL) (Table 2). In the PrL, acute EtOH increased c‐Fos+ cell density 2.6‐fold (p < 0.05), while acute stress increased c‐Fos+ cell density 6.1‐fold (p < 0.05) (Fig. 3 B). However, acute EtOH and acute stress did not further interact to enhance c‐Fos+ cell density beyond the levels caused by stress alone (Fig. 3 B). Results were similar in the IL (Table 3). We found plasma CORT and PrL c‐Fos were significantly correlated across groups (r = 0.695, p < 0.001). Plasma endotoxin and PrL CD11b+ IR were also significantly correlated across groups (r = 0.495, p = 0.019). However, plasma CORT and PrL CD11b did not correlate, and plasma endotoxin and PrL c‐Fos did not correlate. This suggests PrL neuronal activation is related to the CORT response, whereas PrL microglial activation is related to the endotoxin response rather than the CORT response. Overall, these data suggest acute EtOH and acute stress interact to increase mPFC CD11b+ IR, but not mPFC neuronal c‐Fos.
Table 2.
Effects of Acute Ethanol (EtOH) and Acute Stress on CD11b Expression Across Brain Regions (% control)
| Brain region | CON | E | S | E+S |
|---|---|---|---|---|
| Prefrontal cortex | ||||
| PrL | 100 ± 19a | 100 ± 23a,b | 94 ± 10a | 167 ± 23b |
| IL | 100 ± 21a | 91 ± 18a,b | 74 ± 6a | 149 ± 23b |
| Nucleus accumbens | ||||
| Core | 100 ± 8a | 109 ± 30a | 127 ± 22a | 195 ± 19b |
| Shell | 100 ± 8a | 97 ± 20a | 126 ± 21a,b | 169 ± 13b |
| Bed nucleus of the stria terminalis | ||||
| dBNST | 100 ± 14a | 171 ± 56a,b | 202 ± 25a,b | 283 ± 30b |
| vBNST | 100 ± 10a | 138 ± 29a,b | 215 ± 27a,b | 247 ± 32b |
| Hypothalamus | ||||
| PVN | 100 ± 28a | 126 ± 36a | 551 ± 70b | 361 ± 20b |
| Amygdala | ||||
| BLA | 100 ± 16a | 112 ± 15a | 129 ± 13a | 207 ± 18b |
| CeA | 100 ± 8a | 108 ± 10a | 99 ± 9a | 153 ± 13b |
BLA, basolateral amygdala; CeA, central nucleus of the amygdala; dBNST, dorsal bed nucleus of the stria terminalis; IL, infralimbic cortex; PVN, paraventricular nucleus of the hypothalamus; PrL, prelimbic cortex; vBNST, ventral bed nucleus of the stria terminalis.
Rats were treated simultaneously with acute EtOH and a 2‐hour restraint/water immersion stressor. The rats were then sacrificed immediately following the conclusion of the stressor. Brain regions were stained for CD11b to assess microglial activation. n = 4 to 10 per group. Data are presented as mean ± SEM. Statistically significant differences are indicated by different letters; for example, means with superscripts “a” and “b” are statistically different (Tukey's post hoc test), while means with superscripts “a” and “a” are not statistically different.
Table 3.
Effects of Acute Ethanol (EtOH) and Acute Stress on c‐Fos Expression Across Brain Regions (cells/mm2)
| Brain region | CON | E | S | E+S |
|---|---|---|---|---|
| Prefrontal cortex | ||||
| PrL | 81 ± 5a | 213 ± 33b | 497 ± 31c | 394 ± 33c |
| IL | 48 ± 8a | 137 ± 32b | 224 ± 33c | 219 ± 31c |
| Nucleus accumbens | ||||
| Core | 219 ± 48a | 316 ± 111a,b | 543 ± 39b | 402 ± 47a,b |
| Shell | 149 ± 40a,b | 122 ± 45a | 312 ± 60b | 139 ± 19a |
| Bed nucleus of the stria terminalis | ||||
| dBNST | 63 ± 7a | 145 ± 40a | 140 ± 11a | 113 ± 7a |
| vBNST | 40 ± 5a | 63 ± 25a | 222 ± 23b | 118 ± 21a |
| Hypothalamus | ||||
| PVN | 95 ± 28a | 260 ± 6b | 503 ± 54c | 418 ± 38c |
| Amygdala | ||||
| BLA | 113 ± 22a | 100 ± 29a | 272 ± 31b | 143 ± 29a |
| CeA | 104 ± 12a | 385 ± 88b | 204 ± 41a | 195 ± 32a |
BLA, basolateral amygdala; CeA, central nucleus of the amygdala; dBNST, dorsal bed nucleus of the stria terminalis; IL, infralimbic cortex; PVN, paraventricular nucleus of the hypothalamus; PrL, prelimbic cortex; vBNST, ventral bed nucleus of the stria terminalis.
Rats were treated simultaneously with acute EtOH and a 2‐hour restraint/water immersion stressor. The rats were then sacrificed immediately following the conclusion of the stressor. Brain regions were stained for c‐Fos to assess neuronal activation. n = 4 to 10 per group. Data are presented as mean ± SEM. Statistically significant differences are indicated by different letters; for example, means with superscripts “a” and “b” are statistically different (Tukey's post hoc test), while means with superscripts “a” and “a” are not statistically different.
Figure 3.
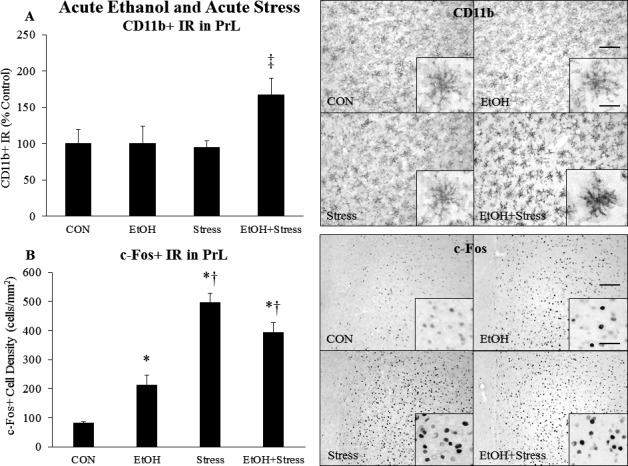
Effects of acute ethanol (EtOH) and acute stress on CD11b+ IR and c‐Fos+ IR in the prelimbic cortex. Rats were treated with EtOH (5.0 g/kg, 25% v/v, i.g.) and/or a 2‐hour restraint/water immersion stressor and sacrificed immediately following the conclusion of the stressor. (A) CD11b+ pixel density in the PrL was assessed in each group. Data are presented as mean ± SEM. ‡ p < 0.05 compared to Stress (Tukey's post hoc test). n = 4 to 10/group. A representative image of CD11b staining from each group is shown. A higher magnification image is displayed in the inset. The scale bar in the low‐magnification image measures 100 microns, and the scale bar in the high‐magnification inset measures 20 microns. (B) c‐Fos+ cell density in the PrL was assessed in each group. Data are presented as mean ± SEM. *p < 0.05 compared to CON, † p < 0.05 compared to EtOH (Tukey's post hoc test). n = 4 to 10/group. A representative image of c‐Fos staining from each group is shown. A higher magnification image is displayed in the inset. The scale bar in the low‐magnification image measures 100 microns, and the scale bar in the high‐magnification inset measures 20 microns.
Effects of Acute EtOH and Acute Stress on Microglia and Neurons in the NAc and Bed Nucleus of the Stria Terminalis
We next investigated microglial and neuronal activation in the NAc, a brain region involved in reward, and the bed nucleus of the stria terminalis (BNST), a brain region involved in stress, fear, and anxiety. Neither acute EtOH nor acute stress increased CD11b+ IR in the NAc, although the combination enhanced CD11b+ IR 95% (p < 0.05) in the NAc core and 69% (p < 0.05) in the NAc shell (Fig. 4 A). Staining for c‐Fos found that acute EtOH did not enhance c‐Fos+ cell density in the NAc, but that acute stress increased c‐Fos+ cell density 148% (p < 0.05) in the NAc core and 100% in the NAc shell (Fig. 4 B). The combination of acute EtOH and acute stress did not impact c‐Fos+ cell density in the NAc core, but returned c‐Fos to control levels in the NAc shell (Fig. 4 B). In the BNST, both acute EtOH and acute stress showed a trend to increase CD11b, with the combination further increasing CD11b (Fig. 4 C). Acute stress increased c‐Fos+ cell density 5.5‐fold (p < 0.05) in the vBNST, with the combination of acute EtOH and acute stress blunting this response (Fig. 4 D). Acute EtOH, acute stress, or the combination did not significantly increase c‐Fos+ cell density in the dBNST, although there were trends for EtOH and stress to increase c‐Fos in this region. Overall, these data suggest that acute EtOH and acute stress interact to enhance microglial CD11b in the NAc, but decrease neuronal c‐Fos in the NAc shell. Also, acute EtOH and acute stress combined tend to increase microglial CD11b in the BNST, while acute EtOH blunts the effects of acute stress on neuronal c‐Fos in the vBNST.
Figure 4.
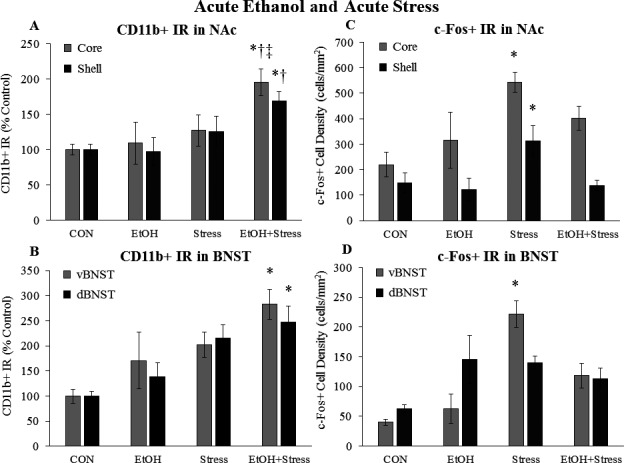
Effects of acute ethanol (EtOH) and acute stress on CD11b+ IR and c‐Fos+ IR in the nucleus accumbens (NAc) and bed nucleus of the stria terminalis (BNST). Rats were treated with EtOH (5.0 g/kg, 25% v/v, i.g.) and/or a 2‐hour restraint/water immersion stressor and sacrificed immediately following the conclusion of the stressor. CD11b+ IR was assessed in (A) the NAc core and shell and (B) the ventral and dorsal BNST. c‐Fos+ cell density in (C) the NAc core and shell and (D) the ventral and dorsal BNST was assessed in each group. Data are presented as mean ± SEM. *p < 0.05 compared to CON, † p < 0.05 compared to EtOH, ‡ p < 0.05 compared to Stress (Tukey's post hoc test). n = 4 to 10/group.
Effects of Acute EtOH and Acute Stress on Microglia and Neurons in the PVN and Amygdala
We also examined microglial and neuronal activation in the PVN and amygdala, key stress‐response brain regions that are also activated by EtOH. Acute EtOH did not increase CD11b+ IR in the PVN, whereas acute stress caused a marked 5.5‐fold (p < 0.05) increase in CD11b+ IR that was not altered by the combination of EtOH and stress (Fig. 5 A). Acute EtOH increased PVN c‐Fos+ cell density 160% (p < 0.05), while acute stress increased c‐Fos+ cell density 5‐fold (p < 0.05) in the PVN (Fig. 5 B). There was no interaction of acute EtOH with acute stress on c‐Fos in the PVN. In the amygdala, neither acute EtOH nor acute stress significantly increased CD11b+ IR; however, the combination increased CD11b+ IR 107% (p < 0.05) in the BLA and a 53% (p < 0.05) in the CeA (Table 2, Fig. 5 C). While acute EtOH did not increase c‐Fos+ cell density in the BLA, acute stress caused a marked 2.7‐fold (p < 0.05) increase in BLA c‐Fos+ cell density (Table 3). Acute EtOH blunted the stress‐induced increase in BLA c‐Fos (Table 3). While acute EtOH caused a robust 3.8‐fold (p < 0.05) increase in c‐Fos+ cell density in the CeA, acute stress had no significant effect (Fig. 5 D). Interestingly, the combination of acute EtOH and acute stress blunted the EtOH‐induced increase in CeA c‐Fos. Overall, these data suggest that acute EtOH and acute stress cause marked increases in PVN c‐Fos, while only acute stress increased PVN CD11b. Furthermore, acute EtOH and acute stress interact to enhance microglial CD11b in the amygdala, whereas the BLA neurons respond to acute stress and CeA neurons respond to acute EtOH.
Figure 5.
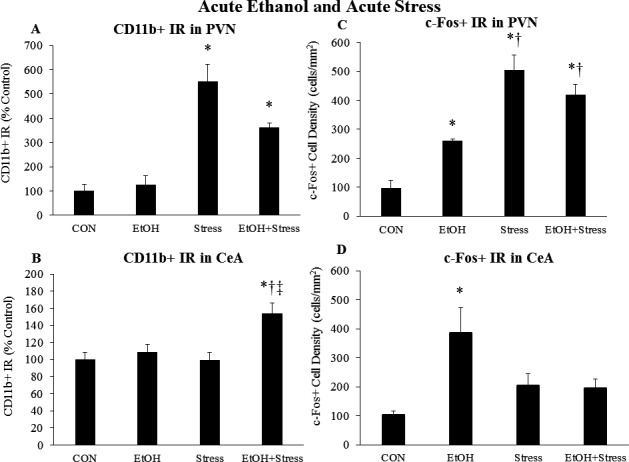
Effects of acute ethanol (EtOH) and acute stress on CD11b+ IR and c‐Fos+ IR in the paraventricular nucleus of the hypothalamus (PVN) and the central nucleus of the amygdala (CeA). Rats were treated with EtOH (5.0 g/kg, 25% v/v, i.g.) and/or a 2‐hour restraint/water immersion stressor and sacrificed immediately following the conclusion of the stressor. CD11b+ IR was assessed in (A) the PVN and (B) the CeA. c‐Fos+ cell density in (C) the PVN and (D) the CeA was assessed in each group. Data are presented as mean ± SEM. *p < 0.05 compared to CON, † p < 0.05 compared to EtOH, ‡ p < 0.05 compared to Stress (Tukey's post hoc test). n = 4 to 10/group.
Persistent Effects of Chronic Intermittent EtOH on the Plasma CORT and Plasma Endotoxin to Acute Stress
A large body of research suggests that chronic EtOH alters brain stress systems (Koob, 2013). However, it is unknown whether chronic EtOH leads to long‐lasting changes in the acute stress response. To investigate this, we administered chronic EtOH (CE) to male Wistar rats in an intermittent 2‐d on/2‐d off pattern for 1 month (Fig. 1 B). Tail blood was drawn to determine BECs. Mean BECs in CE‐treated animals were 167 ± 11 mg/dl after 8 doses and 130 ± 10 mg/dl after the final dose (Fig. 1 B). Following a 40‐day period of abstinence, animals were acutely stressed with a 2‐hour restraint/water immersion stressor or remained in their home cage. The rats were sacrificed 2 hours following the conclusion of the stressor, and plasma CORT and endotoxin were assessed. Acute stress caused a significant increase in plasma CORT levels (p < 0.05) (Fig. 6 A); however, this was not altered by chronic EtOH. Chronic EtOH did not persistently change plasma endotoxin levels, while acute stress showed a trend to increase plasma endotoxin (p = 0.08) (Fig. 6 B). Chronic EtOH did enhance acute stress‐induced plasma endotoxin 2.7‐fold (p < 0.05) (Fig. 6 B). These results suggest that chronic EtOH enhanced stress‐induced plasma endotoxin, without impacting the plasma CORT response.
Figure 6.
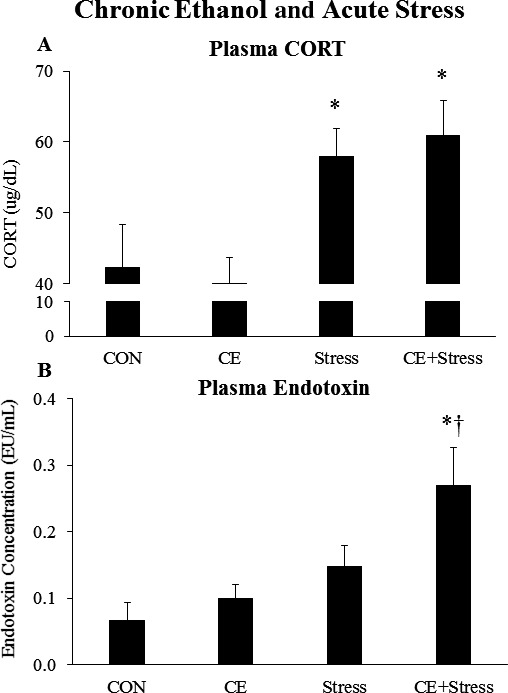
Effects of chronic ethanol (EtOH) and acute stress on plasma corticosterone (CORT) and endotoxin. Rats were treated with chronic EtOH (5.0 g/kg, 20 to 30% v/v, 2 days on, 2 days off from P25 to P54) and/or acutely stressed with a 2‐hour restraint/water immersion stressor following prolonged abstinence on P96/P97. The rats were sacrificed 2 hours following the conclusion of the stressor. (A) The levels of plasma corticosterone were measured. Data are presented as mean ± SEM. *p < 0.05 compared to CON, (Tukey's post hoc test). n = 8 to 10/group. (B) The levels of plasma endotoxin were measured. Data are presented as mean ± SEM. *p < 0.05 compared to CON, † p < 0.05 compared to EtOH (Tukey's post hoc test). n = 8 to 10/group.
Persistent Effects of Chronic Intermittent EtOH on the Microglial and Neuronal Response in the PFC
To determine whether chronic EtOH exposure persistently altered the microglial or neuronal response to acute stress, we treated rats with chronic EtOH as described above and stained for CD11b, c‐Fos, and EGR1 (Tables 4, 5, 6). In the PrL, chronic EtOH caused a persistent 3.6‐fold increase (p < 0.05) in CD11b+ IR, while acute stress increased PrL CD11b+ IR 7‐fold (p < 0.05) (Fig. 7 A), with the combination of the two further increasing CD11b+ IR by 11‐fold (p < 0.05) (Fig. 7 A). A similar pattern was observed in the IL (Table 4). Chronic EtOH and acute stress had similar effects on Iba1 + IR in the PrL and IL (Fig. S3 A,C), but did not increase markers of full microglial activation such as CD68, MHCII, or iNOS (Fig. S4). Neither chronic EtOH nor acute stress changed microglial cell density in the PrL or IL (Fig. S3 B,D). A group of animals was also sacrificed at P55, 24 hours after the last EtOH treatment, to examine the short‐term effects of CE on microglia. Interestingly, CE decreased CD11b+ IR about 50% (p < 0.05) at P55 without changing microglial cell density (Fig. S2). Chronic EtOH had no lasting effect on c‐Fos+ cell density in the PrL, while acute stress increased c‐Fos+ cell density about 2‐fold (p < 0.05) in both control and CE groups (Fig. 7 B). A similar pattern was observed in the IL (Table 5). While stress increased EGR1 + cell density in the PrL and IL, there was no effect of CE (Table 6). Plasma endotoxin and PrL CD11b were correlated (r: 0.508, p: 0.005) across groups, consistent with endotoxin inducing inflammation resulting in microglial activation. Plasma CORT also correlated with PrL CD11b (r: 0.425, p: 0.012) across groups, and plasma endotoxin correlated with PrL c‐Fos (r: 0.422, p: 0.015) across groups. However, plasma CORT and PrL c‐Fos did not correlate. Overall, these data suggest that chronic EtOH persistently sensitizes the microglial CD11b response to acute stress, but not the neuronal c‐Fos response to acute stress, in the PFC.
Table 4.
Effects of Chronic Ethanol (EtOH) and Acute Stress on CD11b Expression Across Brain Regions (% control)
| Brain region | CON | CE | Stress | CE+Stress |
|---|---|---|---|---|
| Prefrontal cortex | ||||
| PrL | 100 ± 29a | 358 ± 105b | 702 ± 95c | 1,115 ± 154d |
| IL | 100 ± 19a | 156 ± 35a | 236 ± 25b | 322 ± 26c |
| Nucleus accumbens | ||||
| Core | 100 ± 11a | 464 ± 183a | 1,664 ± 289b | 2,835 ± 501c |
| Shell | 100 ± 47a | 170 ± 110a | 824 ± 184a | 2,266 ± 664b |
| Bed nucleus of the stria terminalis | ||||
| dBNST | 100 ± 12a | 415 ± 97b | 778 ± 51c | 1,300 ± 139d |
| vBNST | 100 ± 15a | 218 ± 49a | 674 ± 113b | 597 ± 59b |
| Hippocampus | ||||
| CA1 | 100 ± 20a | 249 ± 114a,b | 331 ± 37a,b | 429 ± 80b |
| CA3 | 100 ± 17a | 164 ± 50a,b | 211 ± 16b | 248 ± 35b |
| DG | 100 ± 17a | 172 ± 66a,b | 211 ± 17a,b | 282 ± 35b |
| Hypothalamus | ||||
| PVN | 100 ± 56a | 211 ± 55a | 1,700 ± 244b | 1,700 ± 300b |
| Amygdala | ||||
| BLA | 100 ± 22a | 222 ± 22a | 1,378 ± 144b | 2,489 ± 433c |
| CeA | 100 ± 16a | 277 ± 116a | 488 ± 25b | 610 ± 67b |
| Periaqueductal gray | ||||
| lPAG | 100 ± 15a | 225 ± 39b | 325 ± 11c | 350 ± 41c |
BLA, basolateral amygdala; CeA, central nucleus of the amygdala; dBNST, dorsal bed nucleus of the stria terminalis; DG, dentate gyrus; IL, infralimbic cortex; lPAG, lateral periaqueductal gray; PVN, paraventricular nucleus of the hypothalamus; PrL, prelimbic cortex; vBNST, ventral bed nucleus of the stria terminalis.
Rats were treated with chronic intermittent EtOH and, after a period of prolonged abstinence, were acutely stressed with 2 hours of restraint/partial water immersion. Rats were sacrificed 2 hours following the termination of the stressor. Brain regions were stained for CD11b to assess microglial activation. n = 6 to 10 per group. Data are presented as mean ± SEM. Statistically significant differences are indicated by different letters; for example, means with superscripts “a” and “b” are statistically different (Tukey's post hoc test), while means with superscripts “a” and “a” are not statistically different.
Table 5.
Effects of Chronic Ethanol (EtOH) and Acute Stress on c‐Fos Expression Across Brain Regions (cells/mm2)
| Brain region | CON | CE | Stress | CE+Stress |
|---|---|---|---|---|
| Prefrontal cortex | ||||
| PrL | 142 ± 39a | 144 ± 24a | 302 ± 52b | 297 ± 33b |
| IL | 116 ± 33a | 138 ± 16a | 242 ± 37b | 282 ± 35b |
| Nucleus accumbens | ||||
| Core | 37 ± 9a | 35 ± 8a | 119 ± 27b | 136 ± 16b |
| Shell | 83 ± 23a | 55 ± 9a | 207 ± 27b | 236 ± 24b |
| Bed nucleus of the stria terminalis | ||||
| dBNST | 7 ± 2a | 8 ± 2a | 33 ± 4b | 41 ± 4b |
| vBNST | 26 ± 7a | 37 ± 8a | 97 ± 13b | 123 ± 17b |
| Hippocampus | ||||
| CA1 | 5 ± 1a | 4 ± 1a | 4 ± 1a | 4 ± 1a |
| CA3 | 9 ± 2a | 11 ± 2a | 6 ± 1a | 6 ± 1a |
| DG | 9 ± 1a | 10 ± 1a | 14 ± 2a | 11 ± 1a |
| Hypothalamus | ||||
| PVN | 141 ± 24a | 83 ± 20a | 544 ± 51b | 661 ± 47b |
| Amygdala | ||||
| BLA | 11 ± 2a | 12 ± 2a | 22 ± 3b | 20 ± 3b |
| CeA | 14 ± 2a | 21 ± 3a | 66 ± 7b | 83 ± 9b |
| Periaqueductal gray | ||||
| lPAG | 46 ± 11a | 74 ± 7a | 160 ± 10b | 171 ± 12b |
BLA, basolateral amygdala; CeA, central nucleus of the amygdala; dBNST, dorsal bed nucleus of the stria terminalis; DG, dentate gyrus; IL, infralimbic cortex; lPAG, lateral periaqueductal gray; PVN, paraventricular nucleus of the hypothalamus; PrL, prelimbic cortex; vBNST, ventral bed nucleus of the stria terminalis.
Rats were treated with chronic intermittent EtOH and, after a period of prolonged abstinence, were acutely stressed with 2 hours of restraint/partial water immersion. Rats were sacrificed 2 hours following the termination of the stressor. Brain regions were stained for c‐Fos to assess neuronal activation. n = 6 to 10 per group. Data are presented as mean ± SEM. Statistically significant differences are indicated by different letters; for example, means with superscripts “a” and “b” are statistically different (Tukey's post hoc test), while means with superscripts “a” and “a” are not statistically different.
Table 6.
Effects of Chronic Ethanol (EtOH) and Acute Stress on EGR1 Expression Across Brain Regions (cells/mm2)
| Brain region | CON | AIE | Stress | AIE+Stress |
|---|---|---|---|---|
| Prefrontal cortex | ||||
| PrL | 1,086 ± 133a | 1,197 ± 70a | 1,424 ± 116b | 1,492 ± 63b |
| IL | 701 ± 77a | 733 ± 51a | 1,008 ± 78b | 941 ± 89b |
| Bed nucleus of the stria terminalis | ||||
| dBNST | 115 ± 16a | 102 ± 16a | 231 ± 19b | 253 ± 26b |
| vBNST | 163 ± 29a | 164 ± 23a | 335 ± 28b | 376 ± 25b |
| Hypothalamus | ||||
| PVN | 352 ± 90a | 217 ± 70a | 879 ± 115b | 1,017 ± 97b |
| Amygdala | ||||
| BLA | 271 ± 17a | 203 ± 30a | 244 ± 28a | 271 ± 22a |
| CeA | 395 ± 35a | 232 ± 58b | 581 ± 52c | 413 ± 33a |
| Periaqueductal gray | ||||
| lPAG | 86 ± 16a | 103 ± 14a | 154 ± 18b | 158 ± 17b |
BLA, basolateral amygdala; CeA, central nucleus of the amygdala; dBNST, dorsal bed nucleus of the stria terminalis; IL, infralimbic cortex; lPAG, lateral periaqueductal gray; PVN, paraventricular nucleus of the hypothalamus; PrL, prelimbic cortex; vBNST, ventral bed nucleus of the stria terminalis.
Rats were treated with chronic intermittent EtOH and, after a period of prolonged abstinence, were acutely stressed with 2 hours of restraint/partial water immersion. Rats were sacrificed 2 hours following the termination of the stressor. Brain regions were stained for EGR1 to assess neuronal activation. n = 6 to 10 per group. Data are presented as mean ± SEM. Statistically significant differences are indicated by different letters; for example, means with superscripts “a” and “b” are statistically different (Tukey's post hoc test), while means with superscripts “a” and “a” are not statistically different.
Figure 7.
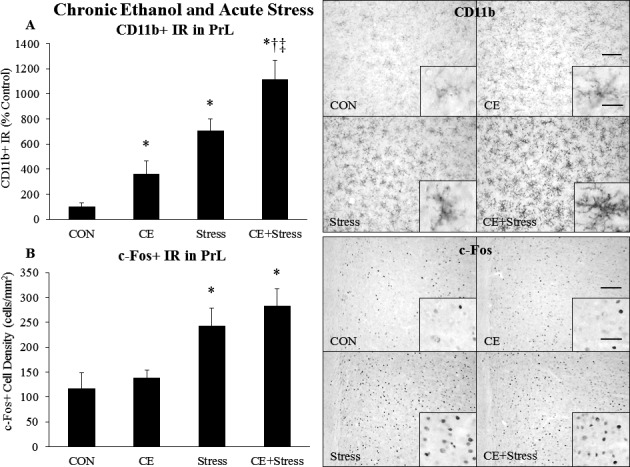
Effects of chronic ethanol (EtOH) and acute stress on CD11b+ IR and c‐Fos+ IR in the prelimbic cortex (PrL). Rats were treated with chronic EtOH (5.0 g/kg, 20 to 30% v/v, 2 days on, 2 days off from P25 to P54) and/or acutely stressed with a 2‐hour restraint/water immersion stressor following prolonged abstinence on P96/P97. The rats were sacrificed 2 hours following the conclusion of the stressor. (A) CD11b+ pixel density in the PrL was assessed in each group. Data are presented as mean ± SEM. *p < 0.05 compared to CON, † p < 0.05 compared to EtOH, ‡ p < 0.05 compared to Stress (Tukey's post hoc test). n = 8 to 10/group. A representative image of CD11b staining from each group is shown. A higher magnification image is displayed in the inset. The scale bar in the low‐magnification image measures 100 microns, and the scale bar in the high‐magnification inset measures 20 microns. (B) c‐Fos+ cell density in the PrL was assessed in each group. Data are presented as mean ± SEM. *p < 0.05 compared to CON (Tukey's post hoc test). n = 8 to 10/group. A representative image of c‐Fos staining from each group is shown. A higher magnification image is displayed in the inset. The scale bar in the low‐magnification image measures 100 microns, and the scale bar in the high‐magnification inset measures 20 microns.
Persistent Effects of Chronic Intermittent EtOH on the Microglial and Neuronal Response in the NAc and Bed Nucleus of the Stria Terminalis
We also examined whether chronic EtOH alters the microglial or neuronal response to acute stress in the NAc or BNST. In the NAc, there was a trend for chronic EtOH to increase CD11b+ IR (Fig. 8 A). Acute stress increased CD11b+ staining 16‐fold (p < 0.05) in the NAc core and 8‐fold in the NAc shell (Fig. 8 A). CD11b+ IR was further increased by the combination of chronic EtOH and acute stress in both the core (p < 0.05) and shell (p < 0.05) (Fig. 8 A). Chronic EtOH did not persistently alter c‐Fos in the NAc core or shell (Fig. 8 B), while acute stress increased c‐Fos+ cell density 3.2‐fold (p < 0.05) in the NAc core and 2.5‐fold (p < 0.05) in the NAc shell (Fig. 8 B). Chronic EtOH and acute stress did not interact to increase c‐Fos in the NAc (Fig. 8 B). In the dBNST, chronic EtOH persistently increased CD11b+ IR 315% (p < 0.05) (Fig. 8 C). Acute stress increased CD11b+ IR 7.8‐fold (p < 0.05) in the dBNST and 6.7‐fold (p < 0.05) in the vBNST (Fig. 8 C). Chronic EtOH and acute stress enhanced CD11b staining 13‐fold in the dBNST (p < 0.05) (Fig. 8 C). Chronic EtOH had no persistent effect on c‐Fos in the BNST, while acute stress increased c‐Fos+ cell density in the dBNST and vBNST (Fig. 8 D). Chronic EtOH and acute stress did not interact to change c‐Fos in the BNST. While stress increased EGR1 + cell density in the NAc and BNST, there was no effect of CE (Table 6). These data suggest that chronic EtOH does not alter the NAc c‐Fos response to acute stress, but enhances the NAc CD11b response to acute stress. Also, chronic EtOH persistently increases CD11b in the dBNST and enhances the BNST CD11b response to acute stress, but not the BNST c‐Fos response.
Figure 8.
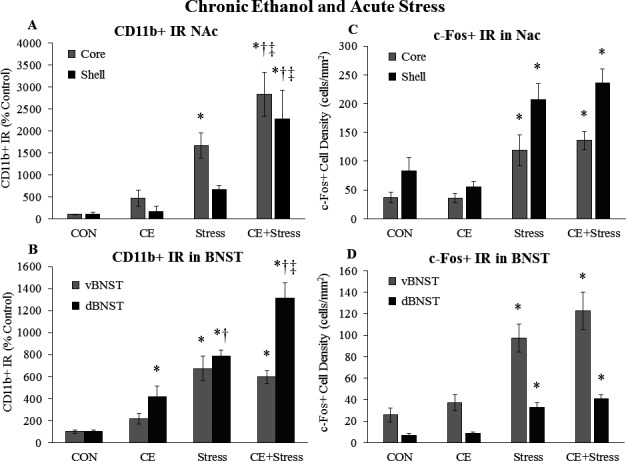
Effects of chronic ethanol (EtOH) and acute stress on CD11b+ IR and c‐Fos+ IR in the nucleus accumbens (NaC) and bed nucleus of the stria terminalis (BDST). Rats were treated with chronic EtOH (5.0 g/kg, 20 to 30% v/v, 2 days on, 2 days off from P25 to P54) and/or acutely stressed with a 2‐hour restraint/water immersion stressor following prolonged abstinence on P96/P97. The rats were sacrificed 2 hours following the conclusion of the stressor. CD11b+ IR was assessed in (A) the NAc core and shell and (B) the ventral and dorsal BNST. c‐Fos+ cell density in (C) the NAc core and shell and (D) the ventral and dorsal BNST was assessed in each group. Data are presented as mean ± SEM. *p < 0.05 compared to CON, † p < 0.05 compared to EtOH, ‡ p < 0.05 compared to Stress (Tukey's post hoc test). n = 8 to 10/group.
Persistent Effects of Chronic Intermittent EtOH on the Microglial and Neuronal Response in the PVN and Amygdala
We also determined the effects of chronic EtOH and acute stress on microglia and neurons in the PVN and amygdala. Chronic EtOH had no persistent effect on c‐Fos or CD11b in the PVN. However, acute stress increased CD11b+ IR 17‐fold (p < 0.05), with no further increase by chronic EtOH (Fig. 9 A). Acute stress also increased c‐Fos+ cell density by 286% (p < 0.05) in the PVN, with no further increase by chronic EtOH (Fig. 9 B). In the amygdala, there was a trend for chronic EtOH to increase CD11b+ IR. Acute stress increased CD11b+ IR 13.8‐fold (p < 0.05) in the BLA and 4.9‐fold (p < 0.05) in the CeA (Table 4, Fig. 9 C). Chronic EtOH plus acute stress increased CD11b+ IR 25‐fold (p < 0.05) in the BLA and 6‐fold in the CeA. Chronic EtOH had no persistent effects on c‐Fos in the CeA or BLA. However, acute stress increased c‐Fos+ cell density 2‐fold (p < 0.05) in the BLA and 4.7‐fold (p < 0.05) in the CeA (Fig. 9 D), with no further effect of chronic EtOH (Fig. 9 D). While stress increased EGR1 + cell density in the PVN and amygdala, there was no effect of CE (Table 6). Overall, these data suggest chronic EtOH does not persistently alter the c‐Fos or CD11b response to acute stress in the PVN or amygdala.
Figure 9.
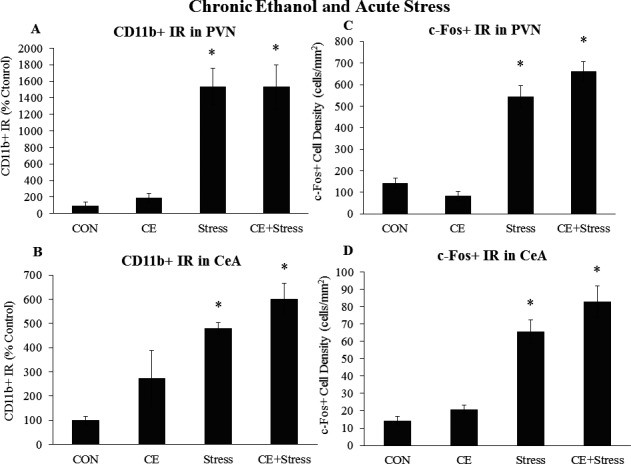
Effects of chronic ethanol (EtOH) and acute stress on CD11b+ IR and c‐Fos+ IR in the paraventricular nucleus of the hypothalamus (PVN) and central nucleus of the amygdala (CeA). Rats were treated with chronic EtOH (5.0 g/kg, 20 to 30% v/v, 2 days on, 2 days off from P25 to P54) and/or acutely stressed with a 2‐hour restraint/water immersion stressor following prolonged abstinence on P96/P97. The rats were sacrificed 2 hours following the conclusion of the stressor. CD11b+ IR was assessed in (A) the PVN and (B) the CeA. c‐Fos+ cell density in (C) the PVN and (D) the CeA was assessed in each group. Data are presented as mean ± SEM. *p < 0.05 compared to CON (Tukey's post hoc test). n = 8 to 10/group.
Discussion
In this study, we examined the effects of acute EtOH and acute stress individually and combined, and the effects of chronic EtOH and acute stress individually and combined on plasma CORT, plasma endotoxin, and brain regional microglial CD11b and neuronal c‐Fos. We used models of heavy binge drinking and intense stress to assure measurable responses. We report that acute binge EtOH increased plasma CORT and c‐Fos + IR in multiple brain regions, specifically the mPFC, PVN, and CeA. Acute stress increased plasma CORT about twice as much as acute EtOH and increased c‐Fos+ IR in the mPFC, NAc, BNST, PVN, and BLA. These results are consistent with previous studies showing that EtOH induces plasma CORT (Ellis, 1966) and c‐Fos across the brain (Cullinan et al., 1995). The combination of acute EtOH and acute stress did not change plasma CORT, but enhanced plasma endotoxin (Fig. 10 A). Acute EtOH and acute stress combined blunted EtOH‐induced c‐Fos in the CeA and blunted stress‐induced c‐Fos in the NAc shell, vBNST, and BLA. The combination of acute EtOH and stress also enhanced microglial CD11b in the mPFC, NAc, and amygdala. We report that chronic AIE exposure had no persistent effects on plasma CORT, plasma endotoxin, or c‐Fos, but increased CD11b in the PrL, dBNST, and lPAG. Chronic EtOH exposure in adolescence led to a persistently enhanced plasma endotoxin response to acute stress, as well as the microglial CD11b response in multiple brain regions, including the PFC, NAc, dBNST, and BLA (Fig. 10 B). These studies suggest acute EtOH exposure enhances the blood endotoxin response to stress as well as increasing microglial CD11b + IR stress responses in frontal cortex, NAc, and amygdala. Chronic AIE exposure resulted in increased adult blood endotoxin responses to stress and increased microglial CD11b + IR stress responses in the PFC, NAc, and dBNST, but not the amygdala or PVN. These findings are consistent with blood endotoxin and microglia contributing to the interactions of stress with alcohol on brain function.
Figure 10.
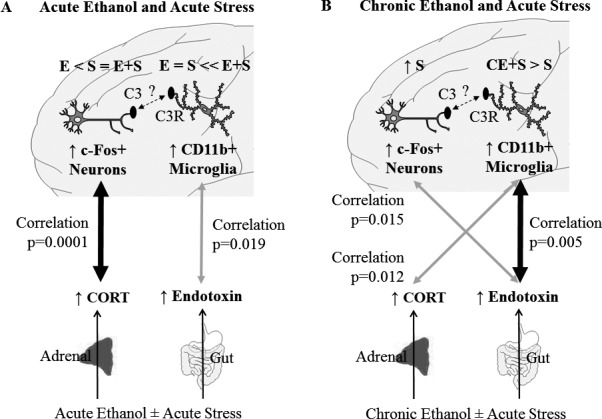
Effects of acute and chronic ethanol (EtOH) on the systemic and central nervous system response to acute stress. (A) Both acute EtOH and acute stress increase plasma CORT and neuronal c‐Fos in the PFC. The magnitude of these effects is represented by E < S = E+S. Furthermore, CORT and PFC c‐Fos levels correlate, suggesting these 2 responses are related. This positive correlation is represented by the thick arrow, and the p‐value for the Pearson correlation is displayed. Furthermore, acute EtOH and acute stress alone have little effect on plasma endotoxin or microglial CD11b in the mPFC. However, acute EtOH and acute stress together interact to enhance plasma endotoxin and microglial CD11b in the PFC. This is represented by E = S << E+S. Plasma endotoxin and PFC CD11b also correlate. This positive correlation is represented by the thin arrow, and the p‐value for the Pearson correlation is displayed. A possible interaction between microglial CR3 and neuronal C3 is also displayed. Increased CD11b, a component of CR3, may contribute to change synaptic remodeling. (B) Acute stress increases plasma CORT and PFC c‐Fos, but this is not altered by prior chronic EtOH, as indicated by ↑S. However, chronic EtOH enhances the plasma endotoxin and PFC CD11b response to acute stress. This is represented by CE + S > S. Plasma endotoxin and microglial CD11b also correlate, consistent with endotoxin‐induced microglial activation. This correlation is represented by the thick arrow, and the p‐value of the Pearson correlation is shown. Interestingly, the plasma endotoxin and PFC c‐Fos response also correlate. This is represented by the thin arrow, and the p‐value for the Pearson correlation is shown. Furthermore, the plasma CORT response correlates with the PrL CD11b response. This is represented by the thin arrow, and the p‐value of the Pearson correlation is shown. A possible interaction between microglial CR3 and neuronal C3 is also displayed. Increased CD11b, a component of CR3, may contribute to change synaptic remodeling.
Exposure to both acute EtOH and acute restraint stress increased blood CORT and endotoxin. To our knowledge, we report for the first time that the combination of stress and alcohol increased the blood endotoxin response, but not the CORT response. A previous study found that EtOH did not enhance plasma CORT after 15 minutes of restraint stress, but did enhance plasma CORT after 60 minutes of restraint stress (Trudeau et al., 1990). We only tested 1 heavy binge EtOH dose, 1 intense stressor, and sacrifice at 1 time point optimum for neuronal c‐Fos, for example, 2 hours after restraint, and did not find the combination of stress and alcohol altered the CORT response. We cannot rule out the possibility that interactions with CORT occur at other EtOH doses, stress intensities, or time, but our findings are consistent with the combination of stress and alcohol increasing blood endotoxin through increased gut permeability. Previous studies have reported acute EtOH binge exposure (Rivera et al., 1998) and acute restraint stress (Ait‐Belgnaoui et al., 2012) increase plasma endotoxin. Elevated plasma endotoxin occurs through enhanced gut leakiness, leading to bacterial translocation into the peripheral circulation (Rao, 2009). Interestingly, treatment with antibiotics to reduce gut bacteria reduces stress‐induced blood endotoxin and CORT as well as the stress‐induced hypothalamic inflammatory cytokine responses and corticotropin‐releasing factor (CRF) (Ait‐Belgnaoui et al., 2012). These findings suggest blood endotoxin contributes to stress‐induced CORT responses. Similarly, acute EtOH exposure–induced endotoxin responses are reduced by antibiotic treatment reducing gut bacteria (Rivera et al., 1998). Increased systemic endotoxin and blood cytokines impact brain cytokines and microglia (Mayfield et al., 2013). It is possible our CORT response to stress is maximal, preventing further increases by endotoxin or other mechanisms. Regardless, the discovery that acute combined exposure to EtOH and stress further enhance plasma endotoxin, and increased microglial responses in key brain regions could contribute to the stress and alcohol interaction increasing risks of AUD. Stress has been implicated in exacerbating binge/heavy drinking, motivation to drink, and the chronic relapsing nature of AUD (Blaine and Sinha, 2017; Stephens and Wand, 2012). We report here that stress and EtOH exposure interact through changes in blood endotoxin and microglial activation. Microglia in brain respond to both stress and alcohol with increases in innate immune signaling molecules such as cytokines. Stress sensitizes brain to endotoxin responses (Frank et al., 2007), and stress hormones appear to enhance brain innate immune stress responses (Frank et al., 2016; Sorrells et al., 2009). Interestingly, this is consistent with studies finding innate immune signaling contributes to the development of AUD (Crews et al., 2017; de Timary et al., 2017). Innate immune signaling cytokines alter EtOH consumption (Marshall et al., 2017, 2017), and cytokine receptors contribute to stress‐induced EtOH consumption (Karlsson et al., 2017). Further, immune signaling molecules are increased in postmortem human alcoholic brain (Crews et al., 2017) and regulate preference for alcohol drinking in animal studies (Mayfield et al., 2013). Cytokines also enhance stressful emotional states following alcohol withdrawal (Breese et al., 2008). These data, taken together with our findings reported here that combined acute EtOH and stress enhance endotoxin and brain microglial CD11B + IR in the PFC, NAc, and CeA, are consistent with microglia within these brain regions contributing to the interaction of EtOH exposure with stress that impact alcohol drinking and increase risks for AUD.
In this study, we assessed microglial responses using CD11b, also known as integrin alpha M, a constitutively expressed microglia‐specific gene up‐regulated when microglia are activated (Kettenmann et al., 2011). CD11b complexes with integrin beta‐2 to form the heterodimeric integrin α M β 2, also known as complement receptor 3 (CR3), and is important for microglial adhesion, migration, and complement‐mediated pruning of synapses (Schafer et al., 2012; Solovjov et al., 2005). Increased CD11b with acute EtOH and acute stress suggests enhanced microglial activation to what has been called a hyperramified state associated with cytokine secretion and increased responsiveness or “priming,” but not the fully activated microglial bushy or phagocytic morphology activation states (Crews and Vetreno, 2016; He and Crews, 2008). Stress and alcohol can impact brain through multiple mechanisms including CORT, systemic cytokines, and endotoxin, as well as direct vagal sensory pathways (Crews et al., 2017). We found acute stress and acute EtOH exposure increase blood endotoxin and the CD11b response in PFC far more than either acute treatment alone. These responses correlate across groups suggesting the endotoxin–microglial PFC CD11b responses may be linked (Fig. 10), although there are multiple possible mechanisms by which stress and alcohol induce changes in brain. These results suggest the intriguing possibility that acute alcohol and acute stress interact increasing microglial‐CD11b “priming” through gut‐derived endotoxin in frontal cortical, limbic, and reward‐associated brain regions.
We also report here that AIE repeated chronic EtOH exposure persistently increases adult CD11b in many examined brain regions, consistent with a persistent long‐lasting “priming” of microglia. These results replicate previous studies showing an intense, 4‐day binge EtOH treatment in rats increased CD11b + IR in the hippocampus immediately following the treatment. This CD11b + IR increase declined over time, but remained persistently increased for at least 28 days (Marshall et al., 2013; McClain et al., 2011). Our studies add to this work by showing that less intense AIE treatment persistently increases CD11b in many adult brain regions. Thus, the intermittent cycles of EtOH exposure and withdrawal may alter synapses and circuits known to be impacted by EtOH by changing microglial CD11b levels. Overall, these results show that chronic EtOH persistently increases microglial CD11b in multiple brain regions and are consistent with microglial signaling and perhaps synapse regulation contributing to long‐lasting changes in brain.
To investigate the persistent effects of EtOH exposure on stress responses, we used a model of adolescent binge drinking, for example, AIE, previously found to increase expression of adult brain neuroimmune signaling cytokines including HMGB1 as well as Toll‐like receptors (TLRs) and RAGE, an HMGB1 receptor (Crews et al., 2013; Vetreno and Crews, 2012). We report here for the first time that AIE persistently increases microglial CD11b + IR in PFC, NAc, dBNST, amygdala, and lPAG. Thus, the intermittent cycles of EtOH exposure and withdrawal may alter synapses and circuits known to be impacted by EtOH by changing microglial CD11b levels. This is consistent with chronic EtOH causing a long‐lasting “priming” of microglia, a process in which an initial stimulus causes a greater response to a secondary stimulus (Perry and Holmes, 2014). Previous studies using 4‐day binge EtOH dependence model in rats find increased CD11b + IR in the hippocampus, just after exposure, and increasing for at least 28 days (Marshall et al., 2013; McClain et al., 2011). Our studies add to this work that AIE persistently increases CD11b + IR 40 days later in adulthood. Previous studies find that AIE increases expression of multiple neuroimmune signaling molecules in PFC that are likely to be in part related to microglial signaling that could alter neurocircuitry and behavior (Vetreno and Crews, 2012; Vetreno et al., 2013). Other recent studies have discovered stress and glucocorticoids sensitize brain neuroimmune responses through induction of HMGB1, microglial TLRs, and induction of proinflammatory cytokines (Frank et al., 2016). Although both chronic EtOH and stress increase brain neuroimmune signaling, there are brain regional differences in responses (Knapp et al., 2016), with some blocked by CRF antagonists (Whitman et al., 2013) further linking stress‐ and alcohol exposure–induced changes in neuroimmune signaling and microglia to allostatic sensitization of stress circuitry (Koob, 2013). We found microglia CD11b + IR persistently increased in brain regions involved in stress responses and reward. Increased microglial CD11b + IR may be related to long‐term changes in synaptic remodeling, as CD11b is a component of the CR3 that is involved in pruning synapses (Schafer et al., 2012) (Fig. 10 A,B). Thus, AIE may alter synapses and circuits by increasing microglial CD11b + IR in stress and EtOH responding brain regions.
Microglia are known to undergo priming, a process in which an initial stimulus causes a greater response to a secondary stimulus (Perry and Holmes, 2014), likely related to the persistent increases in neuroimmune gene expression found after repeated stress or chronic EtOH exposure (Vetreno and Crews, 2014). To our knowledge, the studies reported here for the first time find that adolescent EtOH exposure enhances the adult CD11b microglial response to acute stress in multiple brain regions, including the PrL, IL, dBNST, and BLA. The increase in microglial CD11b likely represents “priming” of microglia. We observed that plasma endotoxin correlated with PrL CD11b across groups in the acute EtOH and acute stress experiment (Fig. 10 A). This suggests a relationship between the plasma endotoxin and the PrL microglial response, consistent with endotoxin inducing an immune response that activates microglia. PrL CD11b correlated with endotoxin and did not correlate with plasma CORT, suggesting the PrL microglia are responding to the inflammatory endotoxin, rather than being suppressed by the anti‐inflammatory CORT. The neuronal c‐Fos responses were robustly increased by stress across lPAG, hypothalamic, BNST, NAc, and PFC brain regions far more than the EtOH response. Plasma CORT correlated with PrL c‐Fos across groups (Fig. 10 A), consistent with acute vagal activation of brain stress pathways and HPA signaling being linked, although this may simply be related to progressive increases in c‐Fos + IR across control, EtOH, stress, and the combination of acute EtOH and stress. Interestingly, acute changes in plasma endotoxin and PrL CD11b also correlated across groups (Fig. 10). The CD11b–endotoxin correlation could also simply be related to increases across groups for both end points; however, systemic cytokines can activate brain microglial responses (Crews et al., 2017), and it is possible the endotoxin–PrL CD11b correlations are directly linked through acute endotoxin–induced increases in blood immune signaling cytokines that increase CD11b and prime microglia in stress responsive brain regions.
To our knowledge, this is the first study on persistent effects of EtOH exposure on stress‐induced changes in blood endotoxin. We find that AIE exposure causes persistent increases in adult stress‐induced plasma endotoxin responses. AIE also persistently increased adult CD11b expression in multiple brain regions, suggesting long‐lasting priming‐like activation of microglia. Chronic EtOH also persistently sensitized the microglial CD11b response to acute stress in multiple brain regions (Fig. 10), consistent with microglial priming and a highly significant correlation of across groups of plasma endotoxin with increases in frontal cortex CD11b + IR. Interestingly, the CORT response following AIE was weakly correlated with CD11b as well. Although this is a weak correlation, it could represent a shift in CORT responses to microglial priming‐like responses (Frank et al., 2016; Sorrells et al., 2009) following chronic AIE exposure. These results are consistent with acute and chronic EtOH enhancing the effects of stress on the neuroimmune system, and may provide novel insight into how EtOH and stress interact to contribute to disease.
We studied c‐Fos neuronal responses in multiple stress and EtOH responsive brain regions. Acute stress induced c‐Fos about 2‐fold or more than acute EtOH in PFC PVN, NAc shell, vBNST, and BLA. Interestingly, the combination of acute stress and EtOH induced c‐Fos similar to acute stress in the brain regions studied, except for the BLA and NAc shell where the combination was less than stress alone. The CeA was particularly unique in that acute EtOH induced a c‐Fos response greater than acute stress and the combination was less than EtOH alone. These c‐Fos responses indicate brain regional differences in the interaction of acute stress with EtOH on neuronal responses. In contrast to microglial CD11b priming, AIE treatment did not alter adult c‐Fos responses to stress in any brain region. Interestingly, we have carried out a similar study of c‐Fos using adult EtOH challenges following AIE treatment (Liu and Crews, 2015). AIE treatments were identical, for example, P28 to P54; however, the acute stress challenge studied in this report was on P96, whereas the EtOH challenge was P80, both adults but slightly different adult ages (Liu and Crews, 2015). The previous studies found AIE changes the adult c‐Fos response to an EtOH challenge, blunting PFC c‐Fos and enhancing NAc c‐Fos consistent with loss of top‐down PFC and enhanced reward responses (Liu and Crews, 2015). These 2 studies might reflect adaptations unique to EtOH or restraint stress. Both EtOH and stress activate stress responsive pathways; however, AIE changed the c‐Fos response to an adult EtOH challenge, and it did not alter the c‐Fos response to adult restraint stress. This is consistent with a specific habituation to the alcohol stressor (Herman, 2013), but not generalization to the restraint stressor. Overall, these results suggest that chronic EtOH causes long‐lasting, EtOH stressor–specific changes in the neuronal c‐Fos response, but does not persistently alter the neuronal stress c‐Fos response.
In the present study, we examined the effects of acute and adolescent intermittent EtOH on the plasma CORT, plasma endotoxin, and microglial and neuronal response to acute stress. We find acute EtOH exposure and acute stress interacted to enhance plasma endotoxin levels, as well as microglial CD11b in multiple brain regions. These findings are consistent with microglial priming by EtOH exposure in stress responsive brain regions that are enhanced when stress and EtOH exposure are combined as well as when adult acute stress occurs following adolescent alcohol exposure. These results provide insight into EtOH and stress effects on endotoxin and microglia that could contribute to how stress and EtOH interact to contribute to AUD.
Sources of Support
This work was supported in part by the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism grants AA011605, AA020024, and AA020023 to FTC and F30AA024027 to TJW. None of the authors have any conflicts of interest to declare.
Supporting information
Fig. S1. Schematic representation of brain regions examined.
Fig. S2. Effects of chronic EtOH on CD11b+ IR and microglial cell density in the mPFC at P55.
Fig. S3. Effects of chronic EtOH and acute stress on Iba1+ IR and cell density in the mPFC.
Fig. S4. Effects of chronic EtOH and acute stress on microglial markers.
Table S1. Table of statistics.
References
- Ahlers KE, Karacay B, Fuller L, Bonthius DJ, Dailey ME (2015) Transient activation of microglia following acute alcohol exposure in developing mouse neocortex is primarily driven by BAX‐dependent neurodegeneration. Glia 63:1694–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait‐Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L, Houdeau E, Fioramonti J, Bueno L, Theodorou V (2012) Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology 37:1885–1895. [DOI] [PubMed] [Google Scholar]
- Beynon SB, Walker FR (2012) Microglial activation in the injured and healthy brain: what are we really talking about? Practical and theoretical issues associated with the measurement of changes in microglial morphology. Neuroscience 225:162–171. [DOI] [PubMed] [Google Scholar]
- Blaine SK, Sinha R (2017) Alcohol, stress, and glucocorticoids: from risk to dependence and relapse in alcohol use disorders. Neuropharmacology 122:136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, Overstreet DH, Navarro M, Wills TA, Angel RA (2008) Repeated lipopolysaccharide (LPS) or cytokine treatments sensitize ethanol withdrawal‐induced anxiety‐like behavior. Neuropsychopharmacology 33:867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Lawrimore CJ, Walter TJ, Coleman LG (2017) The role of neuroimmune signaling in alcoholism. Neuropharmacology 122:56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Nixon K, Wilkie ME (2004) Exercise reverses ethanol inhibition of neural stem cell proliferation. Alcohol 33:63–71. [DOI] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J (2013) High mobility group box 1/Toll‐like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry 73:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Vetreno RP (2016) Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 233:1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan WE, Herman JP, Battaglia DF, Akil H, Watson SJ (1995) Pattern and time course of immediate early gene expression in rat brain following acute stress. Neuroscience 64:477–505. [DOI] [PubMed] [Google Scholar]
- Ellis FW (1966) Effect of ethanol on plasma corticosterone levels. J Pharmacol Exp Ther 153:121–127. [PubMed] [Google Scholar]
- Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, Fioramonti J (2006) Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol 168:1148–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF (2007) Microglia serve as a neuroimmune substrate for stress‐induced potentiation of CNS pro‐inflammatory cytokine responses. Brain Behav Immun 21:47–59. [DOI] [PubMed] [Google Scholar]
- Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF (2016) The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: a role for the NLRP3 inflammasome. Brain Behav Immun 55:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Weber MD, Watkins LR, Maier SF (2015) Stress sounds the alarmin: the role of the danger‐associated molecular pattern HMGB1 in stress‐induced neuroinflammatory priming. Brain Behav Immun 48:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Crews FT (2008) Increased MCP‐1 and microglia in various regions of the human alcoholic brain. Exp Neurol 210:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP (2013) Neural control of chronic stress adaptation. Front Behav Neurosci 7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson C, Schank JR, Rehman F, Stojakovic A, Björk K, Barbier E, Solomon M, Tapocik J, Engblom D, Thorsell A, Heilig M (2017) Proinflammatory signaling regulates voluntary alcohol intake and stress‐induced consumption after exposure to social defeat stress in mice. Addict Biol 22:1279–1288. [DOI] [PubMed] [Google Scholar]
- Kato K, Murai I, Asai S, Matsuno Y, Komuro S, Kaneda N, Iwasaki A, Ishikawa K, Nakagawa S, Arakawa Y, Kuwayama H (1998) Protective role of melatonin and the pineal gland in modulating water immersion restraint stress ulcer in rats. J Clin Gastroenterol 27(Suppl 1):S110–S115. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Harper KM, Whitman BA, Zimomra Z, Breese GR (2016) Stress and withdrawal from chronic ethanol induce selective changes in neuroimmune mRNAs in differing brain sites. Brain Sci 6:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF (2013) Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci 13:3–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Ahmed SH, Boutrel B, Chen SA, Kenny PJ, Markou A, O'Dell LE, Parsons LH, Sanna PP (2004) Neurobiological mechanisms in the transition from drug use to drug dependence. Neurosci Biobehav Rev 27:739–749. [DOI] [PubMed] [Google Scholar]
- Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D (2016) Microglial TNF‐alpha suppresses cocaine‐induced plasticity and behavioral sensitization. Neuron 90:483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Crews FT (2015) Adolescent intermittent ethanol exposure enhances ethanol activation of the nucleus accumbens while blunting the prefrontal cortex responses in adult rat. Neuroscience 293:92–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, Casachahua JD, Rinker JA, Blose AK, Lysle DT, Thiele TE (2016) IL‐1 receptor signaling in the basolateral amygdala modulates binge‐like ethanol consumption in male C57BL/6J mice. Brain Behav Immun 51:258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, McClain JA, Kelso ML, Hopkins DM, Pauly JR, Nixon K (2013) Microglial activation is not equivalent to neuroinflammation in alcohol‐induced neurodegeneration: the importance of microglia phenotype. Neurobiol Dis 54:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, McKnight KH, Blose AK, Lysle DT, Thiele TE (2017) Modulation of binge‐like ethanol consumption by IL‐10 signaling in the basolateral amygdala. J Neuroimmune Pharmacol 12:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J, Ferguson L, Harris RA (2013) Neuroimmune signaling: a key component of alcohol abuse. Curr Opin Neurobiol 23:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Morris SA, Deeny MA, Marshall SA, Hayes DM, Kiser ZM, Nixon K (2011) Adolescent binge alcohol exposure induces long‐lasting partial activation of microglia. Brain Behav Immun 25(Suppl 1):S120–S128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi K, Yuzurihara M, Ishige A, Sasaki H, Chui DH, Tabira T (2000) Chronic stress induces impairment of spatial working memory because of prefrontal dopaminergic dysfunction. J Neurosci 20:1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB (2013) Microglia promote learning‐dependent synapse formation through brain‐derived neurotrophic factor. Cell 155:1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The Rat Brain in Stereotaxic Coordinates Academic Press, San Diego, CA. [Google Scholar]
- Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10:217–224. [DOI] [PubMed] [Google Scholar]
- Qin L, Crews FT (2012) Chronic ethanol increases systemic TLR3 agonist‐induced neuroinflammation and neurodegeneration. J Neuroinflammation 9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block M, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews F (2007) Systemic LPS causes chronic inflammation and progressive neurodegeneration. Glia 55:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R (2009) Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 50:638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera CA, Bradford BU, Seabra V, Thurman RG (1998) Role of endotoxin in the hypermetabolic state after acute ethanol exposure. Am J Physiol 275:G1252–G1258. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 74:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepis TS, Rao U, Yadav H, Adinoff B (2011) The limbic‐hypothalamic‐pituitary‐adrenal axis and the development of alcohol use disorders in youth. Alcohol Clin Exp Res 35:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovjov DA, Pluskota E, Plow EF (2005) Distinct roles for the alpha and beta subunits in the functions of integrin alphaMbeta2. J Biol Chem 280:1336–1345. [DOI] [PubMed] [Google Scholar]
- Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM (2009) The stressed CNS: when glucocorticoids aggravate inflammation. Neuron 64:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens MA, Wand G (2012) Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res 34:468–483. [PMC free article] [PubMed] [Google Scholar]
- Sugama S, Takenouchi T, Fujita M, Conti B, Hashimoto M (2009) Differential microglial activation between acute stress and lipopolysaccharide treatment. J Neuroimmunol 207:24–31. [DOI] [PubMed] [Google Scholar]
- de Timary P, Starkel P, Delzenne NM, Leclercq S (2017) A role for the peripheral immune system in the development of alcohol use disorders? Neuropharmacology 122:148–160. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Aragon CM, Amit Z (1990) Effects of ethanol on locomotor depression and corticosterone release induced by restraint‐stress: support for a stress‐ethanol interaction. Pharmacol Biochem Behav 36:273–278. [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT (2012) Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll‐like receptors in the adult prefrontal cortex. Neuroscience 226:475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT (2014) Current hypotheses on the mechanisms of alcoholism. Handb Clin Neurol 125:477–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Qin L, Crews FT (2013) Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiol Dis 59:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilpoux C, Warnault V, Pierrefiche O, Daoust M, Naassila M (2009) Ethanol‐sensitive brain regions in rat and mouse: a cartographic review, using immediate early gene expression. Alcohol Clin Exp Res 33:945–969. [DOI] [PubMed] [Google Scholar]
- Walter TJ, Crews FT (2017) Microglial depletion alters the brain neuroimmune response to acute binge ethanol withdrawal. J Neuroinflammation 14:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Stone E, McEwen BS (1994) Induction and habituation of c‐fos and zif/268 by acute and repeated stressors. NeuroReport 5:1321–1324. [PubMed] [Google Scholar]
- Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF (2015) Stress induces the danger‐associated molecular pattern HMGB‐1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci 35:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman BA, Knapp DJ, Werner DF, Crews FT, Breese GR (2013) The cytokine mRNA increase induced by withdrawal from chronic ethanol in the sterile environment of brain is mediated by CRF and HMGB1 release. Alcohol Clin Exp Res 37:2086–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Schematic representation of brain regions examined.
Fig. S2. Effects of chronic EtOH on CD11b+ IR and microglial cell density in the mPFC at P55.
Fig. S3. Effects of chronic EtOH and acute stress on Iba1+ IR and cell density in the mPFC.
Fig. S4. Effects of chronic EtOH and acute stress on microglial markers.
Table S1. Table of statistics.