

Abstract
Objective
De novo mutations contribute significantly to severe early‐onset genetic disorders. Even if the mutation is apparently de novo, there is a recurrence risk due to parental germ line mosaicism, depending on in which gonadal generation the mutation occurred.
Methods
We demonstrate the power of using SMRT sequencing and ddPCR to determine parental origin and allele frequencies of de novo mutations in germ cells in two families whom had undergone assisted reproduction.
Results
In the first family, a TCOF1 variant c.3156C>T was identified in the proband with Treacher Collins syndrome. The variant affects splicing and was determined to be of paternal origin. It was present in <1% of the paternal germ cells, suggesting a very low recurrence risk. In the second family, the couple had undergone several unsuccessful pregnancies where a de novo mutation PTPN11 c.923A>C causing Noonan syndrome was identified. The variant was present in 40% of the paternal germ cells suggesting a high recurrence risk.
Conclusions
Our findings highlight a successful strategy to identify the parental origin of mutations and to investigate the recurrence risk in couples that have undergone assisted reproduction with an unknown donor or in couples with gonadal mosaicism that will undergo preimplantation genetic diagnosis.
Short abstract
What's already known about this topic?
De novo mutations contribute significantly to severe early‐onset genetic disorders.
what does this study add?
A novel successful strategy to identify the parental origin of de novo mutations and to investigate the recurrence risk by SMRT sequencing and ddPCR.
1. INTRODUCTION
Over the past few years, it has been demonstrated that de novo mutations play a prominent part in rare and common forms of neurodevelopmental disorders.1, 2 In common clinical practice, a mutation is considered de novo if the mutation can be detected by Sanger sequencing in peripheral blood of the proband, but not in either of the parents. After birth of a child with a genetic disease caused by a de novo mutation, the recurrence risk of the condition in a future offspring will vary depending on the gonadal generation in which the mutation occurred. A mutation in a germline cell early in development can produce a large clone of mutant germ cells and result in germline mosaicism (also called gonadal mosaicism). De novo mutations can also occur in the early stage of embryonic development. Depending on the timing of the postzygotic mutation during development, such mutations may lead to somatic or germline mosaicism or both (for review see Forsberg et al).3 Germline mosaicism is usually only confirmed if parents have more than one child with the same genetic disorder and both parents are not affected or carriers of the condition. Parental germline mosaicism has now been reported for a wide range of disorders.4, 5 This group of disorders comprises various conditions that typically are autosomal dominant but occasionally manifests with an autosomal recessive inheritance where one parent is gonadal mosaic and the other has a germline variant in the same gene.6
Recent studies, using more sensitive technologies such as next generation sequencing and genomic microarrays, have suggested that mosaicism in somatic tissues of apparently healthy parents are more common than previously thought.4 Such somatically mosaic parents might also have germline mosaicism that potentially can cause recurrence. The presence of germline mosaicism complicates genetic counseling and usually an empiric risk is quoted. The recurrence risk for future offspring after birth of an affected child may be as high as 50% if one of the apparently healthy parents is mosaic. It is possible to investigate if the unaffected father is producing a proportion of mutant sperm; however, this is only performed at research basis. Genetic testing of female germline is not feasible and is therefore limited to somatic tissues, e.g. skin fibroblasts or hair root. In common practice, the couples are usually offered prenatal diagnosis in the next pregnancy to identify affected fetuses, if the disease‐causing variant is known. Germline mosaicism is assumed if two or more pregnancies with the same deviations have occurred and preimplantation genetic diagnosis (PGD) may then be available to help these couples identify embryos carrying the genetic disease, thus avoiding affected offspring. For PGD, it is important to know the parental origin of the mutation to increase the chance to identify an unaffected embryo. Mutation detection alone cannot be applied in PGD, as allelic drop out is common when analyzing one or a few cells. PGD is therefore routinely done by linkage analysis and mutation detection.7 The limitations are that several affected fetuses are needed to identify informative markers for linkage analysis and haplotypes can be difficult to define because of the short read length of Sanger sequencing (up to 1000 bp).
In this study, we demonstrate the power of using single‐molecule real time (SMRT) sequencing and Droplet Digital PCR (ddPCR) to identify the parental origin of de novo mutations in two families that had undergone assisted reproduction and PGD, respectively, with the specific aim to investigate the recurrence risk or to facilitate PGD.
In the first family, the couple had undergone assisted reproduction with oocyte donation, and the biological mother was unknown. After birth of the child, Treacher Collins syndrome (TCS; OMIM 154500) was suspected, and a variant of unknown significance (VUS c.3156C>T) (p.Gly1052Gly) was identified in TCOF1 (Treacle Protein). TCS is a rare autosomal dominant disorder characterized by hypoplasia of the zygomatic bones and mandible, downslanting palpebral fissures, colobomata of the lower eyelids, and external ear abnormalities often associated with bilateral conductive hearing loss. The most common gene, involved in approximately 60% of all cases with TCS, is TCOF1,8 but mutations have also been identified in POLR1C and POLR1D1.9, 10 TCS has an extreme inter and intra familial phenotypic variability. Approximately 60% of individuals have the disorder because of a de novo mutation, and approximately 40% of the individuals diagnosed with TCS have an affected parent. A case of suspected germ line mosaicism of a TCOF1 mutation has previously been reported.11
In the second family, the couple had undergone 4 pregnancies, two spontaneous miscarriages, and two terminated pregnancies due to ultrasound aberrations. The two fetuses from the terminated pregnancies were subjected for genetic testing, and a previously reported PTPN11 mutation c.923A>C (p.Asn308Thr)12 associated with Noonan syndrome (NS) was identified in both affected fetuses. NS is a heterogeneous developmental disorder with a variable clinical expression including short stature, congenital heart defect, unusual pectus deformity, and typical facial features.13, 14 It is caused by autosomal dominant activating mutations in genes involved in the RAS‐MAPK signaling pathway,15, 16, 17, 18, 19, 20, 21 and approximately 50% of the cases are associated with mutations in PTPN11.22 Although many individuals with NS have a de novo pathogenic variant, an affected parent is recognized in 30% to 75% of families, and many are diagnosed first after the diagnosis of the affected child. When the parents are clinically unaffected, the recurrence risk appears to be low (<1%).
In both families, carrier analysis of peripheral blood from the available biological parents revealed that none were carrier of the variants. To investigate the families further, we have utilized the advantages of SMRT sequencing by its extensive read lengths and depth. We could uncover the pathogenic effect of the VUS in TCOF1 as well as to determine parental origin and the allele frequencies of the mutated alleles in both PTPN11 and TCOF1. The allele frequencies were also verified by using ddPCR on DNA from sperm.
WHAT'S ALREADY KNOWN ABOUT THIS TOPIC?
De novo mutations contribute significantly to severe early‐onset genetic disorders.
WHAT DOES THIS STUDY ADD?
A novel successful strategy to identify the parental origin of de novo mutations and to investigate the recurrence risk by SMRT sequencing and ddPCR.
2. METHODS
2.1. Ethics approval and consent to participate
The study was approved according to the declaration of Helsinki. An ethical approval (2013‐01‐23, Dnr 2012/321) and informed consent for all individuals included in this project was obtained.
2.2. The first family with Treacher Collins syndrome
2.2.1. Identification of disease causing variants
Genomic DNA was extracted from EDTA blood, and sequencing of TCOF1 (26 coding exons including exon 6A) was performed (Genetics Laboratories, Churchill hospital, Oxford, England). A variant, c.3156C>T (NM_001135244) was identified and validated with PCR on gDNA in both forward and reverse direction (forward primer: 5′‐TTG CTC AGG AGT GGC AGC‐3′ and reverse primer: 5′‐ATC AAA AGC CCC ACT ACG CT‐3′), generating a 698 bp product according to standard conditions, available upon request. Sanger sequencing on 3500XL Genetic Analyzer (Thermo Fisher Scientific, Carlsbad, CA, USA) using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) was performed. Sequence analysis was performed with CodonCode Aligner V.5.0.1 software (CodonCode Corporation, Centerville, MA, USA).
2.2.2. RNA extraction, cDNA synthesis, and PCR
Total RNA was extracted from peripheral blood from the child according to the Tempus Spin RNA Isolation Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) protocol. cDNA synthesis was performed using Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) according to manufacturer's instructions (using oligo(dT) primers) following PCR amplification. Two different reverse primers to capture both isoforms of the gene (inclusion or exclusion of exon 20) were used, with forward primer (exon 18) 5′‐ TAA TCG TAG TCC AGC TGG CC‐3′, reverse primer (exon 21) 5′‐ GCT GCT GTC CTC ACT GTC AT‐3′, and reverse primer (exon 20) 5′‐ CAC TTG AAG GCT GGG TCC TG‐3′ following Sanger sequencing and analysis as previously described.
2.2.3. Separation of TCOF1 alleles with TA cloning
TCOF1 was PCR amplified from 100 ng cDNA synthesized from blood RNA, from the heterozygous child by using AmpliTaq Gold® Polymerase (Thermo Fisher Scientific, Carlsbad, CA, USA), 95°C 5 minutes, 20× (95°C 20 seconds, 65–55°C 30 seconds, 72°C 30 seconds), 25× (95°C 20 seconds, 55°C 30 seconds, 72°C 30 seconds, 72°C 20 minutes). Primers used were forward (exon 18) 5′‐TAA TCG TAG TCC AGC TGG CC‐3′, reverse (exon 21) 5′‐GCT GCT GTC CTC ACT GTC AT‐3′, and reverse (exon 20) 5′‐CAC TTG AAG GCT GGG TCC TG‐3′. TA cloning was performed with 4‐μL PCR product using TOPO® TA Cloning® Kit (Thermo Fisher Scientific, Carlsbad, CA, USA), and transformation was performed using 1‐μL salt solution and 25‐μL One Shot® TOP10 Chemically Competent E. coli (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer's instructions. Fifteen colonies were screened by dilution of the colonies in 50‐μL H2O, 10‐minute incubation at 95°C, 5 minutes centrifuge at 13 000 rpm followed by PCR, Sanger sequencing, and analysis as previously described.
2.3. The second family with Noonan syndrome
2.3.1. Identification of disease causing variant
Genomic DNA from fetus II:2 was Sanger sequenced for mutations in PTPN11 (NM_002834; exon 2–4, 7, 8, 12, and 13), SOS1 (NM_005633; exon 3, 6, 10, 14 and 16), RAF1 (NM_002880; exon 7, 14 and 17), KRAS: NM_004985; exon 2, 3, 4, and 5), and SHOC2 (NM_007373; exon 2). The result revealed the presence of a heterozygous mutation in PTPN11 c.923A>C (p.Asn308Thr, NM_002834.3), later also identified in fetus II:4. Subsequent carrier testing of the parents revealed no PTPN11 mutations, suggesting the presence of gonadal mosaicism in one of the parents. The couple decided to undergo PGD. The mutation was further validated with PCR, Sanger sequencing, and analysis according to standard conditions as previously described on gDNA in both forward and reverse direction (forward primer: 5′‐CAG CCA TTT GAG CTC CCT GA‐3′ and reverse primer: 5′‐CTG GCC TCC TAG CTA CAA CC‐3′) generating a 511 bp product.
2.3.2. Preimplantation genetic diagnosis (PGD)
The couple opted for PGD even though the origin of the mutation had not been determined at that point. PGD was offered and performed with the strategy to perform linkage analysis and transfer embryos that had inherited both the alternative maternal and alternative paternal haplotype as compared with the affected fetuses. In theory, 25% of the embryos would be possible to transfer. Four PGD cycles were conducted from October 2014 until November 2015:
-
Cycle 1
Three embryos, one unaffected, transfer, no pregnancy.
-
Cycle 2
Six embryos, one unaffected, transfer, no pregnancy.
-
Cycle 3
Two embryos, none unaffected.
-
Cycle 4
Two embryos, none unaffected.
For cycle 2, 3, and 4, one additional embryo had been possible to transfer if the carriership had been known at that point, because the embryo had inherited the “healthy” paternal allele.
2.4. Investigation of haplotype ancestry and gonadal mosaicism using SMRT sequencing
To search for variation and to determine haplotype blocks among the father and child (TCS) and the family trio (NS), QIAGEN LongRange PCR (Hilden, Germany) was first performed. For TCS, forward primer: 5′‐GCT TGG GGT CAG GTC TCT G‐3′: and reverse primer: 5′‐CCC ACA GCA CTG TCA ACC TA‐3′ was used to create a 3773 bp product and for NS, forward primer: 5′‐CCA TTC TTT GTG TGT GTC TAT GCA‐3′ and reverse primer: 5′‐GGG CTT CAT TCT TCC TAT CCA CA‐3′ was used creating a 3950 bp product.
SMRTbells™ were constructed and sequenced following the recommended Pacific Biosciences 1 kb template preparation protocol. In brief, DNA amplicons (400 ng) underwent end‐repair and adaptor ligation processes to generate SMRTbell™ libraries for circular consensus sequencing. Libraries were then subjected to exo treatment and Ampure bead wash procedures for clean‐up. SMRTbell™ libraries were quantified using the Qubit assay and library size was confirmed using the Bioanalyzer 12000 kit. Following SMRTbell™ construction, v2 primers and P5 polymerase were annealed and enzyme bound complexes attached to magnetic beads for loading. Two (TS) and three (NS) SMRTbell™ amplicon libraries were loaded on to one SMRT cell and sequenced on the PacBio RS II instrument using C3 chemistry and a 180‐minute movie time.
Mutations in the TCOF1 and PTPN11 gene were identified by aligning the PacBio sequencing reads to the reference sequence (hg19 assembly version) followed by variant calling using the Minor Variant Caller tool available in the SMRT analysis portal. A separate analysis was performed for each sample, and this revealed a total of five mutations in the TCOF1 transcript and one in the PTPN11 transcript. For each sample, high‐quality circular consensus (CCS) reads were then constructed using the Reads Of Insert plug‐in in the SMRT analysis portal. All CCS reads not having perfect matches to the forward and reverse primer sequences were removed in a subsequent filtering step. The filtered CCS reads were then used as input to the CAVA tool (https://github.com/NationalGenomicsInfrastructure/CAVA), a computational method for analysis of mutations from long‐read amplicon sequence data. The CAVA analysis determined the allele frequencies of the previously identified mutations, as well as their clonal composition, in all sequenced samples.
2.5. Frequency estimation using ddPCR
Sperm sample from both fathers were collected, genomic DNA was extracted and analyzed with ddPCR to investigate the frequency of the point mutation and estimate the recurrence risk. The ddPCR was performed using ddPCR™ Supermix for Probes (No dUTP) (Bio‐Rad, Hercules, CA) according to the manufacturer's instructions, with the restriction enzyme HaeIII for the c.923A>C (PTPN11) variant and MseI (New England Biolabs, Ipswich, MA, USA) for the c.3156C>T (TCOF1) variant. Each sample was run in triplicates with a total of 82 to 200 ng of DNA. Primers used for detection of c.3156C>T (TCOF1) were forward: 5′‐CAG CAA GGA GTC CAG TC‐3′ and reverse: 5′‐GGT ACC TGA GTG GCT G‐3′, generating a 63 bp fragment. Detection probe for C (wild‐type) was: 5′‐CCT GTT TCT TgC CAT CTG‐3′ (HEX IowaBlack) and T (mutation): 5′‐CCT CCT GTT TCT TaC CAT CT‐3′ (FAM IowaBlack). Primers used for detection of c.923A>C (PTPN11) were forward: 5′‐ CAA TGA GCC TGT TTC AGA T‐3′ and reverse 5′‐ TGT ATG GTC AGA AAA CAC TG‐3′ creating an 80 bp fragment. Detection probe for A (wild‐type) was: 5′‐ ACC ATG ATG ATA TtT GCA TTG A‐3′ (HEX IowaBlack) and C (mutation): 5′‐ TGA TGA TAT gTG CAT TGA TGT‐3′ (FAM IowaBlack). The reactions were analyzed on QX200™ Droplet Digital™ PCR System.
3. RESULTS
3.1. Treacher Collins syndrome (TCS)
3.1.1. Clinical report
A boy was born using oocyte donation and in vitro fertilization due to Turner syndrome and premature ovarian failure in the woman. A voluntary unpaid donor donated the oocyte (Figure 1A). The pregnancy was complicated with polyhydramniosis, and ultrasound in gestational week 32 + 5 showed that the fetus had malformation of the facial skeleton, cleft palate, and ear malformations. Another three ultrasound examinations were performed during the pregnancy showing persistent polyhydramniosis, but normal kidney, bladder, and ventricle of the fetus. The child was delivered by Caesarean section in the 39th gestational week. Birth weight was 2900 g, length 50 cm, and head circumference 35.5 cm. Apgar 7‐9‐9 (−1 for breathing, color, and tonus). The boy had difficulties in keeping free airway and needed continuous positive airway pressure (CPAP). The boy was born with a broad forehead, and midface hypoplasia, he had bilaterally symmetric hypoplasia of the zygomatic bones, prominent nose, and downward slant of the eyes, and no eyelid coloboma. He had a hypoplastic short mandible and a wide U‐shaped cleft of the hard and soft palate. He had low set malformed external ears with atresia of both the external auditory canals. Ultrasound of the brain showed no malformations, and ultrasound of the heart showed a persistent foramen ovale. The features are consistent with Treacher Collins syndrome.
Figure 1.
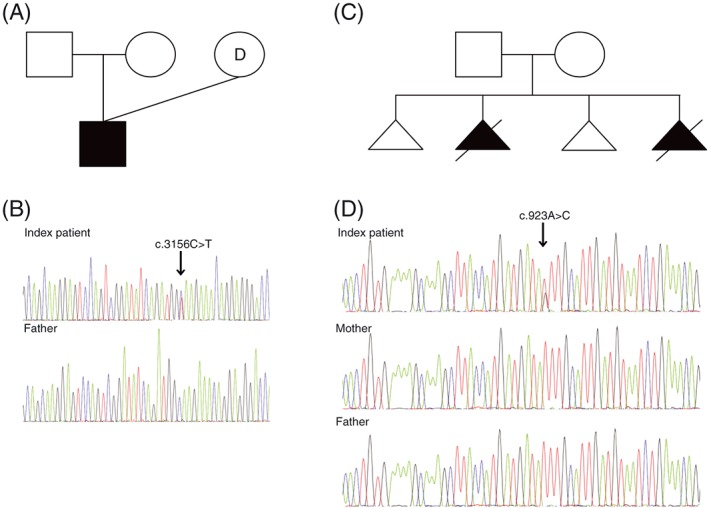
Family history and Sanger sequencing data of predicted pathogenic variants in two developmental disorders, Treacher Collins syndrome and Noonan syndrome. (A) Pedigree of the family with a boy affected with Treacher Collins syndrome, born after ovum donation. (B) Sanger sequencing of the TCOF1 gene revealed a de novo heterozygous variant of uncertain significance (VUS) c.3156C>T (p.Gly1052Gly) in the index patient. The variant was not detected in the father's blood. (C) Pedigree showing two spontaneous miscarriages and two elective abortions following aberrant ultrasound findings in the family consistent with Noonan syndrome. (D) Sanger sequencing of the PTPN11 gene revealed a de novo recurrent heterozygous mutation, c.923A>C, in one affected fetus [Colour figure can be viewed at wileyonlinelibrary.com]
3.1.2. Identification of disease‐causing variant
DNA from the affected child was subjected to Sanger sequencing of the TCOF1 gene. Data revealed six variants (Supplementary Table S1), five were considered as benign, and one heterozygous variant of uncertain significance (VUS) c.3156C>T (p.Gly1052Gly) at position chr5:149769559 (exon 19) in the TCOF1 gene (NM_001135244) was identified (Figure 1B). The variant is predicted to create a new splice donor site and affect the protein structure by causing a premature termination codon. The variant has been implicated as a potential disease‐causing variant in TCS once before; however, its effect on splicing remained to be proven.23 The variant was not identified in the father's blood (Figure 1B) and has not been reported in ExAc (http://exac.broadinstitute.org), gnomAD (http://gnomad.broadinstitute.org), or dbSNP 147 databases. Sanger sequencing of cDNA, synthesized from blood RNA, showed two isoforms for inclusion or exclusion of exon 20 and indicated that four different isoforms were present in the heterozygous index patient. Compared with a control DNA sample, primers spanning exon 19 and 20 showed the presence of one isoforms and two different isoforms when analyzed with primers from exon 19 and 21 (Supplementary Figure S1).
3.1.3. The novel variant affects splicing and creates a truncated protein
PCR and TA cloning on patient cDNA, followed by Sanger Sequencing verified a new splice donor site and four transcripts of exon 19‐21 (TCOF1, NM_001135244). The wild‐type sequence with primers of exon 19‐21 has 2 transcripts, including or excluding exon 20. The c.3156C>T variant creates two additional transcripts. If exon 20 is included, a deletion of 29 bp and a frameshift mutation of 24 amino acids resulting in a stop mutation occur, predicted to result in a truncated protein or no protein. If exon 20 is excluded, a stop gain is immediately introduced (Figure 2 , Supplementary Figure S1).
Figure 2.
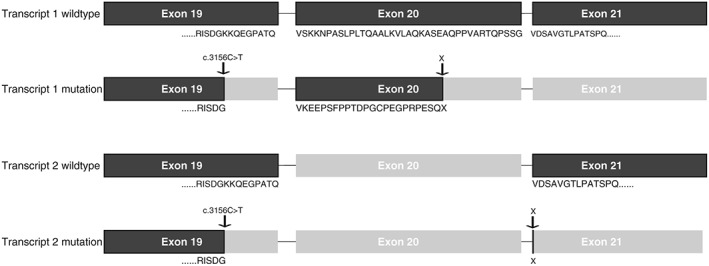
Verification of a new splice donor site in TCOF1. The wild‐type sequence has two transcripts, including or excluding exon 20. The c.3156C>T variant creates two additional transcripts. If exon 20 is included, this leads to a deletion of 29 bp and a frameshift mutation of 24 amino acids resulting in a stop mutation, predicted to result in a truncated protein or no protein. In the transcript where exon 20 is excluded, a stop gain is immediately introduced
3.2. Noonan syndrome
3.2.1. Clinical report
A non‐consanguineous healthy couple (born 1981 and 1980) have experienced two spontaneous miscarriages and two elective abortions following aberrant ultrasound findings (Figure 1C). The first pregnancy ended in a spontaneous miscarriage early in the pregnancy (II:1). In the second pregnancy, ultrasound examination was performed in the 19th gestational week, and cardiac defect and intestinal malrotation in the fetus (II:2) were detected, and the pregnancy was terminated. An autopsy confirmed the ultrasound findings: hygroma of the neck and edema of the scalp, micrognathia, flexion deformed fingers, a right sided clubfoot, a heart malformation with an atrioventricular septal defect (AVSD), dyplastic aortic valve and aortic valve stenosis, and a malrotation of the intestines. Chromosome analysis of amniotic fluid revealed a normal male karyotype, and microarray analysis of lung tissue revealed no abnormalities. In the third pregnancy, a spontaneous miscarriage occurred early in the pregnancy (approximately in the 10th gestational week; II:3). During the fourth pregnancy, a chromosome analysis was performed on Chorionic villus sampling (CVS), which showed a normal male karyotype (II:4). In the 14th gestational week, ultrasound showed hygroma, and the pregnancy was terminated. The following abnormalities were found at autopsy; hygroma of the neck and bilateral flexion deformities of the knees, but no other malformations of any organ were found. Further genetic investigation included microarray analysis, performed on fetus II:4, with no detectable pathogenic duplications or deletions. Based on the presence of hygroma in fetus II:2 and II:4, NS was suspected.
3.2.2. Identification of disease‐causing variant
A recurrent heterozygous mutation in PTPN11 c.923A>C (p.Asn308Thr, NM_002834.3) was identified in fetus II:2 and II:4 by Sanger sequencing, but not in the parental blood (Figure 1D). This mutation is previously described as pathogenic causing NS.
3.3. Investigation of haplotype ancestry and gonadal mosaicism with SMRT sequencing
SMRT sequencing on the PacBio system was performed in both families to investigate the parental origin of the mutations in TCOF1 and PTPN11, by searching for variants located adjacent to the mutation. For the TCS family, only the father's and child's DNA was investigated, whereas for the NS family, the mother, father, and affected child were analyzed. In the TCS family, a total of five variants were identified (rs79012265, rs77558738, c.3156C>T, rs2295223 and rs8004246). A total of 249 (48.1%) vs 269 (51.9%) reads, respectively, defined the child's haplotype, and 1839 reads spanned the father's haplotype a total region of 3.8 kb. The sequence variant c.3156C>T was identified on the father's haplotype (Table 1 ). For the NS family, no informative variants were detected, and therefore no result regarding haplotype origin was obtained. The region analyzed were 3.9 kb in size with a coverage of 1975–4167 X (data not shown).
Table 1.
Single‐molecule real time (SMRT) sequencing of a 3.8 kb region spanning the c.3156C>T variant. Five variants were identified in the heterozygous index patients and father (number of reads spanning 249–1837 X). The variant of uncertain significance (VUS), c.3156C>T, was identified on the father's haplotype. The biological mother's haplotype was unknown because of an oocyte donation
| Sample | rs79012265 | rs77558738 | c.3156C>T | rs2295223 | rs80042046 | Nb of reads | Haplotype % |
|---|---|---|---|---|---|---|---|
| Index patient | G | C | T | C | C | 269 | 51,93 |
| Index patient | A | T | C | T | T | 249 | 48,07 |
| Father | G | C | C | C | C | 1839 | 100% |
SMRT sequencing was performed in duplicates to investigate low‐frequency occurrence of gonadal mosaicism in the father's sperm from both the TCS and NS families. SMRT sequencing on the Pacific Bioscience system generated between 1454 and 3275 reads. For the TCS family, one mutated read, c.3156C>T, was identified in both runs, giving a frequency below detection rate for occurrence. For the NS family, 576 and 1201 reads for each run identified the variant c.923A>C, estimating a frequency of 36.6 to 37.1% in the sperm (Table 2).
Table 2.
SMRT sequencing of sperm samples (replicates) to investigate frequencies of gonadal mosaicism in Treacher Collins syndrome family and Noonan syndrome family revealed presence of the c.923A>C variant. The c.3156C>T variant (TCOF1) was below detection rate with one single read carrying the variant and was therefore designated a negative detection rate. The c.923A>C variant (PTPN11) presented a frequency of 36.6–37.1% and was considered positive
| Mutation | Wild‐type reads | Variant readsa | Nb of reads | Frequency | Detection |
|---|---|---|---|---|---|
| TCOF1 | 1453 | 1 | 1454 | 0.001 | Negative |
| TCOF1 | 3101 | 1 | 3102 | 0 | Negative |
| PTPN11 | 976 | 576 | 1552 | 0.371 | Positive |
| PTPN11 | 2074 | 1201 | 3275 | 0.366 | Positive |
For TCOF1 = c.3156C>T.
For PTPN11 = c.923A>C.
3.4. ddPCR screening for replication and frequency estimation
To confirm the results, estimate recurrence risk and the accuracy of previously obtained allele frequencies from SMRT sequencing, ddPCR was performed in triplicates on all available samples from both families. For the TCS family, the index patient had a total of 49.7% of mutation positive droplets and 50.3% wild‐type positive droplets, which is expected for a heterozygous individual. Blood and sperm from the father had 0% positive droplets likewise a healthy control sperm sample included (Figure 3; Supplementary Table S2). The recurrence risk for the TCS family, using the same oocyte donor, is therefore extremely small, based on that no mutation positive droplets were identified in the father's sperm. For the NS family, the heterozygous index patient had a total of 50.3% of mutation positive droplets and 49.7% wild‐type positive droplets. The blood samples from the mother and father were negative, with 0% mutated droplets identified. However, in the sperm sample from the father, 40% mutated droplets and 60% wild‐type reads were identified. The risk of recurrence was therefore estimated to be 40% (Figure 3; Supplementary Table S2).
Figure 3.
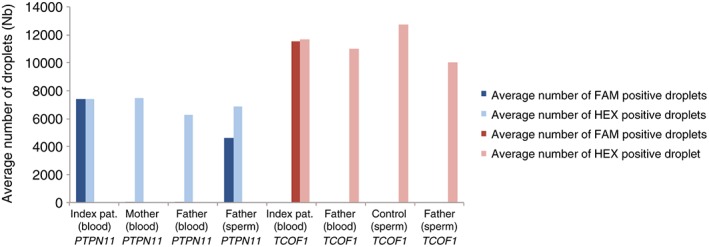
Droplet Digital PCR (ddPCR) to estimate recurrence risk and in triplicates on all samples from the Treacher Collins syndrome and Noonan syndrome families. For the Noonan syndrome family (in blue), the heterozygote index patient had a total of 50.3% of mutation positive droplets (dark blue) and 49.7% wild‐type positive droplets (light blue). The blood samples from the mother and father were negative, with 0% mutated droplets identified. Sperm sample from the father identified 40% mutated droplets (dark blue) and 60% wild‐type reads (light blue). The risk of recurrence was estimated to be 40%. For the TCS family (in red), the index patient had a total of 49.7% of mutation positive droplets (dark red) and 50.3% wild‐type positive droplets (light red). Blood and sperm from the father had 0% positive droplets as well as the healthy control sperm sample included in the study [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
De novo mutations in human disease have many implications for routine genetic testing and clinical practice. They are the cause of disease in a large fraction of patients with severe early‐onset disorders, such as rare congenital malformation syndromes,24, 25 and neurodevelopmental disorders (intellectual disability,26 epilepsy,27 and autism spectrum disorders28). These disorders represent a large proportion of all patients seen at the clinics worldwide.
Current techniques (e.g. standard Sanger sequencing), widely used in diagnostic laboratories have limitations because they are unable to distinguish between haplotype origins and to detect mutation frequencies at a low level. With the introduction of massively parallel sequencing technologies, it is now possible to study mutations at a new level of resolution. Next generation sequencing technologies allow identification at an increased depth and accuracy than previously. SMRT sequencing from Pacific Biosciences has been developed to produce long and highly accurate DNA sequences from individual molecules.29, 30 Its use has previously been mainly implicated in de novo genome assemblies, but is now being increasingly used for clinical analyses such as detection of somatic revertant mosaicism,31 quantification of BCR‐ABL transcripts in chronic myeloid leukemia (CML),32 and studying FMR1 repeat expansions in the fragile X syndrome.33 The PacBio system provides sufficiently long reads to determine haplotype origin and depth to detect mutation frequencies at a level of 1%. In addition, ddPCR system allows for an absolute quantification, high throughput, high sensitivity, and high precision with mutation detection reliability below 1%.34
This paper describes how these massively parallel sequencing technologies can be used in the clinical setting for families that have or will undergo assisted reproduction. Firstly, we demonstrate the usability of SMRT sequencing for haplotyping the parental origin of the variant TCOF1 c.3156C>T (p.Gly1052Gly), which was demonstrated to effect splicing by creating new splice donor site resulting in a frameshift mutation and introduction of a stop codon in two different transcripts, respectively. The premature termination of the TCOF1 protein, causing TCS, leads to a reduction in the amount of functional treacle proteins and rRNA, which triggers apoptosis of cells involved in the early development of the face. An abnormal cell death could explain the disturbed facial development characterizing TCS.35 Secondly, we demonstrate the usability of two different technologies (SMRT sequencing and ddPCR) for detection of gonadal mosaicism in a clinical setting, and lastly, based on above methods, the possibility to estimate the recurrence risk for subsequent pregnancies.
In the NS family, SMRT sequencing could not determine the parental origin of the de novo mutation. The region surrounding the variant was 3.9 kb in size and could have been expanded to 20 kb to increase the possibility of separating the parents haplotype. This was not performed, because the pathogenic variant was identified in the father's sperm. The ability to define the parental origin of a de novo mutation will depend on the haplotype block length between the parents, if they are consanguineous and how close their relationship is. Another factor is the recombination rate for the locus studied.
During recent years, it has been demonstrated that the amounts of de novo mutations in the offspring are associated with increased parental age. A larger proportion of the germline de novo mutations have a paternal origin as paternal germline cells acquire 0.91 mutations per year in comparison to females with 0.24 mutations per year.36 A higher paternal age at conception results in an increased number of de novo mutations in the offspring.36 This is because spermatogonial cells (SSC) continue to divide through life and have the possibility to accumulate mutations during DNA replication and also of failure to repair non‐replicative DNA damage between cell divisions.37 It is also generally accepted that 60% of TCS cases arise from de novo mutations, where mosaicism can account for a number of these cases.11 Interestingly, in NS, it has been proposed that mutated SSC gain a selective advantage by repression of stem cell differentiation signals possible by inhibiting the activity of STAT3. Previous reports suggest that the average age of fathers of NS children is greater than the average age of fathers in the general population, suggesting a paternal age effect (PAE).38 Interestingly, many of these PTPN11 mutations are recurrent with nucleotide substitution rates substantially greater than the genome average. The most common mutation, c.922A>G (p.Asn308Asp) exceeds the genome average A>G mutation frequency by more than 2400‐fold achieved by germ line selection.39 Interestingly, this mutation affects the same codon in SHP‐2 as in the NS family reported here, suggesting a similar underlying mechanism which might explain the high frequency observed for the c.923A>C (p.Asn308Thr) variant.
From a clinical perspective, it is of major relevance to investigate the origin of de novo mutations in order to estimate recurrence risk. This was strongly reflected in the Noonan family, where four PGD cycles had been performed (a total of 13 embryos). At that time, with no knowledge of paternal or maternal origin of the mutation, only two embryos were considered unaffected. Both embryos were transferred where none resulted in a pregnancy. If the carriership had been known at that time, three additional embryos could have been classified as unaffected and transferred, highlighting the importance of rapidly moving next generation sequencing technologies into a clinical setting. With inheritance information and risk assessment of recurrence, the couple is now proceeding with another cycle of PGD. In addition, the TCS family can proceed with transferring their frozen embryos, with a low recurrence risk.
5. CONCLUSIONS
Our results emphasize a strategy to identify the parental origin and investigate the recurrence risk of pathogenic mutations with the introduction of massively parallel sequencing technologies into a clinical setting. These methods will be successful for couples that have undergone assisted reproduction with an unknown donor or in couples with gonadal mosaicism that will undergo PGD.
AUTHORSHIP
All of the authors contributed significantly to this research. MW, CF, and MLB designed and coordinated the study. ELS, HM, MLB, GA, and CF assessed the clinical findings of the cases. MW, JJ, AA, and SG performed the molecular genetic studies and analyzed the data. SG and JJ performed the TA cloning. MW and MLB wrote the draft of the manuscript with input from the other co‐authors. All authors read and approved the final manuscript.
CONFLICT OF INTERESTS
The authors declare no competing financial interests.
Supporting information
Figure S1. Sanger sequencing data showing the introduction of 2 novel transcripts in TCOF1 the index patient affected by Treacher Collins syndrome. A) Sanger data of the index patient and a control sample before cloning, showing 2 transcripts when analyzing exon 19–20 and 4 transcripts when analyzing exon 19–21. B) Sanger sequencing after TA cloning verifying the 2 novel transcripts.
Table S1. Sanger sequencing of TCOF1 gene in the patient diagnosed with Treacher Collins syndrome revealed 6 variants. Five were considered as non‐pathogenic and 1 heterozygous variant of uncertain significance (VUS) c.3156C > T (p.Gly1052Gly) at chr5:149769559 (exon 18) in the TCOF1 gene (NM_001135244) was identified.
Table S2. Mutation detection of PTPN11 and TCOF1 using Droplet Digital PCR (ddPCR) on gDNA extracted from blood and/or sperm of the available family members.
ACKNOWLEDGEMENTS
The authors wish to acknowledge the families for participating in this study. SMRT sequencing using the PacBio system sequencing was performed at the Uppsala node of the National Genomics Infrastructure (NGI). This study was supported by grants from Uppsala University, Faculty of Medicine, for psychiatric and neurological research, Marcus Borgströms Foundation, Magnus Bergvalls Foundation, The Lars Hierta Memorial Foundation, and The Royal Physiographic Society in Lund, The Swedish Society of Medicine and grants from Uppsala University Hospital. MW was supported by grants from the Swedish Society for Medical Research (SSMF). SG was supported by grants from the Sävstaholm Foundation.
Wilbe M, Gudmundsson S, Johansson J, et al. A novel approach using long‐read sequencing and ddPCR to investigate gonadal mosaicism and estimate recurrence risk in two families with developmental disorders. Prenatal Diagnosis. 2017;37:1146–1154. https://doi.org/10.1002/pd.5156
Maria Wilbe and Sanna Gudmundsson are both first authors.
Contributor Information
Maria Wilbe, Email: maria.wilbe@igp.uu.se.
Marie‐Louise Bondeson, Email: marielouise.bondeson@igp.uu.se.
REFERENCES
- 1. Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565‐575. [DOI] [PubMed] [Google Scholar]
- 2. Deciphering developmental disorders S: prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease—clones picking up speed. Nat Rev Genet. 2017;18:128‐142. [DOI] [PubMed] [Google Scholar]
- 4. Campbell IM, Yuan B, Robberecht C, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. 2014;95:173‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14:307‐320. [DOI] [PubMed] [Google Scholar]
- 6. Anazi S, Al‐Sabban E, Alkuraya FS. Gonadal mosaicism as a rare cause of autosomal recessive inheritance. Clin Genet. 2014;85:278‐281. [DOI] [PubMed] [Google Scholar]
- 7. Harton GL, De Rycke M, Fiorentino F, et al. ESHRE PGD consortium best practice guidelines for amplification‐based PGD. Hum Reprod. 2011;26:33‐40. [DOI] [PubMed] [Google Scholar]
- 8. Vincent M, Genevieve D, Ostertag A, et al. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet Med. 2016;18:49‐56. [DOI] [PubMed] [Google Scholar]
- 9. Splendore A, Fanganiello RD, Masotti C, Morganti LS, Passos‐Bueno MR. TCOF1 mutation database: novel mutation in the alternatively spliced exon 6A and update in mutation nomenclature. Hum Mutat. 2005;25:429‐434. [DOI] [PubMed] [Google Scholar]
- 10. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. The Treacher Collins Syndrome Collaborative Group. Nat Genet. 1996;12:130‐136. [DOI] [PubMed] [Google Scholar]
- 11. Shoo BA, McPherson E, Jabs EW. Mosaicism of a TCOF1 mutation in an individual clinically unaffected with Treacher Collins syndrome. Am J Med Genet A. 2004;126A:84‐88. [DOI] [PubMed] [Google Scholar]
- 12. Zenker M, Buheitel G, Rauch R, et al. Genotype‐phenotype correlations in Noonan syndrome. J Pediatr. 2004;144:368‐374. [DOI] [PubMed] [Google Scholar]
- 13. Allanson JE. Noonan syndrome. J Med Genet. 1987;24:9‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007;2:4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schubbert S, Bollag G, Lyubynska N, et al. Biochemical and functional characterization of germ line KRAS mutations. Mol Cell Biol. 2007;27:7765‐7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denayer E, de Ravel T, Legius E. Clinical and molecular aspects of RAS related disorders. J Med Genet. 2008;45:695‐703. [DOI] [PubMed] [Google Scholar]
- 17. Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cordeddu V, Di Schiavi E, Pennacchio LA, et al. Mutation of SHOC2 promotes aberrant protein N‐myristoylation and causes Noonan‐like syndrome with loose anagen hair. Nat Genet. 2009;41:1022‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cirstea IC, Kutsche K, Dvorsky R, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010;42:27‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martinelli S, De Luca A, Stellacci E, et al. Heterozygous germline mutations in the CBL tumor‐suppressor gene cause a Noonan syndrome‐like phenotype. Am J Hum Genet. 2010;87:250‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aoki Y, Niihori T, Banjo T, et al. Gain‐of‐function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93:173‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP‐2, cause Noonan syndrome. Nat Genet. 2001;29:465‐468. [DOI] [PubMed] [Google Scholar]
- 23. Bowman M, Oldridge M, Archer C, et al. Gross deletions in TCOF1 are a cause of Treacher‐Collins‐Franceschetti syndrome. Eur J Hum Genet. 2012;20:769‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoischen A, van Bon BW, Rodriguez‐Santiago B, et al. De novo nonsense mutations in ASXL1 cause Bohring‐Opitz syndrome. Nat Genet. 2011;43:729‐731. [DOI] [PubMed] [Google Scholar]
- 26. Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109‐1112. [DOI] [PubMed] [Google Scholar]
- 27. Epi KC, Epilepsy Phenome/Genome P , Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iossifov I, O'Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eid J, Fehr A, Gray J, et al. Real‐time DNA sequencing from single polymerase molecules. Science. 2009;323:133‐138. [DOI] [PubMed] [Google Scholar]
- 30. Hestand MS, Van Houdt J, Cristofoli F, Vermeesch JR. Polymerase specific error rates and profiles identified by single molecule sequencing. Mutat Res. 2016;784‐785:39‐45. [DOI] [PubMed] [Google Scholar]
- 31. Gudmundsson S, Wilbe M, Ekvall S, et al. Revertant mosaicism repairs skin lesions in a patient with keratitis‐ichthyosis‐deafness (KID) syndrome by second‐site mutations in Connexin 26. Hum Mol Genet. 2017;26:1070‐1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cavelier L, Ameur A, Haggqvist S, et al. Clonal distribution of BCR‐ABL1 mutations and splice isoforms by single‐molecule long‐read RNA sequencing. BMC Cancer. 2015;15:45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ardui S, Race V, Zablotskaya A, et al. Detecting AGG interruptions in male and female FMR1 premutation carriers by single‐molecule sequencing. Hum Mutat. 2017;38:324‐331. [DOI] [PubMed] [Google Scholar]
- 34. Hindson BJ, Ness KD, Masquelier DA, et al. High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604‐8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dixon J, Jones NC, Sandell LL, et al. Tcof1/treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006;103:13403‐13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goldmann JM, Wong WS, Pinelli M, et al. Parent‐of‐origin‐specific signatures of de novo mutations. Nat Genet. 2016;48:935‐939. [DOI] [PubMed] [Google Scholar]
- 37. Gao Z, Wyman MJ, Sella G, Przeworski M. Interpreting the dependence of mutation rates on age and time. PLoS Biol. 2016;14(1):e1002355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tartaglia M, Cordeddu V, Chang H, et al. Paternal germline origin and sex‐ratio distortion in transmission of PTPN11 mutations in Noonan syndrome. Am J Hum Genet. 2004;75:492‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yoon SR, Choi SK, Eboreime J, Gelb BD, Calabrese P, Arnheim N. Age‐dependent germline mosaicism of the most common noonan syndrome mutation shows the signature of germline selection. Am J Hum Genet. 2013;92:917‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sanger sequencing data showing the introduction of 2 novel transcripts in TCOF1 the index patient affected by Treacher Collins syndrome. A) Sanger data of the index patient and a control sample before cloning, showing 2 transcripts when analyzing exon 19–20 and 4 transcripts when analyzing exon 19–21. B) Sanger sequencing after TA cloning verifying the 2 novel transcripts.
Table S1. Sanger sequencing of TCOF1 gene in the patient diagnosed with Treacher Collins syndrome revealed 6 variants. Five were considered as non‐pathogenic and 1 heterozygous variant of uncertain significance (VUS) c.3156C > T (p.Gly1052Gly) at chr5:149769559 (exon 18) in the TCOF1 gene (NM_001135244) was identified.
Table S2. Mutation detection of PTPN11 and TCOF1 using Droplet Digital PCR (ddPCR) on gDNA extracted from blood and/or sperm of the available family members.