

Abstract
Macrocycles play an increasing role in drug discovery, but their synthesis is often demanding. Computational tools that suggest macrocyclization based on a known binding mode and that estimate the binding affinity of these macrocycles could have a substantial impact on the medicinal chemistry design process. For both tasks, we established a workflow with high practical value. For five diverse pharmaceutical targets we show that the effect of macrocyclization on binding can be calculated robustly and accurately. Applying this method to macrocycles designed by LigMac, a search tool for de novo macrocyclization, our results suggest that we have a robust protocol in hand to design macrocycles and prioritize them prior to synthesis.
Keywords: drug design, free energy calculations, macrocycles, molecular dynamics, molecular modeling
Introduction
Improving the binding affinity of a ligand binding to a target protein is a main optimization parameter in drug design. In contrast to experiments, computational approaches have the advantage of not being dependent on the actual physical existence of a certain ligand. Hence, these approaches can be used to estimate the binding affinity and prioritize compounds prior to synthesis. Significant progress in calculating binding affinities has been made in recent years.1 Thanks to substantially increased computational performance of modern graphic cards, these simulation‐based techniques are now also fast enough to match drug design project timelines.
It has been shown that ligands with macrocyclic structure can provide several advantages such as diverse functionality, high binding affinity and selectivity.2 Especially for challenging targets, macrocycles can lead to well‐suited drug candidates.3 Nevertheless, broad interest in synthetic macrocycles is a relatively young phenomenon.4
To the best of our knowledge, this is the first study to investigate whether or not accurate and reliable computational estimates of binding affinities of macrocyclic ligands can be obtained and whether these estimates are robust with respect to changes of protein or ligand conformations. This study uses a version of the Schrödinger Suite5 which contains an improved version of FEP+ which allows for ring opening and closing.5e, 6 Furthermore, we figure and point out chances and challenges of designing macrocycles based on non‐macrocyclic ligands using “LigMac” (see Supporting Information), a novel tool in this area of research.
We examine five diverse pharmaceutical targets: the receptor tyrosine kinase anaplastic lymphoma kinase (ALK),7 the serine protease factor 7 (FVII),8 farnesyltransferase (FTase),9 the phosphohydrolase MTH1,10 and the bromodomain‐containing protein 4 (BRD4).11
Results
The comparison of experimental and calculated binding affinity for all targets is shown in Figure 1. Nearly all simulated values agree with experiment within 1 kcal mol−1. Furthermore, the estimates of equivalent simulations are in general very similar. Simulations a) and b) are run to study the robustness of the simulation protocol with respect to small changes in the initial protein structure. Simulation c) mimics the design situation where the ligand's conformation inside the protein binding pocket is not known a priori. An overview of the different simulations is given in Table 1. Detailed information about the calculated binding affinities can be found in Tables 2–6.
Figure 1.
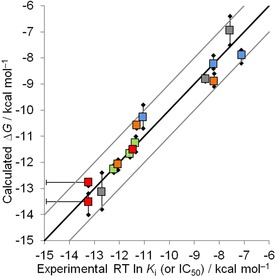
Calculated versus experimental binding affinities for all targets. Mean (squares), minimum and maximum (black diamonds) calculated affinities for the three equivalent FEP+5e simulations with different initial conditions (see Table 1) are shown. ALK is plotted in red, FVII is blue, FTase is green, MTH1 is grey, and BRD4 is orange. The black line represents the ideal estimate, and the grey lines enclose the area of an error smaller than 1 kcal mol−1. Please note that calculated ΔΔG values were transformed into ΔG values by taking the mean of all experimental data points per target as reference. This implies that only the correlation within one target (one color) is meaningful but not the overall correlation (all colors). Error bars on experimental data are used for ALK to indicate the experimental data points labeled with “<”.
Table 1.
Setup for simulations a), b), and c).[a]
| Simulation | Protein structure | Ligand | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| a) | PNL | NL | ML | 3L |
| b) | PML | NL | ML | 3L |
| c) | PNL | NL | DL | 3L |
[a] NL: non‐macrocyclic ligand, PNL: associated protein crystal structure. ML is a macrocyclic ligand similar to NL and PML its native protein crystal structure. DL is obtained by docking ML into PNL, and 3L is a third ligand needed to calculate the cycle‐closure hysteresis. For ALK and BRD4, all structures are taken from PDB files. For all other targets, 3L was modelled because there was no similar ligand available in the PDB. More details on starting coordinates and protein structures are listed in Supporting Information Table S1.
Table 2.
Calculated binding affinities for ALK.[a]
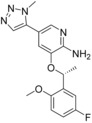
|
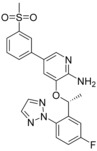
|
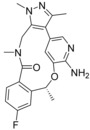
|
|||
|---|---|---|---|---|---|
| 6 b (4CNH) | 6 i (4CMT) | 8 a (4CMU) | |||
| Compd in Lit.7 | Exp. IC50 [nm] | Exp. RT ln IC50 | Calc. RT ln IC50 | ||
| a) | b) | c) | |||
| 6 b | 4 | −11.5 | −11.5 | −11.5 | −11.5 |
| 6 i | <0.2 | <−13.2 | −13.3 | −14 | −13.2 |
| 8 a | <0.2 | <−13.2 | −12.7 | −12.9 | −12.7 |
[a] Related PDB codes are given in parentheses. The starting coordinates in a) and c) of compound 8 a have an RMSD of 0.72 Å. All RT ln IC50 values are given in kcal mol−1.
Table 6.
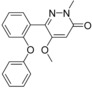
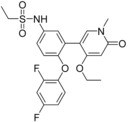
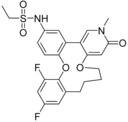
Calculated binding affinities for BRD4.[a]
|
|
|
|||
|---|---|---|---|---|---|
| 11 (5UEZ) | 25 e (5UEY) | 61 a (5UEX) | |||
| Compd in Lit.11 | Exp. IC50 [nm] | Exp. RT ln IC50 | Calc. RT ln IC50 | ||
| a) | b) | c) | |||
| 11 | 970 | −8.2 | −9 | −8.6 | −9.1 |
| 25 e | 5.1 | −11.3 | −10.5 | −10.7 | −10.5 |
| 61 a | 1.5 | −12 | −12 | −12.3 | −11.9 |
| R 2 | 0.88 | 0.93 | 0.88 | ||
| MUE | 1 | 0.7 | 1.2 | ||
[a] The starting coordinates in a) and c) of compound 61 a have an RMSD of 0.17 Å. All RT ln IC50 values are given in kcal mol−1.
A necessary, yet not sufficient condition for convergence is cycle closure hysteresis. In this study we observed hystereses ranging from 0.1 to 1.7 kcal mol−1 (see Table S1). The best converged results were obtained for the smallest protein in the set BRD4 (cycle closure hysteresis smaller than 0.5 kcal mol−1 for all simulations), whereas the largest protein, FTase, showed the slowest convergence (hysteresis up to 1.7 kcal mol−1) suggesting that more precise results could be obtained for this target by prolonging the simulation time.
Besides computationally demanding methods like FEP+,1c there are alternative approaches to calculate binding affinities such as MM‐GBSA.12 Because it makes more approximations, it is faster than FEP+. The results obtained by MM‐GBSA are displayed in Supporting Information Figure S1. Comparing with the FEP+ results (Figure 1) it can be seen that the higher computational investment does lead to substantially improved estimates.
The following results are obtained using LigMac, a tool to find possible macrocycles for a given, non‐macrocyclic ligand in the context of the binding site. A description of LigMac can be found in the Supporting Information. In this study, we used carbon‐only linkers. We applied LigMac to one structure per target. The structures used were 4CNH7 (ALK), 4ZXX8 (FVII), 1S639b (FTase), 5ANT10 (MTH1) and 5UEY11 (BRD4). LigMac returns different numbers of possible macrocycles in different conformations. Figure 2 displays an example macrocycle designed with LigMac and the original ligand 27 (5ANT, Table 3).
Figure 2.
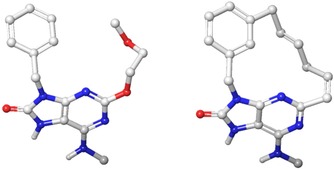
Ligand 27 from PDB 5ANT (left) and one derived LigMac result (right).
Table 3.
Calculated binding affinities for MTH1.[a]
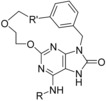
| |||||||
|---|---|---|---|---|---|---|---|
| Compd in Lit.10 | R | R′ | Exp. IC50 [nm] | Exp. RT ln IC50 | Calc. RT ln IC50 | ||
| a) | b) | c) | |||||
| 27 (5ANT) | CH3 | (open chain) | 536 | −8.6 | −8.8 | −8.9 | −8.7 |
| 15 (5ANU) | CH3 | −CH2‐O− | 0.5 | −12.7 | −13.2 | −12.4 | −13.8 |
| 26 | H | (open chain) | 2809 | −7.6 | −6.9 | −7.5 | −6.4 |
| R 2 | 0.99 | 0.99 | 0.98 | ||||
| MUE | 0.8 | 0.5 | 1.5 | ||||
[a] The starting coordinates in a) and c) of compound 15 have an RMSD of 0.13 Å. All RT ln IC50 values are given in kcal mol−1.
Due to the computational complexity of FEP+, it was not possible to run it for all LigMac suggestions. To rank all resulting ligands, we considered different metrics. One strategy was to investigate whether LigMac generates macrocycles that are close to the known literature macrocycle. To this end we compared all LigMac macrocycles to the known macrocycle crystal structure by the RMSD of the coordinates of the linker atoms (metric 1, see experimental section for details). This strategy is, however, only possible if one has such a macrocycle crystal structure available. Our aim, however, is to apply LigMac especially to examples, where there is no known macrocycle. We therefore tried MM‐GBSA for all LigMac results. A favorable MM‐GBSA score, however, often seemed to be coupled with large, hydrophobic carbon loops that probably do not come with the advantages mentioned in the introduction.2 A way to improve this was to calculate the energetic efficiency of the added structure by dividing the MM‐GBSA score by the length of the linker (metric 2).
For each target we performed one FEP+ simulation including the non‐macrocyclic ligand using its crystal structure starting coordinates and the best results from LigMac in metrics 1 and 2, respectively. The protein structure is taken from the non‐macrocyclic ligand. Our results are illustrated in Figure 3.
Figure 3.
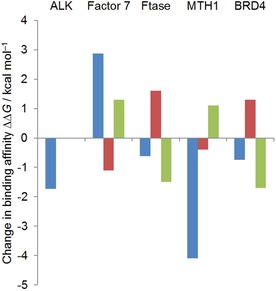
Change in binding affinity going from the five non‐macrocyclic ligands we used for LigMac to different macrocycles. Blue indicates experimentally observed binding affinity changes when going from an open‐chain molecule to published macrocycles 8 a (4CMU,7 ALK), 4 (4ZXY,8 FVII), 66 (1LD7,9a FTase), 15 (5ANU,10 MTH1), and 61 a (5UEX,11 BRD4). For each target the open‐chain reference molecules are 6 b (ALK), 1 (FVII), 2 (FTase), 27 (MTH1), and 25 e (BRD4). Red and green indicate calculated (FEP+) binding affinity changes for the best ligands according to metrics 1 and 2, respectively. All simulated LigMac macrocycles can be found in Supporting Information Table S2. A negative change indicates stronger binding. For ALK, there is one ligand scoring highest in both metrics. Its calculated binding affinity is identical to the binding affinity of the non‐macrocyclic ligand 6 b (4CNH, Table 2), and therefore the change is 0. Please note that the indicated experimental binding affinity difference for ALK is an upper bound.
Discussion
One key insight of our work is that differences in experimental binding affinities can be reproduced very well for macrocyclic and non‐macrocyclic ligands. Comparing Figure 1 and Figure S1 it is also clear, that the higher computational demand of FEP+ leads to more accurate results than the rather simple and fast version of MM‐GBSA used in this study. Additionally, FEP+ results are very robust regarding changes in the initial protein structure unlike MM‐GBSA where the values of simulation a) and b) differ significantly. We will therefore focus on data obtained by FEP+ in the following discussion.
We discuss all examples individually starting with ALK. For this target, all computed binding affinities match the experimentally obtained numbers very well: The calculated data fit the experimental data with an error of less than 1 kcal mol−1. Simulations a) and c) have nearly the identical results (difference ≤0.1 kcal mol−1) indicating that the simulations are very robust with respect to slightly different ligand starting conformations. Results from simulation b) are also similar to experimental results meaning there is no strong dependence of the initial protein structure used on the calculated affinities. The mean unsigned error (MUE) and R 2 could not be calculated for ALK as precise experimental binding affinity measurements were only available for compound 6 b.
For MTH1, all simulations return data very close to experimental results (R 2≥0.98). Interestingly, the mean unsigned error in simulation c) is higher than in a) although ligand starting coordinates differed only by 0.13 Å. Therefore, the difference in MUE is likely not due to the difference in ligand starting conformation but an indication of the intrinsic variation (precision) of these calculations.
FVII results show a similar behavior: While R 2 is very close to 1 for all simulations, the mean unsigned error in c) is relatively high (MUE=1.5 kcal mol−1) compared to a) and b) (MUE=0.7 and 0.9 kcal mol−1, respectively).
Very robust (i.e., independent of the initial protein and/or ligand starting coordinates) and accurate (i.e., in agreement with experiment) results could also be obtained for FTase and BRD4. All calculated binding affinities matched the experimental values with MUEs of ≤0.4 and ≤1.2 kcal mol−1 and R 2 of ≥0.84 and ≥0.88, respectively.
Our results are very encouraging, but we would like to point out that FEP+ was not applicable to all targets we planned to include in this study. There were four further data sets of interest. β‐Secretase 113 and Heat shock protein 9014 could not be included due to atom mapping problems of FEP+. The problems have been reported to the developers and will be fixed in forthcoming versions. Two further systems had to be excluded because the experimental data was not suited upon closer inspection: For casein kinase 2,15 the protein constructs for crystallization and biochemical assay came from different species and for cyclin‐dependent kinase 216 the crystal structure was lacking the cyclin whereas the assay was run with cyclin E.
The aim of LigMac is to find strongly binding macrocycles on the basis of a given, non‐macrocyclic ligand. All LigMac compounds (including the original non‐macrocyclic basis) for which we performed FEP+ calculations are shown in Supporting Information Table S2.
For ALK, there is a well binding macrocycle 8 a (4CMU, exp. IC50<0.2 nm, see Table 2) and a less strongly binding non‐macrocyclic ligand 6 b (4CNH, exp. IC50 4 nm, Table 2) that, besides the actual cycle, only differs in one five membered ring being a pyrazole in compound 8 a and a triazole in compound 6 b. A further computational FEP+ study (see Supporting Information Table S3) suggests that macrocyclization alone does not lead to a potency improvement but is only advantageous in combination with this pyrazole. Due to the fact that LigMac builds all macrocycles on the basis of 6 b having triazole, finding a macrocycle that was estimated to be as potent as 8 a was rather unlikely. However, the LigMac ligand scoring highest in both metrics (i.e. closest to literature macrocycle 8 a in terms of linker RMSD and top ranked by MM‐GBSA) performs as well as the non‐macrocyclic ligand 6 b and is identical to 8 a with respect to linker length and distribution of sp2 and sp3 centers.
In the FVII data set, macrocycle 4 (4ZXY, Table 4) has a very weak binding affinity (IC50=920 nm) relative to the non‐macrocyclic ligand 1 (4ZXX, Table 4), whose IC50 is 8 nm.8 Additional FEP+ calculations (see Table S4) suggest that this loss of affinity is due to the lack of a sulfone group in the macrocycle and that macrocyclization with a linker as in 4 does not have an influence on potency. For the LigMac generated macrocycles we observe mixed results: The best ligand in metric 1 is suggested to be better binding than ligand 1 and the best ligand from metric 2 is calculated to be worse binding than 1.
Table 4.
Calculated binding affinities for FVII.[a]
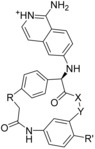
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd in Lit.8 | R | R′ | X−Y | Exp. IC50 [nm] | Exp. RT ln IC50 | Calc. RT ln IC50 | ||
| a) | b) | c) | ||||||
| 1 (4ZXX) | (open chain) |
|
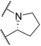
|
8 | −11.1 | −10.7 | −10.3 | −9.8 |
| 4 (4ZXY) | −(CH2)2− | H | −N‐CH2− | 920 | −8.2 | −7.9 | −8.4 | −8.4 |
| 3 | −(CH2)3− | H | −N‐CH2− | 6100 | −7.1 | −7.8 | −7.7 | −8.2 |
| R 2 | 0.95 | 1 | 0.98 | |||||
| MUE | 0.7 | 0.9 | 1.5 | |||||
[a] The starting coordinates in a) and c) of compound 4 have an RMSD of 0.1 Å. All RT ln IC50 values are given in kcal mol−1.
All three chosen FTase literature ligands are in the same range of binding affinity (IC50 from 1.1 to 4.9 nm),9a even though the structure of macrocycle 66 (1LD7, Table 5) differs from the open‐chain ligand 2 (1S63, Table 5) more than in other data sets. This scaffold difference is the reason why LigMac macrocycles are not easily comparable with the literature macrocycle. Nevertheless the macrocycle suggested by metric 2 is the best ligand we simulated for this target, so our calculations suggest that is possible to improve binding to FTase by macrocyclization.
Table 5.
Calculated binding affinities for FTase.[a]
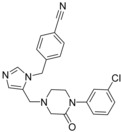
|
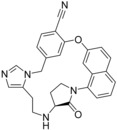
|
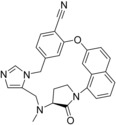
|
|||
|---|---|---|---|---|---|
| 2 (1S63) | 66 (1LD7) | 51 | |||
| Compd in Lit.9a | Exp. IC50 [nm] | Exp. RT ln IC50 | Calc. RT ln IC50 | ||
| a) | b) | c) | |||
| 2 | 3.3 | −11.6 | −11.5 | −11.7 | −11.8 |
| 66 | 1.1 | −12.2 | −12.1 | −12.3 | −12.4 |
| 51 | 4.9 | −11.3 | −11.6 | −11.2 | −11 |
| R 2 | 0.84 | 0.97 | 0.86 | ||
| MUE | 0.3 | 0.2 | 0.4 | ||
[a] The starting coordinates in a) and c) of compound 66 have an RMSD of 0.05 Å. All RT ln IC50 values are given in kcal mol−1.
Following Kettle et al.,10 the literature macrocycle for MTH1 15 (5ANU, Table 3) combines two advantages: It has a very polar linker and locks the polar centers in a favorable position. Because we use LigMac with generic carbon linkers only, none of LigMac's results is able to close the macrocycle with a hydrophilic linker. It is therefore not surprising that the estimated binding affinities of the LigMac designed macrocycles are much weaker than that of macrocycles 15.
Concerning the BRD4 dataset, the two ligands proposed by metric 1 and 2 (see Supporting Information Table S2) are very similar to the literature macrocycle 61 a (5UEX, Table 6). Similar to the results obtained for FTase, the BRD4 results suggest that potent macrocycles can be identified by our procedure. The LigMac designed macrocycle resulting from metric 2 is calculated to be a stronger binder than the literature macrocycle 61 a (see Figure 3, green bar). On the other hand, macrocyclic structures are not necessarily better, which can be seen looking at the LigMac suggestion closest to the literature macrocycle 61 a (in terms of linker atoms RMSD, that is, metric 1) suggested to be worse (see Figure 3, red bar) than the non‐macrocyclic ligand 25 e (5UEY, Table 6).
In future work we plan to extend the generic linker library by non‐generic, hydrophilic linkers. The evaluation of further metrics, such as for example, estimated ligand‐lipophilicity efficiency,17 will help to decrease the number of proposed macrocycles to a number manageable for FEP+.
Conclusions
We show that the effect of macrocyclization on binding affinity can be reproduced by calculations with good accuracy. Moreover, the results obtained are robust with respect to changes of the initial protein structure on the one hand and changes in the ligands’ starting conformations on the other hand. We also observe good agreement of calculated and experimental affinities when using docked macrocycles as input conformations. Hence we conclude that we have a robust protocol to evaluate the binding affinity of self‐designed macrocycles in a prospective setting.
Our results in macrocycle design using LigMac suggest that LigMac in combination with binding affinity calculations allows designing highly potent macrocycles. Future work will extend the linker library which is an important step toward opening the way to a larger chemical space.
Experimental Section
The platform for our work was Schrödinger's Maestro (version 2017‐1).5a All crystal structures were taken from the PDB18 and were preprocessed using the Protein Preparation Wizard5b as follows. No waters were deleted. In case of missing loops or side chains, the missing parts were filled in using Prime.5c For structures containing several chains, we chose chain A and deleted all other chains and associated ligands. Furthermore, non‐water solvent molecules stemming from the crystallization buffer were deleted. Protonation states were assigned corresponding to pH 7. The hydrogen bonding network was optimized, followed by restrained minimization using the OPLS3 force field.19
For docking jobs with Glide,5f grids were prepared using default settings. Docking was done in standard precision with flexible ligand sampling. Nitrogen inversions and ring conformations were sampled. Ligand input conformations were canonicalized. A core constraint was applied using the largest common substructure shared by all three ligands and the non‐macrocyclic ligand (Ligand 1, see Tables 1 and S1) as reference. By default, the tolerance of this core constraint was 0.1 Å. If this setting did not lead to any docked result, the procedure was retried with 1 Å tolerance. Only the binding pose with the best docking score was used during the following calculations.
Prime MM‐GBSA5c was used in combination with the OPLS319 force field. The protein was allowed to relax within 5 Å of the ligand. To make MM‐GBSA values comparable with FEP+ results, we added an offset to the calculated affinities (ΔG values) such that the mean of all calculated values for each simulation and target matched the experimental values.
To prepare the ligands for FEP+,5e, 6 missing torsional parameters were calculated using the Force Field Builder.19 The FEP Mapper was then used to set up the simulation. An improved version of FEP+ was used that allows ring opening and closing.6 For all data sets, all possible perturbations were calculated. The simulation time was set to 20 ns. For simulations of the protein–ligand complexes, the buffer width was set to 10 Å. For ligand‐in‐solvent simulations, the buffer was increased to 15 Å. The non‐bonded interaction cutoff radius was increased to 13 Å only in solvent. This larger cutoff was necessary in version 2017‐1 to ensure that if non‐macrocyclic ligands adopted an extended conformation in solvent, no (dummy) bond exceeded the cutoff radius. Default parameters were used for all other FEP+ simulation parameters.1c The optimal estimates of the relative free energies were obtained from the calculated free energy differences as detailed in Appendix A of the report by Wang et al.20 The relative free energies (ΔΔG values) were then transformed into absolute free energies (ΔG values) by taking the mean of all experimental data points per target as reference.
The program LigMac was used to generate macrocycles based on one non‐macrocyclic ligand for each of the five targets. LigMac first generates a conformer ensemble of linker fragments. These linkers are then used to connect two distant atoms within the ligand to form a macrocycle. In a last step macrocycles that clash with the protein or do not fulfill certain geometrical parameters are discarded. LigMac was applied to the PDB file of the non‐macrocyclic ligand crystal structure with two different linker libraries. The first consisted of sp2/sp3‐carbon linkers. The second library was built up similarly, but only with sp3‐carbon atoms. Additionally, the two‐command line parameters −one and −cut 0.1 were used. Only for MTH1 we chose −cut 0.3, because a lower cut would not return any macrocycles. More information about the LigMac algorithm can be found in Supporting Information.
RMSD calculations for the docked macrocycle in simulation c) were performed by superposition of all atoms. Finding the generated macrocycle closest to the literature one was done in several steps. First, the linker in the literature macrocycle was identified. Then, all heavy non‐carbon atoms were changed to carbon, keeping the original hybridization. The RMSD was then calculated without further superposition taking into account only the atoms of the generated dummy linker.
Abbreviations
BRD4, bromodomain‐containing protein 4; FVII, factor 7; FTase, farnesyltransferase; MTH1, mutT human homologue 1; MM‐GBSA, molecular mechanics energies combined with generalized Born and surface area continuum solvation method; MUE, mean unsigned error; SMARTS, SMILES arbitrary target specification; SMILES, simplified molecular input line entry specification; NL, non‐macrocyclic ligand; ML macrocyclic ligand; PNL, protein of the non‐macrocyclic ligand; PML, protein of the macrocyclic ligand; DL, docked ligand; 3L, third ligand.
Supporting Information
MM‐GBSA results for all simulation setups. Detailed protein and ligand starting coordinates for all simulations. Depiction of all selected macrocycles designed by LigMac. Setup and results of additional simulations for ALK and FVII. Description of the “LigMac” program.
Conflict of interest
The authors declare the following competing financial interest(s): V.W., L.J., H.B., and C.C. are or have been employees and/or are stockholders of Bayer AG.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Joseph Goose, Robert Abel, Lingle Wang, Daniel Cappel, and Thomas Steinbrecher for technical support and helpful discussions.
V. Wagner, L. Jantz, H. Briem, K. Sommer, M. Rarey, C. D. Christ, ChemMedChem 2017, 12, 1866.
References
- 1.
- 1a. Christ C. D., Fox T., J. Chem. Inf. Model. 2014, 54, 108–120; [DOI] [PubMed] [Google Scholar]
- 1b. Homeyer N., Stoll F., Hillisch A., Gohlke H., J. Chem. Theory Comput. 2014, 10, 3331–3344; [DOI] [PubMed] [Google Scholar]
- 1c. Wang L., Wu Y., Deng Y., Kim B., Pierce L., Krilov G., Lupyan D., Robinson S., Dahlgren M. K., Greenwood J., Romero D. L., Masse C., Knight J. L., Steinbrecher T., Beuming T., Damm W., Harder E., Sherman W., Brewer M., Wester R., Murcko M., Frye L., Farid R., Lin T., Mobley D. L., Jorgensen W. L., Berne B. J., Friesner R. A., Abel R., J. Am. Chem. Soc. 2015, 137, 2695–2703; [DOI] [PubMed] [Google Scholar]
- 1d. Aldeghi M., Heifetz A., Bodkin M. J., Knapp S., Biggin P. C., Chem. Sci. 2016, 7, 207–218; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Gathiaka S., Liu S., Chiu M., Yang H., Stuckey J. A., Kang Y. N., Delproposto J., Kubish G., J. B. Dunbar, Jr. , Carlson H. A., Burley S. K., Walters W. P., Amaro R. E., Feher V. A., Gilson M. K., J. Comput.-Aided Mol. Des. 2016, 30, 651–668; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1f. Hu Y., Sherborne B., Lee T. S., Case D. A., York D. M., Guo Z., J. Comput.-Aided Mol. Des. 2016, 30, 533–539; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Abel R., Wang L., Harder E. D., Berne B. J., Friesner R. A., Acc. Chem. Res. 2017, 50, 1625–1632; [DOI] [PubMed] [Google Scholar]
- 1h.A. S. J. S. Mey, J. Juárez-Jiménez, J. Michel, bioRxiv 2017, 150474, DOI: https://doi.org/10.1101/150474;
- 1i. Mobley D. L., Gilson M. K., Annu. Rev. Biophys. 2017, 46, 531–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Driggers E. M., Hale S. P., Lee J., Terrett N. K., Nat. Rev. Drug Discovery 2008, 7, 608–624; [DOI] [PubMed] [Google Scholar]
- 2b. Mallinson J., Collins I., Future Med. Chem. 2012, 4, 1409–1438; [DOI] [PubMed] [Google Scholar]
- 2c. Marsault E., Peterson M. L., J. Med. Chem. 2011, 54, 1961–2004; [DOI] [PubMed] [Google Scholar]
- 2d. Wessjohann L. A., Ruijter E., Garcia-Rivera D., Brandt W., Mol. Diversity 2005, 9, 171–186. [DOI] [PubMed] [Google Scholar]
- 3. Giordanetto F., Kihlberg J., J. Med. Chem. 2014, 57, 278–295. [DOI] [PubMed] [Google Scholar]
- 4. Vendeville S., Cummings M. D., Annu. Rep. Med. Chem. 2013, 48, 371–386. [Google Scholar]
- 5.
- 5a.Schrödinger Release 2017-1: Maestro, Schrödinger, LLC, New York, NY, 2017;
- 5b.Schrödinger Release 2017-1: Schrödinger Suite 2017-1 Protein Preparation Wizard;
- 5c.Schrödinger Release 2017-1: Prime, Schrödinger, LLC, New York, NY, 2017;
- 5d.Schrödinger Release 2017-1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2017. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2017;
- 5e.Schrödinger Release 2017-1: FEP+, Schrödinger, LLC, New York, NY, 2017;
- 5f.Schrödinger Release 2017-1: Glide, Schrödinger, LLC, New York, NY, 2017.
- 6. Wang L., Deng Y., Wu Y., Kim B., LeBard D. N., Wandschneider D., Beachy M., Friesner R. A., Abel R., J. Chem. Theory Comput. 2017, 13, 42–54. [DOI] [PubMed] [Google Scholar]
- 7. Johnson T. W., Richardson P. F., Bailey S., Brooun A., Burke B. J., Collins M. R., Cui J. J., Deal J. G., Deng Y. L., Dinh D., Engstrom L. D., He M., Hoffman J., Hoffman R. L., Huang Q., Kania R. S., Kath J. C., Lam H., Lam J. L., Le P. T., Lingardo L., Liu W., McTigue M., Palmer C. L., Sach N. W., Smeal T., Smith G. L., Stewart A. E., Timofeevski S., Zhu H., Zhu J., Zou H. Y., Edwards M. P., J. Med. Chem. 2014, 57, 4720–4744. [DOI] [PubMed] [Google Scholar]
- 8. Priestley E. S., Cheney D. L., DeLucca I., Wei A., Luettgen J. M., Rendina A. R., Wong P. C., Wexler R. R., J. Med. Chem. 2015, 58, 6225–6236. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Bell I. M., Gallicchio S. N., Abrams M., Beese L. S., Beshore D. C., Bhimnathwala H., Bogusky M. J., Buser C. A., Culberson J. C., Davide J., Ellis-Hutchings M., Fernandes C., Gibbs J. B., Graham S. L., Hamilton K. A., Hartman G. D., Heimbrook D. C., Homnick C. F., Huber H. E., Huff J. R., Kassahun K., Koblan K. S., Kohl N. E., Lobell R. B., J. J. Lynch, Jr. , Robinson R., Rodrigues A. D., Taylor J. S., Walsh E. S., Williams T. M., Zartman C. B., J. Med. Chem. 2002, 45, 2388–2409; [DOI] [PubMed] [Google Scholar]
- 9b. Reid T. S., Long S. B., Beese L. S., Biochemistry 2004, 43, 9000–9008. [DOI] [PubMed] [Google Scholar]
- 10. Kettle J. G., Alwan H., Bista M., Breed J., Davies N. L., Eckersley K., Fillery S., Foote K. M., Goodwin L., Jones D. R., Kack H., Lau A., Nissink J. W., Read J., Scott J. S., Taylor B., Walker G., Wissler L., Wylot M., J. Med. Chem. 2016, 59, 2346–2361. [DOI] [PubMed] [Google Scholar]
- 11. Wang L., Pratt J. K., Soltwedel T., Sheppard G. S., Fidanze S. D., Liu D., Hasvold L. A., Mantei R. A., Holms J. H., McClellan W. J., Wendt M. D., Wada C., Frey R., Hansen T. M., Hubbard R., Park C. H., Li L., Magoc T. J., Albert D. H., Lin X., Warder S. E., Kovar P., Huang X., Wilcox D., Wang R., Rajaraman G., Petros A. M., Hutchins C. W., Panchal S. C., Sun C., Elmore S. W., Shen Y., Kati W. M., McDaniel K. F., J. Med. Chem. 2017, 60, 3828–3850. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Huang N., Kalyanaraman C., Irwin J. J., Jacobson M. P., J. Chem. Inf. Model. 2006, 46, 243–253; [DOI] [PubMed] [Google Scholar]
- 12b. Rapp C., Kalyanaraman C., Schiffmiller A., Schoenbrun E. L., Jacobson M. P., J. Chem. Inf. Model. 2011, 51, 2082–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Moore K. P., Zhu H., Rajapakse H. A., McGaughey G. B., Colussi D., Price E. A., Sankaranarayanan S., Simon A. J., Pudvah N. T., Hochman J. H., Allison T., Munshi S. K., Graham S. L., Vacca J. P., Nantermet P. G., Bioorg. Med. Chem. Lett. 2007, 17, 5831–5835; [DOI] [PubMed] [Google Scholar]
- 13b. Lindsley S. R., Moore K. P., Rajapakse H. A., Selnick H. G., Young M. B., Zhu H., Munshi S., Kuo L., McGaughey G. B., Colussi D., Crouthamel M. C., Lai M. T., Pietrak B., Price E. A., Sankaranarayanan S., Simon A. J., Seabrook G. R., Hazuda D. J., Pudvah N. T., Hochman J. H., Graham S. L., Vacca J. P., Nantermet P. G., Bioorg. Med. Chem. Lett. 2007, 17, 4057–4061. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Miura T., Fukami T. A., Hasegawa K., Ono N., Suda A., Shindo H., Yoon D. O., Kim S. J., Na Y. J., Aoki Y., Shimma N., Tsukuda T., Shiratori Y., Bioorg. Med. Chem. Lett. 2011, 21, 5778–5783; [DOI] [PubMed] [Google Scholar]
- 14b. Suda A., Koyano H., Hayase T., Hada K., Kawasaki K., Komiyama S., Hasegawa K., Fukami T. A., Sato S., Miura T., Ono N., Yamazaki T., Saitoh R., Shimma N., Shiratori Y., Tsukuda T., Bioorg. Med. Chem. Lett. 2012, 22, 1136–1141. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Nie Z., Perretta C., Erickson P., Margosiak S., Almassy R., Lu J., Averill A., Yager K. M., Chu S., Bioorg. Med. Chem. Lett. 2007, 17, 4191–4195; [DOI] [PubMed] [Google Scholar]
- 15b. Nie Z., Perretta C., Erickson P., Margosiak S., Lu J., Averill A., Almassy R., Chu S., Bioorg. Med. Chem. Lett. 2008, 18, 619–623. [DOI] [PubMed] [Google Scholar]
- 16. Lücking U., Siemeister G., Schäfer M., Briem H., Krüger M., Lienau P., Jautelat R., ChemMedChem 2007, 2, 63–77. [DOI] [PubMed] [Google Scholar]
- 17. Leeson P. D., Springthorpe B., Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- 18. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J. Y., Wang L., Lupyan D., Dahlgren M. K., Knight J. L., Kaus J. W., Cerutti D. S., Krilov G., Jorgensen W. L., Abel R., Friesner R. A., J. Chem. Theory Comput. 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- 20. Wang L., Deng Y., Knight J. L., Wu Y., Kim B., Sherman W., Shelley J. C., Lin T., Abel R., J. Chem. Theory Comput. 2013, 9, 1282–1293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary