

Abstract
Chalcogen bonding is a noncovalent interaction based on electrophilic chalcogen substituents, which shares many similarities with the more well‐known hydrogen and halogen bonding. Herein, the first application of selenium‐based chalcogen bond donors in organocatalysis is described. Cationic bifunctionalized organoselenium compounds activate the carbon–chlorine bond of 1‐chloroisochroman in a benchmark reaction. While imidazolium‐based derivatives showed no noticeable activation, benzimidazolium backbones yielded potent catalysts. In all cases, syn‐isomers were markedly more active, presumably due to bidentate coordination, which was confirmed by DFT calculations. Comparison experiments with the corresponding non‐selenated as well as the non‐cationic reference compounds clearly indicate that the catalytic activity can be ascribed to chalcogen bonding. The rate acceleration by the catalyst—compared to the non‐selenated derivative—was about 10 fold.
Keywords: chalcogen bonding, Lewis acids, noncovalent interactions, nucleophilic substitution, organocatalysis
In noncovalent organocatalysis, the majority of applications are based on hydrogen bonding (HB).1 However, in the last years, interactions like anion– 2 and halogen bonding (XB)3 have begun to emerge as viable alternatives to hydrogen bonding in several fields, including anion recognition and said organocatalysis. Another type of interaction that is closely related to halogen bonding is chalcogen bonding (ChB), that is, the attraction between an electrophilic chalcogen substituent Ch in compounds R−Ch−R′ (Ch=S, Se, or Te) and Lewis bases (LB; Figure 1).4 In this context, the organochalcogen compound is called a chalcogen bond donor even though it acts as a Lewis acid (electron acceptor).
Figure 1.
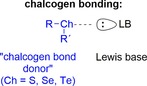
Definition of chalcogen bonding (R, R′=substituents, LB=Lewis base).
Similarly to halogen bonding, chalcogen bonding is highly directional, with an R−Ch⋅⋅⋅LB angle of approximately 180°.5, 6 Its electronic origins are based on electrostatics7 and orbital interactions,5 both of which explain the demand for electronegative substituents R to obtain stronger binding.8
Most applications of chalcogen bonding concern solid state structures and supramolecular assemblies like nanotubes,9a, 9b nanosheets,9c and macrocycles.9d Its use in solution is still very rare and is limited to a few fundamental studies on anion binding10a–10c and related applications; seleno‐ and telluriumtriazol(ium) motifs, for instance, were used by Beer et al. in anion‐binding rotaxanes.10d In addition, Matile et al. showed that sulfur‐based ChB donors with a dithienothiophene (DTT) core unit are suitable for anion transport.11
The same group also reported the first use of ChB Lewis acids as organocatalysts using DTT derivatives for the catalytic hydrogenation of quinoline to the corresponding dihydroquinolines.12 Even though these findings clearly indicate the potential of organosulfur compounds for organocatalysis, selenium as the more polarizable element should yield even stronger ChB donors. Consequently, in a recent publication, selenium‐based ChB donors were shown to activate the carbon–bromine bond of benzhydryl bromide in a solvolysis reaction.13 In this case, however, the organoselenium compounds had to be used in stoichiometric amounts.
Herein, we present, to the best of your knowledge, the first catalytic application of selenium‐based ChB donors in organocatalysis. As a benchmark reaction for the activation of a carbon–chlorine bond, the substitution of 1‐chloroisochroman 1 with a silyl ketene acetal (2) was chosen (Scheme 1), since it had already successfully served as test case for hydrogen bonding and halogen bonding organocatalysis.14a, 14b As backbones for suitable catalyst candidates, bisimidazolium structures as well as the already successfully employed bisbenzimidazolium moieties were considered. With selenium as the chalcogen, two to three different alkyl groups as second substituent were used to obtain a first idea on the role of this parameter. In the bisbenzimidazolium compounds, an additional trifluoromethyl group was placed on the central benzene core. This served different purposes, namely a) as a marker for 19F NMR monitoring, b) as further electron‐withdrawing group, and c)—most importantly—as a barrier to rotation of the benzimidazolium groups, enabling the separation of syn‐ and anti‐atropisomers.
Scheme 1.
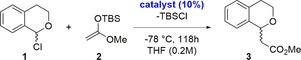
Benchmark reaction of 1‐chloroisochroman (1) with silyl ketene acetal 2 in presence of ChB (chalcogen bonding) catalysts. TBS=tert‐butyldimethylsilyl. A slight excess of 2 (1.5 equiv) was used.
The bisimidazolium‐based compounds were synthesized according to Scheme 2, starting from compound 4 (for further details see the Supporting Information).
Scheme 2.
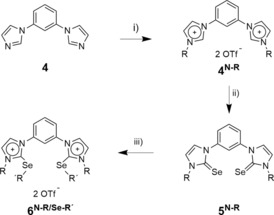
Reagents and conditions: i) R‐OTf (3 equiv.; R=methyl (Me), octyl (Oct)) CH2Cl2 (0.3 m), rt, 24 h; ii) Se (2.5 equiv.), Cs2CO3 (2.5 equiv.), MeOH (0.2 m), reflux, 24 h;15 iii) R‐OTf (4 equiv.; R=Me, Oct), CH2Cl2 (0.03 m), rt, 24 h. Selected yields: 90 % of 4N‐Oct, 95 % of 4N‐Me, 49 % of 5N‐Oct, 22 % of 5N‐Oct, 75 % of 6N‐Oct/Se‐Me, 56 % of 6N‐Me/Se‐Oct.
The bisbenzimidazolium compounds that were envisaged as catalyst candidates or reference compounds (see Figure 2 for structures and nomenclature) were synthesized as recently published by our group.13
Figure 2.
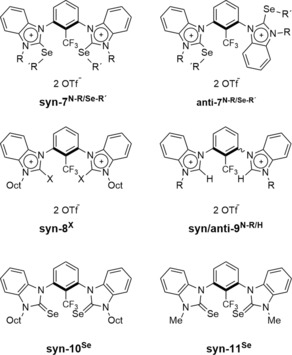
Overview of all benzimidazolium‐based catalyst candidates. R′=methyl (Me), octyl (Oct), or isopropyl (iPr); X=I or Br; OTf=trifluoromethanesulfonate. In case of syn /anti‐9N‐R/H, an inseparable mixture of both isomers was used.
Prior to the actual catalysis studies with ChB donors, some initial experiments were performed, most importantly to rule out activation by anion–π interactions of the cationic moieties or by other effects not related to chalcogen bonding (Table 1).
Table 1.
Overview of initial experiments; background activity and reference compounds to rule out activation by other effects than chalcogen bonding.
| No. | Catalyst | Mol % | Yield of 3 [%]a,b |
|---|---|---|---|
| 1 | – | 0 | ≤5 |
| 2 | syn /anti‐9N‐Me/H | 10 | ≤5 |
| 3 | syn /anti‐9N‐Oct/H | 10 | 9 |
[a] Reaction time was 118 h. [b] Yield of 3 according to 1H NMR analysis (see the Supporting Information).
It had already been shown that several possible impurities—especially traces of acid—do not act as catalyst in this reaction.14a, 16 In the absence of any additive, no background reaction was observed at −78 °C even after 118 h (Table 1, Entry 1). The non‐selenated reference compounds (and HB donors) syn /anti‐9N‐Me/H and syn /anti‐9N‐Oct/H showed little to no activity, indicating that the cationic moieties in themselves (as well as the counterions) are not capable of catalysis via anion–π interactions or other effects. The inactivity of the corresponding imidazolium‐derived hydrogen bond donors (4N‐R) had already been demonstrated.14a Hence, any activity by the selenated derivatives must be caused by the additional alkylselanyl group and, thus, by chalcogen bonding.
Based on these results, the reaction was then performed in the presence of various cationic chalcogen bond donors and comparison compounds (Table 2). All reactions were monitored by 19F NMR to check catalyst stability and all compounds proved to be stable under the reaction conditions.
Table 2.
Overview of all tested activating reagents, the catalyst load, and the determined yields of 3.[a]
| No. | Catalyst | Mol % | Yield of 3 [%][b] |
|---|---|---|---|
| 1 | syn ‐8I | 10 | ≥95[c] |
| 2 | syn ‐8Br | 10 | 40 |
| 3 | 6N‐Oct/Se‐Me | 10 | ≤5 |
| 4 | 6N‐Me/Se‐Oct | 10 | ≤5 |
| 5 | syn ‐10Se | 10 | ≤5 |
| 6 | syn ‐11Se | 10 | 26 |
| 7 | anti ‐7N‐Oct/Se–iPr | 10 | 42 |
| 8 | anti ‐7N‐Oct/Se‐Me | 10 | 19 |
| 9 | anti ‐7N‐Me/Se‐Oct | 10 | 24 |
| 10 | syn ‐7N‐Oct/Se–iPr | 10 | 74 |
| 11 | syn ‐7N‐Oct/Se‐Me | 10 | 66 |
| 12 | syn7N‐Oct/Se‐Oct | 10 | 92 |
[a] Reactions were performed at least two times. [b] Yield of 3 according to 1H NMR analysis (see the Supporting Information). [c] After 24 h.
Bisimidazolium‐based chalcogen bond donors 6N‐Oct/Se‐Me and 6N‐Me/Se‐Oct were inactive as catalysts (Table 2, Entries 3 and 4). Possibly, the imidazolium groups do not provide sufficient polarization of the selenium centers. In addition, the free rotation of the chalcogen bonding substituents does not allow the formation of strong ChBs due to the loss of entropy upon binding (especially compared to the locked benzimidazolium structures in Figure 2). Unfortunately, all attempts to synthesize locked atropisomers of the bisimidazolium compounds have failed so far.
In contrast, all tested bisbenzimidazolium compounds showed catalytic activity, with the syn isomers (Table 2, Entries 10–12) reaching up to 92 % yield of product 3. The corresponding anti analogues (Table 2, Entries 7–9) were markedly less active for all three derivatives. This indicates that while the anti isomers effectively act as twofold monodentate chalcogen bond donors, the syn isomers seem to bind in an at least partially bidentate fashion to chloride. Orientating DFT calculations were performed to demonstrate the feasibility of such a bidentate coordination of a bis(benzimidazolium)‐based model chalcogen bond donor (with methyl groups on nitrogen and selenium) to chloride (for details, see the Supporting Information). The complex is shown in Figure 3.
Figure 3.
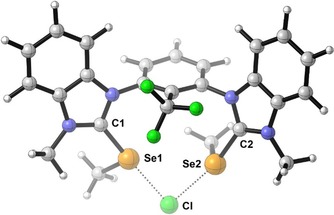
DFT calculation of the complex of a model bidentate chalcogen bond donor with chloride. Plot by CYLview[17].
Comparably weak activity was observed for the corresponding neutral selenourea compounds syn ‐10Se and syn ‐11Se (Table 2, Entries 5 and 6). As these nonalkylated species are expected to be much weaker chalcogen bond donors, this further corroborates the mode of activation by chalcogen bonding—and it also stresses the need for cationic core structures for catalytic activity.
The dependency of the activity of chalcogen bond donors syn ‐7N‐R/Se‐R′ on the alkyl group R′ on selenium is slightly different for the anti (iPr>Oct>Me) and the syn (Oct>iPr>Me) isomer. In general, bulkier groups seem to induce more activity than methyl ones for currently unknown reasons.
A direct comparison of ChB with XB (Table 2, Entries 1 and 2) showed that the iodinated Lewis acid syn ‐8I is markedly more active than even the best chalcogen bond donor. In contrast, the XB donor with the element of the same period (syn ‐8Br) is somewhat less active than ChB donors syn ‐7N‐R/Se‐R′. This is in agreement with the relative performance of these compounds as activators in a solvolysis reaction.13
To obtain further insights into the relative activity of chalcogen bonding versus halogen bonding, kinetic measurements with the strongest ChB donor syn ‐7N‐Me/Se‐Oct, the corresponding brominated XB donor syn ‐8Br, and the neutral selenourea syn ‐11Se as reference compound were performed (Figure 4). The background reaction was unsuitable as a reference, since, even after 118 h, less than 5 % of product 3 were formed.
Figure 4.
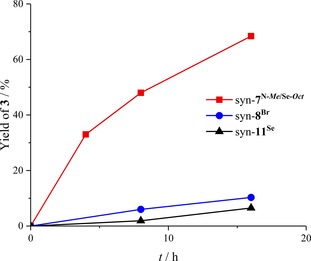
Yield‐versus‐time profile of selected reactions of Table 2. Yields were determined by 1H NMR spectroscopy (see the Supporting Information).
Compared to selenourea syn ‐11Se (k rel=1.0), XB donor syn ‐8Br (k rel=1.5) was slightly more active. In contrast, ChB donor syn ‐7N‐Me/Se‐Oct (k rel=10.8) was still seven times more active than the brominated compound. These results illustrate the fact that the difference between ChB and XB donors with elements of the same period is somewhat underestimated by the yields in Table 2.
Finally, 1H NMR titration experiments18 were performed with ChB donors 6N‐Oct/Se‐Me, 6N‐Me/Se‐Oct, and syn ‐7N‐Oct/Se–iPr to correlate their binding strength to halides with their catalytic activity. Unfortunately, the obvious choice of chloride as guest with THF as solvent was not feasible due to slight decomposition of the hosts and due to precipitation of the complexes after addition of less than one equivalent of chloride. Thus, only bromide as guest in CD3CN as solvent proved to be a suitable titration system at room temperature (Table 3).
Table 3.
Titration experiments for selected ChB donors with tetraoctylammonium bromide at room temperature.
| No. | Catalyst | Solvent | Anion | K [M−1][a] |
|---|---|---|---|---|
| 1 | 6N‐Oct/Se‐Me | CD3CN | Br‐ | 317 |
| 2 | 6N‐Me/Se‐Oct | CD3CN | Br‐ | 351 |
| 3 | syn ‐7N‐Oct/Se–iPr | CD3CN | Br‐ | 341 |
[a] K=binding constant.
The bindings constants obtained for all three ChB donors were relatively similar at around 300 m −1. While the data of 6N‐Oct/Se‐Me and 6N‐me/Se‐Oct (317 m −1 and 351 m −1) may be in line with their inactivity in the test reaction, the virtually identical binding constant of syn ‐7N‐Oct/Se–iPr (341 m −1) is evidently in stark contrast to the catalysis study. While this likely indicates that, inter alia, the relative binding strengths to bromide in CD3CN are different from the ones to chloride in THF, the data still provides a rough first estimate of the chalcogen bonding strength for these systems.
In conclusion, the first application of selenium‐based ChB donors as Lewis acidic organocatalysts was presented. While bisimidazolium‐derived ChB donors were inactive, bisbenzimidazolium‐based ones provided up to 92 % yield in the benchmark reaction of 1‐chloroisochroman with a silyl ketene acetal. Comparison experiments with the analogous HB donors ruled out other possible modes of activation next to chalcogen bonding. The syn‐atropisomers were reproducibly more active than the related anti‐isomers, pointing towards an at least partially multidentate binding of chloride by the most active ChB donors. Compared to a brominated XB donor, the rate acceleration by the XB donor was about 7 fold stronger. All these findings provide a solid basis for the further development of chalcogen bonding organocatalysis.
Even though the interaction may be generally weaker than HB or XB, it also features some unique advantages. Maybe the most notable one with respect to the catalysis presented herein is the presence of a second substituent on the electrophilic atom that is orientated at a 90° angle to the electrophilic axis. This close proximity to the substrate may offer additional control not possible with HB or XB. Studies towards the utilization of this effect are currently underway.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
P.W. and S.M.H. gratefully acknowledge funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 638337). The authors also thank the Deutsche Forschungsgemeinschaft (Cluster of Excellence RESOLV, EXC 1069) and the Fonds der Chemischen Industrie for financial support.
P. Wonner, L. Vogel, F. Kniep, S. M. Huber, Chem. Eur. J. 2017, 23, 16972.
References
- 1.See, for example:
- 1a. Taylor S. M., Jacobsen E. N., Angew. Chem. Int. Ed. 2006, 45, 1520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1550; [Google Scholar]
- 1b. Doyle A. G., Jacobsen E. N., Chem. Rev. 2007, 107, 5713; [DOI] [PubMed] [Google Scholar]
- 1c. Schreiner P. R., Chem. Soc. Rev. 2003, 32, 289; [DOI] [PubMed] [Google Scholar]
- 1d. Connon J., Chem. Eur. J. 2006, 12, 5418;16514689 [Google Scholar]
- 1e. Alemán J., Parra A., Jiang H., Jørgensen K. A., Chem. Eur. J. 2011, 17, 6890; [DOI] [PubMed] [Google Scholar]
- 1f. Zhang Z., Bao Z., Xing H., Org. Biomol. Chem. 2014, 12, 3151. [DOI] [PubMed] [Google Scholar]
- 2.See, for example:
- 2a. Zhao Y., Cotelle Y., Sakai N., Matile S. J., J. Am. Chem. Soc. 2016, 138, 4270; [DOI] [PubMed] [Google Scholar]
- 2b. Giese M., Albrecht M., Rissanen K., Chem. Commun. 2016, 52, 1778. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G., Terraneo G., Chem. Rev. 2016, 116, 2478; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Erdélyi M., Chem. Soc. Rev. 2012, 41, 3547. [DOI] [PubMed] [Google Scholar]
- 4. Murray J. S., Lane P., Clark T., Politzer P., J. Mol. Model. 2007, 13, 1033. [DOI] [PubMed] [Google Scholar]
- 5. Rosenfield R. E., Parthasarathy R., Dunitz J., J. Am. Chem. Soc. 1977, 99, 4860. [Google Scholar]
- 6. Guru Row T. N., Parthasarathy R., J. Am. Chem. Soc. 1981, 103, 477. [Google Scholar]
- 7.See, for example:
- 7a. Bleiholder C., Werz D. B., Köppel H., Gleiter R., J. Am. Chem. Soc. 2006, 128, 2666; [DOI] [PubMed] [Google Scholar]
- 7b. Nakanishi W. in Handbook of Chalcogen Chemistry: New Perspective in Sulfur, Selenium and Tellurium (Ed.: F. A. Devillanova), 2007, RSC, London, pp. 644–668; [Google Scholar]
- 7c. Cozzolino A. F., Elder P. J. W., Vargas-Baca I., Coord. Chem. Rev. 2011, 255, 1426. [Google Scholar]
- 8.
- 8a. Iwaoka M., Tomoda S., J. Am. Chem. Soc. 1996, 118, 8077; [Google Scholar]
- 8b. Iwaoka M., Komatsu H., Katsuda T., Tomoda S., J. Am. Chem. Soc. 2002, 124, 1902. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Werz D. B., Gleiter R., Rominger F., J. Am. Chem. Soc. 2002, 124, 10638; [DOI] [PubMed] [Google Scholar]
- 9b. Gleiter R., Werz D. B., Rausch B. J., Chem. Eur. J. 2003, 9, 2676; [DOI] [PubMed] [Google Scholar]
- 9c. Yi Y., Fa S., Cao W., Zeng L., Wang M., Xu H., Zhang X., Chem. Commun. 2012, 48, 7495; [DOI] [PubMed] [Google Scholar]
- 9d. Ho P. C., Szydlowski P., Sinclair J., Elder P. J. W., Kübel J., Gendy C., Lee L. M., Jenkins H., Britten J. F., Morim D. R., Vargas-Baca I., Nat. Commun. 2016, 7, 11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Zhao H., Gabbaï F. P., Nat. Chem. 2010, 2, 984; [DOI] [PubMed] [Google Scholar]
- 10b. Garrett G. E., Gibson G. L., Straus R. N., Seferos D. S., Taylor M. S., J. Am. Chem. Soc. 2015, 137, 4126; [DOI] [PubMed] [Google Scholar]
- 10c. Garrett G. E., Carrera E. I., Seferos D. S., Taylor M. S., Chem. Commun. 2016, 52, 9881; [DOI] [PubMed] [Google Scholar]
- 10d. Lim J. Y. C., Marques I., Thompson A. L., Christensen K. E., Felix V., Beer P. D., J. Am. Chem. Soc. 2017, 139, 3122. [DOI] [PubMed] [Google Scholar]
- 11. Benz S., Macchione M., Verolet Q., Mareda J., Sakai N., Matile S., J. Am. Chem. Soc. 2016, 138, 9093. [DOI] [PubMed] [Google Scholar]
- 12. Benz S., López-Andarias J., Mareda J., Sakai N., Matile S., Angew. Chem. Int. Ed. 2017, 56, 812; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 830. [Google Scholar]
- 13. Wonner P., Vogel L., Düser M., Gomes L., Kniep F., Mallick B., Werz D. B., Huber S. M., Angew. Chem. Int. Ed. 2017, 56, 12009–12012; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12172–12176. [Google Scholar]
- 14.
- 14a. Jungbauer S. H., Huber S. M., J. Am. Chem. Soc. 2015, 137, 12110; [DOI] [PubMed] [Google Scholar]
- 14b. Reisman S. E., Doyle A. G., Jacobsen E. N., J. Am. Chem. Soc. 2008, 130, 7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhabak K. B., Sathesshkumar K., Jayavelu S., Mugesh G., Org. Biomol. Chem. 2011, 9, 7343. [DOI] [PubMed] [Google Scholar]
- 16. Kniep F., Jungbauer S. H., Zhang Q., Walter S. M., Schindler S., Schapperelle I., Herdtweck E., Huber S. M., Angew. Chem. Int. Ed. 2013, 52, 7028; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7166. [Google Scholar]
- 17.C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org.
- 18.Even though we focused on 1H NMR in this study to obtain the binding constants in an efficient manner, additional valuable information could also be gained by 77Se NMR and 13C NMR (by the chemical shift changes of the carbon atoms bound to Se).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary