

Abstract
Selective oxyfunctionalizations of inert C−H bonds can be achieved under mild conditions by using peroxygenases. This approach, however, suffers from the poor robustness of these enzymes in the presence of hydrogen peroxide as the stoichiometric oxidant. Herein, we demonstrate that inorganic photocatalysts such as gold–titanium dioxide efficiently provide H2O2 through the methanol‐driven reductive activation of ambient oxygen in amounts that ensure that the enzyme remains highly active and stable. Using this approach, the stereoselective hydroxylation of ethylbenzene to (R)‐1‐phenylethanol was achieved with high enantioselectivity (>98 % ee) and excellent turnover numbers for the biocatalyst (>71 000).
Keywords: Biocatalysis, oxyfunctionalization, peroxygenases, photocatalysis, TiO2
The selective oxyfunctionalization of (non‐)activated C−H bonds still represents one of the major challenges in organic synthesis. Heme‐dependent oxygenases are valuable catalysts for this task as they feature highly reactive FeIVO species in the sterically well‐defined active site of an enzyme.1 Today, mostly P450 monooxygenases are used as biocatalysts but peroxygenases (E.C. 1.11.2.1) represent a practical alternative especially owing to their ease of application. Instead of relying on complex electron supply chains providing the enzymes with reducing equivalents as in the case of P450 monooxygenases, peroxygenases directly use hydrogen peroxide (H2O2) to form the catalytically active oxyferryl species (Compound I).2
H2O2, however, also inactivates heme enzymes as it induces an oxidative decomposition of the prosthetic group. In situ generation of H2O2 in low concentrations is the preferred approach to alleviate this problem.1b Generally, this is achieved through the in situ reduction of O2 to H2O2, posing questions with regard to the nature of the electron donor used for this reaction. Aside from electrochemical methods,1b oxidations of stoichiometric amounts of cosubstrates, such as EDTA, amino acids, alcohols, and other reductants,1b have been investigated. Today, the most common system for in situ H2O2 generation is certainly the glucose/glucose oxidase one. The poor atom efficiency of this system (glucose is oxidized only once to the corresponding lactone, generating one equivalent of H2O2), together with the pH shift that is due to gluconic acid accumulation, poses significant technological challenges (especially at preparative scales; see the Supporting Information, Table S5 for further details). Therefore, we recently developed an enzymatic cascade to fully oxidize methanol to CO2 and utilized the reduction equivalents liberated for H2O2 generation to promote peroxygenase reactions (Scheme 1).3 However, a rather complicated cascade process comprising four enzymes and one cofactor was required. Despite the success of this reaction system, we asked ourselves whether a simpler and more elegant in situ H2O2 generation method would be possible.
Scheme 1.
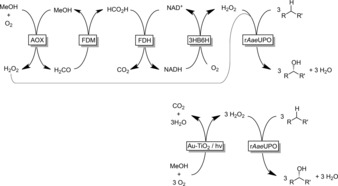
Comparison with the previously reported in situ H2O2 generation method to promote peroxygenase‐catalyzed hydroxylations of alkanes using the recombinant peroxygenase from Agrocybe aegerita (rAaeUPO). Top: The previously reported multienzyme cascade comprising alcohol oxidase (AOx), formaldehyde dismutase (FDM), formate dehydrogenase (FDH), 3‐hydroxybenzoate‐6‐hydroxylase (3HB6H), as well as the nicotinamide cofactor (NADH/NAD+).3 Bottom: Photochemical oxidation of methanol using Au‐loaded TiO2 (Au‐TiO2).
Inspired by recent work by Choi and Tada,4 we set out to evaluate gold‐loaded TiO2 (Au‐TiO2) as a plasmonic photocatalyst for the oxidation of methanol and the reductive activation of molecular oxygen to promote peroxygenase‐catalyzed oxyfunctionalization reactions (Scheme 1).
To test our hypothesis, we synthesized Au‐loaded TiO2 (rutile phase)5 as a methanol oxidation catalyst (see the Supporting Information for details), and employed it in the selective hydroxylation of ethylbenzene to (R)‐1‐phenylethanol catalyzed by the recombinant evolved peroxygenase from Agrocybe aegerita (rAaeUPO).6
Pleasingly, the proof‐of‐concept reaction proceeded smoothly to full conversion (Figure 1). Overall 10.7 mm of (R)‐1‐phenylethanol (98.2 % ee) were obtained within 72 h, which corresponds to a turnover number (TON=molproduct×molcatalyst −1) of more than 71 000 for the biocatalyst. Traces of acetophenone originating from the overoxidation of the product by rAaeUPO (commencing upon depletion of the starting material) were detected as the only side product. Omitting the biocatalyst resulted in the generation of small amounts (<0.15 mm) of racemic 1‐phenylethanol. In the absence of the photocatalyst or when the reaction was performed in the dark, the product was not detected. In the absence of methanol, some product formation was observed, which we attributed to Au‐TiO2‐catalyzed water oxidation (Figure S30).
Figure 1.
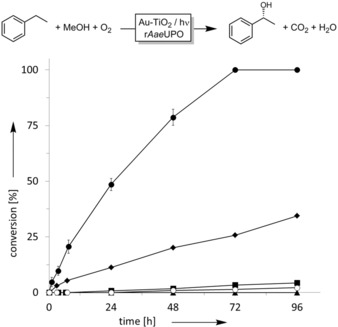
Photochemoenzymatic hydroxylation of ethylbenzene to (R)‐1‐phenylethanol with Au‐TiO2 as the photocatalyst for in situ H2O2 generation and rAaeUPO for the stereospecific hydroxylation reaction (•). Negative controls without enzyme (▪), light (▴), methanol (⧫), or rutile Au‐TiO2 (○). Reaction conditions: [methanol]=250 mm, [Au‐TiO2]=5 mg mL−1, [rAaeUPO]=150 nm, and [ethylbenzene]=15 mm in 60 mm phosphate buffer (pH 7.0) under illumination.
It should be mentioned that evaporation of the reagents can be a challenge for the current reaction setup. In particular, reactions with volatile reagents suffered from poor mass balances when exposed to the ambient atmosphere. Optimized setups, particularly closed vessels, circumvent this apparent limitation (Table S2).
Next, we systematically investigated the influence of the various reagents on the rate of the photoenzymatic hydroxylation reaction (Table 1 and Figures S17–S25). The concentration of MeOH had a significant effect on the initial rate, which steadily increased with increasing [MeOH] (Table 1, entries 1–6), and correlated well with the increasing formation rate and steady‐state concentration of H2O2. Au‐TiO2 is known to also oxidize H2O2 to O2, thereby preventing its continuous accumulation in the reaction mixture.4a, 7 Hence, both H2O2 and MeOH compete for oxidation at the catalyst surface, which explains the higher steady‐state concentration of H2O2 in the presence of methanol. At MeOH concentrations exceeding approximately 250 mm, the photocatalyst surface appeared to be fully saturated as no further increase in the product formation rate was observed. It is also worth mentioning that the addition of MeOH not only increased the overall reaction rate but also positively influenced the robustness of the process (Figure S31 and Table S3).
Table 1.
Photochemical in situ H2O2 generation to promote peroxygenase‐catalyzed oxyfunctionalization reactions.[a]
| Entry | Electron | [rAaeUPO] | [Electron | [Au‐TiO2] | Initial rate [mm h−1] | Steady‐state | [(R)‐1‐phenyl‐ | GC yield | TON (rAaeUPO) | |
|---|---|---|---|---|---|---|---|---|---|---|
| donor | [nm] | donor] [mm] | [g L−1] | Product | H2O2 [b] | [H2O2] [μm][b] | ethanol] [mm][c] | [%][d] | ×10−3[e] | |
| 1 | MeOH | 150 | 0 | 5 | 0.17 | 0.37 | 42 | 2.9 | 26 | 19 |
| 2 | MeOH | 150 | 5 | 5 | 0.20 | 0.56 | 55 | 3.3 | 24 | 22 |
| 3 | MeOH | 150 | 50 | 5 | 0.26 | 0.28 | 128 | 5.9 | 71 | 39 |
| 4 | MeOH | 150 | 100 | 5 | 0.24 | 0.56 | 231 | 6.4 | 76 | 42 |
| 5 | MeOH | 150 | 250 | 5 | 0.45 | 0.52 | 156 | 10.7 | >99 | 71 |
| 6 | MeOH | 150 | 500 | 5 | 0.46 | n.d. | n.d. | 10.4 | 97 | 69 |
| 7 | MeOH | 50 | 250 | 5 | 0.27 | 0.52 | 156 | 2.8 | 36 | 55 |
| 8 | MeOH | 350 | 250 | 5 | 0.47 | 0.52 | 156 | 10.7 | 97 | 31 |
| 9 | MeOH | 150 | 250 | 10 | 0.46 | 1.05 | 160 | 11.9 | >99 | 79 |
| 10 | MeOH | 150 | 250 | 20 | 0.29 | 0.44 | 97 | 10.1 | >99 | 67 |
| 11 | HCHO | 150 | 250 | 5 | 0.73 | 1.01[f] | 1050[f] | 13.7 | >99 | 91 |
| 12 | NaHCO2 | 150 | 250 | 5 | 0.58 | 0.98[f] | 193[f] | 12.6 | 99 | 84 |
| 13 | EtOH | 150 | 250 | 5 | 0.20 | 0.32 | 154 | 3.8 | 33 | 25 |
| 14 | iPrOH | 150 | 250 | 5 | 0.26 | 0.36 | 122 | 5.3 | 46 | 35 |
[a] Reaction conditions: [ethylbenzene]=15 mm in 60 mm phosphate buffer (pH 7.0) at 30 °C for 72 h under illumination. [b] As determined in comparative experiments by illuminating Au‐TiO2 in the reaction buffer without enzyme (Figures S11, S14, S18, and S21); n.d.=not determined. [c] Product with 98 % ee was obtained unless indicated otherwise. [d] GC yield: [(R)‐1‐phenylethanol]final×([(R)‐1‐phenylethanol]final+[ethylbenzene]final)−1. [e] TON: [(R)‐1‐phenylethanol]final×[rAaeUPO]−1. [f] Determined at 100 mm of the sacrificial reductant.
In terms of the photocatalyst concentration, a value of approximately 10 g L−1 was found to be optimal with respect to the rate of the photoenzymatic hydroxylation reaction (Table 1, entries 5, 9, and 10). This observation makes sense when considering the decreasing optical transparency of the reaction mixture with increasing photocatalyst loading (Figure S26). Hence, the increase in H2O2 generation activity with increasing photocatalyst concentration is counteracted by the decreasing transparency of the reaction mixture. Again, there was a good correlation between the overall rate and the steady‐state H2O2 concentration.
Increasing the enzyme concentration to greater than 150 nm resulted in no further increase in the overall reaction rate (Table 1, entries 5, 7, and 8). A plausible explanation is that above this value, the system is entirely H2O2‐limited, that is, almost every H2O2 molecule generated is consumed productively by the enzyme. As the H2O2 formation rate was measured to be 0.52 mm h−1 under these conditions and the initial enzymatic product formation rate was 0.45 mm h−1, the efficiency of the enzymatic H2O2 utilization was approximately 87 %. On the contrary, when the enzyme concentration was decreased to a third of this value, the reaction rate was approximately halved, indicating that H2O2 was no longer the (sole) limiting factor. Under these conditions, the H2O2 utilization efficiency dropped to 52 % as not all of the peroxide was consumed by the enzyme anymore and the excess was degraded by the photocatalyst and other unproductive processes.
The photon flux inside the reaction vessel, determined by ferrioxalate actinometry,8 was 2851 mE L−1 h−1. Consequently, under the standard conditions (150 nm UPO, 250 mm methanol), the photonic efficiencies of hydrogen peroxide and (R)‐1‐phenylethanol formation were 0.036 % and 0.032 %, respectively. Assuming that only the fraction of light that corresponds to the band gap of the rutile photocatalyst (≥3 eV/≤413 nm, 0.7 % of the lamp intensity; Figure S7) was responsible for the activity, photonic efficiencies of 5.2 % for hydrogen peroxide generation and 4.5 % for the enzymatic conversion product can be estimated. In view of the previously reported photonic efficiency of only 1 % for TiO2,9 this may suggest that the photocatalyst used here could also harvest some of the visible light as well, presumably via the gold plasmonic resonance at approximately 550–600 nm (Figure S6).
1H NMR analysis revealed that the Au‐TiO2‐catalyzed oxidation of methanol did not stop at the formaldehyde level but also produced formic acid and, presumably, CO2 (Figures S27 and S28). To further investigate this (desired) overoxidation of methanol, a set of experiments were conducted by substituting methanol with formaldehyde and formate, respectively, under otherwise identical conditions (Table 1, entries 11 and 12). Formaldehyde and formate gave approximately 32 % and 18 % higher reaction rates than methanol, respectively. This can be readily explained by the higher hydrogen peroxide formation rates observed for these compounds, both showed about 75 % higher H2O2 formation rates. Formaldehyde also suppressed H2O2 degradation, which resulted in a higher steady‐state concentration of H2O2. The fact that the increase in peroxide formation was somewhat diminished in the enzymatic reaction rate might be explained by two effects. On the one hand, the response of the enzyme to a higher H2O2 formation rate is non‐linear as at some point, the enzyme approaches its maximum turnover rate. On the other hand, the experiments with methanol are automatically superimposed by the higher reaction rates observed with formaldehyde and formate as they are formed during the reaction. This will be more pronounced in the photoenzymatic experiments than in the photocatalytic H2O2 formation owing to the longer timescale of the experiments, which allows for a higher fraction of the methanol to be converted. Nevertheless, especially formate may represent an attractive alternative to methanol as a sacrificial electron donor (Figures S24 and 25).
Other alcohols such as ethanol or isopropanol could also be used as sacrificial electron donors to promote the overall reaction but they were less effective than methanol (Table 1, entries 13 and 14). The relative rates found with ethanol and isopropanol correlate well with the steady‐state concentration and formation rate of H2O2 and roughly correlate with the oxidation potentials of the alcohols.10
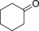
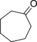
Finally, we also evaluated the substrate scope of the proposed photochemobiocatalytic reaction sequence (Table 2). In line with the reported substrate scope of rAaeUPO,11 a range of (cyclo)alkanes and alkyl arenes were converted into the corresponding alcohols. The regio‐ and enantioselectivities were essentially the same as in previous studies. The only side reaction observed was a minor overoxidation to the corresponding ketone as described above. On the one hand, this may be due to Au‐TiO2‐catalyzed oxidation; on the other hand, also rAaeUPO is capable of this overoxidation reaction.
Table 2.
Preliminary substrate scope of the photochemobiocatalytic hydroxylation reaction.[a]

| Entry | Product | mm | ee [%] | Side product | mm | GC yield [%][b] | TON (rAaeUPO) ×10−3 |
|---|---|---|---|---|---|---|---|
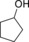
| 1 |
|
6.6 | – |
|
0.5 | 92.4 | 43.9 |
| 2 |
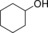
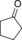
|
9.2 | – |
|
0.3 | >99 | 61.5 |
| 3 |
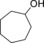
|
4.3 | – |
|
0.4 | 55.7 | 28.6 |
| 4 |
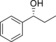
|
6.9 | >99 |
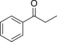
|
1.6 | 72 | 45.8 |
| 5 |
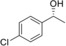
|
8.9 | 95.0 |
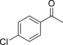
|
1.6 | 91.2 | 59.6 |
| 6 |
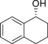
|
8.0 | 93.3 |
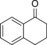
|
1.3 | 83.5 | 53.5 |
| 7 |
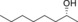
|
1.0 | 89 |
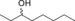
|
1.6 | 67.8 | 17.5 |
[a] Reaction conditions: [substrate]=10.0 mm, [rutile Au‐TiO2]=10 g L−1, [rAaeUPO]=150 nm, [MeOH]=250 mm in phosphate buffer (pH 7.0, 60 mm), T=30 °C, 70 h, under illumination. [b]=[alcohol]final×([ketone]final+[starting material]final)−1.
Very pleasingly, high turnover numbers could be achieved throughout these experiments that compare well with the numbers reported thus far with more complicated in situ H2O2 generation systems.1b Hence, we are optimistic that further optimization of the reaction setup may well lead to an economically attractive oxyfunctionalization reaction. Indeed, a preparative‐scale hydroxylation reaction of ethylbenzene yielded more than 100 mg of essentially enantiopure product (75 % conversion, 51 % yield of isolated product). Further optimization is currently underway.
As mentioned above, methanol addition not only accelerated the overall reaction but also contributed to its robustness (Figures S29 and S31). In the absence of methanol, rAaeUPO lost its catalytic activity almost instantaneously under illumination whereas in the presence of methanol, the enzyme activity was retained for several hours (Figure S31). We suspected that reactive oxygen species formed by the photocatalysts are responsible for this, which was qualitatively confirmed by EPR spectroscopy (Figure 2 A).13 More quantitatively, the coumarin method14 showed that hydroxyl radicals were formed in significant amounts only in the absence of methanol (Figure 2 B). Upon addition of methanol (250 mm), the hydroxyl radical formation rate dropped to only 0.6 % of the original value.
Figure 2.
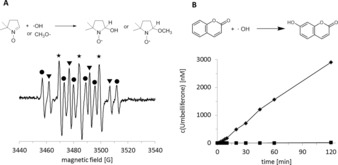
Qualitative and quantitative determination of radicals formed during the photocatalytic process. A) EPR spectra recorded during the illumination of rutile Au‐TiO2 in water with methanol for 20 min. Signals marked with an asterisk (★) belong to the oxidation product of DMPO, 5,5‐dimethyl‐2‐oxopyrroline‐1‐oxyl (DMPOX).12 Signals marked with triangles (▾) belong to the spin adduct .DMPO–OH, and signals marked with circles (•) belong to the spin adduct .DMPO–CH2OH from methanol.13 Reaction conditions: [Au‐TiO2]=5 g L−1, [DMPO]=30 mm, [methanol]=100 mm, RT, under illumination. B) Time course of the photocatalytic umbelliferone generation from coumarin as a specific detection method for .OH radicals. Reaction conditions: 60 mm phosphate buffer (pH 7), [Au‐TiO2]=5 g L−1, [coumarin]=0.1 mm, [methanol]=0 (⧫) or 250 mm (▪), T=30 °C, under illumination.
Apparently, methanol oxidation occurs significantly faster than water oxidation, which makes sense considering the redox potentials of the oxidation of water to hydroxyl radicals (+2.8 V)15 and the oxidation of methanol to methanol radicals (+1.2 V).16 Moreover, owing to the strongly reducing nature of the methanol radical (−1.3 V), it can readily inject an electron into TiO2, which leads to formaldehyde formation and results in up to two conduction band electrons per reactive photon, an effect also known as current doubling (Figure S32).17 Hence, methanol oxidation not only accelerated H2O2 generation but also prevented the formation of ROS from water oxidation (Figure S32 and Table S3 for further details).18
Overall, we have demonstrated the application of methanol as a sacrificial reductant for in situ H2O2 generation from O2 to promote selective, peroxygenase‐catalyzed oxyfunctionalization reactions. Admittedly, the productivities reported here do not reach preparatively useful values yet. Also the very high turnover numbers for rAaeUPO reported previously have not been reached yet. Future efforts will therefore focus on optimizing the light penetration into the reaction medium and increasing the H2O2 generation rate, for example, by using photochemical flow‐chemistry setups19 or wireless‐powered internal illumination.20
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
F.H and W.Z. gratefully acknowledge financial support by the European Research Council (ERC Consolidator Grant No. 648026). B.O.B. and J.Z.B. are grateful for financial support from the German Research Foundation (DFG, Grant No. BL 1425/1‐1). We thank Ben Norder (Delft University of Technology) for XRD, Dr. Wiel H. Evers (Delft University of Technology) for TEM, and Prof. Fred Hagen (Delft University of Technology) for EPR measurements.
W. Zhang, B. O. Burek, E. Fernández-Fueyo, M. Alcalde, J. Z. Bloh, F. Hollmann, Angew. Chem. Int. Ed. 2017, 56, 15451.
Contributor Information
Dr. Jonathan Z. Bloh, Email: bloh@dechema.de.
Dr. Frank Hollmann, Email: f.hollmann@tudelft.nl.
References
- 1.
- 1a. Wang Y., Lan D., Durrani R., Hollmann F., Curr. Opin. Chem. Biol. 2017, 37, 1–9; [DOI] [PubMed] [Google Scholar]
- 1b. Bormann S., Gomez Baraibar A., Ni Y., Holtmann D., Hollmann F., Catal. Sci. Technol. 2015, 5, 2038–2052. [Google Scholar]
- 2. Hofrichter M., Ullrich R., Curr. Opin. Chem. Biol. 2014, 19, 116–125. [DOI] [PubMed] [Google Scholar]
- 3. Ni Y., Fernández-Fueyo E., Baraibar A. G., Ullrich R., Hofrichter M., Yanase H., Alcalde M., van Berkel W. J. H., Hollmann F., Angew. Chem. Int. Ed. 2016, 55, 798–801; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 809–812. [Google Scholar]
- 4.
- 4a. Moon G. H., Kim W., Bokare A. D., Sung N. E., Choi W., Energy Environ. Sci. 2014, 7, 4023–4028; [Google Scholar]
- 4b. Teranishi M., Hoshino R., Naya S.-I., Tada H., Angew. Chem. Int. Ed. 2016, 55, 12773–12777; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12965–12969. [Google Scholar]
- 5. Priebe J. B., Radnik J., Lennox A. J. J., Pohl M. M., Karnahl M., Hollmann D., Grabow K., Bentrup U., Junge H., Beller M., Brückner A., ACS Catal. 2015, 5, 2137–2148. [Google Scholar]
- 6. Molina-Espeja P., Ma S., Mate D. M., Ludwig R., Alcalde M., Enzyme Microb. Technol. 2015, 73–74, 29–33. [DOI] [PubMed] [Google Scholar]
- 7. Li X. Z., Chen C. C., Zhao J. C., Langmuir 2001, 17, 4118–4122. [Google Scholar]
- 8. Hatchard C. G., Parker C. A., Proc. R. Soc. London Ser. A 1956, 235, 518–536. [Google Scholar]
- 9. Kormann C., Bahnemann D. W., Hoffmann M. R., Environ. Sci. Technol. 1988, 22, 798–806. [DOI] [PubMed] [Google Scholar]
- 10. Schneider J., Matsuoka M., Takeuchi M., Zhang J., Horiuchi Y., Anpo M., Bahnemann D. W., Chem. Rev. 2014, 114, 9919–9986. [DOI] [PubMed] [Google Scholar]
- 11. Peter S., Kinne M., Ullrich R., Kayser G., Hofrichter M., Enzyme Microb. Technol. 2013, 52, 370–376. [DOI] [PubMed] [Google Scholar]
- 12. Bilski P., Reszka K., Bilska M., Chignell C. F., J. Am. Chem. Soc. 1996, 118, 1330–1338. [Google Scholar]
- 13. Dvoranová D., Barbieriková Z., Brezová V., Molecules 2014, 19, 17279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J., Nosaka Y., J. Phys. Chem. C 2013, 117, 1383–1391. [Google Scholar]
- 15. Wardman P., J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar]
- 16. Koppenol W. H., Rush J. D., J. Phys. Chem. 1987, 91, 4429–4430. [Google Scholar]
- 17. Schneider J., Bahnemann D. W., J. Phys. Chem. Lett. 2013, 4, 3479–3483. [Google Scholar]
- 18. Kuwahara S., Katayama K., Phys. Chem. Chem. Phys. 2016, 18, 25271–25276. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Cambié D., Bottecchia C., Straathof N. J. W., Hessel V., Noël T., Chem. Rev. 2016, 116, 10276–10341; [DOI] [PubMed] [Google Scholar]
- 19b. Gemoets H. P. L., Su Y., Shang M., Hessel V., Luque R., Noel T., Chem. Soc. Rev. 2016, 45, 83–117. [DOI] [PubMed] [Google Scholar]
- 20.B. O. Burek, A. Sutor, D. W. Bahnemann, J. Z. Bloh, Catal. Sci. Technol 2017, https://doi.org/10.1039/c7cy01537b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary